

Abstract
Glioblastoma Multiforme (GBM), a malignant brain tumor, is among the most lethal of all cancers. Temozolomide is the primary chemotherapy treatment for patients diagnosed with GBM. The methylation status of the promoter or the enhancer regions of the O6− methylguanine methyltransferase (MGMT) gene may impact the efficacy and sensitivity of temozolomide, and hence may affect overall patient survival. Microscopic genetic changes may manifest as macroscopic morphological changes in the brain tumors that can be detected using magnetic resonance imaging (MRI), which can serve as noninvasive biomarkers for determining methylation of MGMT regulatory regions. In this research, we use a compendium of brain MRI scans of GBM patients collected from The Cancer Imaging Archive (TCIA) combined with methylation data from The Cancer Genome Atlas (TCGA) to predict the methylation state of the MGMT regulatory regions in these patients. Our approach relies on a bi-directional convolutional recurrent neural network architecture (CRNN) that leverages the spatial aspects of these 3-dimensional MRI scans. Our CRNN obtains an accuracy of 67% on the validation data and 62% on the test data, with precision and recall both at 67%, suggesting the existence of MRI features that may complement existing markers for GBM patient stratification and prognosis. We have additionally presented our model via a novel neural network visualization platform, which we have developed to improve interpretability of deep learning MRI-based classification models.
Keywords: Deep learning, convolutional neural networks, MRI data, network visualization, Glioblastoma Multiforme
1. Introduction
Glioblastoma multiforme (GBM) is an aggressive brain cancer, with a median survival of only 15 months.1 The efficacy of the first-line chemotherapy treatment, temozolomide, is in part dependent on the methylation status of the O6-methylguanine methyltransferase (MGMT) regulatory regions (promoter and/or enhancer). MGMT removes alkyl groups from compounds and is one of the few known proteins in the DNA Direct Reversal Repair pathway.2 Loss of the MGMT gene, or silencing of the gene through DNA methylation, may increase the carcinogenic risk after exposure to alkylating agents. Similarly, high levels of MGMT activity in cancer cells create a resistant phenotype by blunting the therapeutic effect of alkylating agents and may be an important determinant of treatment failure.3 Thus, methylation of MGMT increases efficacy of alkylating agents such as temozolomide.1
As such, methylation status of MGMT regulatory regions has important prognostic implications and can affect therapy selection in GBM. Currently, determining the methylation status is done using samples obtained from fine needle aspiration biopsies, which is an invasive procedure. However, several studies have demonstrated that some genetic changes can manifest as macroscopic changes, which can be detected using magnetic resonance imaging (MRI).4,5 Previous approaches have constructed models to predict MGMT status from imaging and clinical data.6,7 However, these models typically rely on hand curated features with classifiers such as SVM and random forests, and using neural networks may enable the discovery of novel biological features and increase the ease of implementation of such models.
Recently, convolutional neural networks (CNNs), a class of deep, feed-forward artificial neural networks, have emerged to be effective for autonomous feature extraction and have excelled at many image classification tasks.8 A CNN consists of one or more convolutional layers, each layer composed of multiple filters. The architecture of a CNN captures different features (edges, shapes, texture, etc.) by leveraging the 2-dimensional spatial structure of an image using these filters. On the other hand, recurrent neural networks have shown a lot of promise to analyze ordered sequences of words or image frames, such as sentences or videos, for tasks such as machine translation, named entity recognition and classification.9 Using fixed weight matrices (often termed, memory units) and vectorial representations for each sequence item (e.g. a word or a frame), an RNN can capture the temporal context in a dataset. While developing and implementing these neural network models may be inherently difficult, they can directly work on atomic features (e.g. pixels of an image, words in a sentence), and do not require exhaustive feature curation, as required in conventional machine learning methods.
Since MRI scans are 3-dimensional reconstruction of the human brain, they can be treated as volumetric objects or videos. Volumetric objects and sequences of image frames can be analyzed effectively by combining convolutional and recurrent neural networks.10,11 However, few methods that combine CNNs with RNNs using end-to-end learning have been applied to radio-genomic analyses. Constructing an architecture that combines CNN and RNN for powerful image analysis while maintaining information transfer between image slices may reveal novel features that are associated with MGMT methylation.
In this work, we present an approach using a bi-directional convolutional recurrent neural network (CRNN) architecture on brain MRI scans to predict the methylation status of MGMT. We use a dataset of 5,235 brain MRI scans of 262 patients diagnosed with glioblastoma multiforme from The Cancer Imaging Archive (TCIA).12,13 Genomics data corresponding to these patients is retrieved from The Cancer Genome Atlas (TCGA).14 The CNN and RNN modules in the architecture are jointly trained in an end-to-end fashion. We evaluate our model using accuracy, precision, and recall. We also develop an interactive visualization platform to visualize the output of the convolutional layers in the trained CRNN network. The results of our study, as well as the visualizations of the MRI scans and the CRNN pipeline can be accessed at http://onto-apps.stanford.edu/m3crnn/.
1.1. Deep learning methods over biomedical data
Recently, several variations of deep learning architectures (neural networks, CNNs, RNNs, etc.) have been introduced for the analysis of imaging data, -omics data and biomedical literature.15,16 A recent study by Akkus et al. has used CNNs to extract features from MRI images and predict chromosomal aberrations.17 2-dimensional and 3-dimensional CNN architectures have been used to determine the most discriminative clinical features and predict Alzheimer’s disease using brain MRI scans.10,18 Poudel et al. have developed a novel recurrent fully-connected CNN to learn image representations from cardiac MRI scans and leverage inter-slice spatial dependences through RNN memory units. The architecture combines anatomical detection and segmentation, and is trained end-to-end to reduce computational time.11 For tumor segmentation, Stollenga et al. developed a novel architecture, PyramidL-STM, to parallelize multi-dimensional RNN memory units, and leverage the spatial-temporal context in brain MRI scans that is lost by conventional CNNs.19 Chen et al. developed a transferred-RNN, which incorporates convolutional feature extractors and a temporal sequence learning model, to detect fetal standard plane from ultrasound videos. They implement end-to-end training and knowledge transfer between layers to deal with limited training data.20 Kong et al. combined an RNN with a CNN, and designed a new loss function, to detect the end-diastole and end-systole frames in cardiac MRI scans.21
2. Methods
2.1. Dataset and Features
We used the brain MRI scans of glioblastoma multiforme (GBM) patients from The Cancer Imaging Archive (TCIA) and the methylation data, for those corresponding patients, from The Cancer Genome Atlas (TCGA).
2.1.1. Preprocessing of Methylation Data
We downloaded all methylation data files from GBM patients available via TCGA. The methylation consisted of 423 unique patients, with 16 patients having duplicate samples. We extracted methylation sites that are located in the minimal promoter and enhancer regions shown to have maximal methylation activity and affect MGMT expression.22–24 Specifically, these methylation sites are cg02941816, cg12434587, and cg12981137. These are the same sites used in previous MGMT methylation studies that use TCGA data.25 Similar to Alonso et al., we considered a methylation beta value of at least 0.2 to be a positive methylation site. As methylation of either the minimal promoter or the enhancer were shown to decrease transcription, we considered a patient to have a positive methylation status if any of the three sites were positive.
2.1.2. Preprocessing of the MRI scans
We downloaded 5,235 MRI scans for 262 patients diagnosed with GBM from TCIA. Each brain MRI scan can be envisioned as a 3-dimensional reconstruction of the brain (Figure 1). Each MRI scan consists of a set of image frames captured at a specific slice thickness and pixel spacing (based on the MRI machine specifications). The raw dataset contained a total of 458,951 image frames. From these, we selected ‘labeled’ T1/T2/Flair axial MRI scans for those patients for whom we had corresponding methylation data.
Fig. 1. MRI scan.
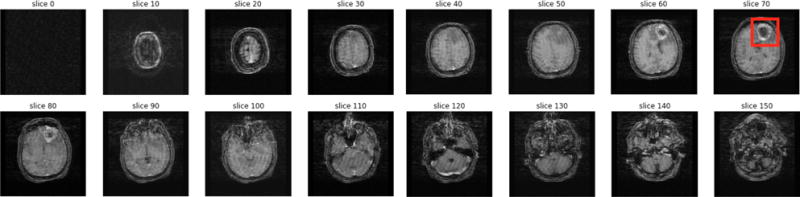
A visualization of different MRI image frames in one MRI scan, with the GBM tumor highlighted in red on slice 70.
These image frames are made available in a DICOM format (Digital Imaging and Communications in Medicine), a non-proprietary data interchange protocol, digital image format, and file structure for biomedical images and image-related information.26 The image frames are grayscale (1-channel) and the DICOM format allows storage of other patient-related meta-data (sex, age, weight, etc.) as well as image-related metadata (slice thickness, pixel spacing etc.). As these image frames may be generated by different MRI machines with varying slice thickness (range: 1 to 10) and pixel spacing, we normalize these attributes across different MRI scans by resampling to a uniform slice thickness of 1.0 and pixel spacing of [1, 1].
MRI image frames are grayscale, and instead of RGB channel values, each pixel is assigned a numerical value termed the Hounsfield Unit (HU), which is a measure of radiodensity. We filter out those image frames that are “noisy” by looking at the distribution of Hounsfield Units in the pixels. When removing noisy images, we used mean and standard deviation thresholds of 20 HU to determine image validity. An example of the distributions and the images are shown in Figure 2. We further limit our MRI scans to only those slices that contain the tumor to the nearest 10th slice. This was achieved by annotating the MRI scans through our visualization platformb. Finally, we resize all images to 128 × 128 dimensions.
Fig. 2. Removing noisy images.
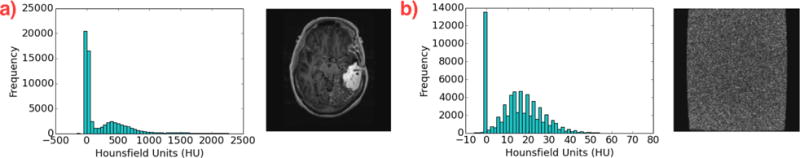
We use the distributions of Hounsfield units (which vary drastically) to determine if an image is a valid MRI scan (a), or has only noisy pixels (b).
2.2. Data Augmentation
For our CRNN, we used data augmentation to increase the size of our dataset and to help combat overfitting. Specifically, we applied image rotation and MRI scan reversal, so that the methylation status and location of the tumor is preserved. Images were rotated every 4 degrees from -90 to +90 degrees, and were flipped such that in the RNN, the MRI scans were represented from superior to inferior and vice versa. This resulted in a 90 fold increase in the number of MRI scans.
2.3. Training and Evaluation
Given that our MRI scans are similar to video objects with a variable number of frames, we implemented a bi-directional convolutional recurrent neural network (CRNN) architecture (Figure 3). Each image frame of the MRI scan is first input into a CNN. Multiple convolutional layers extract essential features (e.g. shape, edges, etc.) from the image. The image is then processed through two fully connected neural network layers, so that the output from each image is a vector of length 512. All frames from one MRI scan are then represented by a series of vectors, which are input into a many-to-one bi-directional RNN. The bi-directional RNN is dynamic and can adjust for variable-length sequences, an advantage over using 3-dimensional CNN, which requires uniform volumes. Padding and bucketing of MRI scans of similar length was carried out for efficient computation. The RNN analyzes the sequence of MRI image frames and outputs a binary classification of methylation status per MRI scan. The entire architecture was developed using the Tensorflow Python libraryc.
Fig. 3. CRNN Architecture Overview.
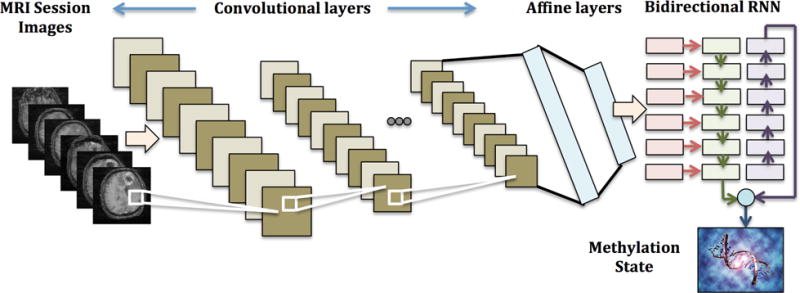
Combining CNN and RNN to predict the methylation state from MRI scan images.
We split our the MRI scans into a 70% training set, 15% validation set, and 15% test set. As MRI scans of the same patient are highly correlated, we split our data such that all MRI scans pertaining to each patient are in the same set. We randomized the order of the training data based on the number of frames, bucketing MRI scans with similar frame numbers. We padded MRI scans within each bucket so all MRI scans in each batch had the same number of frames, while the number of frames differed across batches. We trained using softmax cross entropy as our loss function using the Adam optimizer with learning rates ranging from 5e-6 to 5e-1. We applied L2 regularization, with coefficients from 0.001 to 0.1 and dropout with keep probabilities ranging from 0.5 to 1. We varied the number of filters between 8 and 16, and trained our model until it converged, for ten epochs.
For comparison, we also implemented a random forest classifier, to evaluate how our CRNN performs in comparison to alternative, more conventional machine learning algorithms that do not capture spatial information. For our random forest classifier, each frame was considered one sample, where each pixel was one feature. Each MRI scan was treated as an ensemble of individual frames, where we averaged the prediction across all frames for each scan.
When assessing our results, we calculated the area under the receiver operator characteristic curve (AUC), accuracy, precision, and recall at the patient and MRI scan levels. We calculated methylation status probability as the proportion of positive individual MRIs. Out of these metrics, we used patient level accuracy in the validation set to tune our architecture and hyperparameters. The CRNN was then evaluated using the independent test set.
3. Results
3.1. Data Statistics
Our training dataset consisted of 344 positive MRI scans and 351 negative scans, which corresponded to 117 patients. Our validation dataset consisted of 21 patients, with 73 positive scans and 62 negative scans. Our test set also had 21 patients, with 62 positive and 62 negative scans. After data augmentation, this resulted in 62,550 examples in the training set, and 12,150 in the validation set, and 11,160 in the test set. After preprocessing, we had an average of 45.9 frames per scan in the training set, 52.7 frames per scan in the validation set, and 43.2 frames per scan in the test set.
3.2. Architecture and Hyperparameters
The specific architecture of our CRNN is detailed in Table 1. Our architecture consisted mainly of alternating convolutional and pooling layers, using the rectified linear unit (ReLU) as our activation function. We used batch normalization, and we implemented L2 regularization and drop out layers to limit overfitting. We then followed these layers with fully connected (FC) layers to create the output for the RNN, which contained 512 neurons. We implemented a bi-directional RNN with gated recurrent units (GRU), with a state size of 256. We then followed the RNN with an additional FC layer before using the softmax classifier to predict methylation status. We used the Adam optimizer, with a learning rate of 1e-5.
Table 1. Bi-directional CRNN Architecture.
Convolutional layers followed by fully connected layers and a many-to-one bi-directional RNN.
| Layers | Hyperparmaeters |
|---|---|
| [5×5 Conv-ReLU-BatchNorm-Dropout-2×2 Max Pool] × 2 [5×5 Conv-ReLU-BatchNorm-Dropout] × 1 [5×5 Conv-ReLU-BatchNorm-Dropout-2×2 Max Pool] × 1 |
L2 Regularization: 0.05 Dropout Keep Probability: 0.9 Number of Filters: 8 |
| FC-ReLU-BatchNorm-Dropout | Number of Neurons: 1024 L2 Regularization: 0.05 Dropout Keep Probability: 0.9 |
| FC-ReLU-BatchNorm-Dropout | Number of Neurons: 512 L2 Regularization: 0.05 Dropout Keep Probability: 0.9 |
| Bi-directional GRU with ReLU-Dropout | State Size: 256 L2 Regularization: 0.05 Dropout Keep Probability: 0.9 |
| FC-ReLU | Number of Neurons: 256 L2 Regularization: 0.05 Dropout Keep Probability: 0.9 |
| Softmax |
3.3. Evaluation
For CRNN, our test set results are shown in Table 2. At the patient level, the test data yielded an accuracy of 0.61, with a precision of 0.67 and recall of 0.67. ROC curves are shown at the MRI scan and the patient level in Figure 4a. The training data obtained accuracies of 0.97 for MRI scans and at the patient level. Though we observe overfitting, increasing the dropout probability, increasing the L2 regularization coefficient, and decreasing model complexity did not result in significant gains in validation accuracy during model tuning. In comparison, our random forest classifier achieved an AUC of 0.56 on the validation set and 0.44 on the test set at the patient level.
Table 2.
CRNN Performance Metrics for test, validation, and training sets at the patient and MRI scan level.
| Set | Level | AUC | Accuracy | Precision | Recall |
|---|---|---|---|---|---|
| Test | Patient | 0.61 | 0.62 | 0.67 | 0.67 |
| MRI Scan | 0.73 | 0.63 | 0.72 | 0.42 | |
|
| |||||
| Validation | Patient | 0.66 | 0.67 | 0.67 | 0.73 |
| MRI Scan | 0.54 | 0.53 | 0.57 | 0.55 | |
Fig. 4. Evaluation of the CRNN method.
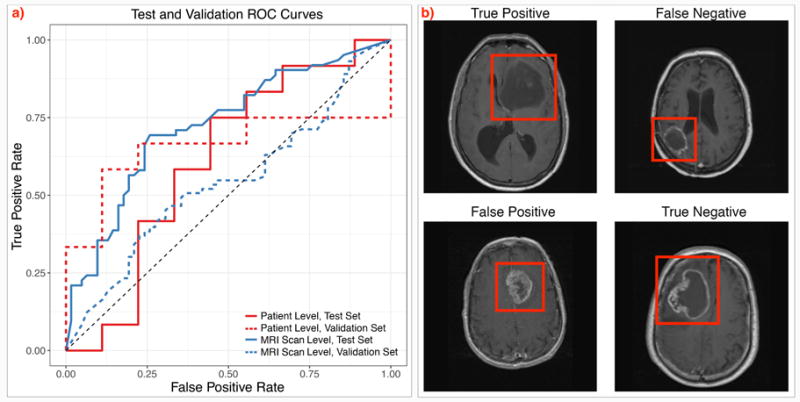
a) ROC curves depicting results at the patient and MRI scan levels in the validation and held-out test set, and b) Classifier prediction examples. True positive, true negative, and misclassified false positive and false negative examples from our test set. The tumors are highlighted in the red boxes.
We examined our classifier predictions in the test set, and show examples of true and false positives and negatives in Figure 4b. In particular, it appears that our classifier tends to classify lesions with ring enhancement as having a negative methylation status, and tumors with less clearly defined borders as positive. Predicted positive tumors also tended to have a more heterogeneous texture in appearance. Tumor location varied, and did not appear to be correlated with methylation status prediction.
3.4. Visualization
Deep learning methods, especially convolutional and recurrent neural networks, are thought to be less interpretable and clinically reliable, as compared to standard machine learning models. To provide a more visual perspective on how our model perceives the input MRI scan, we have developed an interactive, online visualization interface deployed at http://onto-apps.stanford.edu/m3crnn/. The domain user (e.g. a radiologist or a biomedical researcher) can select an MRI scan from a list and load it through the pre-trained CRNN pipeline (Figure 5a). Once the pipeline completes the computation, the user can visualize the original MRI scan, click on each filter in each CRNN layer to see the output from each filter in each convolutional layer (Figure 5b). The user can also visualize the output after applying the ReLU activation function. Each visualization (either MRI scan, filter output or ReLU output) opens up in its own separate dialog window that can be dragged around the browser. Hence, multiple visualizations can be compared with each other (Figure 5c–e). Finally, the predicted output, the probability score as well as the actual methylation status, are also presented for the domain user to determine features and flaws of our model.
Fig. 5. CRNN Visualization Interface.
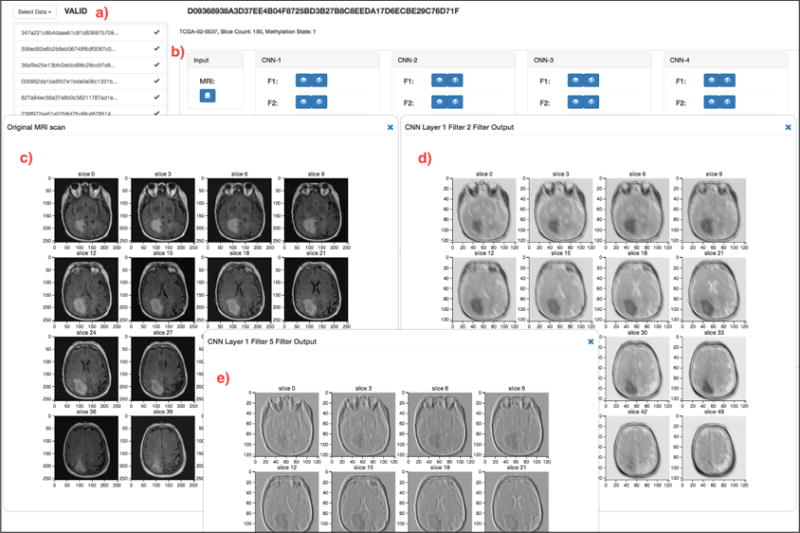
(a) The domain user can select any MRI scan to load into the Tensorflow CRNN pipeline. (b) After the pipeline completes the computation to predict the MGMT methylation status, the user can visualize the original MRI Scan (c), the output from any filter, in each convolutional layer (d, e), as well as the output after ReLU activation.
The output from two filters in the first convolutional layer are visualized (Figure 5d,e). As with many CNN architectures, the first layer places a heavy emphasis on edge detection, and we can clearly see the outline of the cranium and the tumor in each of these filters. Each filter also appears to show the brain slice at different contrasts. As specific tissues attenuate signal differently, in some sense these filters may be attempting to highlight different tissue types by varying the contrast. To the best of our knowledge, this is the first example of an online, interactive interface that can execute a deep learning pipeline over any selected MRI scan and can visualize intermediate layer outputs. It is very flexible, in the sense that the interface can easily be configured for variable number of convolutional layers and filters.
4. Discussion
In this work, we constructed a jointly trained, bi-directional convolutional recurrent neural network in order to predict the methylation status of MGMT from brain MRI scans. We explore macroscopic MRI features that may be correlated with MGMT methylation status to gain insight into GBM pathology. We use the publicly available data in TCGA and TCIA, where few studies, if any, have combined imaging data with -omics data using a deep learning framework. In addition, we present a generalizable platform for visualizing the different filters and layers of deep learning architectures for brain MRI scans to aid model interpretability for clinicians and biomedical researchers.
Our CRNN obtains modest patient level accuracies of 0.67 and 0.62 on the validation and test data, respectively, and on the test data, the precision and recall were both 0.67. Our data contained approximately equal proportions of positive and negative patients, indicating that our classifier is making predictions to balance precision and recall, and not relying on label distributions. Though the patient level performance does decrease from the validation to the test data set, the general similarity in performance indicates there are likely a subset of features that are correlated with MGMT methylation, as has been found in previous studies.27,28 In comparison, the random forest model had an AUC of 0.57 in the validation set and 0.44 in the test set (versus CRNN with a validation AUC of 0.66 and test AUC of 0.61). This suggests that there is some useful information encoded in the individual pixels, but that reproducibility and performance are likely improved by using a method that can better capture spatial information.
We focused primarily on patient level results, leveraging multiple MRI scans per patient to obtain a prediction in an ensemble style. We secondarily assessed MRI scan results, as being able to predict methylation status from a single MRI scan would be highly relevant to clinicians and patients. The results at the MRI scan level were comparable to the patient level in the test set, but we see a decrease in performance in the validation set. This is likely due to our classifier being less confident at the MRI scan level, resulting in greater variability in results and prediction probabilities further from 0 or 1.
The difference in confidence between the patient level and MRI scan results suggests that combining information from multiple MRI scans is beneficial for MGMT methylation prediction. Deep learning models have been able to successfully learn multiple representations of the same object in other classification tasks.8,9 However, we believe that combining different representations of the same tumor to reach a prediction per patient is more robust and clinically relevant. We accomplish this using majority voting. Incorporating additional layers into our model to combine MRI scans may also lead to further improvement in performance.
With a training set accuracy of nearly 1.0, our classifier is overfitted to the training set. To combat overfitting, we implemented L2 regularization, dropout layers, and data augmentation. Regularization had only a modest effect at curtailing overfitting and improving performance, and further increases in regularization resulted in decreasing validation set performance. Even though data augmentation was able to greatly decrease the speed of model overfitting, we still reach nearly perfect classification given enough training epochs. Data augmentation also substantially increased the number and variability of images for training, improving the robustness and performance of our model. However, due to the limited availability of publicly accessable patient data with both imaging and -omics measurements, our overall dataset of 159 patients can still be considered to be very small. The incorporation of additional patient data holds potential for further reduction of model variance and overfitting.
Currently, methylation status is not readily discernible by a human radiologist from MRI scans, even though multiple previous studies have attempted to correlate features to discover imaging-based biomarkers.6,27–29 These studies typically require extensive manual feature curation, and may incorporate clinical data along with imaging features for classification. In comparison, our work is primarily focused on using raw MRI frames, which combines feature extraction and classification as one problem. Though we do manually annotate subsections of each MRI, we note that our method can work on full MRIs, and thus has the potential to be completely automated. While the validation accuracy of full MRI scans is simiar to the results in Table 2, training the CRNN on full scans requires additional computational time and resources. Additionally, though we have formulated our prediction task as binary classification, it is possible to use regression with CRNNs to predict methylation activity, which may be more informative. As we are interested in discovering MRI features independent of demographic or patient characteristics, we chose not to incorporate additional clinical data (e.g. age of onset or sex). However, these clinical data may provide additional signal from a classification standpoint.
When assessing our classifier predictions, our model had a tendency to assign positive methylation status to heterogeneous, larger tumors with poorly defined margins (Figure 4b). Furthermore, many of our classification predictions are in concordance with previous results from Drabeyz et al.30 and Eoli et al..31 These studies discovered that ring-enhanced lesions were associated with negative MGMT promoter methylation. Hence, our model is able to autonomously determine some clinically relevant features correlated to MGMT methylation, without manual curation or predefined feature engineering as required in previous methods.
Deep learning methods have become powerful tools in image analysis and in the biomedical domain.15,16 However, these methods typically are not easily interpretable, and it can be challenging for a clinician or researcher to understand the model’s reasoning. Hence, these methods are often infamously termed “black-box models”. To address this challenge, we have developed a visualization platform that allows the domain user to select each MRI scan, load it through the CRNN computational pipeline, and interactively view and compare different filters and layers of our model. Our platform is generalizable, and can be easily extended for use with additional MRI prediction tasks and with different model architectures (e.g. variational number of filters and convolutional layers). For example, though we are primarily focused on the GBM tumor and its MGMT methylation status in this work, one may visualize whole brain MRI scans, in different orientations (e.g. saggital), for tasks such as risk stratification or lesion diagnosis. Moreover, any similar deep learning pipeline, that may use other type of MRI scans (e.g. cardiac) or other volumetric biomedical data (e.g. ultrasound), can be deployed with ease. Through the platform, we also visualize all the classifier predictions for our test set, and group them into four distinct sets — true and false positives and negatives. The domain users can browse and capture additional clinical features used by our model for prediction, or flaws in our model, that we may have not discussed here. We envision the visualization platform to be used in other relevant research and hence, we have released the source code.d
5. Conclusions
In this work, we implemented a convolutional recurrent neural network (CRNN) architecture to predict MGMT regulator methylation status using axial brain MRI scans from glioblastoma multiforme patients. Based on this model, we constructed a generalizable visualization platform for exploring the filtered outputs of different layers of our model architecture. Our CRNN achieved a test set accuracy of 0.62, with a precision of 0.67 and recall of 0.67. Using our predictions, we highlight macroscopic features of tumor morphology which may provide additional insight into the effects of MGMT methylation in glioblastoma multiforme. Though modest, our results support the existence of an association between MGMT methylation status and tumor characteristics, which merits further investigation using a larger cohort.
Acknowledgments
Funding
We would like to thank Fei-Fei Li, Justin Thompson, Serena Yeung and other staff members of the Stanford CS231N course (Convolutional Neural Networks for Visual Recognition) for their constructive feedback on this project. This work used the XStream computational resource, supported by the NSF Major Research Instrumentation program (ACI-1429830). LH is funded by NIH F30 AI124553. The results shown here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. We dedicate this work to the memory of Rajendra N. Kamdar, father of Maulik R. Kamdar, who passed away during the course of this research.
Footnotes
References
- 1.Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, Villano JL. Cancer Epidemiology and Prevention Biomarkers. 2014;23 doi: 10.1158/1055-9965.EPI-14-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Margison GP, Povey AC, Kaina B, et al. Carcinogenesis. 2003 Apr;24:625. doi: 10.1093/carcin/bgg005. [DOI] [PubMed] [Google Scholar]
- 3.Hegi ME, Diserens AC, et al. New England Journal of Medicine. 2005 Mar;352:997. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 4.Ellingson BM. Current Neurology and Neuroscience Reports. 2015 Jan;15:506. doi: 10.1007/s11910-014-0506-0. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto S, Maki DD, et al. American Journal of Roentgenology. 2012 Sep;199:654. doi: 10.2214/AJR.11.7824. [DOI] [PubMed] [Google Scholar]
- 6.Korfiatis P, Kline TL, Coufalova L, Lachance DH, Parney IF, Carter RE, Buckner JC, Erickson BJ. Medical Physics. 2016 May;43:2835. doi: 10.1118/1.4948668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levner I, Drabycz S, Roldan G, et al. Proceedings of the 12th International Conference on Medical Image Computing and Computer-Assisted Intervention. 2009;522 doi: 10.1007/978-3-642-04271-3_64. [DOI] [PubMed] [Google Scholar]
- 8.Krizhevsky A, et al. Advances in neural information processing systems. 2012;1097 [Google Scholar]
- 9.Yue-Hei Ng J, Hausknecht M, Vijayanarasimhan S, Vinyals O, Monga R, Toderici G. Proceedings of the IEEE conference on computer vision and pattern recognition. 2015;4694 [Google Scholar]
- 10.Payan A, Montana G. arXiv. 2015 Feb; [Google Scholar]
- 11.Poudel RPK, Lamata P, Montana G. arXiv. 2016 Aug; [Google Scholar]
- 12.Scarpace L, Mikkelsen T, Cha S, et al. The Cancer Imaging Archive. 2016 [Google Scholar]
- 13.Clark K, Vendt B, Smith K, Freymann J, Kirby J, Koppel P, Moore S, Phillips S, Maffitt D, Pringle M, Tarbox L, Prior F. Journal of Digital Imaging. 2013 Dec;26:1045. doi: 10.1007/s10278-013-9622-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinstein JN, Collisson EA, Mills GB, et al. Nature Publishing Group. 2013;45 [Google Scholar]
- 15.Litjens G, Kooi T, Bejnordi BE, et al. arXiv. 2017 Feb; [Google Scholar]
- 16.Min S, Lee B, Yoon S. Briefings in Bioinformatics. 2016 Jul;:bbw068. doi: 10.1093/bib/bbw068. [DOI] [PubMed] [Google Scholar]
- 17.Akkus Z, Ali I, Sedlar J, et al. arXiv. 2016 Nov; [Google Scholar]
- 18.Sarraf S, Tofighi G. arXiv. 2016 Mar; [Google Scholar]
- 19.Stollenga MF, Byeon W, Liwicki M, Schmidhuber J. arXiv. 2015 Jun; [Google Scholar]
- 20.Chen H, Dou Q, Ni D, Cheng JZ, et al. Proceedings of the 18th International Conference on Medical Image Computing and Computer-Assisted Intervention. 2015;507 [Google Scholar]
- 21.Kong B, Zhan Y, Shin M, Denny T, Zhang S. Proceedings of the 19th International Conference on Medical Image Computing and Computer-Assisted Intervention. 2016;264 [Google Scholar]
- 22.Harris LC, Remack JS, Brent TP. Nucleic Acids Research. 1994;22:4614. doi: 10.1093/nar/22.22.4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris LC, Potter PM, Tano K, et al. Nucleic Acids Research. 1991;19:6163. doi: 10.1093/nar/19.22.6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawachi T, Soejima H, Urano T, et al. Oncogene. 2003;22:8835. doi: 10.1038/sj.onc.1207183. [DOI] [PubMed] [Google Scholar]
- 25.Alonso S, Dai Y, Yamashita K, et al. Oncotarget. 2015;6:3420. doi: 10.18632/oncotarget.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mildenberger P, Eichelberg M, Martin E. European Radiology. 2002 Apr;12:920. doi: 10.1007/s003300101100. [DOI] [PubMed] [Google Scholar]
- 27.Moon WJ, Choi JW, Roh HG, Lim SD, Koh YC. Neuroradiology. 2012;54:555. doi: 10.1007/s00234-011-0947-y. [DOI] [PubMed] [Google Scholar]
- 28.Gupta A, Omuro AMP, Shah AD, Graber JJ, Shi W, Zhang Z, Young RJ. Neuroradiology. 2012 Jun;54:641. doi: 10.1007/s00234-011-0970-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanas VG, Zacharaki EI, Thomas GA, Zinn PO, Megalooikonomou V, Colen RR. Computer Methods and Programs in Biomedicine. 2017;140:249. doi: 10.1016/j.cmpb.2016.12.018. [DOI] [PubMed] [Google Scholar]
- 30.Drabycz S, Roldán G, de Robles P, Adler D, McIntyre JB, Magliocco AM, Cairncross JG, Mitchell JR. NeuroImage. 2010;49:1398. doi: 10.1016/j.neuroimage.2009.09.049. [DOI] [PubMed] [Google Scholar]
- 31.Eoli M, Menghi F, Bruzzone MG, De Simone T, Valletta L, Pollo B, Bissola L, Silvani A, Bianchessi D, D’Incerti L, Filippini G, et al. Clinical Cancer Research. 2007;13:2606. doi: 10.1158/1078-0432.CCR-06-2184. [DOI] [PubMed] [Google Scholar]