

Abstract
Described herein are the synthesis and photophysical characterization of a library of aryl-substituted oxazole- and thiazole-based dipeptidomimetic analogues, and their incorporation into position 66 of green fluorescent protein (GFP) in lieu of the natural fluorophore. These fluorescent analogues resemble the fluorophore formed naturally by GFP. As anticipated, the photophysical properties of the analogues varied as a function of the substituents at the para position of the phenyl ring. The fluorescence emission wavelength maxima of compounds in the library varied from ~365 nm (near-UV region) to ~490 nm (visible region). The compounds also exhibited a large range of quantum yields (0.01–0.92). The analogues were used to activate a suppressor tRNACUA and were incorporated into position 66 of GFP using an in vitro protein biosynthesizing system that employed engineered ribosomes selected for their ability to incorporate dipeptides. Four analogues with interesting photophysical properties and reasonable suppression yields were chosen, and the fluorescent proteins (FPs) containing these fluorophores were prepared on a larger scale for more detailed study. When the FPs were compared with the respective aminoacyl-tRNAs and the actual dipeptide analogues, the FPs exhibited significantly enhanced fluorescence intensities at the same concentrations. Part of this was shown to be due to the presence of the fluorophores as an intrinsic element of the protein backbone. There were also characteristic shifts in the emission maxima, indicating the environmental sensitivity of these probes. Acridon-2-ylalanine and oxazole 1a were incorporated into positions 39 and 66 of GFP, respectively, and were shown to form an efficient Förster resonance energy transfer (FRET) pair, demonstrating that the analogues can be used as FRET probes.
Graphical abstract
Fluorescent labeling of proteins and peptides can provide a powerful tool for monitoring their behavior in vitro as well as in vivo.1 Generally, fluorescent labeling is achieved by employing a derivative of the fluorophore that reacts specifically with certain functional groups present in the biomolecule.2 Numerous fluorescent probes that possess a wide range of properties and functionalities to meet the requirements of the labeling technique and the technical goals of specific studies are currently available. There are, however, certain caveats associated with the use of synthetic fluorophores.2 For example, they should be small in size and chemically stable under physiological conditions; they should not perturb the structure or function of the peptides/proteins being labeled, and when used in intact cells or organisms, they should not be cytotoxic and should be permeable to cell membranes. In addition, when they are used to measure protein conformational changes, the covalent linkage between the synthetic probe and the specific residue in the target molecule should provide the probe with a range of motion that is small relative to the change in protein conformation being monitored.3
Following the successful cloning of green fluorescent protein4 (GFP) and its utilization as a marker for gene expression,5 this application for GFP and its variants has been used widely.6 There are certain advantages of genetic tagging over chemical labeling of expressed proteins.7 For example, because the fluorescent protein is fused to the target protein, they are colocalized with absolute specificity, which obviates the problem of nonspecific postsynthetic labeling. The fluorophore is embedded in the protein backbone and sheltered by the β-barrel structure, imparting a controlled environment for the probe. Nonetheless, there are significant limitations of using fluorescent proteins as labels, including their need for posttranslational activation, their limited stability, and their large size.
Over a period of several years, we have reported the reengineering of Escherichia coli ribosomes, allowing them to incorporate α-D-amino acids8,9 and β-amino acids,10,11 while still incorporating α-L-amino acids with good fidelity. More recently, we have extended this strategy to the selection of ribosomes capable of incorporating dipeptides and dipeptide analogues from an activated suppressor tRNA into a specific site in a protein in a single ribosomal bond-forming event by suppression of a nonsense codon.12,13 This has added importantly to the repertoire of amino acid analogues potentially amenable to incorporation, because the modified ribosomes apparently fail to recognize the amide bond connecting the two incorporated amino acids. Accordingly, the modified ribosomes were capable of incorporating a thiodipeptide, as well as fluorescent oxazole (1a) and thiazole (8a) heterocycles (Figure 1) substituted with NH2 and COOH groups at distances that approximated those between such functional groups in a dipeptide.12,13
Figure 1.
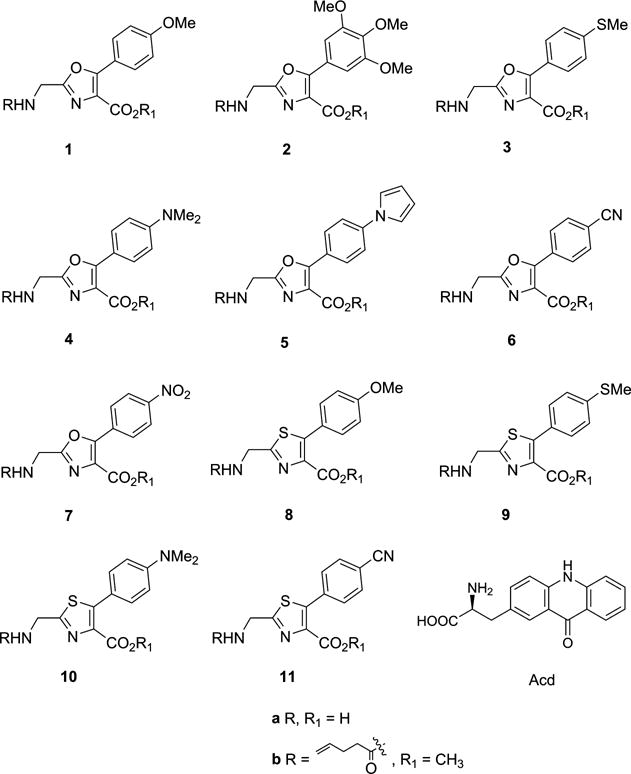
Structures of the dipeptidomimetic analogues synthesized (1–11), and of FRET acceptor L-acridon-2-ylalanine (Acd).
The structures of the dipeptidomimetic analogues incorporated were particularly interesting because they resemble the GFP fluorophore and are also fluorescent. When dipeptidomimetic analogues 1a and 8a were incorporated into position 66 of GFP by suppression of a UAG codon introduced into that position in lieu of the normal tyrosine codon, the resulting FPs had fluorescence intensities significantly stronger than that of wild-type GFP.12,13 These dipeptidomimetic analogues do not require activation by posttranslational modification; further, they are completely stable chemically both as the free “dipeptides” and also as constituents of proteins. The success of these studies12,13 suggested that it should be possible to identify additional fluorescent dipeptidomimetic analogues having diverse photophysical properties. Described in this report are the synthesis and photophysical properties of a library of dipeptidomimetic analogues and their ribosomal incorporation into position 66 of Aequorea victoria GFP. Selected fluorophores with interesting properties were made in greater amounts and characterized with regard to their environmental sensitivity and ability to act as a FRET donor following incorporation into the β-barrel structural domain of GFP, using acridon-2-ylalanine (Acd)14 as the acceptor.
MATERIALS AND METHODS
All experiments requiring anhydrous conditions were conducted in flame-dried glassware fitted with a rubber septum under a positive pressure of dry nitrogen or dry argon. Reactions were performed at room temperature unless otherwise indicated. Analytical thin layer chromatography was performed using glass plates precoated with silica gel (0.25 mm, 60 Å pore size, 230–400 mesh, Silicycle) impregnated with a fluorescent indicator (254 nm). TLC plates were visualized by exposure to ultraviolet (UV) light. Flash column chromatography was performed employing silica gel (60 Å pore size, 40–63 μm, standard grade, Silicycle). An acetone cooling bath was used for the appropriate low-temperature reactions by the addition of portions of dry ice.
1H NMR and 13C NMR spectra were recorded on Varian INOVA 400 (400 MHz) and Varian INOVA 500 (500 MHz) spectrometers at 25 °C. Proton chemical shifts are expressed in parts per million (δ scale) and are referenced to residual protium in the NMR solvent (CDCl3, DMSO-d6, or CD3OD). Splitting patterns are designated as follows: s, singlet; br s, broad singlet; d, doublet; dd, doublet of doublets; t, triplet; q, quartet; m, multiplet. High-resolution mass spectra were recorded at the Arizona State University CLAS High Resolution Mass Spectrometry Facility or the Michigan State University Mass Spectrometry Facility. HPLC purification was performed with a Waters 600 pump coupled with a Varian ProStar 340 detector and a Grace Econosil C18 column (250 mm × 10 mm, 5 μm). The tetra-n-butylammonium (TBA) salt of pdCpA was prepared using Dowex 50W X8 (200–400 mesh) activated in its TBA form.
The chemicals used for synthesis were purchased from Aldrich Chemical Co., Sigma Chemical Co., Fisher Scientific, Oakwood Chemicals or Combi-Blocks. THF was distilled under argon using sodium-benzophenone ketyl; CH2Cl2 was distilled under argon using calcium hydride.
Synthesis of pdCpA Derivatives of Dipeptidomimetic Analogues
The route employed for the synthesis of the pdCpA derivative of dipeptidomimetic analogue 5a and its attachment to suppressor tRNACUA-COH is outlined in Scheme 1. The syntheses of the pdCpA derivatives of dipeptidomimetics 2a–4a, 6a, 7a, and 9a–11a are outlined in Schemes S1–S8. The syntheses of the pdCpA derivatives of the dipeptidomimetic analogues 1a and 8a and acceptor Acd have been reported previously.12–14 The experimental procedures and compound characterizations are provided in the Supporting Information.
Scheme 1.
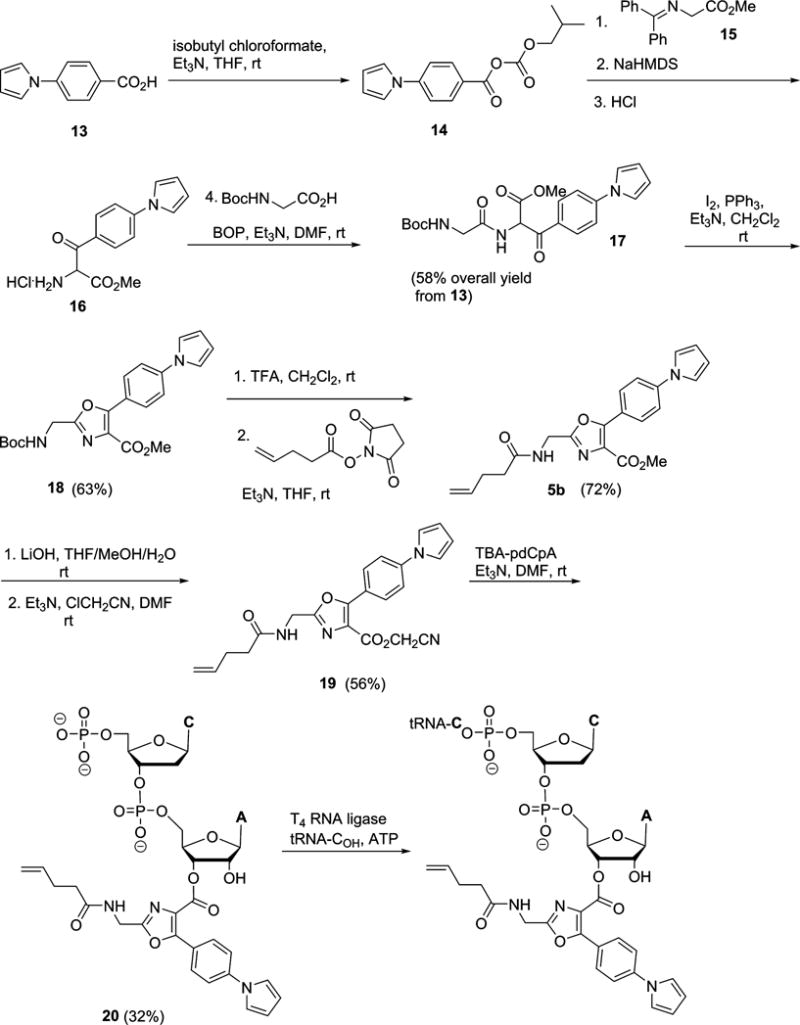
Synthetic Route Employed for the Preparation of the pdCpA Derivative of Dipeptidomimetic Analogue 5a and Its Attachment to a Suppressor tRNACUA
Methyl 3-[4-(1H-Pyrrol-1-yl)phenyl]-2-{2-[(tert-butoxycarbonyl)amino]acetamido}-3-oxopropanoate (17)
To a solution of 0.83 g (4.41 mmol) of acid 13 in 25 mL of THF were added 1.59 mL (1.16 g, 18.6 mmol) of Et3N and 0.74 mL (0.78 g, 9.31 mmol) of isobutyl chloroformate. The reaction mixture was stirred at room temperature for 2 h to obtain the acid anhydride 14, which was used as a crude material in the next step without further purification.
To a solution of 1.17 g (4.63 mmol) of imine ester 15 in 100 mL of THF was added 4.63 mL (4.63 mmol) of a 1 M NaHMDS solution in THF at −78 °C. After 30 min, crude acid anhydride 14 was added to the reaction mixture, which was stirred at −78 °C for 2 h. The reaction was quenched by the addition of a concentrated HCl solution until a pH of ~2 was reached. The reaction mixture was concentrated under diminished pressure to obtain amine salt 16 as a colorless solid, which was used in the next reaction without further purification.
To a solution of 0.85 g (4.85 mmol) of Boc-Gly-OH in 30 mL of DMF were added 2.14 g (4.85 mmol) of BOP reagent and 0.67 mL (0.48 g, 4.85 mmol) of Et3N. After 2 min, amine salt 16 dissolved in 20 mL of DMF was added to the reaction mixture, which was stirred overnight at room temperature. The reaction mixture was diluted with 300 mL of water and extracted with two 50 mL portions of EtOAc. The organic phase was dried (MgSO4) and concentrated under diminished pressure. The residue was purified by chromatography on a silica gel column (10 cm × 4 cm). Elution with 1:1 hexanes/ethyl acetate gave desired product 17 as a colorless foam: 1.06 g yield (58% overall yield from acid 13); silica gel TLC Rf = 0.50 (1:1 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.44 (s, 9H), 3.71 (s, 3H), 3.93 (d, 2H, J = 4.0 Hz), 5.39 (br s, 1H), 6.21 (d, 1H, J = 8.0 Hz), 6.38–6.39 (m, 2H), 7.16–7.17 (m, 2H), 7.47 (d, 2H, J = 12.0 Hz) 7.60 (d, 1H, J = 8.0 Hz), 8.17 (d, 2H, J = 8.0 Hz); 13C NMR (CDCl3) δ 28.5, 44.3, 53.5, 57.9, 80.5, 111.3, 119.1, 119.2, 119.4, 123.3, 130.8, 131.7, 131.8, 145.2, 150.7, 156.2, 167.2, 169.9, 189.9; mass spectrum (APCI) m/z 416.1824 (M + H)+ (C21H26N3O6 requires m/z 416.1822).
Methyl 5-[4-(1H-Pyrrol-1-yl)phenyl]-2-{[(tert-butoxycarbonyl)amino]methyl}oxazole-4-carboxylate (18)
To a stirred solution of 0.23 g (0.88 mmol) of triphenylphosphine and 0.22 g (0.88 mmol) of iodine in 50 mL of CH2Cl2 was added 0.24 mL (0.17 g, 1.75 mmol) of Et3N. The dark yellow solution was stirred for 5 min, and 0.18 g (0.44 mmol) of ketoamide 17 was added to the reaction mixture, which was stirred at room temperature for 2 h. The reaction mixture was concentrated under diminished pressure, and the residue was purified by chromatography on a silica gel column (10 cm × 1 cm). Elution with 1:1 hexanes/ethyl acetate gave desired product 18 as a colorless solid: 0.11 g yield (63%); silica gel TLC Rf = 0.50 (1:1 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.45 (s, 9H), 3.91 (s, 3H), 4.51 (d, 2H, J = 8.0 Hz), 5.34 (br s, 1H), 6.34–6.35 (m, 2H), 7.11–7.13 (m, 2H), 7.42–7.45 (m, 2H), 8.10–8.13 (m, 2H); 13C NMR (CDCl3) δ 28.5, 38.1, 53.6, 80.5, 111.4, 119.1, 119.3, 119.8, 120.2, 123.7, 123.7, 126.6, 128.1, 129.9, 142.0, 155.4, 155.7, 159.7, 162.6; mass spectrum (APCI) m/z 398.1714 (M + H)+ (C21H24N3O5 requires m/z 398.1716).
Methyl 5-[4-(1H-Pyrrol-1-yl)phenyl]-2-(pent-4-enamidomethyl)oxazole-4-carboxylate (5b)
To a solution of 0.20 g (0.55 mmol) of Boc-protected amine 18 in 10 mL of CH2Cl2 was added 10 mL of CF3COOH. The reaction mixture was stirred overnight and concentrated under diminished pressure to afford the deprotected amine, which was used in the next step without purification.
To the solution of deprotected amine in 15 mL of THF was added 0.11 g (0.55 mmol) of 4-pentenoyloxysuccinimide followed by 0.14 mL (0.10 g, 1.00 mmol) of Et3N. The reaction mixture was stirred overnight at room temperature and then concentrated under diminished pressure. The residue was purified by chromatography on a silica gel column (10 cm × 1 cm). Elution with 1:10 MeOH/ethyl acetate gave desired product 5b as a colorless solid: 0.14 g yield (72%); silica gel TLC Rf = 0.75 (1:10 MeOH/ethyl acetate); 1H NMR (CDCl3) δ 2.31–2.38 (m, 4H), 3.86 (s, 3H), 4.59 (d, 2H, J = 4.0 Hz), 4.92–5.03 (m, 2H), 5.76–5.78 (m, 1H), 6.31–6.32 (m, 2H), 6.82–6.85 (m, 1H), 7.08–7.09 (m, 2H), 7.38–7.41 (m, 2H), 8.04–8.06 (m, 2H); 13C NMR (CDCl3) δ 29.5, 35.5, 36.6, 52.4, 111.4, 115.8, 119.0, 119.2, 119.3, 119.6, 122.9, 123.5, 126.4, 129.9, 131.6, 136.9, 142.0, 155.3, 159.4, 162.4, 172.8; mass spectrum (APCI) m/z 380.1607 (M + H)+ (C21H22N3O4 requires m/z 380.1610).
Cyanomethyl 5-[4-(1H-Pyrrol-1-yl)phenyl]-2-(pent-4-enamidomethyl)oxazole-4-carboxylate (19)
To a solution of 0.12 g (0.31 mmol) of ester 5b in 6 mL of a 1:1 MeOH/THF mixture was added 0.46 mL (0.46 mmol) of 1 M aqueous LiOH. The reaction mixture was stirred overnight at room temperature and then concentrated under diminished pressure to obtain the free acid.
To a solution of the acid in 15 mL of DMF was added 0.06 mL (0.07 g, 0.93 mmol) of chloroacetonitrile followed by 0.22 mL (0.16 g, 1.54 mmol) of Et3N. The reaction mixture was stirred overnight at room temperature and then concentrated under diminished pressure. The residue was purified by chromatography on a silica gel column (10 cm × 1 cm). Elution with 1:10 MeOH/ethyl acetate gave desired product 19 as a yellow solid: 70.0 mg yield (56%); silica gel TLC Rf = 0.75 (1:10 MeOH/ethyl acetate); 1H NMR (CDCl3) δ 2.37–2.44 (m, 4H), 4.66 (d, 2H, J = 8.0 Hz), 4.95 (s, 2H), 4.97–5.10 (m, 2H), 5.80–5.84 (m, 1H), 6.27 (br s, 1H), 6.38–6.39 (m, 2H), 7.15–7.16 (m, 2H), 7.48–7.51 (m, 2H), 8.11–8.14 (m, 2H); 13C NMR (CDCl3) δ 29.6, 35.7, 36.8, 49.1, 111.8, 114.3, 116.1, 119.2, 119.9, 122.9, 124.7, 130.0, 130.3, 137.0, 142.7, 157.4, 159.7, 160.6, 170.1, 172.7; mass spectrum (APCI) m/z 405.1563 (M + H)+ (C22H21N4O4 requires m/z 405.1563).
5-[4-(1H-Pyrrol-1-yl)phenyl]-2-(pent-4-enamidomethyl)-oxazole-4-carboxylic Acid pdCpA Ester (20)
To a stirred solution containing 8.50 mg (6.37 μmol) of pdCpA tetrabutylammonium salt15 in 100 μL of a 9:1 anhydrous DMF/Et3N mixture was added 6.40 mg (15.9 μmol) of cyanomethyl ester 19. The reaction mixture was sonicated for 6 h. The reaction mixture was purified by C18 reversed phase HPLC (250 mm × 10 mm) using a gradient of 1 to 65% acetonitrile in 50 mM ammonium acetate (pH 4.5) over a period of 1 h. The retention time of the desired product was 24.6 min. The fractions containing the product were lyophilized to afford 20 as a colorless solid: 2.0 mg yield (32%); mass spectrum (ESI) m/z 982.2283 (M – H)− (C39H42N11O16P2 requires m/z 982.2286).
Measurement of the Photophysical Properties of Fluorescent Dipeptidomimetics
The UV/vis absorption spectra (220–400 nm) were recorded using a Cary 60 UV/vis spectrophotometer. Fluorescence quantum yields (Table 1) were determined using the gradient method.16 Dipeptidomimetic analogues were dissolved in methanol. Solutions of each compound were made such that the UV absorptions at the wavelength maxima were within the range of 0.01–0.1. Anthracene (ΦF = 0.27; λex = 340 nm) was used as a reference standard to calculate the fluorescence quantum yields of the dipeptidomimetic analogues according to the formula Φx = Φs × (Gradx × nx2)/(Grads × ns2), where Grad is the gradient of the plot of integrated intensity versus absorbance, n is the refractive index of the solvent, s is the standard of known ΦF, and x is the tested sample.16
Table 1.
Photophysical Properties of the Dipeptidomimetic Analoguesa
| Analogue | Structure | λabs (nm) | λm (nm)b | ε (M−1cm−1) | ΦF | Brightness |
|---|---|---|---|---|---|---|
| 1b |
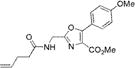
|
296±2 | 389±2 | 17600±1000 | 0.90±0.02 | 15800±1300 |
| 2b |
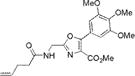
|
302±2 | 425±2 | 14300±900 | 0.72±0.02 | 10300±930 |
| 3b |
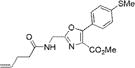
|
319±3 | 419±2 | 21000±1000 | 0.92±0.03 | 19300±1500 |
| 4b |
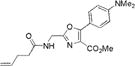
|
351±2 | 458±2 | 20000±1200 | 0.60±0.02 | 12000±1100 |
| 5b |
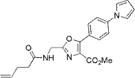
|
314±2 | 401±3 | 27900±1000 | 0.03±0.001 | 840±60 |
| 6b |
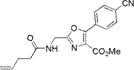
|
285±2 | 365±3 | 13800±1100 | 0.26±0.01 | 3600±400 |
| 7b |
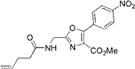
|
309±3 | 410±4 | 16200±1000 | 0.01±0.001 | 160±25 |
| 8b |
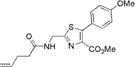
|
300±2 | 420±3 | 11000±1000 | 0.24±0.03 | 2600±500 |
| 9b |
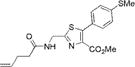
|
313±2 | 447±2 | 13100±900 | 0.91±0.04 | 12000±1400 |
| 10b |
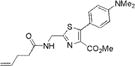
|
351±2 | 490±2 | 7500±500 | 0.04±0.001 | 300±30 |
| 11b |
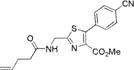
|
290±3 | 389±5 | 9500±500 | 0.01±0.001 | 95±15 |
Determined for the N-pentenoyl-protected methyl esters in MeOH.
Following excitation at the absorption maxima.
Ligation of Suppressor tRNA-COH with Dipeptidomimetics and Deprotection of the N-Pentenoyl Group
The activation of suppressor tRNACUA and tRNACCCG was conducted as described previously.17,18 Briefly, a 100 μL reaction mixture of 100 mM Na Hepes (pH 7.5) contained 1.0 mM ATP, 15 mM MgCl2, 100 μg of suppressor tRNACUA-COH or tRNACCCG-COH, 0.5 A260 unit of N-pentenoyl-protected aminoacyl-pdCpA, 15% DMSO, and 100 units of T4 RNA ligase. The reaction mixture was incubated at 37 °C for 1.5 h and the reaction quenched by the addition of 0.1 volume of 3 M NaOAc (pH 5.2). The N-protected aminoacylated tRNA was precipitated with 3 volumes of cold EtOH. The efficiency of ligation was estimated by 8% polyacrylamide–7 M urea gel electrophoresis (pH 5.0).19 The N-pentenoyl-protected amino-acyl-tRNACUAs were deprotected by treatment with 5 mM aqueous I2 at 25 °C for 15 min. The solution was centrifuged, and the supernatant was adjusted to 0.3 M NaOAc and then treated with 3 volumes of cold EtOH to precipitate the aminoacylated tRNA. The tRNA pellet was collected by centrifugation, washed with 70% aqueous EtOH, air-dried, and dissolved in 10 μL of RNase free H2O.
Preparation of S-30 Extracts from Cells Having Modified Ribosomes
Aliquots (5–10 μL) from liquid stocks of E. coli BL-21(DE-3) cells, harboring plasmids with a modified rrnB gene, were placed on LB agar supplemented with 100 μg/mL ampicillin and grown at 37 °C for 16–18 h. One colony was picked from each agar plate and transferred into 3 mL of LB medium supplemented with 100 μg/mL ampicillin and 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG).
The cultures were grown at 37 °C for 3–6 h in a thermostated shaker until an OD600 of ~0.15–0.3 was reached, then diluted with LB medium supplemented with 100 μg/mL ampicillin, 1 mM IPTG, and 3 μg/mL erythromycin (for selectively enhancing the modified ribosome fraction) until an OD600 of 0.01 was reached, and then grown at 37 °C for 12–18 h. The optimal concentration of the final cultures was an OD600 of 0.5–1.0. Cells were harvested by centrifugation (5000g at 4 °C for 10 min) and washed three times with S-30 buffer [1 mM Tris-OAc (pH 8.2) containing 1.4 mM Mg(OAc)2, 6 mM KOAc, and 0.1 mM DTT] supplemented with β-mercaptoethanol (0.5 mL/L) and once with S-30 buffer having 0.05 mL/L β-mercaptoethanol.
The weight of the wet pellet was estimated, and 1.27 mL of S-30 buffer was added to suspend each gram of cells. The volume of the suspension was measured and used for estimating the amounts of the other components. The preincubation mixture (0.3 mL) [0.29 M Tris (pH 8.2) containing 9 mM Mg(OAc)2, 13 mM ATP, 84 mM phosphoenolpyruvate, 4.4 mM DTT, and 5 μM amino acid mixture], 15 units of pyruvate kinase, and 10 μg of lysozyme were added per milliliter of cell suspension, and the resulting mixture was incubated at 37 °C for 30 min. The incubation mixture was then frozen at −80 °C (~30 min), melted (37 °C, 30 min), and again frozen and melted at room temperature (~30 min). Ethylene glycol tetraacetic acid (EGTA) was then added to a final concentration of 2.5 mM, and the cells were incubated at 37 °C for 30 min. The same molar concentration of CaCl2 was added, mixed well, and frozen (−80 °C, 30 min). The frozen mixture was centrifuged (15000g at 4 °C for 1 h), and the supernatant was stored in aliquots at −80 °C.
In Vitro Protein Translation
Protein translation reactions were conducted in 15–1700 μL of incubation mixture containing 0.3 μL/μL of S-30 system, 170 ng/μL plasmid, 35 mM Tris acetate (pH 7.4), 190 mM potassium glutamate, 30 mM ammonium acetate, 2 mM DTT, 0.2 mg/mL total E. coli tRNA, 3.5% PEG 6000, 20 μg/mL folinic acid, 20 mM ATP and GTP, 5 mM CTP and UTP, 100 μM amino acid mixture, 0.5 μCi/μL of [35S]methionine (for 15 μL reaction mixtures), and 1 μg/mL rifampicin. In the case of plasmids having a gene with a TAG codon, an activated suppressor tRNA was added to a concentration of 1.5 μg/μL. In the case of the plasmid having a gene with TAG and CGGG codons to permit the incorporation of two unnatural amino acids, two aminoacyl-tRNAs (aminoacyl-tRNACUA and aminoacyl-tRNACCCG) were added. Reactions were conducted at 37 °C for 1 h (for 15 μL reaction mixtures) to 1.5 h (for 1700 μL reaction mixtures) and terminated by chilling the mixtures on ice. Aliquots from in vitro translation mixtures were analyzed by SDS–PAGE followed by quantification of the radioactive bands by phosphorimager analysis (for 15 μL reaction mixtures).
Purification of GFP
Samples of GFP were diluted with 50 mM Tris-HCl (pH 8.2) and applied to a 100 μL Ni-NTA agarose column that had been equilibrated with the same buffer. The column was washed with 1 mL of the same buffer, and GFP was eluted with 500 μL of 50 mM Tris-HCl (pH 8.2) containing 250 mM imidazole. A 50 μL column of DEAE-Sepharose was equilibrated with two 500 μL portions of 25 mM Tris-HCl (pH 7.4). Samples of GFP purified by Ni-NTA chromatography were diluted 3-fold in the same buffer and applied to the resin. The column was washed with four 500 μL portions of the same buffer. GFP was eluted from the resin with 25 mM Tris-HCl (pH 7.4) containing 0.25 M NaCl. The fractions were analyzed on a 15% polyacrylamide gel and stained using Coomassie R-250, and protein-containing fractions were combined and concentrated by the use of an Amicon Ultra 10K filter device. The concentrations of all modified proteins were estimated by comparison with those of the corresponding wild-type proteins, which had been prepared in vivo and purified using the same protocols. The concentrations of the reference proteins were determined on the basis of OD280 determination and the extinction coefficient for the protein. The wild-type protein of known concentration was then used as a reference standard at several concentrations in an SDS–PAGE experiment visualized by Coomassie staining in comparison with the modified proteins.
Protein Fluorescence Measurements
The fluorescence spectra of modified GFPs containing compounds 1a, 3a, 4a, and 8a at position 66, as well as compound 1a at position 66 and Acd at position 39, were measured using a Varian Cary Eclipse fluorescence spectrophotometer with the excitation slit set at 10 nm and the emission slit at 10 nm in 25 mM Tris-HCl (pH 7.4) containing 0.5 M NaCl.
The emission spectra of modified GFPs containing compounds 1a, 3a, 4a, and 8a in position 66 were measured after excitation at appropriate wavelengths (300 nm for 1a and 8a, 320 nm for 3a, and 360 nm for 4a). The emission spectra of the GFP analogue having Acd at position 39 and compound 1a at position 66 were measured after excitation at two wavelengths (296 and 370 nm), corresponding to excitation maxima for compounds 1a and Acd, respectively.
RESULTS
Syntheses of the Fluorescent Dipeptidomimetic Analogues and Their pdCpA Esters
The synthesis of the aminoacylated pdCpA derivative of dipeptidomimetic analogue 5a (Figure 1) began with the conversion of commercially available 4-(1H-pyrrol-1-yl)benzoic acid (13) to activated intermediate 14 by treatment with isobutylchloroformate and triethylamine (Scheme 1). This anhydride was condensed with the monoanion of the diphenylmethylene-glycine methyl ester (15), followed by imine hydrolysis using ~6 N HCl to afford amine salt 16, which was used in the next step without further purification.20 Amine salt 16 was coupled with Boc-Gly-OH using BOP21 as a coupling agent to form α-amido-β-ketoester 17 in 58% overall yield from acid 13. Cyclodehydration of 17 using triphenylphosphine in the presence of iodine and triethylamine in CH2Cl2 afforded oxazole 18 in 63% yield.20 Subsequent removal of the Boc group in 18 using CF3COOH, followed by treatment with pentenoyloxysuccinimide, gave amide 5b in 72% yield.18 The methyl ester moiety of 5b was saponified using an ~1 N LiOH solution, and the corresponding acid was activated as cyanomethyl ester 19 in a 56% yield for two steps.18,22 Treatment of cyanomethyl ester 19 with the tris(tetrabutylammonium) salt of pdCpA (TBA-pdCpA) in anhydrous DMF gave corresponding aminoacylated pdCpA ester 20 in 32% yield.15 Finally, activated pdCpA derivative 20 was ligated to an abbreviated suppressor tRNACUA-COH transcript using T4 RNA ligase, providing the respective misacylated suppressor tRNA transcript.
The syntheses of the aminoacylated pdCpA derivative of dipeptidomimetics 1a and 8a have been reported previously.12,13 The synthesis of the pdCpA derivative of compound 2a is described in Scheme S1 of the Supporting Information. The synthesis began with the condensation of commercially available 3,4,5-trimethoxybenzoyl chloride with the monoanion of the diphenylmethylene-glycine methyl ester (15), followed by imine hydrolysis using concentrated HCl to afford the hydrochloride salt of amine 21. The latter was coupled directly with CBz-Gly-OSu in the presence of triethylamine to form α-amido-β-ketoester 22 in 79% overall yield from imine 15. The remaining steps for the synthesis of the pdCpA derivative of compound 2a were similar to those described in Scheme 1.
The synthesis of the aminoacylated pdCpA derivative of oxazole 3a (Scheme S2 of the Supporting Information) and the corresponding thiazole 9a (Scheme S6 of the Supporting Information) began from known glycine derivative 26.23 Thus, fully protected glycine 26 was deprotonated using 1 M NaHMDS and condensed with 4-thiomethylbenzoyl chloride to obtain ketoester 27. The Boc groups were removed from 27 using CF3COOH followed by coupling with Boc-Gly-OH using the BOP reagent21 to obtain α-amido-β-ketoester 28 in 72% overall yield from 26. Cyclodehydration of 28 using triphenylphosphine in the presence of iodine and triethylamine in CH2Cl2 gave oxazole 29 in 62% yield (Scheme S2 of the Supporting Information).24 Treatment of 28 with the Lawesson’s reagent gave corresponding thiazole 47 in 87% yield (Scheme S6 of the Supporting Information).20 The remaining steps for the synthesis of pdCpA derivatives of oxazole dipeptidomimetic 3a and corresponding thiazole dipeptidomimetic 9a were similar to those described in Scheme 1.
Similar reaction schemes were employed for the syntheses of the aminoacylated pdCpA derivatives of the oxazole dipeptidomimetics 4a, 6a, and 7a, and these are described in Schemes S3–S5, respectively, of the Supporting Information. Likewise, the reaction schemes for the syntheses of the aminoacylated pdCpA derivatives of thiazole dipeptidomimetics 10a and 11a are shown in Schemes S7 and S8, respectively, of the Supporting Information.
Photophysical Properties of the Fluorescent Dipeptidomimetic Analogues
We characterized the photophysical properties of the library of oxazole- and thiazole-based dipeptidomimetic analogues, including molar absorptivities, quantum yields, and emission and absorption spectra of the corresponding N-pentenoyl methyl ester derivatives in methanol (Table 1 and Figure S1). Ideally, the photophysical data should have been measured in aqueous buffer to allow direct correlations with the same compounds present within a protein attached to a tRNA. However, this was not possible because of the very limited solubility of some of the fluorophores in aqueous media. The absorbance maxima of the analogues ranged from 285 to 351 nm, while the emission maxima ranged from 365 to 490 nm. The photophysical properties of 1b and 8b have been described previously using samples having purity considered routine for synthetic products, although the careful purification of the compounds by reversed phase HPLC has now provided samples with reevaluated quantum yields.12,13 The UV absorption and fluorescence emissions of 4b and 10b were the most red-shifted of those of the analogues studied. The emission peak wavelength of 10b (490 nm) was closest to the emission peak wavelength of wild-type GFP (509 nm) with a large Stokes shift (139 nm). Nonetheless, 10b proved to be a poor fluorescent probe because it has a very low quantum yield (0.04). We verified (UV, HPLC analysis) that compound 10b was unaltered chemically as a consequence of its excitation. Compound 4b has quite favorable properties as a fluorescent probe. It has a high quantum yield (0.60) and a high molar absorptivity (20000 M−1 cm−1), emits in the blue region (458 nm) of the spectrum, and has a large Stokes shift (107 nm) compared with that of BFP (75 nm). Compounds 3b and 9b were the best oxazole/thiazole pair in terms of quantum yield; both exhibited very high quantum yields, 0.92 and 0.91, respectively. Interestingly, the other oxazole/thiazole pairs differed significantly from each other in quantum yields; the thiazoles had quantum yields significantly lower than those of the corresponding oxazoles. This likely reflects the electron donating and withdrawing properties of the substituents by which these analogues differ. Apart from this, 3b and 9b emit in the visible region (419 and 447 nm, respectively) and have large Stokes shifts (100 and 134 nm, respectively) and large molar absorptivities (21000 and 13100 M−1 cm−1, respectively), which make them efficient fluorescent probes. Although the absorption maximum of 2b was similar to that of 1b, the emission maximum was red-shifted by ~35 nm. The molar absorptivity of 2b (14300 M−1 cm−1) was roughly 80% of that of 1b (17600 M−1 cm−1), such that the brightness of 2b (10300 M−1 cm−1) was lower than that of 1b (15800 M−1 cm−1). In addition to 3b and 9b, which were quite bright (19300 and 12000 M−1 cm−1, respectively), analogue 4b also exhibited a favorable brightness (12000 M−1 cm−1). Compounds 5b, 7b, 10b, and 11b exhibited one or more photophysical properties that diminish their brightness, and thus their potential utility as fluorescent probes.
Activation of Suppressor tRNACUA and Synthesis of Modified GFPs
The individual N-pentenoyl-protected aminoacylated pdCpA derivatives were ligated to an abbreviated suppressor tRNACUA-COH using T4 RNA ligase. The N-pentenoyl groups were removed using 5 mM aqueous I2 at room temperature for 15 min. The free aminoacyl-tRNAs so obtained were then utilized in a cell free coupled transcription–translation system containing an E. coli S-30 preparation and pETGFP66 plasmid. Plasmid pETGFP66 contained a modified GFP gene having a TAG codon at position 66. As shown in Figure 2 and Table 2, the dipeptidomimetic analogues were all incorporated into GFP but showed varying suppression efficiencies. In replicate experiments, the lowest suppression yield was ~3%, observed for compound 5a, and the highest suppression yield was ~12%, observed for compound 4a. Among the four oxazole/thiazole pairs (1a/8a, 3a/9a, 4a/10a, and 6a/11a), the best incorporation efficiency was shown by the 4a/10a pair. Compound 4a was incorporated in ~12% yield and compound 10a in ~10% yield. For the 1a/8a and 3a/9a pairs, the suppression yields realized with the oxazoles were reproducibly higher than those observed for the corresponding thiazoles. Compounds 6a and 11a displayed roughly the same incorporation efficiencies, ~5% yields. Compounds 2a, 5a, and 7a were incorporated in ~6, ~3, and ~11% yields, respectively. Five of the analogues had suppression yields of ≥10%, which should permit quite reasonable quantities of proteins containing these analogues to be prepared. It may be noted that oxazole 1a has previously been incorporated into DHFR in suppression yields of up to 15%.12
Figure 2.
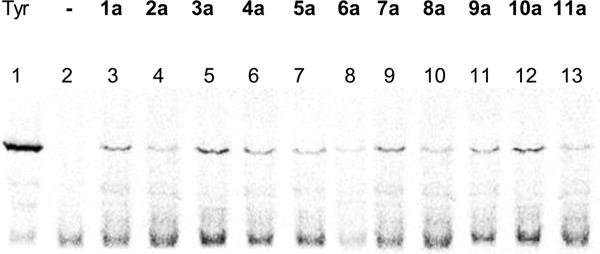
Autoradiogram of a 15% SDS–polyacrylamide gel depicting the translation of modified GFPs from GFP mRNA, containing a UAG codon in position 66, in the presence of dipeptidomimetic (1a–11a) tRNACUA and an S-30 system prepared from ribosomal clone 010328R4 (lanes 3–13). Lanes 1 and 2 demonstrate the synthesis of full length GFP from the same mRNA in the presence of tyrosyl-tRNACUA and nonacylated tRNACUA, respectively. Phosphorimager analysis was performed using an Amersham Biosciences Storm 820 instrument equipped with ImageQuant version 5.2 from Molecular Dynamics.
Table 2.
Summary of Incorporation of Compounds 1a–11a into Position 66 of GFP in Comparison with the Incorporation of Tyrosine
| average yield of full size protein in the presence of tRNA, containing fluorescent compounds (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Tyra | 1a | 2a | 3a | 4a | 5a | 6a | 7a | 8a | 9a | 10a | 11a |
| 100 | 11 ± 1 | 6 ± 0.2 | 10 ± 1 | 12 ± 2 | 3 ± 1 | 6 ± 1 | 11 ± 2 | 8 ± 1 | 6 ± 1 | 10 ± 1 | 4 ± 2 |
The efficiency of full length GFP synthesis in the presence of tyrosyl-tRNA was ~40–60% of that of wild-type GFP synthesis under the same conditions.
Fluorescent Properties of the Oxazoles and Thiazoles, as tRNA Esters and Incorporated in Proteins and Peptides
GFP analogues having compounds 1a, 3a, 4a, and 8a at position 66 were prepared on a larger scale and purified by Ni-NTA agarose chromatography. The fluorescence emission spectra of these modified GFPs were compared after excitation of each at the absorption maxima of the corresponding free fluorophores. The fluorescence emission maxima of proteins containing 1a, 3a, 4a, and 8a were observed at wavelengths (377, 410, 445, and 406 nm, respectively) shorter than the emission maxima observed for the individual dipeptide analogues (cf. Tables 1 and 3). It must be noted, however, that the monomeric fluorophores were investigated in MeOH, while the protein and activated tRNAs containing the same fluorophores were studied as aqueous solutions.
Table 3.
Comparison of Emission Wavelengths (λmax) of Free Dipeptidomimetic Compounds and the Same Compounds, Esterified to tRNA and Incorporated into Position 66 of GFPa
| emission wavelength maximum (λmax) (nm)
|
|||
|---|---|---|---|
| compound | free compound | esterified to tRNA | position 66 of GFP |
| 1a | 389 ± 2 | 411 ± 3 | 377 ± 3 |
| 2a | 425 ± 2 | 450 ± 2 | |
| 3a | 419 ± 2 | 445 ± 2 | 410 ± 2 |
| 4a | 458 ± 2 | 460 ± 2 | 445 ± 2 |
| 5a | 401 ± 3 | 420 ± 3 | |
| 6a | 365 ± 3 | 370 ± 3 | |
| 7a | 410 ± 4 | 420 ± 4 | |
| 8a | 420 ± 3 | 435 ± 2 | 406 ± 2 |
| 9a | 447 ± 2 | 470 ± 2 | |
| 10a | 490 ± 2 | 490 ± 2 | |
| 11a | 389 ± 5 | 405 ± 4 | |
The emission spectra of the free oxazoles/thiazoles 1b–11b were determined in MeOH, while those of the activated tRNAs and GFPs were measured in 25 mM Tris-HCl (pH 7.4) containing 0.25 M NaCl.
These emission wavelengths were characterized in more detail, by comparing the fluorescence emission spectra of analogue 1a, 3a, 4a, or 8a present either at position 66 of GFP or at the 3′-end of a suppressor tRNA. As shown in Figure 3, in which the concentrations of the activated tRNAs and GFPs had been adjusted to afford emission spectra of roughly comparable intensities, the emission maxima for the species containing 1a, 3a, and 8a differed by ~30–35 nm. In comparison, the emission spectrum of the activated tRNA and GFP containing 4a differed by only 15 nm, and the emission spectrum of the activated tRNA was clearly narrower than that of the modified GFP.
Figure 3.
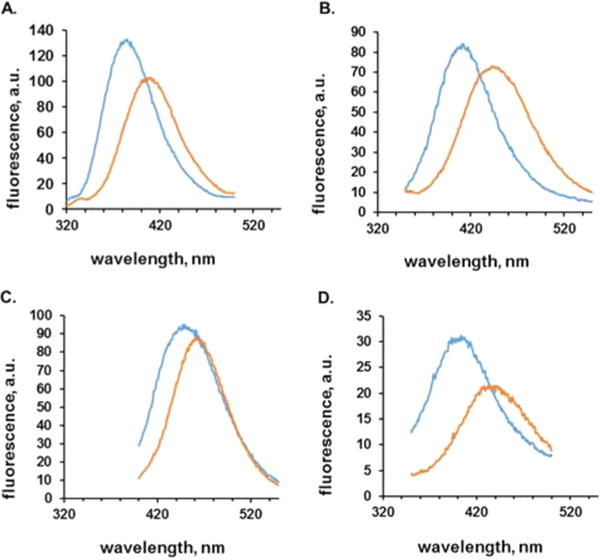
Comparison of emission profiles of oxazoles 1a, 3a, and 4a (A–C, respectively) and thiazole 8a (D), esterified to tRNACUA (orange) vs incorporation in position 66 of GFP (blue). The concentrations of the activated tRNA solutions were adjusted to be roughly 3–4-fold greater than those of the proteins, to afford emission spectra of comparable intensity.
The effect of environment on the fluorescence emission intensity of dipeptidomimetic analogue 1a was studied by comparing the emission spectra of the free oxazole with those of the corresponding aminoacyl-tRNA, and the modified GFP containing 1a at position 66 (Figure 4). As shown, 100 nM free oxazole 1a (green trace) and the same concentration of the suppressor tRNA activated with oxazole 1a (red trace) exhibited emission spectra of comparable intensity, with the emission maximum of the spectrum due to the activated tRNA being at longer wavelengths (cf. Table 3). In comparison, the modified GFP having 1a at position 66 emitted at a much shorter wavelength (377 nm) with several-fold greater intensity (Figure 4). The effect of solvent on the fluorescence emission wavelength and intensity of oxazole 1a was also studied (Figure S2), and it was found that much of the observed difference in emission wavelength was due to the solvent. The slightly longer wavelength emission of the fluorophores attached to the 3′-end of tRNAs (e.g., Figure S2) presumably reflects a more polar environment induced by the tRNA, which contains multiple charged and polar functional groups.
Figure 4.
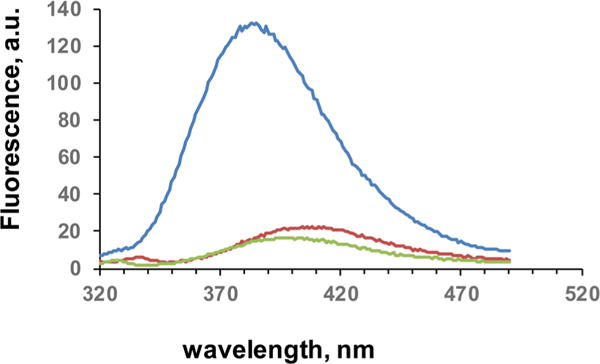
Fluorescence emission spectra of free oxazole 1a (green), oxazole 1a present in position 66 of GFP (blue), and oxazole 1a esterified to tRNACUA (red) after excitation at 300 nm. All samples were present at a concentration of 100 nM in 25 mM Tris-HCl (pH 7.4) containing 0.25 M NaCl.
The source of the enhanced fluorescence emission intensity observed for the modified GFP having 1a at position 66 was explored by embedding oxazole 1a in the tetrapeptide Gly-Gly-Gly-Gly to determine whether the simple presence of the fluorophore within a peptide backbone would influence its photophysical properties. The route used for the synthesis of requisite peptide 12 is shown in Scheme 2, and that for the preparation of the requisite photophysical control (N-Boc 1a methyl ester, 63) is provided in Scheme S9. As noted in Table S1, the photophysical properties of Boc-deprotected peptide 12 differed in interesting ways from those of the analogously deprotected oxazole 63 (1a methyl ester). While both had the same absorption maxima when measured in MeOH (300 nm), the emission maximum of Boc-deprotected 12 was at a shorter wavelength (381 vs 391 nm), analogous to the shifts observed when the oxazoles and thiazoles were present at position 66 of GFP (cf. Table 3). Further, while their quantum yields were similar (0.88 and 0.92 for deprotected 12 and 63, respectively), compound 12 may have exhibited molar absorptivity somewhat greater than, and consequently a brightness greater than, that of 63 (Table S1), although these differences were not statistically significant. The emission spectra of 12 and 63 were also recorded in MeOH and reflected the same trends (Figure S3). When the same two Boc-deprotected compounds were studied in 25 mM Tris-HCl (pH 7.4), containing 0.5 M NaCl as had been done for the modified GFPs (Table 3), the overall differences in their photophysical properties were more pronounced (Table 4). The molar absorptivities and quantum yields decreased for both compounds, but the decrease in quantum yield was much greater for deprotected oxazole 63. As a consequence, deprotected 12 was more than 3-fold brighter than deprotected 63 (Table 4). Once again, these trends were reflected in the fluorescence emission spectra of 63 and 12 themselves taken in the same buffer (Figure 5). Thus, the several-fold differences in fluorescence intensity noted in Figure 4 must be due in part to the presence of the fluorophore as an intrinsic structural constituent of the protein backbone (rather than as the usual electronically isolated substituent attached to the peptide backbone), and in part to its additional presence within the β-barrel structure of GFP.
Scheme 2.
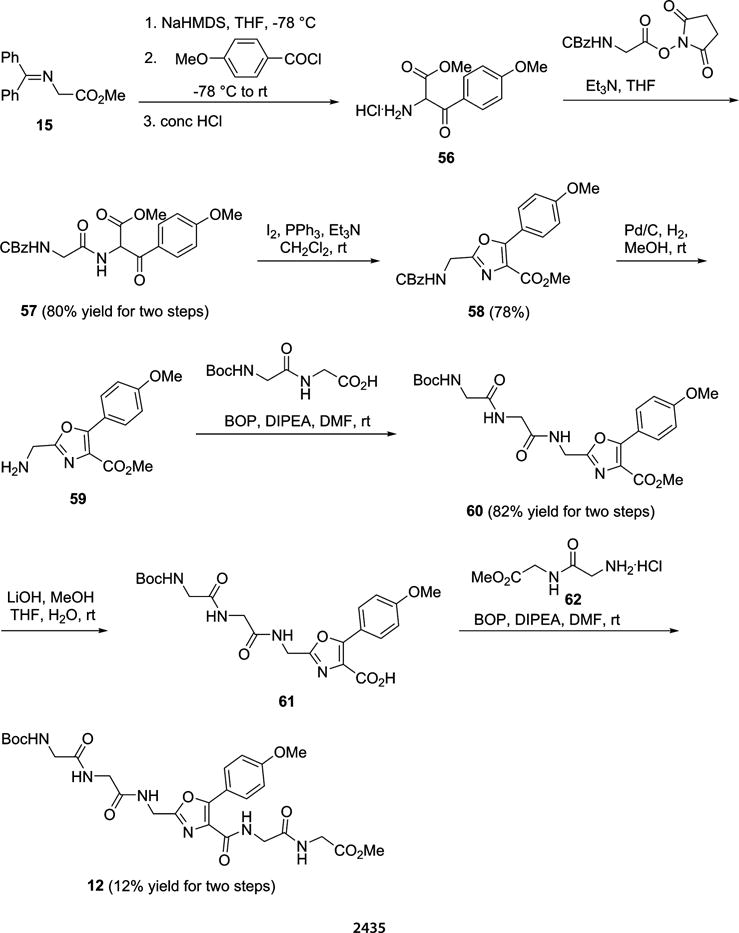
Synthetic Route Employed for the Preparation of the N-Boc Methyl Ester Derivative of Fluorescent Peptide 12
Table 4.
Photophysical Properties of the Free Amines Derived from Compounds 12 and 63a
| |||||
|---|---|---|---|---|---|
|
| |||||
| compound | λabs (nm) | λem (nm) | ε (M−1 cm−1) | ΦF | brightness |
| 12 | 295 ± 2 | 399 ± 2 | 14100 ± 1000 | 0.69 ± 0.04 | 9700 ± 1250 |
| 63 | 295 ± 2 | 407 ± 3 | 12500 ± 1000 | 0.25 ± 0.02 | 3100 ± 500 |
Determined in 25 mM Tris-HCl (pH 7.4) containing 0.5 M NaCl. Both compounds were treated with CF3COOH to remove the Boc protecting groups prior to the acquisition of photophysical data.
Figure 5.
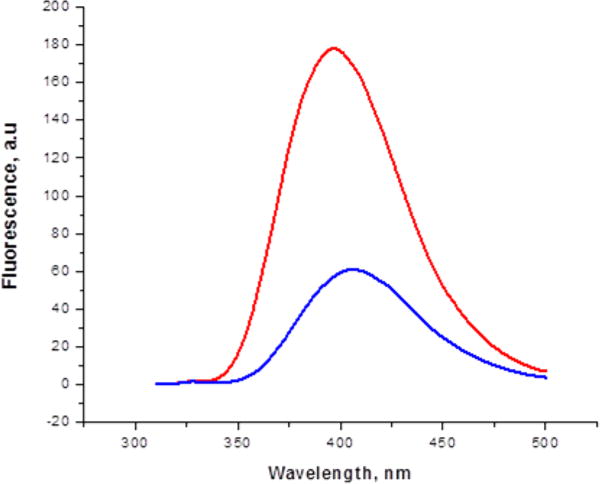
Fluorescence emission spectra of Boc-protected oxazole derivatives 63 (blue) and 12 (red). Both samples were present at a concentration of 1.0 μM in 25 mM Tris-HCl (pH 7.4) containing 0.5 M NaCl.
To explore the capability of the dipeptidomimetic analogues as FRET probes, we selected compound 1a as the donor. The emission peak wavelength of 1a after incorporation into position 66 of GFP was at 377 nm (Table 3). As the fluorescence acceptor we chose acridon-2-ylalanine (Acd), which has absorption peaks at 388 and 407 nm in water as a monomer and emission peaks at ~420 and 450 nm. As position 66 is within the β-barrel structure of GFP, it was chosen for donor 1a, whereas position 39, outside of the β-barrel, was chosen for acceptor Acd. The spectral overlap between the fluorescence emission of oxazole 1a and the absorption spectrum of Acd is shown in Figure S4. The predicted R0 value for these two fluorophores was calculated25 to be 32.8 Å, while the distance between the α-carbon atoms of residues 39 and 66 is 17.0 Å based on X-ray crystallography (Figure 6B). By decoding the four-base codon CGGG with the acridon-2-ylalanyl-tRNACCCG and the nonsense codon UAG with the oxazolyl-tRNACUA, we incorporated Acd and compound 1a into GFP at positions 39 and 66, respectively. The incorporation yield was 5% compared to wild-type GFP. After purification, the doubly modified GFP was excited at 296 nm. As demonstrated in Figure 6A, the modified GFP containing fluorescence acceptor Acd at position 39 and compound 1a at position 66 displayed a FRET signal at ∼450 nm. The theoretical maximal intensity of the FRET peak is defined by the orange curve in Figure 6A, which shows the emission spectrum of Acd following its direct excitation by irradiation at 370 nm. A control experiment involving a GFP having glycine at position 66 and Acd at position 39 gave only weak emission at 450 nm upon being irradiated at 296 nm (Figure S5).
Figure 6.
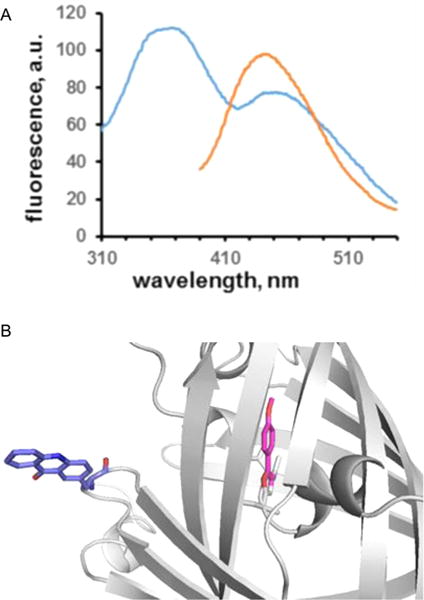
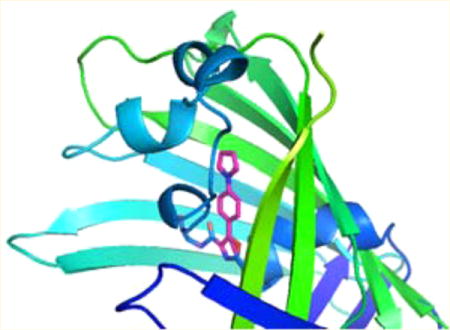
(A) Förster resonance energy transfer (FRET) between oxazole 1a at position 66 of GFP and Acd at position 39 of the same protein. The sample was excited at 296 nm, affording a FRET signal at ~450 nm (blue). A control sample having 1a at position 66, but no acceptor, exhibited no FRET upon being irradiated at 300 nm (Figure 4). Irradiation of the same sample at the excitation wavelength of Acd (370 nm) produced fluorescence emission centered at ~450 nm (orange). The sample was present at a concentration of ~20 nM in 25 mM Tris-HCl (pH 7.4) containing 0.25 M NaCl. (B) Model based on the X-ray crystal structure of green fluorescent protein (Protein Data Bank entry 1GFL), showing the placement of oxazole 1a in lieu of Tyr66, and Acd in place of Tyr39.
DISCUSSION
We have recently described the recognition by engineered bacterial ribosomes of suppressor tRNAs activated with dipeptide analogues containing oxazole and thiazole cores, and the incorporation of these heterocyclic species into dihydrofolate reductase and green fluorescent protein.12,13 The incorporations have been verified by mass spectrometric analysis of tryptic digests of the modified proteins. The strongly fluorescent nature of the derived proteins prompted the preparation and study of a library of substituted oxazoles and thiazoles to permit a better understanding of the nature and scope of our initial observations. The synthesis of 1,3-oxazoles and 1,3-thiazoles has been the focus of many studies as a consequence of their widespread occurrence in natural products. Nature biosynthesizes oxazoles through cyclodehydration and oxidation of acylserine derivatives,26 and many synthetic routes for oxazoles follow this general strategy.27,28 Another versatile method for the synthesis of 1,3-oxazoles involves the cyclodehydration of α-acylaminoketones, esters, or amides;29 the oxazole synthesis described in this report falls into this category. The α-amido-β-ketoesters were cyclized directly using a dehydrating mixture of 2 equiv of triphenylphosphine, iodine, and triethylamine,24 and the reactions were typically complete within 15 min at room temperature.
There are also number of methods for the synthesis of 1,3-thiazoles, the most common involving the condensation of substituted α-haloketones and thioamides. In this report, the thiazoles were obtained by the cyclization of α-amido-β-ketoesters using Lawesson’s reagent.30 This method was followed because the α-amido-β-ketoester was a common intermediate for both oxazole and thiazole syntheses, which simplified the overall process of library construction.
The A. victoria GFP chromophore is formed by a dehydrative cyclization, involving the Tyr moiety at position 66 in the nascent polypeptide. The phenolic OH group of the Tyr moiety is particularly important for the photophysical properties of GFP.31 There have been extensive structure–activity studies of the GFP fluorophore. In addition, site-directed mutagenesis has permitted the mutation of the GFP chromophore by Phe analogues with different para substituents in place of Tyr66.32,33 These studies indicated that the wavelength maxima for both absorption and emission peaks of the GFP fluorophore increased with increasing electron donating ability of these substituents. To build a library of the dipeptidomimetic analogues with a wide variety of photophysical properties, several oxazole and thiazole analogues containing either electron-donating (NMe2, SMe, and OMe) or electron-withdrawing (NO2, pyrrole, and CN) substituents were synthesized. The wavelengths of emission peaks for both oxazoles and thiazoles decreased in the following order: NMe2 > SMe > OMe ≈ NO2 ≈ pyrrole > CN (i.e., roughly in the order predicted by studies of the GFP fluorophore). As for the most electron-donating substituent, the presence of NMe2 elicited the maximal red shift of the absorption and emission peaks. Compounds 4b and 10b both had absorption maxima at 351 nm and emission maxima at 458 and 490 nm in methanol, respectively. Nitrile 6b exhibited the shortest wavelength absorption and emission peaks (285 and 365 nm, respectively). Compound 2b can be considered as a more electron dense congener of compound 1b because it contains methoxyl groups at both meta positions in addition to the one at the para position. It was anticipated correctly that compound 2b would exhibit a red shift in absorption and emission peak wavelengths compared to compound 1b, although the shift was not dramatic, presumably because of the placement of the additional OMe groups at the meta positions. In all cases, the emission maxima of the thiazoles were ~20–30 nm red-shifted compared to those of the corresponding oxazoles. The molar absorptivity values for the oxazoles were also greater compared to those of the corresponding thiazoles. Similarly, the quantum yields for the oxazoles were generally greater than for the corresponding thiazoles, with the exception of compounds 3b and 9b, both of which exhibited very high quantum yields. In comparison, the compounds having electron-withdrawing substituents (5b, 6b, 7b, and 11b) displayed very low quantum yields.
To study the basis for the observed photophysical properties of the oxazoles and thiazoles, four compounds (1b, 3b, 4b, and 8b) were used to prepare the respective activated suppressor tRNAs and modified GFPs on a larger scale. As shown in Table 3 and Figure 4, when these compounds were incorporated at the 3′-end of tRNAs as activated esters, the fluorescence emission maxima were generally shifted to longer wavelength, albeit without any significant increase in fluorescence intensity. In contrast, when the four compounds were incorporated into position 66 of GFP, the resulting GFP analogues had fluorescence intensities several-fold stronger than those of the free compounds and the corresponding aminoacyl-tRNACUAs, and the fluorescence emission occurred at shorter wavelengths in each case (Figures 3 and 4).
The oxazoles and thiazoles employed in this study are unique as synthetic protein fluorophores in that the conjugated systems that are responsible for their fluorescent properties actually become part of the polypeptide/protein backbone. Accordingly, it seemed possible that the effects noted in Table 3, and Figures 3 and 4, related to alterations in fluorescence intensity and emission wavelength maxima, might be influenced by the simple incorporation of these fluorophores into a polypeptide. As shown in Table 4 and Figure 5, this is clearly the case. Embedding oxazole 1a within the tetrapeptide Gly-Gly-Gly-Gly caused its fluorescence emission wavelength maximum to shift to a shorter wavelength (from 407 to 399 nm), as also noted for the oxazoles and thiazoles incorporated at position 66 of GFP (cf. Table 3). The embedded oxazole also exhibited a brightness of its fluorescence signal ~3-fold greater that that of the free oxazole when measured in Tris-HCl buffer (Table 4). This increase in brightness was associated with an increased quantum yield, plausibly due to suppression of nonradiative deactivation pathways by the rigid peptide matrix. This observation parallels an earlier finding12 in which we noted that the incorporation of oxazole 1a at position 39 of GFP (outside the β-barrel structure) produced an emission stronger than that observed for free oxazole 1b. Inclusion of the embedded oxazole at position 66 [which is within the β-barrel structure (Figure 6B)] effected a further enhancement of fluorescence emission intensity. More specifically, we found that the absorption extinction coefficient for GFP containing 1a at position 39 was 50200 M−1 cm−1, while that for GFP itself was 25000 M−1 cm−1.12 The quantum yield for the GFP analogues containing 1a at position 39 was 0.84 versus 0.79 for GFP itself. When 1a was present at position 66 of GFP, the absorption extinction coefficient was 90300 M−1 cm−1 and the quantum yield was 0.91. Thus, introducing 1a at position 39 of GFP produced a protein ~2-fold brighter than native GFP, while introducing this fluorophore at position 66 resulted in a protein ~4-fold brighter than wild-type GFP.12 This change was associated with a change in the absorption extinction coefficient of 1b. Embedding oxazole 1a in GFP at position 66 enhanced its brightness ~25–30-fold. It is worth noting that our novel GFP, having 1a at position 66, is also 2–3-fold brighter than a “class 2” GFP34 having an absorption extinction coefficient approximately twice that of the GFP that we employed for modification. While oxazole 1a is roughly isosteric with a dipeptide and has been introduced into the β-barrel structure in lieu of a single amino acid (Tyr66), there appears to be ample room within the β-barrel structure to accommodate this larger molecular entity (Figure 6B). Finally, the much smaller Stokes shift observed for oxazole 1a within the β-barrel of GFP12 may reasonably be attributed to a suppression of solvent relaxation. While the environmental sensitivity of these fluorophores should provide the wherewithal for enhanced definition of the microenvironment of proteins and protein complexes, there will also be a corresponding increase in the complexity of quantification as a consequence of the aforementioned environmental sensitivity.
We conclude that the significant observed changes in emission wavelength and intensity are due both to incorporation of the fluorophore within the protein backbone, and also to the environmental sensitivity of these fluorophores, and that this property may potentially be exploited to utilize such compounds as reporters of their environment, and conformational changes that alter that environment. It may be noted that the introduction of oxazole 1a into position 39 of GFP, which is not within the β-barrel structure, afforded a much more significant increase in the fluorescence intensity of the oxazole12 than did its present inclusion within a tetraglycine peptide. Accordingly, it might be anticipated that alteration of peptide structure, e.g., by the inclusion of aromatic or lipophilic amino acids, would also affect the brightness of the embedded oxazole.
Fluorescent proteins have been used as probes of molecular assembly in FRET experiments,35 although their large size does impose constraints on the utility of this approach. The environmental sensitivity of the oxazoles and thiazoles studied here, as well as their small size, chemical stability, and stronger fluorescence as compared with those of natural GFPs,36 suggests that they may have superior potential as reporter groups. Accordingly, we prepared a modified GFP having a strong oxazole fluorophore at position 66 within the β-barrel structure, and a fluorescence acceptor (Acd) whose absorption spectrum overlaps the emission spectrum of the oxazole outside the β-barrel at position 39. The R0 value of this potential FRET pair was calculated25 to be 32.8 Å, while the distance calculated from an X-ray crystal structure37 was 17.0 Å (Figure 6B). As predicted, when the doubly modified GFP was irradiated at the excitation maximum of the oxazole at position 66 (296 nm), a FRET peak was evident (Figure 6A).
Where spectral overlap exists, the R0 value for a given FRET pair increases in direct proportion to the quantum yield of the donor fluorophore and molar absorptivity of the acceptor. Given the very favorable molar absorptivities and quantum yields of many of the novel fluorophores studied here, as well as their environmental sensitivity and ability to be incorporated into proteins with reasonable suppression efficiencies, it is anticipated that they may find utility as reporter groups in experiments involving protein conformation and binding interactions with other (macro)molecules. Further, the activated tRNAs whose fluorescence emission properties are summarized in Table 3 may plausibly be of utility in reporting on the environment of the aminoacyl moiety during the partial reactions of protein biosynthesis. Presumably, such interactions may be unique to protein biosynthesizing systems in which the ribosomes have been modified to accommodate dipeptidyl-tRNAs as substrates.
It should be noted that these experiments were all conducted in cell free systems, and application of the present artificial fluorescent proteins in a cellular context would require significant extensions of the current technology.
CONCLUSIONS
A library of several fluorescent oxazole- and thiazole-based dipeptidomimetic analogues has been synthesized. The analogues are structurally similar to the GFP chromophore but are stable chemically and do not require activation to exhibit fluorescence. To encompass a variety of photophysical properties, the compounds were designed with different electron-donating and electron-withdrawing substituents at the position analogous to the p-OH group in the GFP fluorophore. The compounds displayed reasonable efficiency of incorporation into position 66 of GFP using modified ribosomes and misacylated suppressor tRNACUAs. In addition, four mutant GFPs each containing a dipeptide analogue with favorable fluorescent properties and formed in good suppression yield were prepared in larger quantities and were characterized in more detail. All of the modified GFPs exhibited fluorescence intensities several-fold higher than those of the corresponding aminoacyl tRNACUAs and were more strongly fluorescent than GFP itself. The fluorescent properties (emission wavelength and intensity) were shown to be responsive to environment, both within the protein backbone and especially within a secondary structure such as a β-barrel. Embedding the oxazole within a simple tetrapeptide also enhanced the intensity of its fluorescence emission. A successful FRET experiment demonstrated the potential utility of the fluorophores as reporter groups to monitor protein conformational changes and molecular interactions.
Supplementary Material
Acknowledgments
Funding
This study was supported by Grant GM 103861 from the National Institute of General Medical Sciences, National Institutes of Health.
ABBREVIATIONS
- APCI
atmospheric-pressure chemical ionization
- ESI
electrospray ionization
- GFP
green fluorescent protein
- FP
fluorescent protein
- HPLC
high-performance liquid chromatography
- TLC
thin layer chromatography
- DMSO
dimethyl sulfoxide
- Rf
ratio of fronts
- THF
tetrahydrofuran
- DMF
dimethylformamide
- TBA
tetrabutylammonium
- BOP
N-[oxytris(dimethylamino)phosphonium]benzotriazole hexafluorophosphate
- EGTA
ethylene glycol tetraacetic acid
- PEG
polyethylene glycol
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- Ni-NTA
nickel nitrilotri-acetic acid
- DIPEA
diisopropylethylamine
- UV
ultraviolet
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.6b00102.
Additional data (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Chen I, Ting AY. Site-specific labeling of proteins with small molecules in live cells. Curr Opin Biotechnol. 2005;16:35–40. doi: 10.1016/j.copbio.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Sahoo H. Fluorescent labeling techniques in biomolecules: a flashback. RSC Adv. 2012;2:7017–7029. [Google Scholar]
- 3.Chen S, Fahmi NE, Wang L, Bhattacharya C, Benkovic SJ, Hecht SM. Detection of dihydrofolate reductase conformational change by FRET using two fluorescent amino acids. J Am Chem Soc. 2013;135:12924–12927. doi: 10.1021/ja403007r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci U S A. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 6.Walker CL, Lukyanov KA, Yampolsky IV, Mishin AS, Bommarius AS, Duraj-Thatte AM, Azizi B, Tolbert LM, Solntsev KM. Fluorescence imaging using synthetic GFP chromophores. Curr Opin Chem Biol. 2015;27:64–74. doi: 10.1016/j.cbpa.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Marks KM, Nolan GP. Chemical labeling strategies for cell biology. Nat Methods. 2006;3:591–596. doi: 10.1038/nmeth906. [DOI] [PubMed] [Google Scholar]
- 8.Dedkova LM, Fahmi NE, Golovine SY, Hecht SM. Enhanced D-amino acid incorporation into protein by modified ribosomes. J Am Chem Soc. 2003;125:6616–6617. doi: 10.1021/ja035141q. [DOI] [PubMed] [Google Scholar]
- 9.Dedkova LM, Fahmi NE, Golovine SY, Hecht SM. Construction of modified ribosomes for incorporation of D-amino acids into proteins. Biochemistry. 2006;45:15541–15551. doi: 10.1021/bi060986a. [DOI] [PubMed] [Google Scholar]
- 10.Dedkova LM, Fahmi NE, Paul R, del Rosario M, Zhang L, Chen S, Feder G, Hecht SM. β-Puromycin selection of modified ribosomes for in vitro incorporation of β-amino acids. Biochemistry. 2012;51:401–415. doi: 10.1021/bi2016124. [DOI] [PubMed] [Google Scholar]
- 11.Maini R, Nguyen DT, Chen S, Dedkova LM, Chowdhury SR, Alcala-Torano R, Hecht SM. Incorporation of β-amino acids into dihydrofolate reductase by ribosomes having modifications in the peptidyltransferase center. Bioorg Med Chem. 2013;21:1088–1096. doi: 10.1016/j.bmc.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Maini R, Dedkova LM, Paul R, Madathil MM, Roy Chowdhury S, Chen S, Hecht SM. Ribosome-mediated incorporation of dipeptides and dipeptide analogues into proteins in vitro. J Am Chem Soc. 2015;137:11206–11209. doi: 10.1021/jacs.5b03135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roy Chowdhury S, Maini R, Dedkova LM, Hecht SM. Synthesis of fluorescent dipeptidomimetics and their ribosomal incorporation into green fluorescent protein. Bioorg Med Chem Lett. 2015;25:4715–4718. doi: 10.1016/j.bmcl.2015.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talukder P, Chen S, Liu CT, Baldwin EA, Benkovic SJ, Hecht SM. Tryptophan-based fluorophores for studying protein conformational changes. Bioorg Med Chem. 2014;22:5924–5934. doi: 10.1016/j.bmc.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson SA, Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG. The use of 5′-phospho-2 deoxyribocytidylylriboadenosine as a facile route to chemical aminoacylation of tRNA. Nucleic Acids Res. 1989;17:9649–9660. doi: 10.1093/nar/17.23.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lakowicz JR. Principles of Fluorescence Spectroscopy. Plenum Press; New York: 1983. [Google Scholar]
- 17.Lodder M, Golovine S, Hecht SM. A chemical deprotection strategy for the elaboration of misacylated transfer RNA’s. J Org Chem. 1997;62:778–779. [Google Scholar]
- 18.Lodder M, Golovine S, Laikhter AL, Karginov VA, Hecht SM. Misacylated transfer RNAs having a chemically removable protecting group. J Org Chem. 1998;63:794–803. doi: 10.1021/jo971692l. [DOI] [PubMed] [Google Scholar]
- 19.Varshney U, Lee CP, RajBhandary UL. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem. 1991;266:24712–24718. [PubMed] [Google Scholar]
- 20.Sanz-Cervera JF, Blasco R, Piera J, Cynamon M, Ibanez I, Murguia M, Fustero S. Solution versus fluorous versus solid-phase synthesis of 2,5-disubstituted 1,3-azoles. Preliminary antibacterial activity studies. J Org Chem. 2009;74:8988–8996. doi: 10.1021/jo9016265. [DOI] [PubMed] [Google Scholar]
- 21.Castro B, Dormoy JR, Evin G, Selve C. Peptide coupling reagents. IV. N-[Oxytris(dimethylamino)phosphonium]-benzotriazole hexafluorophosphate. Tetrahedron Lett. 1975;16:1219–1222. [Google Scholar]
- 22.Robertson SA, Ellman JA, Schultz PG. A general and efficient route for chemical aminoacylation of transfer RNAs. J Am Chem Soc. 1991;113:2722–2729. [Google Scholar]
- 23.Kometani M, Ihara K, Kimura R, Kinoshita H. Stereoselective syntheses of (E)-α,β-didehydroamino acid and peptide containing its residue utilizing oxazolidinone derivative. Bull Chem Soc Jpn. 2009;82:364–380. [Google Scholar]
- 24.Wipf P, Miller CP. A new synthesis of highly functionalized oxazoles. J Org Chem. 1993;58:3604–3606. [Google Scholar]
- 25.Speight LC, Muthusamy AK, Goldberg JM, Warner JB, Wissner RF, Willi TS, Woodman BF, Mehl RA, Petersson EJ. Efficient synthesis and in vivo incorporation of acridon-2-ylalanine, a fluorescent amino acid for lifetime and Förster resonance energy transfer/luminescence resonance energy transfer studies. J Am Chem Soc. 2013;135:18806–18814. doi: 10.1021/ja403247j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li YM, Milne JC, Madison LL, Kolter R, Walsh CT. From peptide precursors to oxazole and thiazole-containing peptide antibiotics: microcin B17 synthase. Science. 1996;274:1188–1193. doi: 10.1126/science.274.5290.1188. [DOI] [PubMed] [Google Scholar]
- 27.Sakakura A, Kondo R, Umemura S, Ishihara K. Dehydrative cyclization of serine, threonine and cysteine residues catalyzed by molybdenum(VI) oxo compounds. Tetrahedron. 2009;65:2102–2109. [Google Scholar]
- 28.Meyers AI, Tavares FX. Oxidation of oxazolines and thiazolines to oxazoles and thiazoles. Application of the Kharasch-Sosnovsky reaction. J Org Chem. 1996;61:8207–8215. doi: 10.1021/jo9613491. [DOI] [PubMed] [Google Scholar]
- 29.Robinson R. New synthesis of oxazole derivatives. Proc Chem Soc, London. 1910;25:295–296. [Google Scholar]
- 30.Thomsen I, Pedersen U, Rasmussen PB, Yde B, Andersen TP, Lawesson SO. Studies on organophosphorus compounds. XLIV. Novel and convenient methods for the preparation of substituted thiophenes, thiazoles, and 1,3,4-thiadiazole-2(3H)-thiones from bifunctional substrates. Chem Lett. 1983;12:809–810. [Google Scholar]
- 31.Tolbert LM, Baldridge A, Kowalik J, Solntsev KM. Collapse and recovery of green fluorescent protein chromophore emission through topological effects. Acc Chem Res. 2012;45:171–181. doi: 10.1021/ar2000925. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Xie J, Deniz AA, Schultz PG. Unnatural amino acid mutagenesis of green fluorescent protein. J Org Chem. 2003;68:174–176. doi: 10.1021/jo026570u. [DOI] [PubMed] [Google Scholar]
- 33.Kajihara D, Hohsaka T, Sisido M. Synthesis and sequence optimization of GFP mutants containing aromatic non-natural amino acids at the Tyr66 position. Protein Eng, Des Sel. 2005;18:273–278. doi: 10.1093/protein/gzi033. [DOI] [PubMed] [Google Scholar]
- 34.Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 35.Piston DW, Kremers GJ. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci. 2007;32:407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Shaner NC, Patterson GH, Davidson MW. Advances in fluorescent protein technology. J Cell Sci. 2007;120:4247–4260. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- 37.Yang F, Moss LG, Phillips GN., Jr The molecular structure of green fluorescent protein. Nat Biotechnol. 1996;14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.