

Key Points
Acalabrutinib preserves Src family kinase activity and avoids dysfunctional platelet thrombus formation caused by ibrutinib therapy.
Platelet thrombus formation is enhanced by intermediate purity factor VIII.
Abstract
The Bruton tyrosine kinase (Btk) inhibitor ibrutinib induces platelet dysfunction and causes increased risk of bleeding. Off-target inhibition of Tec is believed to contribute to platelet dysfunction and other side effects of ibrutinib. The second-generation Btk inhibitor acalabrutinib was developed with improved specificity for Btk over Tec. We investigated platelet function in patients with non-Hodgkin lymphoma (NHL) receiving ibrutinib or acalabrutinib by aggregometry and by measuring thrombus formation on collagen under arterial shear. Both patient groups had similarly dysfunctional aggregation responses to collagen and collagen-related peptide, and comparison with mechanistic experiments in which platelets from healthy donors were treated with the Btk inhibitors suggested that both drugs inhibit platelet Btk and Tec at physiological concentrations. Only ibrutinib caused dysfunctional thrombus formation, whereas size and morphology of thrombi following acalabrutinib treatment were of normal size and morphology. We found that ibrutinib but not acalabrutinib inhibited Src family kinases, which have a critical role in platelet adhesion to collagen that is likely to underpin unstable thrombus formation observed in ibrutinib patients. We found that platelet function was enhanced by increasing levels of von Willebrand factor (VWF) and factor VIII (FVIII) ex vivo by addition of intermediate purity FVIII (Haemate P) to blood from patients, resulting in consistently larger thrombi. We conclude that acalabrutinib avoids major platelet dysfunction associated with ibrutinib therapy, and platelet function may be enhanced in patients with B-cell NHL by increasing plasma VWF and FVIII.
Visual Abstract
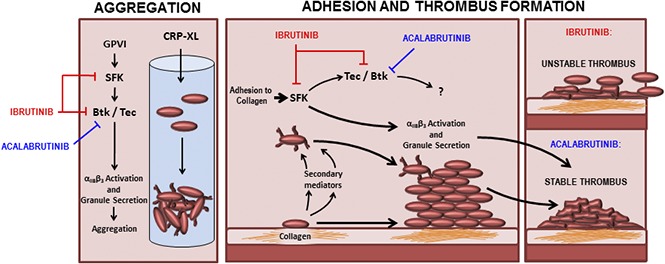
Introduction
The Bruton tyrosine kinase (Btk) inhibitor ibrutinib was the first of a new class of drug for the treatment of indolent non-Hodgkin lymphomas (NHLs).1 Treatment with ibrutinib has proven to be efficacious but is associated with side effects, including increased risk of major bleeding.2 Bleeding associated with ibrutinib is potentially explained by off-target inhibition of other kinases in addition to Btk; a study of patients with X-linked agammaglobulinemia (XLA), who have Btk deficiency, reported no increased risk of bleeding3 although genetic deficiency of Btk was found to increase time occlusion in a mouse model of carotid artery injury.4 Furthermore, studies performed using genetically modified mice demonstrated redundancy between Btk and Tec in platelet signaling because ablation of both kinases was required to render platelets fully insensitive to collagen in aggregation assays.5 The effect of simultaneous genetic ablation of Btk and Tec on platelet adhesion under shear has not been explored in vivo or in vitro. The off-target effects of ibrutinib on platelet function have added complications to its use, such as risks associated with concurrent treatment with anticoagulant or antiplatelet medication, which is common among patients with chronic lymphocytic leukemia (CLL).6 Ibrutinib therapy is interrupted before surgery to reduce the risk of bleeding, but prolonged dose interruption or dose reduction may reduce efficacy and lead to a flare in symptoms.7 Acalabrutinib is a second-generation Btk inhibitor and exhibits greater selectivity over kinases that are important for platelet function such as Src family kinases (SFKs) and Tec.8 No major bleeding events were reported during a phase 2 trial of acalabrutinib for treatment of CLL,8 but it is not yet clear if the selectivity of acalabrutinib is sufficient to fully avoid the platelet dysfunction associated with ibrutinib.
Ibrutinib causes platelet dysfunction downstream of the GPVI receptor, GPIb, and integrin αIIbβ3.9-11 Although XLA is not associated with increased risk of bleeding, deficient expression or function of GPVI,12-14 GPIb,15 or integrin αIIbβ316 is associated with bleeding phenotypes in mice or humans. The precise reason for the discrepancy between bleeding risk in patients with XLA and patients receiving ibrutinib has not been characterized, but is thought to relate to off-target inhibition of Tec.8 However, ibrutinib may also inhibit other important platelet kinases such as SFK that phosphorylate several signaling molecules in the GPVI signaling pathway.17 Btk and Tec are themselves dependent on SFK-mediated tyrosine phosphorylation to become active.18,19 Inhibition of SFK is known to cause hemostatic dysfunction because the Src inhibitor, dasatinib, is associated with increased bleeding risk.20-22 Comparison of ibrutinib and acalabrutinib therapy therefore represents an important opportunity to characterize their relative effects and understand how improved kinase specificity affects platelet dysfunction.
Thrombocytopenia is frequently associated with CLL and may be a contributing factor to bleeding events in combination with drug-induced platelet dysfunction.23,24 Treatment options for patients that are at high risk of bleeding are currently limited and the clinical efficacy of platelet transfusion has not been established.2 Evidence that platelet transfusion may reverse platelet dysfunction caused by ibrutinib is based on in vitro data11 with few studies investigating this approach clinically.25 Furthermore, the benefit of platelet transfusion during treatment with other antiplatelet medication is controversial and may lead to poorer outcomes.26 Investigating alternatives to platelet transfusion to treat or prevent bleeding during ibrutinib or acalabrutinib therapy is therefore a priority. One alternative to platelet transfusion for bleeding patients, or those undergoing surgery and at risk of bleeding from platelet dysfunction, is administration of desmopressin,27-29 which stimulates secretion of the contents of Weibel-Palade bodies and results in an acute increase in plasma von Willebrand factor (VWF) and factor VIII (FVIII) levels of two- to sixfold.30 Indeed, successful administration of desmopressin to stop bleeding in patients suffering from thrombocytopenia has also been reported.31 It may be possible to administer desmopressin to treat bleeding or reduce bleeding risk during treatment with Btk inhibitors, either as an alternative for or adjunct to platelet transfusion. However, because ibrutinib inhibits GPIb-mediated platelet function,11 it has remained unclear if this approach might be beneficial.
Methods
Materials
Type I collagen was obtained from Nycomed (Munich, Germany) and collagen-related peptide (CRP-XL) from Professor Richard Farndale (University of Cambridge, Cambridge, United Kingdom). Adenosine 5′-diphosphate (ADP), arachidonic acid, and U46619 were from Sigma (Gillingham, United Kingdom), and TRAP-6 epinephrine was from Labmedics (Salford, United Kingdom). Acalabrutinib, ibrutinib, and dasatinib were from Selleckchem (Munich, Germany). Antiphosphotyrosine antibody 4G10 was obtained from Millipore (Burlington, MA). PKC substrate antibody and phospho-PLCγ2 Y759 was from New England Biolabs (Hitchin, United Kingdom). Phospho-Src 418 antibody and Fura-2 am was from Life Technologies (Paisley, United Kingdom). Phospho-Btk Y223, phospho-Lyn Y396, and phospho-β3 Y773 antibodies were from Abcam (Cambridge, United Kingdom). Actin 11C antibody was from Santa Cruz Biotechnology (Heidelberg, Germany). Greiner 96-well plates and DiOC6 were from Thermo Fisher Scientific (Dartford, United Kingdom).
Patients
Trough blood samples were obtained from patients with CLL or mantle cell leukemia and receiving either ibrutinib (6 patients) or acalabrutinib (8 patients; Table 1). Our control group comprised Bkt-inhibitor naïve CLL patients (5 patients). All patients provided informed consent in accordance with the declaration of Helsinki. Ethics approval for this study was covered under the Oxford Radcliffe Biobank research tissue bank ethics, HTA License Number 12217, Oxfordshire C REC:09/H0606/5+5, project approval code 17/A016.
Table 1.
Patient information
| Acalabrutinib 100 mg BD (n = 8) | Ibrutinib 480 mg OD (n = 6) | Btk inhibitor naïve (n = 5) | |
|---|---|---|---|
| Age, median (range), y | 68 (53-80) | 68 (60-77) | 78 (60-80) |
| Sex, n (%) | |||
| Female | 2 (25) | 3 (50) | 3 (60) |
| Male | 6 (75) | 3 (50) | 2 (40) |
| Diagnosis, n (%) | |||
| CLL | 7 (87.5) | 6 (100) | 5 (100) |
| Mantle cell lymphoma | 1 (12.5) | — | — |
| Laboratory parameters | |||
| Hemoglobin, median (range), g/L | 139 (113-155) | 131 (109-147) | 140 (117-148) |
| Platelet count, median (range), ×109/L | 148.5 (35-228) | 114 (58-197) | 118 (109-274) |
| Clinical details, n (%) | |||
| Concurrent antiplatelet drugs | — | — | — |
| Concurrent anticoagulants | 1 (13)* | — | — |
| Inherited bleeding disorders | — | — | — |
| Duration of Btk inhibitor, median (range), mo | 21.5 (19-32) | 19 (2-39) | — |
| Active bleeding at time of recruitment | — | — | — |
| Bleeding on Btk inhibitor, n (%)† | |||
| None | 3 (38) | 5 (83) | — |
| Grade 1 | 2 (25) | 1 (17) | — |
| Grade 2 | 3 (38) | — | — |
| Response to Btk inhibitor, n (%)‡ | |||
| Complete remission | 5 (63) | 3 (50) | — |
| Partial remission | 3 (38) | 3 (50) | — |
| Stable or progressive disease | — | — | — |
BD, twice daily; OD, once daily.
Dabigatran 150 mg twice daily.
Common Terminology Criteria for Adverse Events grading scale.
International Workshop on Chronic Lymphocytic Leukemia criteria for chronic lymphocytic leukemia; positron emission tomography computed tomography criteria for mantle cell lymphoma.
Platelet preparation
Blood samples were obtained from healthy donors that had given informed consent and using procedures approved by the University of Reading Research Ethics Committee or from patients and collected into vacutainers containing 3.8% (w/v) sodium citrate. Platelet-rich plasma (PRP) was prepared by centrifuging whole blood at 100g for 20 minutes. Washed platelets were prepared by adding acid citrate dextrose to PRP and centrifuging at 350g for 20 minutes and resuspending the platelet pellet at 4 × 108 cells/mL in Tyrode’s buffer as previously described.9
Plate-based aggregometry
For measurement of aggregation using PRP from patients or healthy donors, 96-well plates (Greiner) containing the following freeze-dried platelet agonists at a range of concentrations: ADP, CRP-XL, epinephrine, U46619, collagen, and TRAP-6 were prepared in advance and stored in vacuum-sealed bags for no more than 2 months. PRP isolated from the blood of healthy donors or patients was loaded onto plates and shaken at 1200 rpm for 5 minutes at 37°C using a plate shaker (Quantifoil Instruments), as described by Lordkipanidzé et al; absorption of 405 nm light was measured using a Multiskan Ascent 354 Microplate Reader (LabSystems). Aggregation of washed platelets was performed in 96-well plates as previously described.9
Thrombus formation under flow
Thrombus formation was performed using blood from healthy donors using microfluidic flow chips (Vena8, CellixLtd, Dublin, Ireland) coated with 100 µg/mL type I collagen and measured in real time following incubation of blood from healthy donors with ibrutinib, acalabrutinib, or vehicle for 10 minutes, or fixed with 10% formyl saline after 8 minutes of perfusion at a shear rate of 1000 s−1 and imaged at a later time.
Protein phosphorylation studies
Washed platelets at 4 × 108 cells per milliliter in the presence of inhibitors to prevent aggregation (100 µM MRS2179, 1 µM cangrelor, 10 µM indomethacin, and 1 mM EGTA) were incubated with ibrutinib or acalabrutinib at concentrations specified in individual figures or vehicle for 5 minutes at 37°C before addition of 1 µg/mL CRP-XL. Samples were incubated for 3 minutes with stirring at 37°C using an aggregometer (Helena) before addition of reducing Laemmli sample treatment buffer.
Phospho-Tec ELISA
Platelets were prepared and stimulated as detailed previously but lysed in NP40 buffer (300 mM NaCl, 20 mM Tris base, 2 mM EGTA, 2 mM EDTA, 1 mM PMSF, 10 µg/mL aprotinin, 10 µg/mL leupeptin, 0.7 μg/mL pepstatin A, 2 mM sodium orthovanadate, 2% v/v NP-40; pH 7.3) and analyzed using a human phosphotyrosine Tec enzyme-linked immunosorbent assay (ELISA kit, RayBio). Absorption at 405 nm was measured using a NOVOstar plate reader (BMG LabTech, Aylesbury, United Kingdom).
Ca2+ measurement
The [Ca2+]i assay was performed at 37°C using fura-2 loaded washed platelets in black 96-well plates (Greiner). Dual excitation was measured using a Novostar spectrofluorometer (BMG Labtech) previously described.9
Platelet adhesion to collagen
Washed platelets at 2 × 107 cells/mL were exposed to collagen (100 µg/mL)-coated, 96-well assay plates, and allowed to adhere for 45 minutes at 37°C. Nonadherent platelets were washed off before, fixed with 10% formyl saline for 10 minutes. The wells were then washed and labeled with DiOC6. Fluorescence images of adherent platelets were captured with the 20× objective lens of an ImageXpress Nano high content imaging system and counted using CellReporterXpress software (Molecular Devices, Winnersh, United Kingdom).
Fluorescence-activated cell sorting measurement of fibrinogen binding and p-selectin exposure
Measurements of fibrinogen binding and p-selectin exposure were performed using washed platelets pretreated with inhibitors as described in figure legends and stimulated with 1 µg/mL CRP in the presence of fluorescein isothiocyanate–conjugated polyclonal rabbit anti-fibrinogen antibody (Agilent Technologies LDA UK Limited, Cheadle, United Kingdom) and PE-Cy5-conjugated mouse anti-P-selectin (CD62P) antibody (BD, Berkshire, United Kingdom), and then incubated for 20 minutes in the dark. Platelets were then fixed by addition of filtered formyl saline (0.2% formaldehyde in 0.15M NaCl) and median fluorescence intensities were measured for 5000 platelets per sample on an Accuri C6 Flow Cytometer (BD Biosciences, Berkshire, United Kingdom).
Statistical methods
Statistical testing as described in figure legends and the results section were performed using GraphPad Prism Software (GraphPad, La Jolla, CA).
Results
Acalabrutinib and ibrutinib cause dysfunction of GPVI-mediated platelet aggregation
Platelet aggregation to GPVI agonists has previously been shown to be inhibited in blood samples taken from patients receiving ibrutinib,10,11 but the effect of acalabrutinib on platelet function has not been reported. Aggregation of PRP was used to characterize platelet function of patients treated with ibrutinib or acalabrutinib (Table 1). One patient receiving acalabrutinib was excluded from this aspect of the study because of a low platelet count (<50 × 109/L). The aggregation assay included ADP, U46619, epinephrine, thrombin receptor activator peptide-6 (TRAP-6), CRP-XL, and collagen was adapted from the method developed and validated by Lordkipanidzé et al.32 We used a range of agonist concentrations to enable comparison of patient sensitivity to each agonist by estimation of the log half maximal effective concentration (logEC50) by nonlinear regression analysis. Results were compared with a group of healthy donors and Btk inhibitor naïve CLL patients (Figure 1A). Aggregation stimulated by ADP, U46619, epinephrine, and TRAP-6 was not significantly different in either patient group compared with healthy controls. Responses to collagen were completely ablated in all patients tested for both patient groups receiving Btk inhibitors, even at the highest concentration of collagen (3 µg/mL) (Figure 1A-B). Responses to the GPVI receptor agonist CRP-XL were either less potent (ibrutinib,2 patients; acalabrutinib, 3 patients) or ablated (ibrutinib, 4 patients; acalabrutinib, 4 patients) relative to responses of healthy controls (logEC50 = −7.7M ± 0.30; Figure 1C) and Btk inhibitor naïve CLL patients (logEC50 = −7.3M ± 0.38). Experiments in which CRP-XL was titrated against ibrutinib and acalabrutinib indicated that 10 µM acalabrutinib or 1 µM ibrutinib was required to ablate platelet aggregation of washed platelets from healthy donors (Figure 1D-E).
Figure 1.

Acalabrutinib and ibrutinib therapy cause dysfunctional GPVI-mediated platelet aggregation. PRP from patients receiving ibrutinib (Ibr; n = 6), acalabrutinib (Acal; n = 7), healthy donors (n = 9), or Btk inhibitor naïve CLL patients (n = 5) was loaded into 96-well microtiter plates containing lyophilized platelet agonists. Plates were shaken for 5 minutes at 37°C and aggregation was measure by light transmission to give an end point measurement. (A) Scatter plot of EC50 values calculated from concentration response curves for ADP, U46619, epinephrine, TRAP-6, CRP-XL, and collagen. Each point represents the response of a patient receiving acalabrutinib (red triangle) or ibrutinib (green square); healthy donor (gray circle) or Btk inhibitor naïve CLL patient (white diamond). NR, patients that did not respond to an agonist at the highest concentration tested. Concentration response curves for GPVI agonists (B) CRP-XL and (C) collagen; each point represents the mean % aggregation values ± standard error of the mean (SEM). Significant differences relative to healthy donors were tested by 2-way ANOVA with the Turkey multiple comparisons test. Aggregation of washed platelets from healthy donors pre-treated with a range of (D) acalabrutinib or (E) ibrutinib concentrations and then stimulated with 3 to 0.01 µg/mL CRP-XL.
Acalabrutinib is selective for SFK but not Tec
Both ibrutinib and acalabrutinib have been screened for activity against a panel of kinases in cell-free, in vitro assays.8,33 However, the estimated potency of the drugs in such assays does not correspond closely with measurements performed in cell-based assays. In addition to this, kinase regulation in the platelet is highly complex and involves positive and negative feedback mechanisms.34 To elucidate the mechanistic differences between ibrutinib and acalabrutinib that underpin differences in bleeding risk, we characterized the effects of the drugs on phosphorylation events downstream of GPVI in platelets from healthy donors (Figure 2). Phosphorylation of Btk Y223, the autophosphorylation site of Btk that correlates with its kinase activity, was inhibited with equal potency by ibrutinib (log half maximal inhibitory concentration [logIC50] = −7.2M ± 0.30) and acalabrutinib (logIC50 = −7.1M ± 0.21; Figure 2A-B). Inhibition of Tec was harder to quantify because of the lack of site-specific phospho antibodies, but total phosphotyrosine levels can be measured by ELISA. Total Tec phosphotyrosine levels are likely to include the Tec autophosphorylation site Y20635 as well as tyrosine residues phosphorylated by other kinases such as SFK. We estimated that Tec was more potently inhibited by ibrutinib (logIC50 = −6.4M) than by acalabrutinib (logIC50 = −5.5M; Figure 2A-B); however, the concentration-response profiles of both ibrutinib and acalabrutinib were complex, involving both positive and negative components and differential regulation of multiple sites. Phosphorylation of the autophosphorylation site Y418 of Src appeared to be potentiated by both drugs at lower concentrations. However, at higher concentrations, ibrutinib also inhibited Src Y418 phosphorylation (logIC50 = −5.7M ± 0.18), whereas acalabrutinib did not inhibit Src activity and actually potentiated activation even at high concentrations (logEC50 = −7.5M ± 0.46; Figure 2A-B). Because our data indicated that platelet Btk and Tec were likely to be inhibited by physiological concentrations of both ibrutinib and acalabrutinib, we further investigated inhibition of SFK by comparing both drugs to the SFK inhibitor dasatinib. Phosphorylation of Lyn Y396 (Figure 2D) was ablated by dasatinib (logIC50 = −7.1M ± 0.05) and ibrutinib (logEC50 = −5.8M ± 0.08) but not acalabrutinib, which only inhibited phosphorylation to 70% of vehicle-treated platelets at the highest concentration tested (10 µM). Total phosphotyrosine levels (Figure 2E) followed a similar pattern whereby dasatinib caused near ablation of CRP-XL–evoked tyrosine phosphorylation (logIC50 = −6.9M ± 0.24), whereas ibrutinib was less potent (logIC50 = −5.1M ± 0.29) and acalabrutinib only inhibited partially (71.7% of vehicle) at 10 µM.
Figure 2.

SFK activation and adhesion to collagen is spared by acalabrutinib but not ibrutinib. Washed human platelets from healthy donors were preincubated with a range of (A) acalabrutinib or (B) ibrutinib and then stimulated with 1 µg/mL CRP-XL for 3 minutes at 37°C; tyrosine phosphorylation of Src (Y418) and Btk (Y223) were measured by western blot using site-specific antibodies and total levels of Tec tyrosine phosphorylation were measure by ELISA. Points represent mean levels of tyrosine phosphorylation relative to vehicle-treated controls ± SEM. (C) Representative images of phosphoblots. Washed platelets treated with a range of acalabrutinib, ibrutinib, or dasatinib concentrations and stimulated as previously and blotted for (D) tyrosine phosphorylation of Lyn (Y396) or (E) total phosphotyrosine using a site-specific antibody or 4G10, respectively. Points represent mean levels of tyrosine phosphorylation relative to vehicle-treated controls ± SEM. (F) Aggregation 1 µg/ml CRP-XL stimulated of washed platelets was performed in 96-well plates after treatment with a range of concentrations of acalabrutinib, ibrutinib, or dasatinib concentration response curves are mean platelet aggregation ± SEM relative to vehicle-treated control. (G) Acalabrutinib, ibrutinib, or dasatinib-treated washed platelets from healthy donors were allowed adhere to collagen-coated wells of a 96-well plate after 45 minutes at 37°C; graphs plot concentration response curves to mean numbers of adhered platelets ± SEM relative to vehicle treatment. (H) Representative images of phosphoblots for platelets treated with tyrosine kinase inhibitors.
Ibrutinib but not acalabrutinib potently inhibits platelet adhesion to collagen
Aggregation of washed platelets evoked by 1 µg/mL CRP-XL was completely ablated by acalabrutinib (logIC50 = −5.3M ± 0.06) and with significantly greater potency by ibrutinib (logIC50 = −6.2M ± 0.28) and dasatinib (logIC50 = −6.6M ± 0.6; Figure 2F). We have previously reported that discrepancies are present in the way that ibrutinib inhibits platelet aggregation to immobilized collagen-coated surfaces.9 Adhesion of platelets to collagen (Figure 2G) was partially inhibited (∼40% of vehicle) by ibrutinib (logIC50 = −5.9M ± 0.18) and dasatinib (logIC50 = −6.9M ± 0.09), which closely matched their ability to inhibit Lyn activity. Acalabrutinib was a weak inhibitor of adhesion to collagen and only reduced the number of adhered platelet to 60% ± 9.4 of vehicle.
Acalabrutinib inhibits signaling downstream of GPVI less potently than ibrutinib
To understand the consequences of the differences in kinase specificity of the inhibitors, signaling events downstream of the kinases were measured. We found that phosphorylation of the Btk substrate, Y759 of PLCγ2, was inhibited by both ibrutinib (logIC50 = −6.7M ± 0.18) and acalabrutinib (logIC50 = −6.7 ± 0.26; Figure 3B). However, acalabrutinib did not completely ablate phosphorylation of PLCγ2 Y759 even at the highest concentration of acalabrutinib tested. Phosphorylation of PKC substrates was inhibited less potently by acalabrutinib (logIC50 = −6.2 M ± 0.19) than by ibrutinib (logIC50 = −6.5 ± 0.24, P < .05, Student t test; Figure 3B). Acalabrutinib inhibited PKC activity by 52% ± 4.5 relative to vehicle, significantly less than ibrutinib, which inhibited by 72% ± 3.3 (P < .05, Student t test) at the highest concentration tested. Release of Ca2+ from intracellular stores into the cytosol is stimulated downstream of PLCγ2 and was also inhibited less potently by acalabrutinib (logIC50 = −6.1M ± 0.08) than ibrutinib (logIC50 = −6.7 ± 0.07, P < .05, Student t test).
Figure 3.

Acalabrutinib inhibits signal transduction downstream of GPVI with lower potency than ibrutinib. Washed human platelets from healthy donors were preincubated with a range of acalabrutinib or ibrutinib concentrations and then stimulated with 1 µg/mL CRP-XL for 3 minutes at 37°C and phosphorylation of (A) PLCγ2 (Y759) and (B) PKC substrates (S/T) was measured by western blotting. (C) Representative western blot images for PLCγ2 (Y759) and S/T phosphorylated PKC substrates. (D) Cytosolic Ca2+ following stimulation with 1 µg/mL CRP-XL was measured in fura-2 loaded platelets in real time for 5 minutes. Points represent the mean response relative to vehicle ± SEM. Representative [Ca2+]i traces following incubation with (E) acalabrutinib or (F) ibrutinib.
Acalabrutinib inhibits granule secretion and integrin αIIbβ3 activation less potently than ibrutinib
Secretion of α-granules was investigated by measuring exposure of P-selectin on the platelet surface following stimulation with CRP-XL. P-selectin exposure was more potently inhibited by ibrutinib (logIC50 = −6.5M ± 0.04) than by acalabrutinib (logIC50 = −5.9M ± 0.11; Figure 4A). Binding of fluorescently labeled fibrinogen was used as measure of integrin αIIbβ3 activation following stimulation with CRP-XL (Figure 4B). Fibrinogen binding was inhibited more potently by ibrutinib (logIC50 = −6.5M ± 0.04) than by acalabrutinib (logIC50 = −5.8M ± 0.07). Phosphorylation of the integrin β3 subunit is a critical event in the initiation of integrin αIIbβ3 outside-in signaling. Phosphorylation of β3 Y773 (corresponding to Y747 in mouse) was potently inhibited by both ibrutinib (logIC50 = −6.7M ± 0.13) and acalabrutinib (logIC50 = −6.6M ± 0.11. Figure 4C). Acalabrutinib appeared to be only a partial inhibitor of β3 Y773 phosphorylation, because even at the highest concentration tested, a reduction to only 27% ± 2.5 relative to vehicle was achieved, whereas ibrutinib caused inhibition to 6% ± 1.0 of vehicle.
Figure 4.

Acalabrutinib is a less potent inhibitor of integrin αIIbβ3activation and granule secretion than ibrutinib. Washed human platelets from healthy donors were preincubated with a range of acalabrutinib or ibrutinib concentrations and then stimulated with 1 µg/mL CRP-XL for 20 minutes. (A) P-selectin and (B) fibrinogen binding was measured by fluorescence-activated cell sorting. (C) Platelets were stimulated with 1 µg/mL CRP-XL for 3 minutes at 37°C. Phosphorylation of β3 (Y773) was measured by western blotting. Points represent mean responses relative to vehicle ± SEM. (D) Representative β3 (Y773) blots.
Acalabrutinib avoids dysfunctional thrombus formation caused by ibrutinib
Given the differences in the effects of the drugs on adhesion to collagen compared with GPVI-mediated aggregation, we compared the effects of the drugs on platelet adhesion and thrombus formation in collagen-coated flow chambers under arterial shear conditions. After treating whole blood from healthy donors with either 1 µM ibrutinib or acalabrutinib for 20 minutes, ibrutinib caused reduced thrombus formation (1 µM ibrutinib: 383AU ±4 0.1, vehicle 585AU ± 67.6, P < .05, Student t test), whereas in the presence of vehicle or acalabrutinib, thrombus formation was not significantly different (Figure 5A-B). The most striking difference between ibrutinib and acalabrutinib treatment was the lack of stable, retracted thrombi after ibrutinib treatment (Figure 5A). In the presence of vehicle or acalabrutinib, platelets accumulated on collagen fibers while continuously contracting into dense thrombi, whereas ibrutinib prevented retraction of platelet aggregates, which consequently remained unstable, appeared loose, and were prone to disaggregation (supplemental Video 1). The effects of 1 µM ibrutinib and acalabrutinib on several important signaling and functional measurements are summarized in Figure 5B.
Figure 5.

Ibrutinib, but not acalabrutinib, causes thrombus instability. Whole blood from healthy donors was incubated with vehicle, 1 µM ibrutinib, and 1 µM acalabrutinib for 20 minutes and flowed through collagen-coated microfluidic flow chambers at a shear rate of 1000 s−1 for 6 minutes. (A) Representative images of thrombi formed under each of the conditions tested at 2-minute intervals. (B) A summary of effects of 1 µM ibrutinib and 1 µM acalabrutinib on thrombus formation on collagen and other platelet signaling and functional measurements relative to vehicle-treated control. Bars represent the mean ± SD; P values were calculated using multiple tests with Holm-Sidak correction for multiple comparisons.
To test whether experiments performed using blood from healthy donors matched the effects of drugs at therapeutic concentrations, we performed measurements of thrombus formation on collagen under arterial shear using blood samples from healthy donors, Btk-inhibitor naïve patients, or patients receiving ibrutinib or acalabrutinib (Table 1). Blood from acalabrutinib patients formed morphologically normal thrombi on collagen (Figure 6A), although heterogeneity in platelet counts strongly influenced thrombus volume (Figure 6C). Thrombus volume in acalabrutinib patients with platelet counts >100 × 109/L (78 895 µm3 ± 16 258) were not significantly different to those of Btk-inhibitor naïve patients (75 608 µm3 ± 19 995, P > .05, Student t test). In contrast, thrombus volume for ibrutinib patients was highly variable and did not correlate with platelet count (Figure 6D). Images of thrombi measured in samples from patients with platelet counts >150 × 109/L are presented in Figure 6A. Samples from 2 of the ibrutinib patients exhibited very pronounced dysfunction whereby platelets formed a loose covering of the majority of the surface of the flow chamber without forming stable thrombi (“large”; Figure 6A), whereas others formed small thrombi (“small”; Figure 6A).
Figure 6.

Thrombus formation on collagen correlates with platelet count during treatment with acalabrutinib but not with ibrutinib. (A) Blood from healthy donors (n = 6), Btk-inhibitor naïve CLL patients (n = 5), patients receiving ibrutinib (n = 6) or acalabrutinib (n = 8) was perfused through collagen-coated microfluidic flow chambers at room temperature at a shear rate of 1000 s−1 before being fixed with 10% formyl saline. Fixed samples were stained with DioC6 and z-stack images acquired to enable estimation of thrombus volume. (A) Representative images of thrombi in blood from a healthy donor and patients in the presence or absence of Haemate P (intermediate factor VIII) added ex vivo. Thrombus volume plotted against platelet count for (B) Btk inhibitor naïve CLL patients; patients treated with (C) acalabrutinib or (D) ibrutinib with or without added Haemate P. Correlation between platelet count and thrombus volume is significant in the presence or absence of Haemate P for acalabrutinib patients (significantly nonzero, P ≤ .05) but nonsignificant for ibrutinib patients (P > .05).
Addition of Haemate P ex vivo to patients’ blood improves platelet function
Thrombus formation assays using patients’ blood were performed in the presence or absence of 2 units/mL of Haemate P. Correlation of thrombus volume with platelet count was not significant within the Btk-inhibitor naïve patient group because platelet counts were all >100 × 109/L and thrombus volume was fairly uniform. The correlation was significant in patients treated with acalabrutinib in the presence and absence of Haemate P (Figure 6B), but not in patients treated with ibrutinib (Figure 6C), despite half of the patients having platelet counts <100 × 109/L, which strongly influences thrombus volume. Addition of Haemate P significantly increased thrombus volume in healthy donors and patients treated with acalabrutinib or ibrutinib (P < .05, 2-way analysis of variance (ANOVA) with repeated measures and the Sidak multiple comparisons test; Figure 7).
Figure 7.

Platelet function is improved by ex vivo addition of Haemate P. Comparison of the volume of thrombi formed in the presence or absence of Haemate P added ex vivo to blood from healthy donors (n = 6), Btk inhibitor naïve CLL patients (n = 5), or patients receiving ibrutinib (n = 6) or acalabrutinib (n = 8, as described in Figure 7). Two-way ANOVA with repeated measures and Sidak multiple comparisons test.
Discussion
Ibrutinib has proven to be an effective treatment of low-grade NHL but is associated with an increased risk of bleeding, whereas the effects of the second-generation Btk inhibitor, acalabrutinib, on platelet signaling and function have not been studied in detail. Ibrutinib has been shown to inhibit platelet aggregation evoked by GPVI receptor agonists,9-11 but the effects of acalabrutinib therapy on platelet aggregation have not been reported. We found that aggregation evoked by collagen and CRP-XL was either partially or completely inhibited in all patients receiving ibrutinib or acalabrutinib (Figure 1). Peak plasma concentrations of ibrutinib and acalabrutinib have been estimated at approximately 300 nM and 1.7 µM, respectively, in vivo,8,36 but our characterization of the concentration-response profile of both drugs in vitro (Figure 1D-E) indicated that a concentration of acalabrutinib corresponding to 3 to 10 µM would be required to achieve the level of inhibition of CRP-XL evoked signaling observed in patients. For this reason, we concluded that the apparent potency of acalabrutinib when measured by treatment of platelets in vitro was lower than that observed in patients. We hypothesize that accumulation of the drugs may occur in platelets because of their irreversible mode of actions and lack of protein turnover in platelets.
Although we found that ibrutinib and acalabrutinib inhibited Btk activity with similar potency, our study of kinase activation downstream of GPVI identified differences in the way the drugs affected SFK and Tec (Figure 2). Although the specificity of both kinases has been characterized in vitro8,33 and ibrutinib has been shown to inhibit SFK and Btk in platelets,9,11 a comprehensive comparison of ibrutinib and acalabrutinib has not previously been reported. We found that activation of Src was potentiated by acalabrutinib and that the concentration-response profile mirrored the inhibition curve for Btk Y223, suggesting that Btk mediates negative feedback regulation on SFK when active. At intermediate concentrations of acalabrutinib, Tec activation was similarly increased, likely as a direct result of the potentiation of Src, which is known to regulate activation of Tec family kinases.37 Ibrutinib caused potentiation over a narrow concentration range because direct inhibition of Tec and Src occurred at intermediate and high concentrations.
Our in vitro experiments indicate that only concentrations of acalabrutinib that inhibit Tec activation (3-10 µM; Figure 2) are able to ablate platelet aggregation evoked by CRP-XL (Figure 1D). Therefore, our observation that CRP-XL–evoked aggregation was strongly inhibited during acalabrutinib therapy (Figure 1) suggested that therapeutic doses of acalabrutinib must inhibit Tec, despite the improvement in selectivity relative to ibrutinib. However, we went on to demonstrate both in vitro and with blood samples from patients receiving ibrutinib or acalabrutinib that only ibrutinib causes severe platelet dysfunction measured by thrombus formation on collagen under conditions of arterial shear (Figure 7). Taken together, these data suggest that Tec family kinase activity is required for CRP-XL–mediated platelet aggregation but is redundant for adhesion and thrombus formation on collagen, which is instead dependent on SFK. We believe that this important distinction underlies the absence of severe platelet dysfunction in patients receiving acalabrutinib and that greater selectivity for Tec family kinases over SKF is likely to be the cause of this difference. Our observation that acalabrutinib did not inhibit SFK, and in fact caused potentiation of SFK activation (Figure 2), may be an important factor in compensating for the loss of Btk kinase activity.
We found that GPVI-mediated platelet aggregation and signaling still occurs when Btk activity is absent, because several functional and signaling events occur at near vehicle-treated levels following treatment with 1 µM acalabrutinib, a concentration that ablates Btk activity (Figure 5). Our data suggest that this may be a direct result of potentiation of signaling through Tec and SFK, which enable Tec to mediate normal levels Ca2+ signaling, PKC and integrin αIIbβ3 activation, and α-granule secretion despite being expressed at approximately eightfold lower levels than Btk in platelets.38 This potentiation mechanism has not previously been reported and may also explain why bleeding risk is not increased in Btk-deficient XLA patients.3
We have previously reported that ibrutinib inhibits integrin αIIbβ3 outside-in signaling, clot retraction, and thrombus stability, attributing this observation to loss of Btk-mediated PLCγ2 activation downstream of integrin αIIbβ3.9 However, in the present study, we found that downstream of GPVI, phosphorylation of β3 Y773, which is the first step in outside-in signaling, was potently inhibited by ibrutinib and acalabrutinib and therefore is likely mediated, at least partially, by Btk (Figure 4). Inhibition of β3 Y773 phosphorylation occurs at concentrations of acalabrutinib that allow almost normal levels of [Ca2+]i signaling, fibrinogen binding, and aggregation, indicating that this is a direct consequence of Btk inhibition and not reduced integrin activation. Additionally, the inhibition profile of β3 Y773 matches that of PLCγ2 Y759, a known Btk substrate.39 However, we found that β3 Y773 phosphorylation was not sensitive to ibrutinib or acalabrutinib downstream of GPCR stimulation (unpublished observation). Mice with dual mutation of the corresponding tyrosine residue Y747 and also Y759 in the cytoplasmic tail of β3 are prone to rebleeding in tail bleeding experiments 16; therefore, inhibition of integrin αIIbβ3 signaling may contribute to the bleeding side effects caused by ibrutinib.
In healthy donors, only ibrutinib caused a reduction in platelet adhesion to collagen (Figure 5) and inhibited retraction of thrombi (supplemental Video 1). In samples from some patients, ibrutinib caused a striking lack of clot retraction, resulting in the formation of a layer of loosely packed platelets, whereas in others the size of the thrombi appeared reduced relative to Btk-inhibitor naïve patients with similar platelet counts (Figure 6). The reason for the variability between ibrutinib patients was unclear but may relate to differences in drug concentration or patient physiology; however, thrombus volume did not seem to correlate with sensitivity to CRP-XL measured by aggregometry. Treatment with acalabrutinib did not affect thrombus formation or retraction, however, and platelet count was the major determinant of thrombus volume in these patients (Figure 6).
We studied ex vivo addition of Haemate P to blood from patients with CLL or mantle cell leukemia receiving ibrutinib or acalabrutinib to approximate the increase in levels of VWF and FVIII that might be achieved following administration of desmopressin to a patient that expresses functional VWF, and measured thrombus formation on collagen (Figure 7). We found that Haemate P increased platelet adhesion to collagen in all patients, regardless of treatment type or platelet count. This finding suggests that agents that increase plasma levels of VWF may improve primary hemostasis in CLL patients at increased risk of bleeding either from thrombocytopenia or drug-induced platelet dysfunction. Platelet transfusion has been investigated as a means to overcome platelet dysfunction during ibrutinib therapy in patients at risk of bleeding.11,25 However, the safety and efficacy of platelet transfusion to reduce bleeding risk in other contexts is controversial and investigation of alternatives is ongoing.31 Desmopressin has been proven effective at improving primary hemostasis in a number of contexts, including platelet dysfunction27,30 and may act both via increasing VWF levels in plasma as well as directly affecting platelets.40 Although ibrutinib has been shown to inhibit adhesion to VWF-coated flow chambers,11 our experiments demonstrate that VWF still contributes to and enhances thrombus formation on collagen; therefore, our data suggest that this could still be a potentially efficacious treatment of patients treated with ibrutinib. Investigation of platelet function in patients that are currently experiencing bleeding during ibrutinib therapy as well as evaluation to improve platelet function could offer further insights into Btk-inhibitor induced platelet dysfunction and potential treatments. Safety improvements of successive generations of Btk inhibitors combined with a better understanding of treatment options for bleeding may further improve outcomes patients with NHL.
In conclusion, we found that Btk activity is redundant in the context of GPVI-mediated platelet aggregation if Tec is functional, but is lost if both tyrosine kinases are simultaneously inhibited. However, Btk and Tec are redundant for platelet adhesion and thrombus formation on collagen under flow, whereas SFK play a critical role and are inhibited by ibrutinib but not acalabrutinib. Loss of SFK function and inhibition of stable thrombus formation is likely to contribute to bleeding risk in ibrutinib treated patients, but we found evidence that platelet function could be improved by agents that increase plasma VWF levels.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors acknowledge the contribution to this study made by the Oxford Centre for Histopathology Research and the Oxford Radcliffe Biobank, which are supported by the National Institutes of Health Research Oxford Biomedical Research Centre.
Authorship
Contribution: A.P.B. designed research, performed experiments, analyzed data, and wrote the manuscript; A.J.U., N.K., S.H.N., and T.S. performed experiments; C.A.T.H., N.A., and D.B. coordinated collection of clinical samples, analyzed data, and revised the manuscript; and M.J.D., C.E.H., and J.M.G. analyzed data, revised the manuscript, and designed research.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jonathan M. Gibbins, Institute for Cardiovascular and Metabolic Research, School of Biological Sciences, Harborne Building, University of Reading, Whiteknights, Reading RG6 6AS, United Kingdom; e-mail: j.m.gibbins@reading.ac.uk.
References
- 1.Ponader S, Burger JA. Bruton’s tyrosine kinase: from X-linked agammaglobulinemia toward targeted therapy for B-cell malignancies. J Clin Oncol. 2014;32(17):1830-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shatzel JJ, Olson SR, Tao DL, McCarty OJT, Danilov AV, DeLoughery TG. Ibrutinib-associated bleeding: pathogenesis, management and risk reduction strategies. J Thromb Haemost. 2017;15(5):835-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Futatani T, Watanabe C, Baba Y, Tsukada S, Ochs HD. Bruton’s tyrosine kinase is present in normal platelets and its absence identifies patients with X-linked agammaglobulinaemia and carrier females. Br J Haematol. 2001;114(1):141-149. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Fitzgerald ME, Berndt MC, Jackson CW, Gartner TK. Bruton tyrosine kinase is essential for botrocetin/VWF-induced signaling and GPIb-dependent thrombus formation in vivo. Blood. 2006;108(8):2596-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood. 2003;102(10):3592-3599. [DOI] [PubMed] [Google Scholar]
- 6.Jones JA, Hillmen P, Coutre S, et al. . Use of anticoagulants and antiplatelet in patients with chronic lymphocytic leukaemia treated with single-agent ibrutinib. Br J Haematol. 2017;178(2):286-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barr PM, Brown JR, Hillmen P, et al. . Impact of ibrutinib dose adherence on therapeutic efficacy in patients with previously treated CLL/SLL. Blood. 2017;129(19):2612-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrd JC, Harrington B, O’Brien S, et al. . Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bye AP, Unsworth AJ, Vaiyapuri S, Stainer AR, Fry MJ, Gibbins JM. Ibrutinib inhibits platelet integrin αIIbβ3 outside-in signaling and thrombus stability but not adhesion to collagen. Arterioscler Thromb Vasc Biol. 2015;35(11):2326-2335. [DOI] [PubMed] [Google Scholar]
- 10.Kamel S, Horton L, Ysebaert L, et al. . Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015;29(4):783-787. [DOI] [PubMed] [Google Scholar]
- 11.Levade M, David E, Garcia C, et al. . Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood. 2014;124(26):3991-3995. [DOI] [PubMed] [Google Scholar]
- 12.Moroi M, Jung SM, Okuma M, Shinmyozu K. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J Clin Invest. 1989;84(5):1440-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi H, Moroi M. Antibody against platelet membrane glycoprotein VI in a patient with systemic lupus erythematosus. Am J Hematol. 2001;67(4):262-267. [DOI] [PubMed] [Google Scholar]
- 14.Kojima H, Moroi M, Jung SM, et al. . Characterization of a patient with glycoprotein (GP) VI deficiency possessing neither anti-GPVI autoantibody nor genetic aberration. J Thromb Haemost. 2006;4(11):2433-2442. [DOI] [PubMed] [Google Scholar]
- 15.López JA, Andrews RK, Afshar-Kharghan V, Berndt MC. Bernard-Soulier syndrome. Blood. 1998;91(12):4397-4418. [PubMed] [Google Scholar]
- 16.Law DA, DeGuzman FR, Heiser P, Ministri-Madrid K, Killeen N, Phillips DR. Integrin cytoplasmic tyrosine motif is required for outside-in alphaIIbbeta3 signalling and platelet function. Nature. 1999;401(6755):808-811. [DOI] [PubMed] [Google Scholar]
- 17.Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124(13):2013-2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wahl MI, Fluckiger AC, Kato RM, Park H, Witte ON, Rawlings DJ. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton’s tyrosine kinase via alternative receptors. Proc Natl Acad Sci USA. 1997;94(21):11526-11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z, Wahl MI, Eguinoa A, Stephens LR, Hawkins PT, Witte ON. Phosphatidylinositol 3-kinase-gamma activates Bruton’s tyrosine kinase in concert with Src family kinases. Proc Natl Acad Sci USA. 1997;94(25):13820-13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quintás-Cardama A, Kantarjian H, Ravandi F, et al. . Bleeding diathesis in patients with chronic myelogenous leukemia receiving dasatinib therapy. Cancer. 2009;115(11):2482-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quintás-Cardama A, Han X, Kantarjian H, Cortes J. Tyrosine kinase inhibitor-induced platelet dysfunction in patients with chronic myeloid leukemia. Blood. 2009;114(2):261-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gratacap MP, Martin V, Valéra MC, et al. . The new tyrosine-kinase inhibitor and anticancer drug dasatinib reversibly affects platelet activation in vitro and in vivo. Blood. 2009;114(9):1884-1892. [DOI] [PubMed] [Google Scholar]
- 23.Byrd JC, Furman RR, Coutre SE, et al. . Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125(16):2497-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kazianka L, Drucker C, Skrabs C, et al. . Ristocetin-induced platelet aggregation for monitoring of bleeding tendency in CLL treated with ibrutinib. Leukemia. 2017;31(5):1117-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seiter K, Stiefel MF, Barrientos J, et al. . Successful treatment of ibrutinib-associated central nervous system hemorrhage with platelet transfusion support. Stem Cell Investig. 2016;3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baharoglu MI, Cordonnier C, Al-Shahi Salman R, et al. ; PATCH Investigators. Platelet transfusion versus standard care after acute stroke due to spontaneous cerebral haemorrhage associated with antiplatelet therapy (PATCH): a randomised, open-label, phase 3 trial. Lancet. 2016;387(10038):2605-2613. [DOI] [PubMed] [Google Scholar]
- 27.Desborough MJ, Oakland KA, Landoni G, et al. . Desmopressin for treatment of platelet dysfunction and reversal of antiplatelet agents: a systematic review and meta-analysis of randomized controlled trials. J Thromb Haemost. 2017;15(2):263-272. [DOI] [PubMed] [Google Scholar]
- 28.American Society of Anesthesiologists Task Force on Perioperative Blood Management. Practice guidelines for perioperative blood management: an updated report by the American Society of Anesthesiologists Task Force on Perioperative Blood Management*. Anesthesiology. 2015;122(2):241-275. [DOI] [PubMed] [Google Scholar]
- 29.Frontera JA, Lewin JJ III, Rabinstein AA, et al. . Guideline for reversal of antithrombotics in intracranial hemorrhage: a statement for healthcare professionals from the Neurocritical Care Society and Society of Critical Care Medicine. Neurocrit Care. 2016;24(1):6-46. [DOI] [PubMed] [Google Scholar]
- 30.Svensson PJ, Bergqvist PB, Juul KV, Berntorp E. Desmopressin in treatment of haematological disorders and in prevention of surgical bleeding. Blood Rev. 2014;28(3):95-102. [DOI] [PubMed] [Google Scholar]
- 31.Desborough MJ, Smethurst PA, Estcourt LJ, Stanworth SJ. Alternatives to allogeneic platelet transfusion. Br J Haematol. 2016;175(3):381-392. [DOI] [PubMed] [Google Scholar]
- 32.Lordkipanidzé M, Lowe GC, Kirkby NS, et al. UK Genotyping and Phenotyping of Platelets Study Group. Characterization of multiple platelet activation pathways in patients with bleeding as a high-throughput screening option: use of 96-well Optimul assay. Blood. 2014;123(8):e11-e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Honigberg LA, Smith AM, Sirisawad M, et al. . The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107(29):13075-13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunster JL, Mazet F, Fry MJ, Gibbins JM, Tindall MJ. Regulation of early steps of GPVI signal transduction by phosphatases: a systems biology approach. PLOS Comput Biol. 2015;11(11):e1004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nore BF, Mattsson PT, Antonsson P, et al. . Identification of phosphorylation sites within the SH3 domains of Tec family tyrosine kinases. Biochim Biophys Acta. 2003;1645(2):123-132. [DOI] [PubMed] [Google Scholar]
- 36.Advani RH, Buggy JJ, Sharman JP, et al. . Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000;19(49):5651-5661. [DOI] [PubMed] [Google Scholar]
- 38.Burkhart JM, Vaudel M, Gambaryan S, et al. . The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73-e82. [DOI] [PubMed] [Google Scholar]
- 39.Kim YJ, Sekiya F, Poulin B, Bae YS, Rhee SG. Mechanism of B-cell receptor-induced phosphorylation and activation of phospholipase C-gamma2. Mol Cell Biol. 2004;24(22):9986-9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colucci G, Stutz M, Rochat S, et al. . The effect of desmopressin on platelet function: a selective enhancement of procoagulant COAT platelets in patients with primary platelet function defects. Blood. 2014;123(12):1905-1916. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.