

SUMMARY
Diverse pathways drive resistance to BRAF/MEK inhibitors in BRAF-mutant melanoma, suggesting that durable control of resistance will be a challenge. By combining statistical modeling of genomic data from matched pre-treatment and post-relapse patient tumors with functional interrogation of over 20 in vitro and in vivo resistance models, we discovered that major pathways of resistance converge to activate the transcription factor, c-MYC (MYC). MYC expression and pathway gene signatures were suppressed following drug treatment, then rebounded during progression. Critically, MYC activation was necessary and sufficient for resistance, and suppression of MYC activity using genetic approaches or BET bromodomain inhibition was sufficient to resensitize cells and delay BRAFi resistance. Finally, MYC-driven, BRAFi-resistant cells are hypersensitive to the inhibition of MYC synthetic lethal partners, including SRC family and c-KIT tyrosine kinases as well as glucose, glutamine, and serine metabolic pathways. These insights enable the design of combination therapies that select against resistance evolution.
Keywords: Melanoma, cancer therapeutics, therapeutic resistance, signaling networks, MYC, metabolism, synthetic lethality
eTOC Blurb
Diverse pathways drive resistance to BRAF/MEK inhibitors in BRAF-mutant melanoma, but by combining statistical modeling of tumor data with functional interrogation of resistance models, Singleton et al. show that these pathways converge to activate MYC. BRAFi-resistant cells are hypersensitive to the inhibition of MYC synthetic lethal partners, informing therapies that select against resistance.
INTRODUCTION
Recently, the treatment of metastatic BRAF mutant melanoma has been revolutionized by two major new therapeutic modalities: targeted therapies (e.g., BRAF and MEK inhibitors, BRAFi/MEKi) and immune checkpoint blockade (e.g., PD-1/PD-L1 and CTLA-4 inhibitors) (Wargo et al., 2014). Therapy with BRAFi/MEKi yields high objective response rates, but diverse mechanisms of acquired resistance limit therapeutic duration (Alcala and Flaherty, 2012; Robert et al., 2015; Solit and Rosen, 2014; Wargo et al., 2014). In contrast, checkpoint inhibitors yield lower response rates, but are often durable (Larkin et al., 2015; Wargo et al., 2014). Ongoing clinical trials are investigating combinations of BRAFi/MEKi with checkpoint inhibitors, hoping to improve their durations and rates of response, respectively. However, emerging evidence suggests that mechanisms driving resistance to BRAFi/MEKi may also drive cross-resistance to checkpoint blockade through the suppression of tumor immune surveillance, underscored by observations that patients who fail first-line treatment with BRAFi/MEKi appear to respond poorly to subsequent checkpoint blockade (Ackerman et al., 2014; Frederick et al., 2013; Hugo et al., 2015; Peng et al., 2015; Wargo et al., 2014) (Puzanov et al., 2015, Pigment Cell Melanoma Res., abstract; Ramanujam et al., 2015, Pigment Cell Melanoma Res., abstract; Simeone et al., 2015, Pigment Cell Melanoma Res., abstract). These findings suggest that innovative and robust strategies for preventing resistance to BRAFi/MEKi may not only increase the durability of responses to first-line therapy but also improve the activity of emerging checkpoint blockade and combination strategies.
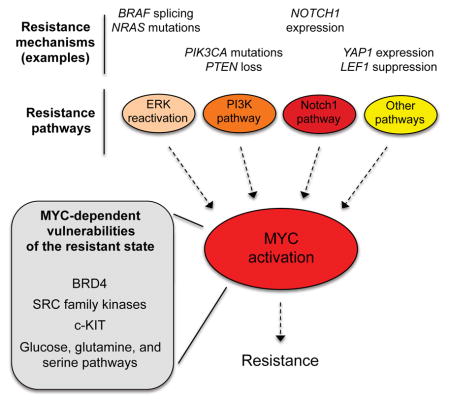
Extensive studies have identified diverse mechanisms of resistance to BRAFi/MEKi in BRAF-mutant melanomas, including those that function by modulating the initial adaptive tumor response (intrinsic resistance) and those selected for over time (acquired resistance). Many resistance mechanisms, including activating mutations in NRAS, MEK, and ERK, NF1 loss, and amplification or alternative splicing of mutant BRAF, result in reactivation of the ERK pathway in the presence of BRAFi/MEKi (Alcala and Flaherty, 2012; Corcoran et al., 2010; Hugo et al., 2015; Maertens et al., 2013; Nazarian et al., 2010; Poulikakos et al., 2011; Rizos et al., 2014; Shi et al., 2014b; Shi et al., 2012; Solit and Rosen, 2014; Van Allen et al., 2014; Whittaker et al., 2013). Additionally, bypass mechanisms, including activation of the phosphoinositide-3-kinase (PI3K) pathway through mutations or altered expression of IGF-1R, PIK3CA, PTEN, and AKT, as well as through microenvironmental changes, can drive resistance (Alcala and Flaherty, 2012; Fedorenko and Smalley, 2015; Hugo et al., 2015; Paraiso et al., 2011; Rizos et al., 2014; Shi et al., 2014a; Shi et al., 2014b; Solit and Rosen, 2014; Villanueva et al., 2010). Similarly, bypass signaling through the Notch1 pathway via altered expression of Notch1 and other pathway members drives resistance in a third, distinct subset of patients (Martz et al., 2014). Importantly, these three pathways—ERK, PI3K and Notch1—have been shown to drive resistance to both single agent and combined BRAFi/MEKi, and together they appear to account for ~75% of acquired resistance cases while also playing important roles in intrinsic resistance (Hugo et al., 2015; Martz et al., 2014; Moriceau et al., 2015; Wagle et al., 2014). Finally, a “long tail” of alternative, rare resistance mutations and non-genomic (transcriptional) alterations, including those affecting the WNT/LEF1 and Hippo/YAP pathways, are likely to play roles in resistance, although the fractions of tumors affected have yet to be defined (Hugo et al., 2015; Lin et al., 2015; Van Allen et al., 2014). In sum, the presence of multiple distinct pathways of resistance, sometimes within the same patient or even the same tumor (Shi et al., 2014b), suggests that the therapeutic inhibition of individual resistance pathways is likely to have only limited clinical value, and that robust therapies may require simultaneous inhibition of multiple resistance pathways (Robert et al., 2015; Solit and Rosen, 2014). Unfortunately, such higher order combination therapies are expected to frequently produce unacceptable toxicities in patients, necessitating the development of conceptually new approaches to circumvent resistance.
One such approach is based on the hypothesis that the seemingly distinct pathways driving resistance to BRAFi/MEKi may actually converge on one or more “common effectors”: downstream signaling targets that are required for the development and maintenance of resistance. This hypothesis is consistent with the notion of oncogene addiction (Settleman, 2012), which asserts that cancers develop dependencies on signaling downstream of driver oncogenes, implying that resistance may require the reactivation of these key downstream target(s). In melanoma, the ‘common effector’ hypothesis is also consistent with the observation that BRAFi/MEKi-resistant tumors driven by diverse upstream signaling alterations exhibit highly recurrent transcriptional programs (Hugo et al., 2015). Finally, this hypothesis is supported by recent data from our group and others demonstrating that distinct mechanisms of resistance to receptor tyrosine kinase inhibitors in lung and colorectal cancers converge on a single downstream signaling axis, the targeting of which can forestall resistance (Hrustanovic et al., 2015; Misale et al., 2015; Tricker et al., 2015).
In this study, we integrate genomic and biochemical data from two cohorts of BRAF mutant melanoma patients with acquired resistance to BRAF/MEK pathway blockade, alongside diverse cellular and animal models of BRAFi resistance, to identify the transcription factor MYC as a convergent downstream effector of multiple major resistance pathways that is both necessary and sufficient for resistance. By leveraging this insight alongside the concept of synthetic lethality, we define combination therapies that, by selectively targeting the MYC-activated, BRAFi/MEKi-resistant state, have the unique property of selecting against resistance evolution and thereby represent promising strategies to durably control resistance.
RESULTS
MYC is Commonly Reactivated in Patients with Acquired Resistance to BRAF inhibitors
To search a potential convergent effector of resistance, we began by reasoning that such an effector should follow two rules: (1) it should be regulated downstream of the driver oncogene and (2) it should rebound to at least pre-treatment expression or activation states at resistance. To identify genes that obey (1), we used a nonlinear classification model that allows for the inference of differential gene expression by estimating effect sizes (Crawford et al., 2016) to identify a set of genes, termed the BRAF response signature, that are most transcriptionally altered by BRAFi/MEKi treatment in cell lines and human tumors from recent published studies (local false sign rate (LFSR) < 0.01; see SI Text for details) (Nazarian et al., 2010; Pratilas et al., 2009; Rizos et al., 2014; Stephens, 2016). Gene Set Enrichment Analysis (GSEA) of these 68 genes revealed numerous enriched signatures associated with the canonical RAS-RAF-MEK-ERK cascade (Table S1, Tabs 1–2). Next, we winnowed the signature to genes that return to their pre-treatment states during relapse in a panel of 29 human melanoma tumors sampled before and after the development of resistance (Rizos et al., 2014). We used Bayes Factors (BF; see SI Text for details) as a metric describing a gene’s return toward its pre-treatment state. The BF is a Bayesian alternative to classical likelihood ratio testing. Here, a large BF indicates signature genes useful for classifying a relapsed sample, whereas a log10BF less than or equal to zero indicates a gene that does not discriminate between treated and resistant tumors. We considered all signature genes with log10BF greater than zero to be potential convergent effectors of resistance. Thirty-three genes were associated with positive log10BF values (Figure 1A and Table S1, Tab 3). We observed that MYC returned the highest log10BF value. Correspondingly, GSEA of the 33-gene panel revealed numerous MYC-related, enriched gene sets (Table S1, Tab 4). By comparing the change in MYC mRNA levels in matched pre-treatment and post-relapse tumors with the expected change in the same following BRAFi treatment, we calculated the probability of MYC rebound in each resistant tumor (see SI Text for details). Strikingly, this analysis revealed ≥90% probability of MYC rebound in 23 of 29 resistant tumors, and ≥50% probability of MYC rebound in all 29 tumors despite the fact that resistance in these tumors was putatively driven by a range of mechanisms including activation of the ERK, PI3K, and Notch1 pathways (Figure 1B and Table S2) (Martz et al., 2014; Rizos et al., 2014). At the protein level, immunohistochemical analysis of formalin-fixed tumor samples from an independent cohort of BRAF mutant melanoma patients showed a similar pattern of MYC suppression on treatment followed by reactivation upon progression (Figure 1C–D).
Figure 1. MYC is a potential convergent effector of diverse resistance pathways in BRAF mutant melanoma tumors that have progressed on treatment with BRAF/MEK inhibitors.

A) Bayes Factor (BF) values for BRAF response signature genes. B) Probability of MYC rebound to pretreatment or greater expression levels in each of 29 patient tumors with acquired resistance to vemurafenib. C) Matched patient samples pre-treatment (PRE), on treatment (ON) with RAF and/or MEK inhibitor, or after progression on treatment (POST). Nuclear MYC staining scored 0: none to 3: maximum (left panel) and percent nuclei staining positive for MYC (right panel). D) Representative images from the data shown in C) at 40X magnification. E) MYC mRNA transcript levels in cell lines with evolved resistance. Data are means of 3 experiments in clonal cell lines normalized to β-actin transcript levels and parental MYC levels. Parental cells were treated with 1μM PLX4720 or DMSO for 72 hrs. F) MYC protein levels shown by immunoblotting. Parental lines (P) were treated with DMSO or 1 μM PLX4720 for 24 hrs and resistant lines (annotated as R(A), PI3K-AKT-mTOR-driven resistance; R(N), Notch1-driven resistance; R(M), ERK reactivation-driven resistance; R(O), other/unknown resistance mechanism; R(Mi), evolved resistance to MEK inhibitor, AZD6244; R(Ei), evolved resistance to ERK inhibitor, VX-11E) were treated with 3 μM PLX4720. Quantification of MYC protein normalized to total protein is shown in the bottom panel and total protein staining is shown in Figure S1C. *P < 0.05. See also Figure S1 and Table S1, Tabs 1–4.
To assess the functional role of MYC activation in BRAFi-resistance, we used step-wise selection in several BRAF mutant melanoma cell lines to establish a panel of clonal derivatives with acquired resistance to BRAF pathway blockade. Functionally, these lines exhibited an array of resistance mechanisms including ERK re-activation and bypass of ERK signaling through PI3K or Notch1 pathway activation. Additionally, the resistance mechanism of one resistant cell line could not be determined (Figure S1A) (Martz et al., 2014). Like their human tumor counterparts, MYC mRNA and protein levels in these models were decreased following initial BRAFi treatment, then rebounded to pre-treatment or greater levels on resistance despite the continued presence of BRAFi (Figures 1E–F and S1B–C). Together, these data suggest that MYC may play an important role in the development of resistance.
MYC Expression is Necessary and Sufficient for Resistance in Diverse Models
To determine whether MYC activation is functionally driving resistance or simply a marker of the proliferative state, we first ectopically expressed a degradation-resistant MYCT58A mutant in sensitive (treatment naïve) cells, revealing that MYC expression alone is sufficient to confer resistance to BRAFi, MEKi, ERKi, and BRAFi+MEKi (Figure 2A and S2A). Next, to determine whether MYC activation is necessary for resistance, we partially suppressed MYC expression with two independent shRNAs in a panel of resistant cell lines (Figure S2B). In each case, MYC knockdown sensitized cells to BRAFi at growth inhibition-50% (GI50) values near or below those of parental, sensitive cells (Figure 2B). By comparison, MYC knockdown had modest effects on BRAFi sensitivity in parental cells (Figure S2C). Also, MYC levels in parental and resistant lines were uncoupled from doubling time (Figure S2D), consistent with the observation that resistant cells, which have higher MYC levels that their parental counterparts, also tend to grow more slowly. To explore whether resistant cells could be sensitized to BRAFi/MEKi using pharmacological methods, we used JQ1, a small molecule BET bromodomain inhibitor that suppresses MYC expression alongside other targets (Figure S2E) (Delmore et al., 2011). JQ1 treatment phenocopied genetic MYC suppression (Figure 2C and S2E–G). As JQ1 blocks the endogenous transcription of multiple genes, we tested its MYC-specific effects using a virally-expressed MYC construct lacking its native promoter (Zuber et al., Nature, 2011). We found that while JQ1 could sensitize control, luciferase expressing cells to BRAFi, it lost this ability upon ectopic expression of MYC, indicating that JQ1’s effects are largely due to on-target inhibition of MYC expression (Figure 2D). Additionally, JQ1 could re-sensitize cell lines with evolved resistance to the ERKi VX-11E (Figure 2E). Finally, consistent with a shift away from BRAF-dependence and toward MYC-dependence, we found that evolved resistant lines were always more sensitive to JQ1 monotherapy than their parental counterparts, and that the sensitivity of parental cell lines to JQ1 was inversely related to their basal sensitivity to BRAFi (Figure 2F). Together, these data demonstrate that MYC activation is both necessary and sufficient for resistance to ERK pathway inhibition in diverse models and suggest that BRAF mutant melanoma cell lines exist along a continuum between BRAF- and MYC-driven states.
Figure 2. MYC activation is necessary and sufficient for resistance to BRAF/MEK pathway inhibitors in diverse BRAF-mutant melanoma cell lines.

A) Fold change in GI50 values for cells expressing MYCT58A and treated with either PLX4720, AZD6244, VX-11E, or the combination of PLX4720 and AZD6244 at a 1:1 dose ratio as compared to cells expressing an empty vector control (pBABE-puro). B) Fold change in GI50 values for PLX4720 between cell lines expressing shGFP compared to each of two independent shRNAs targeting MYC. P values denote significance between the response of shGFP and each shMYC expressing cell line. Parental lines are shaded blue, resistant lines, red/yellow. C) Clonogenic growth assay of A375 line derivatives treated with DMSO, 100 nM, 300 nM or 1 μM PLX4720 +/− 300 nM JQ1. D) WM793 cells expressing ectopic MYC or luciferase control were treated with DMSO or 100nM JQ1 and their PLX4720 GI50 value was determined. JQ1+PLX4720 treated cells are normalized to the viability of cells treated with JQ1 alone to account for nonspecific toxicity. P values denote significance between DMSO and JQ1 treatment in each cell line. E) Clonogenic growth of SK-MEL-5 cells with resistance to VX-11E (VX) in DMSO or 100 nM JQ1. JQ1+VX-11E treated cells are normalized to the viability of cells treated with JQ1 alone to account for nonspecific toxicity. P values denote significance between DMSO and JQ1 treatment. F) JQ1 GI50 values for the indicated PLX-resistant lines compared to matched parental controls. Significance is between the parental cell line and each matched resistant derivative. Data are means (SD) from 3 experiments. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001. See also Figure S2.
MYC Functions Downstream of Common Resistance Pathways
Although the finding that MYC knockdown could reverse resistance in evolved cell lines exhibiting diverse resistance mechanisms suggests that MYC functions as a convergent downstream effector of resistance, we sought to further investigate this in a controlled genetic system. We stably expressed lentiviral cDNAs constitutively activating the ERK, PI3K, and Notch1 resistance pathways in a treatment-naïve, BRAFi-sensitive cell line, UACC-62. Activation of the ERK pathway (through expression of activated KRASG12V, activated HRASG12V, or activated MEK1(S218D/S222D) (MEK1 DD)), the PI3K pathway (through expression of activated, myristoylated AKT1), and the Notch1 pathway (through expression of the Notch1 intracellular domain (ICD)) each increased MYC protein levels relative to cells expressing a negative control construct (luciferase) or activators of pathways that fail to confer resistance (the Janus kinase-Signal Transducer and Activator of Transcription (JAK-STAT) pathway (JAK2V617F) and Hedgehog pathway (truncated Gli2)) (Figures 3A and S3A–B). We next investigated the mechanisms by which each resistance pathway was causing increased MYC expression. It has previously been demonstrated that the ERK and PI3K pathways can increase MYC levels by enhancing protein stability through phosphorylation (Wang et al., 2011). Consistent with these findings, expression of MEK1 DD and myrAKT1 increased MYC protein levels, but not MYC mRNA levels, relative to luciferase expressing cells (Fig. 3A–C and S3B). Further, MEK1 DD and myrAKT1 expression increased MYC stability in a cycloheximide chase assay (Fig. 3C and S3C). Separately, Notch1 is known to activate MYC through transcriptional upregulation (Palomero et al., 2006), consistent with our findings that cells expressing the Notch1 intracellular domain exhibit increased MYC mRNA levels without evidence of increased MYC protein stability (Fig. 3A–C and S3B–C). Resistance caused by ERK pathway reactivation can be reversed with MEKi and/or ERKi treatment. Consistent with this, we found that both ERK pathway activating constructs and evolved cell line models with ERK pathway reactivation lost MYC expression upon treatment with MEKi+ERKi, while AKT-driven resistance models retained partial MYC expression (Figure S3D). Resistance driven by each of these upstream pathways was MYC-dependent, as both shRNA-mediated MYC knockdown and treatment with JQ1 were sufficient to fully reverse resistance (Figures 3D–E and S3E). Finally, cells with forced activation of resistance pathways displayed increased sensitivity to JQ1 relative to controls (Figure 3F). Collectively, these findings demonstrate that major pathways of resistance function through convergent, downstream MYC activation and suggest that resistant cells, which have shifted their dependencies from BRAF to MYC, may be selectively sensitive to therapeutic strategies that disrupt MYC function.
Figure 3. MYC activation drives resistance to BRAF pathway inhibition downstream of common resistance pathways.

A) Immunoblot of MYC levels in UACC-62 cells expressing luciferase (luc) or the indicated pathway activating constructs. Vinculin levels are shown as a loading control. Quantification shown in Figure S3B. B) MYC mRNA transcript levels in UACC-62 cells expressing the indicated constructs. Data are means of 3 experiments normalized to TUBB3 transcript levels and UACC-62-luc MYC levels. C) Immunoblot of MYC levels in UACC-62 cells expressing luc or the indicated pathway activating constructs at the indicated times post treatment with 20 μg/mL cycloheximide. Quantification of MYC protein normalized to total protein is shown in the bottom panel and total protein staining is shown in Figure S3C. D) Change in sensitivity to PLX4720 for lines in A) by expression of two independent shRNAs targeting MYC. P values are the comparison of shGFP to each shMYC-expressing cell line. E) Sensitization to PLX4720 for lines in A) by the addition of 100 nM JQ1. JQ1+PLX4720 treated cells are normalized to the viability of cells treated with JQ1 alone to account for nonspecific toxicity. P values indicate difference between DMSO and JQ1 treatment. F) JQ1 GI50 values of lines in A) that do not (blue shades) or do (red shades) provide resistance to PLX4720. P values indicate significance relative to luc-expressing cells. All data are means (SD) from 3 experiments. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001. See also Figure S3.
BRAF Mutant Cell Lines with Intrinsic Resistance to BRAFi Rapidly Upregulate MYC Upon Treatment
Previously, we and others have described BRAF mutant melanoma cell lines that, like a minority of patients, exhibit intrinsic resistance to BRAFi/MEKi (Konieczkowski et al., 2014; Martz et al., 2014; Wood et al., 2012). Given our finding that evolved resistant cell lines have increased MYC expression relative to their parental counterparts, we expected that intrinsically resistant lines would have similarly elevated MYC levels. Surprisingly, however, we found no clear correlation between MYC expression level and intrinsic resistance status (Figure 4A–C and S4A–B). However, in contrast to sensitive cells, which tend to lose MYC expression following short-term treatment with PLX4720 (24h), intrinsically resistant cell lines increase MYC expression following the same treatment (Figure 4A–C). Accordingly, sensitivity to JQ1 could be potentiated in intrinsically resistant cell lines, but not in sensitive cell lines, following a brief, 4-day pre-treatment with PLX4720 (Figure 4D). To further investigate MYC dynamics, we chronically cultured both a sensitive cell line, A375, and an intrinsically resistant line, WM793, in 1 μM PLX4720, measuring JQ1 and PLX4720 GI50 values over time. A375 gradually developed resistance over the course of 60 days while concurrently developing increased sensitivity to JQ1 (Figure 4E), changes that correlated with MYC expression over time and are consistent with the hypothesis of selection for MYC-driven clones (Figures 4F and S4C). In contrast, the intrinsically resistant cell line exhibited rapid decreases in JQ1 GI50 values that were already apparent at the first measurable time point (3d) and remained stable thereafter, while modestly increasing in PLX4720 resistance (Figure 4G). Changes in drug sensitivity were mirrored by a rapid up-regulation of MYC expression over 24 hours of PLX4720 exposure, which then remained constant for the remaining duration of the experiment (Figures 4H and S4D). Additionally, MYC expression remained stably elevated following removal of PLX4720 (Figure S4E). MYC knockdown could reverse resistance to BRAFi in the intrinsically resistant cell lines (Figure 4I), a result that could be phenocopied with JQ1 (Figure S4F). Finally, we recently reported that these intrinsically resistant cell lines could be re-sensitized to ERKi using combinations of inhibitors directed against the upstream PI3K and Notch1 pathways, with minor contributions from the estrogen receptor alpha (ERα) pathway (Figure 4J) (Martz et al., 2014). Consistent with the hypothesis that MYC is a convergent downstream effector of these resistance pathways, MYC overexpression fully rescued the sensitizing effects caused by upstream inhibition of these pathways (Figure 4K). Together, these findings demonstrate that intrinsic resistance, like evolved resistance, is driven by MYC activation, that a key distinguishing feature of intrinsically resistant lines is their ability to rapidly upregulate MYC expression in response to BRAFi, and that dynamic MYC upregulation in these cells results in corresponding changes in sensitivity to pharmacological MYC suppression with JQ1.
Figure 4. Melanoma cell lines with intrinsic resistance to BRAFi derive resistance by rapidly upregulating MYC following drug treatment.

A) The indicated cell lines were incubated with DMSO or 1 μM PLX4720 for 24 hrs, then immunoblotted for MYC. Total protein staining is shown in Figure S4A. B) As in A). Total protein staining is shown in Figure S4B. C) Quantification of immunoblots shown in A) and B) showing MYC protein levels normalized to total protein. D) JQ1 GI50 values for the indicated lines following pre-incubation with either DMSO or 1 μM PLX4720 for four days. P values indicate significance between DMSO and PLX4720 pre-treatment. E) GI50 values in A375 cells for either PLX4720 (solid line) or JQ1 (dashed line), measured at intervals during continuous culture in 1 μM PLX4720. F) MYC levels in A375 at several time points while in culture with 1 μM PLX4720. Quantification shown in Figure S4C. G) As in E), for the intrinsically resistant line, WM793. H) As in F) for WM793. Quantification shown in Figure S4D. I) Sensitization of the indicated lines to PLX4720 by expression of two shRNAs targeting MYC. Differences between shGFP and shMYC are denoted by P values. J) Signaling pathways controlling intrinsic resistance to BRAFi/MEKi in intrinsically resistant cell lines and their convergence on downstream MYC activation. As indicated, previous findings demonstrate that inhibition of the PI3K-AKT-mTOR and NOTCH1 pathways can sensitize these lines to ERK pathway inhibition. K) Sensitivity of indicated lines to the ERKi VX-11E following expression of luc or MYC and treatment DMSO or a combination of inhibitors (200 nM BEZ235 (PI3K/mTOR inhibitor) for the PI3K-AKT-mTOR pathway and shNotch1 for the Notch1 pathway for Hs 294T cells; 200 nM BEZ235, shNotch1, and 1 μM fulvestrant for Lox IMVI, A2058 and WM793 cells). Viabilities of cells treated with pathway inhibitors plus VX-11E are normalized to the viability of the same cells treated with pathway inhibitors alone to account for nonspecific toxicity. All data are means (SD) from 3 experiments. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001; #, the upper bound of the assay was reached. See also Figure S4.
Combined BRAFi and MYC Suppression Delays the Emergence of Resistance in vitro and in vivo
The finding that multiple pathways converge to drive resistance through MYC suggests that pairing BRAFi/MEKi with MYC suppression may delay the emergence of resistance. To test this hypothesis, we stably expressed shRNAs against either MYC or a negative control (GFP) in A375 cells, then cultured the cells in 3μM PLX4720 with weekly counting until acquired resistance was observed, as indicated by the recovery of exponential growth. As in Figure 4E–F, cells expressing shGFP began proliferating in PLX4720 after three weeks, at which time they also began expressing MYC (Figures 5A and S5A). In contrast, the growth of cells expressing shMYC in PLX4720 was delayed until week 6 of culture, despite the fact that these cells proliferated normally in the absence of drug (Figures 5A and S5A). Given the possibility that clones with incomplete MYC knockdown could have been selected to drive resistance, we measured MYC expression over the experimental time course. Indeed, cells expressing shMYC began to exhibit high MYC expression around week 6, in concordance with their acquisition of exponential grow in PLX4720 (Figures 5A). To determine if these results could be extended to pharmacologic inhibition of MYC expression, we serially passaged A375 cells in PLX4720, JQ1, or the combination. Cells treated with JQ1 alone quickly regained nearly normal proliferation rates, while cells treated with the combination began proliferating at 7–9 weeks, not recovering a population size equal to that of initial seeding until 12 weeks (Figures 5B and S5B). Further, as in the case of shRNA-mediated MYC suppression, cells that emerged with resistance to BRAFi + JQ1 escaped JQ1-mediated MYC suppression (Figure 5C and S5C).
Figure 5. MYC suppression delays the emergence of resistance to BRAF inhibition in vitro and in vivo.

A) Lines indicate cell numbers calculated from weekly measured growth of A375 cells expressing shGFP or shMYC and cultured in 3μM PLX4720 (left axis). Bars indicate quantification of MYC levels in extracts made at the indicated times normalized to β-actin levels (right axis). B) Cell numbers calculated from growth rates of A375 cells cultured in 3 μM PLX4720, 100 nM JQ1, or the combination. C) Immunoblot of MYC in cell lines generated in B), D) and F). D) Cell numbers calculated from growth rates of A375-PLX+JQ1 dual resistant cells cultured in 3 μM PLX4720 or 100 nM JQ1 and 3 μM PLX4720. E) GI50 values of cell lines generated in B) and D) treated with PLX4720 or JQ1. Quantification of MYC protein normalized to total protein and total protein staining is shown in Figure S5C. F) Cell numbers calculated from growth rates of A375-PLX resistant cells cultured in 3 μM PLX4720 and 1 μM AZD6244 (MEKi), 100 nM JQ1, or the combination. G) Growth of A375 xenograft tumors treated with vehicle, JQ1 (45 mg/kg/d), dabrafenib (Dab, 30 mg/kg/d) plus trametinib (Tram, 0.6 mg/kg/d), or the combination over time. Data shown are mean tumor volume +/− SEM, with n=6–8 mice per group. P values were calculated using the one-sided Mann-Whitney test, *p<0.05. H) Waterfall plot showing tumor sizes in the indicated groups from panel G) on day 45 of treatment. For B) and F), dashed line indicates initial seeding number. All data are means (SD) from 3 experiments. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001. See also Figure S5.
To investigate cells in the PLX4720+JQ1 dual resistant state (‘dual cells’), we treated the dual cells with DMSO, PLX4720 or the combination of PLX4720 and JQ1, while measuring the cells’ GI50 values to PLX4720 and JQ1 (Figures 5D–E). Once both drugs were removed, the population gradually shifted back to a BRAFi-sensitive, JQ1-resistant state (Figures 5E). By contrast, if JQ1 was reintroduced after a two-week holiday, cells could be resensitized to the combination, with no outgrowth of resistant clones over 12 weeks (Figure 5D). MYC protein expression was observed in the PLX4720-resistant populations and in the dual resistant cells, suggesting that the dual resistance arises through failure of JQ1 to durably inhibit MYC expression (Figure 5C and S5C). PLX4720-resistant cells acquired resistance to the addition of a MEK inhibitor very quickly, although these cells’ increased dependency on MYC led to eradication of cells when JQ1 was included in the combination (Figure 5F). Further, unlike parental cells, PLX4720-resistant cells were highly sensitive to JQ1 alone (Figure 5F). Both BRAFi- and BRAFi+MEKi-resistant cells exhibited high-level MYC expression, while those treated only with JQ1 lost both their BRAFi resistance and MYC expression (Figure 5C). These results demonstrate that MYC suppression is a viable strategy to delay resistance to BRAFi, and further, are consistent with the hypothesis that resistance evolution requires MYC activation. Although we were able to generate cells resistant to dual BRAF+MYC inhibition, this state was 1) highly unstable, 2) caused by failure of JQ1 to inhibit MYC expression, and 3) reversible with a short JQ1 drug holiday.
In order to investigate the relationship between MYC and resistance in vivo, we established xenografts from A375 cells in immunocompromised mice. After formation of a palpable tumor, mice were treated with either diluent control, the BRAFi/MEKi combination dabrafenib (BRAFi, 30 mg/kg/d) and trametinib (MEKi, 0.6 mg/kg/d), JQ1 (45 mg/kg/d), or the combination of dabrafenib, trametinib and JQ1. While tumors were able to grow similarly to control in the presence of JQ1, their growth was inhibited by the combination of dabrafenib and trametinib, and substantial tumor regressions were observed when JQ1 was added to this combination (Figure 5G–H). Furthermore, while tumors treated with the dabrafenib/trametinib combination began increasing in size after approximately 20 days of treatment, the addition of JQ1 led to sustained tumor regression throughout the experimental time course (Figure 5G). To assess the ability of JQ1 to sensitize a BRAFi-resistant model to BRAFi+MEKi, we established xenografts using a cell line with intrinsic resistance to BRAFi, Hs695T. We observed substantial decreases in tumor size when JQ1 was added to the combination of dabrafenib and trametinib (Figures S5D).
MYC Activation Creates Targetable Vulnerabilities in Resistant Cells
The fact that BRAFi resistant cells exhibit increased MYC dependency suggests that synthetic lethal drugging strategies that target the MYC activated state may be selectively toxic to resistant cells. Numerous potential MYC synthetic lethal partners have been identified, but their applicability to melanomas is unclear given the tissue-specific nature of MYC-driven expression programs (Cermelli et al., 2014; Dang, 2012). Therefore, to target MYC-driven resistance, we leveraged both unbiased and candidate-based methods. First, we screened a library of ~1700 bioactive compounds, including diverse targeted inhibitors, natural products, and chemotherapeutics, to identify those differentially lethal to PLX4720-resistant A375 cells relative to parental cells (Table S3). As expected, compounds targeting the ERK pathway were highly selective for sensitive cells (Figure 6A, p < 10−12 by one-sided Kolmogorov-Smirnov (K-S) test). Conversely, resistant cells exhibited increased sensitivity to many compounds, including annotated inhibitors of SRC family kinases (p = 2 × 10−3), c-KIT (p = 3 × 10−4), the Hedgehog pathway (p = 0.03), and Aurora kinases (p = 9 × 10−6), the latter of which have been previously implicated as MYC synthetic lethal partners (Dang, 2012; den Hollander et al., 2010; Yang et al., 2010) (Figure 6A and S6A–D). Another interesting compound identified in the screen was Tigecycline, whose selectivity could be extended to several BRAFi-resistant models (Figure S6E). In parallel, we examined candidate MYC synthetic lethal partners previously identified in other cancer types using a panel of matched parental and BRAFi-resistant cells (Chipumuro et al., 2014; Goga et al., 2007; Martins et al., 2015; Toledo et al., 2011; Truman et al., 2012; Yang et al., 2010). Of these candidates, the multi-targeted tyrosine kinase inhibitor, dasatinib, was selectively lethal in 6 out of 6 MYC-dependent, PLX4720-resistant clones relative to matched parental cells (Figures 6B and S6F). Given that dasatinib inhibits SRC family and other kinases with nanomolar efficiency (Steinberg, 2007), we examined this compound in greater detail. The combination of dasatinib and PLX4720 yielded an increase in apoptosis and synergistic growth inhibition in PLX4720-resistant cells (Figures 6C and S6G). Further, by simultaneously targeting both sensitive and resistant cells using PLX4720 and dasatinib, respectively, it was possible to block the emergence of resistance in long-term culture (Figures 6D and S6H). Dasatinib is estimated to have over 50 targets with an IC50 of 1 μM or less. In order to begin to elucidate the contributions of these targets to MYC synthetic lethality, we examined gene expression data for dasatinib targets (“das-genes”) in A375 parental cells, PLX4720-resistant A375 cells, and resistant A375 cells expressing a MYC-targeted shRNA. Twenty-eight of 71 das-genes were upregulated in resistant cells in a MYC-dependent fashion (Table S4 and Figure 6E). Among the agents in the screen of 1,700 small molecules, those targeting das-genes upregulated in MYC-active cells were enriched for selectivity against resistant cells (p = 2 × 10−6), while compounds that inhibit non-upregulated das-genes showed no selectivity (p = 1.0) (Figure 6F). Together, these results: (1) identify families of compounds that selectively target MYC-activated, BRAFi-resistant melanoma cells, (2) identify dasatinib as a high priority polypharmacology-based strategy to target resistant cells, and (3) suggest that the protein products of das-genes with increased expression in MYC-activated cells, including LYN, ERBB2, and PDGFRB, warrant individual examination as potentially specific targets. Finally, to nominate additional candidate targets, we defined expression changes in genes that have been identified as canonical, direct MYC targets by combined ChIP-seq and gene expression analysis (Zeller et al., 2003). We examined the set of genes whose promotor is bound by MYC and whose expression is changed following MYC activation to identify those with changed expression in sensitive versus resistant cell lines. Parental and resistant-shMYC samples clustered together (Figure 6G), and the genes with increased or decreased expression in resistant cells relative to parental and resistant-shMYC cells are listed in Figure S6I. Expression changes for all MYC target genes are listed in Table S5, including those like the dasatinib target ERBB2 that are upregulated in a MYC-dependent fashion in resistant cells and may represent high-priority candidate therapeutic targets.
Figure 6. The BRAF/MEK inhibitor-resistant, MYC-activated state is associated with targetable vulnerabilities.

A) Average differential viability between parental A375 and PLX4720-resistant A375 cells treated at 2 μM and 10 μM with each of a 1753-member compound library. Compounds targeting SRC family members, c-KIT, and ERK pathway members are indicated. B) Fold change in dasatinib GI50 values in the indicated PLX-resistant lines relative to matched parental lines. P values indicate significance between parental and resistant derivatives. C) Fold change compared to DMSO treatment in the annexin V+ cell population fraction in parental or resistant A375 cells treated with 1 μM PLX4720, 100 nM dasatinib or the combination for 3 days. P values indicate significance between each treatment and PLX4720 treatment alone. D) Numbers of A375 cells over time during continuous culture in 3 μM PLX4720, 1 μM dasatinib, or the combination. Dashed line indicates initial seeding number. E) Change in expression of dasatinib targets as measured by RNA-seq in A375 parental cells, PLX4720-resistant A375 cells, and resistant cells expressing shMYC. The approximate IC50 of dasatinib for each target is shown. F) Average differential viability between parental A375 and PLX4720-resistant A375 cells treated at 2 μM and 10 μM with each of a 1753-member compound library. Compounds targeting dasatinib-target genes with differential expression in MYC-active vs. non-MYC-active cells are indicated. G) Change in expression of canonical MYC targets as measured by RNA-seq in A375 parental cells, PLX4720-resistant A375 cells, and resistant cells expressing shMYC. All data are means (SD) from 3 experiments. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001. See also Figure S6 and Tables S3–5.
MYC Activation in Resistant Cells Creates Targetable Metabolic Dependencies
A wide body of work has linked MYC activation, oncogenic progression, and cellular metabolic re-wiring (Stine et al., 2015). Further, our statistical analysis outlined in Figure 1 implicated several metabolic pathways as altered on progression (Table S1). To examine if MYC activation drives metabolic changes as cells develop BRAFi resistance, we performed integrated gene expression profiling, alongside metabolomics, in PLX4720-sensitive A375 cells expressing GFP-targeted shRNA (‘parental cells’), parental A375 cells expressing MYCT58A (‘MYCT58A cells’), PLX4720-resistant A375-shGFP (‘resistant cells’) and PLX4720-resistant A375-shMYC (‘shMYC cells’) (see Materials and Methods). Consistent with the concept that MYC drives global gene expression changes, unbiased hierarchical clustering of expression data showed that shMYC cells clustered more closely with parental cells than resistant, MYC activated cells (Figure S7A). Analysis of steady state metabolite levels using pathway enrichment revealed a diverse set of metabolic pathways altered in a MYC-dependent fashion in resistant cells relative to parental cells, as evidenced by the fact that reciprocal changes between the resistant and shMYC cells were observed (Figure 7A). Of note were alterations in glycolysis, serine, and glutamine metabolism, each of which has been implicated in MYC-driven oncogenesis (Jin et al., 2015; Pavlova and Thompson, 2016; Stine et al., 2015). Glycolysis-related gene sets were enriched in BRAFi/MEKi-resistant cell lines and human patient tumors (Figure 7B), and we observed modest MYC-dependent (as well as MYC-independent) changes in the expression of key early glycolytic enzymes (e.g., hexokinase 2 (HK2)) and in the steady-state levels of glycolytic intermediates, in particular, lactate (Figures 7B and S7B–D). Consistent with increases in lactate production, observed in a wide panel of parental and resistant derivative cell lines (Figure 7B, right panel), there was little change in the incorporation of 13C from glucose into citrate and other TCA cycle intermediates, indicating that the resistant cells are increasing aerobic glycolysis, similar to the switch away from anaerobic glycolysis observed during oncogenesis (Figure S7D). For serine synthesis, which occurs through oxidation of the glycolytic intermediate 3-phospho-D-glycerate (3PG), we observed enrichment of serine-synthesis related gene sets in resistant cell lines and human tumors, increased MYC-dependent expression of enzymes involved in the metabolism of serine into glycine to fuel one carbon metabolism in resistant cells, and corresponding MYC-dependent increases in 13C incorporation from glucose into serine, glycine and the purine precursor, IMP, in resistant cells (Figure 7C) (Locasale, 2013). Lastly, glutaminolysis has been shown to be provide important biosynthetic precursors in cancer cells that are primarily performing aerobic glycolysis (Filipp et al., 2012). We observed increases in glutamine pathway gene sets in resistant cell lines and tumors, increased MYC-dependent expression of key enzymes in glutaminolysis and the TCA cycle, and increased MYC-dependent incorporation of 13C into glutamate, aspartate, duhydroorotate, UTP and TCA cycle intermediates in cells grown in 13C-glutamine (13C-gln) media (but not in 13C-glucose media), indicative of preferential use of glutamine to supply biosynthetic building blocks, particularly those for fatty acid synthesis and pyrimidine synthesis (Figures 7D and S7D). Importantly, the increased activities of the glycolysis, serine, and glutamine pathways created new, MYC-dependent vulnerabilities, as diverse models of resistance were hypersensitive to the glycolysis inhibitor 2-deoxy-D-glucose (2-DG), the thymidylate synthase (TS) inhibitor fluorouracil (5-FU), the serine hydroxymethyltransferase (SHMT) inhibitor methotrexate, and the glutaminase inhibitor BPTES relative to sensitive cell lines (Figures 7E–G and S7E). We hypothesized that inhibition of these pathways may select against the development of resistance to BRAFi. For these experiments, we utilized BPTES and 2-DG, the latter of which was chosen for its potential to suppress both the glycolysis and serine pathways. While alone, 2-DG and BPTES treatment could delay resistance to PLX4720 by 1–2 weeks, their combination completely forestalled resistance (Figures 7H and S7F). This finding, which could be phenocopied using media containing glucose and/or glutamine at 10% of their standard concentrations (Figure 7I and S7G). At these concentrations, 2-DG, BPTES, and low glucose/glutamine media only modestly affected cell growth (Figure S7F–H). (Similarly delayed resistance was observed in cells treated with PLX4720 and low dose methotrexate (10nM) (Figure S7I).) Finally, the combination of 2DG and PLX4720 or BPTES and PLX4720 led to synergistic growth inhibition and apoptosis in PLX4720-resistant cells (Figures 7J and S7J). MYC-dependent metabolic changes described here, and their associated targetable vulnerabilities, are summarized in Figure 7K.
Figure 7. MYC activation in BRAFi resistant cells induces changes in the activities and dependencies associated with targetable metabolic pathways.

A) Top scoring metabolic pathways (Metaboanalyst) enriched in either resistant A375-shGFP (R) relative to parental A375-shGFP (P) cells or enriched in R relative to resistant A375-shMYC (shM) cells. Pathways are ranked by impact score in the former comparison. B) GSEA analysis (left) of differential gene expression in patient tumors and cell lines, in each case comparing resistant samples to their sensitive counterparts. Fold change (middle) in lactate levels in resistant and shMYC cells relative to parental. Fold change (right) in extracellular lactate in a panel of PLX-sensitive (blue) and PLX-resistant (red) cell lines normalized to cell number. C) GSEA analysis as in B) (left). Fold change in mRNA transcript levels for the indicated serine metabolism genes in P, R and shM cells (middle). 13C-glucose incorporation in the indicated metabolites in P, R and shM cells cultured after 24 hours in 13C-glucose (13C-Glc) (right). D) GSEA as in B) (upper left). Fold change in mRNA transcript levels for the indicated glutaminolysis pathways in P, R and shM cells (upper right). Relative abundance of 13C-glutamine (13C-Gln) for glutamate, aspartate and all TCA cycle intermediates (except citrate/isocitrate and dihydroorotate, m+4, and UTP, m+3) in P, R and shM cells cultured for 24 hours in 13C-Gln (lower left and right, respectively). E) GI50 values in multiple cell line models treated with the glycolysis inhibitor, 2-deoxy-D-glucose. F) GI50 values of lines treated with 5-FU. G) GI50 values of lines treated BPTES. Data in E), F) and G) are means of 3 experiments. P values denote significance between parental and each of the resistant groups measured by ordinary one-way ANOVA. PLX4720-sensitive cell lines are in blue and resistant cell lines are in red. H) Projected numbers of A375 cells over time during continuous culture in 3 μM PLX4720, 5 mM 2-DG, 10 μM BPTES, the double combinations, or the triple combination. I) A375 cell numbers during continuous culture in 3 μM PLX4720 in either normal DMEM containing 4.5 g/L glucose and 2mM glutamine, DMEM containing 0.45 g/L glucose and 2 mM glutamine, DMEM containing 4.5 g/L glucose and 200 μM glutamine, or DMEM containing 0.45 g/L glucose and 200 μM glutamine. In H) and I), a dashed line indicates initial seeding number. J) Fold change compared to DMSO in the annexin V+ cell population fraction in parental (P) or resistant (R(A)) A375 cells treated with 1 μM PLX4720, 1 mM 2-DG, 10 μM BPTES or the combinations for 3 days. P values are shown comparing each treatment to PLX4720 treatment. K) Summary of metabolic changes found between P, R and shM cell lines. Red arrows indicate metabolic flux increased in resistant cells and returned to parental levels upon MYC knock down. NADPH: Nicotinamide adenine dinucleotide phosphate GSH: glutathione ATP: Adenosine triphosphate NADH: Nicotinamide adenine dinucleotide G6P: glucose-6-phosphate 3PG: 3-phospho-glycerate 3P pyruvate: 3-phospho-pyruvate 3P ser: 3-phospho-serine Ser: serine Gly: glycine Gln: glutamine Glu: glutamate α-KG: α-ketoglutarate. Relationships are derived from a combination of gene expression, metabolite levels and glucose and glutamine 13C isotope tracing data. All data except E), F) and G) are means (SD) from 3 experiments. *P < 0.05; **P < 0.01; ***P < 0.005; ****P < 0.001. See also Figure S7.
DISCUSSION
The importance of MYC in tumorigenesis has long been appreciated, dating to its initial discovery through the investigation of oncogenic retroviruses (Sheiness et al., 1978). One of the most commonly deregulated oncogenes, MYC’s activity is tightly linked to major oncogenic signaling pathways. For example, the ERK and PI3K pathways are believed to regulate MYC protein levels through direct modulation of protein stability, while MYC activation downstream of Notch1 occurs via transcriptional activation (Dang, 2012; Palomero et al., 2006; Sears, 2004). MYC activation is linked to therapeutic resistance, for example in breast cancer cells treated with PI3K pathway inhibitors and in c-Met-addicted cancers treated with c-Met inhibitors (Ilic et al., 2011; Muellner et al., 2011; Shen et al., 2015). In melanoma, our finding that MYC is a nexus of convergent resistance is consistent with a recent report that used network modeling to nominate MYC as a synergistic target with BRAF, then verified this finding by demonstrating synergy between JQ1 and vemurafenib treatment in a cell line (Korkut et al., 2015). Our findings are also interesting in light of a report suggesting that the eukaryotic initiation factor 4F (eIF4F) complex may act as a point of convergence between the ERK and PI3K resistance pathways in melanoma, as MYC and eIF4F interact in a well-characterized synthetic lethal feedforward loop to support tumorigenesis (Boussemart et al., 2014; Lin et al., 2008; Lin et al., 2012). The molecular mechanisms by which MYC is regulated by the major pathways of resistance in melanoma are therefore important areas for future study.
The finding that MYC is a convergent downstream effector of resistance in melanoma reconciles disparate observations from the literature. First, it provides a mechanistic explanation for the observation that diverse resistance pathways can lead to a phenotypically convergent state and clarifies why certain oncogenic pathways can drive resistance while others seemingly cannot (Hugo et al., 2015; Martz et al., 2014; Rizos et al., 2014). Second, the fact that MYC is regulated both transcriptionally and post-translationally, and its activation downstream of major resistance pathways does not require alteration of its genomic locus, provides an explanation for why it was not uncovered in previous, in-depth genomic investigations of therapeutic resistance (Hugo et al., 2015; Moriceau et al., 2015; Rizos et al., 2014; Shi et al., 2014b; Van Allen et al., 2014; Wagle et al., 2014). Third, the concept of MYC-driven convergent resistance provides a rationale for the recent observations that cells with NRAS-driven resistance exhibit increased glutamine dependence and synergy between BRAF and glycolysis inhibition (Baenke et al., 2016; Hernandez-Davies et al., 2015; Parmenter et al., 2014). Finally, this work demonstrates that cells with intrinsic versus acquired resistance – which are driven by similar pathways (Martz et al., 2014) – are distinguished by their differential abilities to mobilize MYC following BRAF inhibition.
The data here reveal that as melanomas become resistant to BRAFi/MEKi, they become more dependent on MYC, suggesting that combination therapies that simultaneously target both states may have the unique property of selecting against resistance. The observation that combined BRAF/MEK/BET bromodomain inhibition exerts a more potent and durable effect in tumor models in vivo further supports this idea. Although BRD4/BET bromodomain targeting is one promising approach to block MYC activity, we also demonstrated that the MYC-dependent state in resistant melanomas can be targeted by exploiting other co-dependencies. However, it is clear that additional MYC synthetic lethal partners are likely to exist and may include regulators of transcription, nucleotide biosynthesis, the SUMOylation machinery, and the spliceosome, among other targets (Cunningham et al., 2014; Hsu et al., 2015; Kessler et al., 2012; Wang et al., 2015). Systematic studies to define and credential these MYC-dependent synthetic lethal partners in the specific context of BRAFi/MEKi-resistant melanoma are therefore warranted. Finally, it is tempting to speculate that other oncogene-driven cancers, including BRAF-driven non-melanoma tumors, may also exhibit convergent resistance to targeted therapy, and as such, this concept may provide a generalizable framework for designing robust therapies that select against resistance evolution.
EXPERIMENTAL PROCEDURES
Statistical modeling of genomic data
See Extended Experimental Methods for details.
Cell lines
Malme-3M, WM793, WM1745, SK-MEL-28, Lox IMVI, Hs294T, A2058 and RPMI-7951 cell lines were obtained from L. Garraway (Harvard University, Dana-Farber Cancer Institute). All other cell lines were purchased from the American Type Culture Collection. All lines were submitted to STR profiling by the Duke University DNA Analysis Facility to confirm their authenticity. See Extended Experimental Methods for culture conditions.
Chemicals
2-Deoxy-D-glucose (2-DG) and BPTES were purchased from Sigma-Aldrich. JQ1 and dasatinib were purchased from Apexbio Technology (Houston, TX). BS-181 was purchased from MedChem Express (Monmouth Junction, NJ). All other inhibitors were purchased from Selleck Chemicals (Houston, TX). For stock preparations, see Extended Experimental Methods.
GI50 and sensitization assays
In order to measure the GI50 values of specific inhibitors and quantify the effect of resistance pathway inhibition on the sensitivity of cell lines to ERK pathway inhibition, small molecule sensitization assays were performed as previously described (Martz et al., 2014). For details, see Extended Experimental Methods.
Combinatorial index value calculation
In order to assess synergy, the GI50 value of each inhibitor in the indicated cell lines was determined. 5,000 cells were plated per well in 96-well cell culture plates. Twenty-four hours later, the cells were treated with a 2-fold dilution series ranging from 4(GI50) to ¼(GI50) singly and in combination at a fixed ration of GI50 of inhibitor1: GI50 of inhibitor2. After 36 hours of treatment, cell viability relative to control was assessed and CI values were calculated according to the Chou and Talalay method (Chou and Talalay, 1984), using CompuSyn software (ComboSyn, Inc., Paramus, NJ).
Immunoblotting
To measure protein levels, aliquots of cell extracts prepared in lysis buffer (see Extended Experimental Methods) were submitted to SDS-PAGE. After electrophoretic transfer to PDMF, filters were blocked in 5% BSA and probed overnight at 4°C with primary antibodies (see Extended Experimental Methods). After washing in TBS-T, filters were incubated for 1 hour at room temperature with alkaline phosphatase-coupled goat anti-rabbit antibodies (Santa Cruz) and developed with Western Lightning Plus (Perkin Elmer, Inc., Waltham, MA).
Immunohistochemistry Staining
Melanoma patient samples (paraffin embedded slices) were obtained from Harvard (Dennie Frederick) and were then sent to the Duke Pathology Research Immunohistology Lab for c-MYC (Abcam 32072) staining. Samples were then sent to a pathologist (Shannon McCall) to be scored for nuclear c-MYC. Scoring was performed blinded to the staging of the samples.
Lentivirus preparation and DNA constructs
All expression clones were prepared in lentiviral form as previously described (Martz et al., 2014). See Extended Experimental Methods.
In vitro adaption of inhibitor resistant cells
PLX4720 or VX-11E-resistant cell lines were produced by 1 of 2 methods as previously described (Martz et al., 2014). See Extended Experimental Methods.
Clonogenic growth assay
To measure the ability of cell lines to form colonies from a single cell, clonogenic growth assays were performed as previously described (Singleton et al., 2015). Briefly, 100 cells were seeded per well in 12-well plates. After 24 hours, the indicated treatments were added and the assay was incubated for 14 days with the addition of fresh media and inhibitors after 7 days. Cells were fixed and stained with 0.5% (w/v) crystal violet in 6.0% (v/v) glutaraldehyde (Thermo Fisher Scientific, Waltham, MA).
Time to progression assay
To evaluate the relative ability of treatments to delay the reemergence of logarithmic cell growth in vitro (i.e. resistance), cells were plated in triplicate in 15 cm plates at 3,000,000 cells per plate in normal growth media. After 24 hours, the growth media was replaced with the indicated treatment. Seven days later, the cells were lifted with 0.25% trypsin (Life Technologies) and counted using a Z2 Coulter Particle Count and Size Analyzer (Beckman Coulter, Pasadena, CA). All cells up to 3,000,000 were centrifuged at 1200 RPM for 5 minutes and resuspended in 1 mL of media and then plated in a 15 cm plate with fresh treatment. This procedure was repeated weekly for 8–20 weeks, depending on the kinetics of resistance. Weekly growth rates (μ) were calculated from the number of cells plated the previous week (N0) and the number counted the current week (N) according to the formula:
where t = elapsed time or 168 hours. These growth rates were then used to project total cell number as if no cells had been discarded.
Annexin V apoptosis assay
The induction of apoptosis was quantified as described previously (Martz et al., 2014). See Extended Experimental Methods.
Xenograft model
Animal procedures were approved by the Center for Animal and Comparative Medicine in Harvard Medical School in accordance with the NIH Guild for Care and Use of Laboratory Animals and the Animal Welfare Act. Nude mice were inoculated subcutaneously with 3 × 106 human melanoma A375 or Hs695T cells in 200 μL PBS. Tumor volumes were measure twice a week and calculated with the formula [(length) × (width)2 × 0.52]. Once tumor volume reached ~300 mm3, mice were randomly divided into treatment groups and treated daily with BET inhibitor, JQ1 [45 mg/kg/day, intraperitoneal (i.p.) injection]; MEK inhibitor, GSK1120212 [0.6 mg/kg/day, oral gavage (o.g.)] and BRAF inhibitor, GSK2118436 (30mg/kg/day, o.g.); or a combination of JQ1, GSK1120212, and GSK2118436. Mice were treated daily for up to 45 days.
RNA extraction and quantitative real-time PCR
The total RNA of cells was isolated with QIAshredder Homogenizers and the RNEasy Mini kit (Qiagen) according to the manufacture’s instruction after a 72-hr treatment with DMSO or 1μM PLX4720. cDNA was then synthesized from 1μg RNA using iScript cDNA Synthesis kit (BioRad). Quantitative real-time PCR was conducted with iQ SYBR Green Supermix and a CFX384 Touch Real-Time PCR Detection System (BioRad). For PCR primers and conditions, see Extended Experimental Methods.
Gene expression analysis
RNA was isolated from whole cells with QIAshredder Homogenizers and the RNEasy Mini kit (Qiagen). RNA integrity was confirmed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA). RNA [20ng/μL] was submitted to the Duke University Sequencing and Genomic Technologies Core Facility. For sequencing conditions and data processing details, see Extended Experimental Methods.
Patient tumor samples
Serial tumor and blood samples were collected from male and female adult patients with Stage IV BRAF mutant melanoma under an IRB approved protocol at the Massachusetts General Hospital (DFHCC 11-181; PI: Boland). Patients were treated with combined BRAF + MEK inhibition. All patients provided written consent for both tissue acquisition and analysis, including all analyses performed in this study.
Metabolomics analysis
Cells were seeded in 6-well plates at an initial seeding density such that 1×106 cells would be harvested after 48 hours in culture. After 24-hrs growth, cells were rinsed with PBS and RPMI-1640 (Life Technologies), RPMI-1640 with 1g/L D-glucose 13C6 (Cambridge Isotope Laboratories, Inc., Tewksbury, MA) or RPMI-1640 with 300mg/L L-glutamine 13C5 (Sigma-Aldridge) was added in triplicate. After 24 hours, cells were harvested in 80% methanol. Cell extracts were pelleted and dried by speed vacuum. Metabolomics study was performed on liquid chromatography-high resolution mass spectrometer (LC-HRMS) Q Exactive plus (Liu et al., 2014). The peak area was used to present relative abundance and to calculate the isotopologue distribution. The natural abundance correction was done based on previously published method (Yuan et al., 2008).
Chemical Screen
A375 cells or a PLX4720-resistant A375 derivative were plated into a 384 well-plate (Corning 3707) at 200 cells/well using an automated liquid dispenser (BioTek EL406). The next day, 50 nL of bioactive compounds in DMSO were added to the assay plate via pin transfer to give a final compound concentration of 10 and 2 μM, respectively. Three days after compound treatment, cell viability was measured with CellTiter-Glo (Promega). Luminescence values were obtained using a multimode plate reader, and percent viability was calculated by comparison to DMSO-treated cells.
Statistics
All results are shown as means±SD. Unless noted otherwise, P values were determined using unpaired, two-tailed Student’s t tests. Unless otherwise noted, n=3 independent, biological replicates.
Supplementary Material
Related to Figure 1. Refer to Supplementary Information for additional details.
Related to Figure 6. Average differential viability indicates the fractional viability of parental A375 cells minus that of matched, MYC-dependent, evolved resistant A375 cells averaged across two compound doses (2 and 10 μM).
Related to Figure 6. Genes highlighted in blue are expressed more highly in BRAFi-sensitive cells (Parental-shGFP and Resistant-shMYC). Genes highlighted in red are expressed more highly in BRAFi-resistant cells (Resistant-shGFP). Statistics were calculated using the GenePattern module, ComparativeMarkerSelection (v10). Genes are ranked by test statistic (T-Test). See http://software.broadinstitute.org/cancer/software/genepattern/modules/docs/ComparativeMarkerSelection/10 for details.
Related to Figure 6. Genes highlighted in blue are expressed more highly in BRAFi-sensitive cells (Parental-shGFP and Resistant-shMYC). Genes highlighted in red are expressed more highly in BRAFi-resistant cells (Resistant-shGFP). Statistics were calculated using the GenePattern module, ComparativeMarkerSelection (v10). Genes are ranked by test statistic (T-Test). See http://software.broadinstitute.org/cancer/software/genepattern/modules/docs/ComparativeMarkerSelection/10 for details.
Related to Materials and Methods.
Related to Materials and Methods. Comparisons are between: (1) empirical Bayes linear model (limma), and (2) a likelihood ratio test (LRT). Genes were included in a signature if they had a Benjamini-Hochberg corrected p-value (BH q-value) below 0.05. Refer to Supplementary Information for additional details.
Highlights.
Major pathways of resistance in BRAF-mutant melanoma converge to activate MYC.
MYC gene signatures are suppressed on therapy and rebound on progression.
MYC activation is necessary and sufficient for resistance to BRAFi in vitro.
The MYC-activated, BRAFi-resistant state is associated with targetable vulnerabilities.
Acknowledgments
The pBABE-puro and pBABE-puro-MYC-T58A expression vectors were provided by Dr. Christopher Counter. We thank Tim Reddy and Alex Barrera for assistance with genomic data analysis. L.A.C. is the recipient of an NSF Graduate Research Fellowship (DGF-1106401). This work was supported by the Duke University School of Medicine, the NIH Building Interdisciplinary Research Careers in Women’s Health Program (K12HD043446), a Stewart Trust Fellowship, a V Scholar Award from the V Foundation for Cancer Research, a Translational Research Award from the Lloyd Charitable Trust, and NIH award R01CA207083, all to K.C.W. The authors declare no conflicts of interest.
Footnotes
ACCESSION NUMBERS
All sequencing data can be accessed in the Gene Expression Omnibus (GEO), accession number, GSE99923.
AUTHOR CONTRIBUTIONS
Conceptualization, K.R.S., L.C., S.M. and K.C.W., Methodology, K.R.S. and L.C., Software, L.C. and S.M., Validation, K.R.S., H.E.M., E.T., A.N.M., E.M.S., J.P.T., and K.S., Formal Analysis, L.C., X.L. and M.V.L., Investigation, K.R.S., H.E.M., E.T., A.N.M., E.M.S, J.P.T., X.L., and K.S. Resources, M.H., D.J.A., J.W.L., K.C., and K.C.W., Data Curation, L.C., X.L., M.V.L., A.N.M., Writing – Original Draft, K.R.S., L.C., and K.C.W., Writing – Review & Editing, all authors, Visualization, K.R.S., L.C., Supervision, M.H., D.J.A., J.W.L., K.C., S.M., and K.C.W. Funding Acquisition, M.H., D.J.A., K.C., and K.C.W.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackerman A, Klein O, McDermott DF, Wang W, Ibrahim N, Lawrence DP, Gunturi A, Flaherty KT, Hodi FS, Kefford R, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695–1701. doi: 10.1002/cncr.28620. [DOI] [PubMed] [Google Scholar]
- Alcala AM, Flaherty KT. BRAF inhibitors for the treatment of metastatic melanoma: clinical trials and mechanisms of resistance. Clin Cancer Res. 2012;18:33–39. doi: 10.1158/1078-0432.CCR-11-0997. [DOI] [PubMed] [Google Scholar]
- Baenke F, Chaneton B, Smith M, Van Den Broek N, Hogan K, Tang H, Viros A, Martin M, Galbraith L, Girotti MR, et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol Oncol. 2016;10:73–84. doi: 10.1016/j.molonc.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, Thomas M, Basmadjian C, Ribeiro N, Thuaud F, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513:105–109. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- Cermelli S, Jang IS, Bernard B, Grandori C. Synthetic lethal screens as a means to understand and treat MYC-driven cancers. Cold Spring Harb Perspect Med. 2014:4. doi: 10.1101/cshperspect.a014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126–1139. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford L, Wood KC, Zhou X, Mukherjee S. Bayesian approximate kernel regression with variable selection. J Am Stat Assoc. 2017 doi: 10.1080/01621459.2017.1361830. (In press, available online at http://amstat.tandfonline.com/doi/full/10.1080/01621459.2017.1361830) [DOI] [PMC free article] [PubMed]
- Cunningham JT, Moreno MV, Lodi A, Ronen SM, Ruggero D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell. 2014;157:1088–1103. doi: 10.1016/j.cell.2014.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J, Issa G, Lemieux M, Rahl P, Shi J, Jacobs H, Kastritis E, Gilpatrick T, Paranal R, Qi J, et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, Kremer M, Graf N, Scheerer M, Hall MA, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116:1498–1505. doi: 10.1182/blood-2009-11-251074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorenko IV, Smalley KS. The complexity of microenvironment-mediated drug resistance. Genes Cancer. 2015;6:367–368. doi: 10.18632/genesandcancer.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipp FV, Ratnikov B, De Ingeniis J, Smith JW, Osterman AL, Scott DA. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Pigment Cell Melanoma Res. 2012;25:732–739. doi: 10.1111/pcmr.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–1231. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goga A, Yang D, Tward AD, Morgan DO, Bishop JM. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med. 2007;13:820–827. doi: 10.1038/nm1606. [DOI] [PubMed] [Google Scholar]
- Hernandez-Davies JE, Tran TQ, Reid MA, Rosales KR, Lowman XH, Pan M, Moriceau G, Yang Y, Wu J, Lo RS, et al. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J Transl Med. 2015;13:210. doi: 10.1186/s12967-015-0581-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, Okimoto RA, Lin L, Neel DS, Sabnis A, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med. 2015;21:1038–1047. doi: 10.1038/nm.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525:384–388. doi: 10.1038/nature14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162:1271–1285. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci U S A. 2011;108:E699–708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene. 2015;35:3619–3625. doi: 10.1038/onc.2015.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–827. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkut A, Wang W, Demir E, Aksoy BA, Jing X, Molinelli EJ, Babur O, Bemis DL, Onur Sumer S, Solit DB, et al. Perturbation biology nominates upstream-downstream drug combinations in RAF inhibitor resistant melanoma cells. Elife. 2015:4. doi: 10.7554/eLife.04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CJ, Cencic R, Mills JR, Robert F, Pelletier J. c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer Res. 2008;68:5326–5334. doi: 10.1158/0008-5472.CAN-07-5876. [DOI] [PubMed] [Google Scholar]
- Lin CJ, Nasr Z, Premsrirut PK, Porco JA, Jr, Hippo Y, Lowe SW, Pelletier J. Targeting synthetic lethal interactions between Myc and the eIF4F complex impedes tumorigenesis. Cell Rep. 2012;1:325–333. doi: 10.1016/j.celrep.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015;47:250–256. doi: 10.1038/ng.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ser Z, Locasale JW. Development and quantitative evaluation of a high-resolution metabolomics technology. Anal Chem. 2014;86:2175–2184. doi: 10.1021/ac403845u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens O, Johnson B, Hollstein P, Frederick DT, Cooper ZA, Messiaen L, Bronson RT, McMahon M, Granter S, Flaherty K, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013;3:338–349. doi: 10.1158/2159-8290.CD-12-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins MM, Zhou AY, Corella A, Horiuchi D, Yau C, Rakshandehroo T, Gordan JD, Levin RS, Johnson J, Jascur J, et al. Linking tumor mutations to drug responses via a quantitative chemical-genetic interaction map. Cancer Discov. 2015;5:154–167. doi: 10.1158/2159-8290.CD-14-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martz CA, Ottina KA, Singleton KR, Jasper JS, Wardell SE, Peraza-Penton A, Anderson GR, Winter PS, Wang T, Alley HM, et al. Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci Signal. 2014;7:ra121. doi: 10.1126/scisignal.aaa1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale S, Bozic I, Tong J, Peraza-Penton A, Lallo A, Baldi F, Lin KH, Di Nicolantonio F, Nowak MA, Zhang L, et al. Vertical suppression of the EGFR pathway delays onset of resistance in colorectal cancer models. Nat Commun. 2015;6:8305–8314. doi: 10.1038/ncomms9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell. 2015;27:240–256. doi: 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, Craig-Mueller N, Colinge J, Duernberger G, Nijman SM. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787–793. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O’Neil J, Neuberg D, Weng AP, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014;4:423–433. doi: 10.1158/2159-8290.CD-13-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2015 doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965–1977. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- Sears RC. The life cycle of C-myc: from synthesis to degradation. Cell Cycle. 2004;3:1133–1137. [PubMed] [Google Scholar]
- Settleman J. Oncogene addiction. Curr Biol. 2012;22:R43–44. doi: 10.1016/j.cub.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Sheiness D, Fanshier L, Bishop JM. Identification of nucleotide sequences which may encode the oncogenic capacity of avian retrovirus MC29. J Virol. 1978;28:600–610. doi: 10.1128/jvi.28.2.600-610.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A, Wang L, Huang M, Sun J, Chen Y, Shen YY, Yang X, Wang X, Ding J, Geng M. c-Myc alterations confer therapeutic response and acquired resistance to c-Met inhibitors in MET-addicted cancers. Cancer Res. 2015;75:4548–4559. doi: 10.1158/0008-5472.CAN-14-2743. [DOI] [PubMed] [Google Scholar]
- Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, Hugo W, Yu CC, Ng C, Chodon T, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014a;4:69–79. doi: 10.1158/2159-8290.CD-13-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014b;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton KR, Hinz TK, Kleczko EK, Marek LA, Kwak J, Harp T, Kim J, Tan AC, Heasley LE. Kinome RNAi Screens Reveal Synergistic Targeting of MTOR and FGFR1 Pathways for Treatment of Lung Cancer and HNSCC. Cancer Res. 2015;75:4398–4406. doi: 10.1158/0008-5472.CAN-15-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solit DB, Rosen N. Towards a unified model of RAF inhibitor resistance. Cancer Discov. 2014;4:27–30. doi: 10.1158/2159-8290.CD-13-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg M. Dasatinib: a tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and philadelphia chromosome-positive acute lymphoblastic leukemia. Clin Ther. 2007;29:2289–2308. doi: 10.1016/j.clinthera.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Stephens M. False discovery rates: A new deal. bioRxiv. 2016 doi: 10.1093/biostatistics/kxw041. http://biorxiv.org/content/early/2016/2001/2029/038216. [DOI] [PMC free article] [PubMed]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18:721–727. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK, et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer Discov. 2015;5:960–971. doi: 10.1158/2159-8290.CD-15-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman AW, Kristjansdottir K, Wolfgeher D, Hasin N, Polier S, Zhang H, Perrett S, Prodromou C, Jones GW, Kron SJ. CDK-dependent Hsp70 Phosphorylation controls G1 cyclin abundance and cell-cycle progression. Cell. 2012;151:1308–1318. doi: 10.1016/j.cell.2012.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu XW, Gimotty PA, Kee D, et al. Acquired Resistance to BRAF Inhibitors Mediated by a RAF Kinase Switch in Melanoma Can Be Overcome by Cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61–68. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M, Sears RC. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 2011;71:925–936. doi: 10.1158/0008-5472.CAN-10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell. 2015;163:174–186. doi: 10.1016/j.cell.2015.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wargo JA, Cooper ZA, Flaherty KT. Universes collide: combining immunotherapy with targeted therapy for cancer. Cancer Discov. 2014;4:1377–1386. doi: 10.1158/2159-8290.CD-14-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE, Garraway LA. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3:350–362. doi: 10.1158/2159-8290.CD-12-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KC, Konieczkowski DJ, Johannessen CM, Boehm JS, Tamayo P, Botvinnik OB, Mesirov JP, Hahn WC, Root DE, Garraway LA, et al. MicroSCALE screening reveals genetic modifiers of therapeutic response in melanoma. Sci Signal. 2012;5:rs4. doi: 10.1126/scisignal.2002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci U S A. 2010;107:13836–13841. doi: 10.1073/pnas.1008366107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Bennett BD, Rabinowitz JD. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat Protoc. 2008;3:1328–1340. doi: 10.1038/nprot.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller KI, Jegga AG, Aronow BJ, O’Donnell KA, Dang CV. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figure 1. Refer to Supplementary Information for additional details.
Related to Figure 6. Average differential viability indicates the fractional viability of parental A375 cells minus that of matched, MYC-dependent, evolved resistant A375 cells averaged across two compound doses (2 and 10 μM).
Related to Figure 6. Genes highlighted in blue are expressed more highly in BRAFi-sensitive cells (Parental-shGFP and Resistant-shMYC). Genes highlighted in red are expressed more highly in BRAFi-resistant cells (Resistant-shGFP). Statistics were calculated using the GenePattern module, ComparativeMarkerSelection (v10). Genes are ranked by test statistic (T-Test). See http://software.broadinstitute.org/cancer/software/genepattern/modules/docs/ComparativeMarkerSelection/10 for details.
Related to Figure 6. Genes highlighted in blue are expressed more highly in BRAFi-sensitive cells (Parental-shGFP and Resistant-shMYC). Genes highlighted in red are expressed more highly in BRAFi-resistant cells (Resistant-shGFP). Statistics were calculated using the GenePattern module, ComparativeMarkerSelection (v10). Genes are ranked by test statistic (T-Test). See http://software.broadinstitute.org/cancer/software/genepattern/modules/docs/ComparativeMarkerSelection/10 for details.
Related to Materials and Methods.
Related to Materials and Methods. Comparisons are between: (1) empirical Bayes linear model (limma), and (2) a likelihood ratio test (LRT). Genes were included in a signature if they had a Benjamini-Hochberg corrected p-value (BH q-value) below 0.05. Refer to Supplementary Information for additional details.