

Key Points
Blockade of CD7 expression with a novel method, combined with a second-generation CAR, results in highly potent anti-CD7 CAR T cells.
This practical strategy provides a new treatment option for patients with high-risk T-cell malignancies, including ETP-ALL.
Abstract
Effective immunotherapies for T-cell malignancies are lacking. We devised a novel approach based on chimeric antigen receptor (CAR)–redirected T lymphocytes. We selected CD7 as a target because of its consistent expression in T-cell acute lymphoblastic leukemia (T-ALL), including the most aggressive subtype, early T-cell precursor (ETP)–ALL. In 49 diagnostic T-ALL samples (including 14 ETP-ALL samples), median CD7 expression was >99%; CD7 expression remained high at relapse (n = 14), and during chemotherapy (n = 54). We targeted CD7 with a second-generation CAR (anti-CD7–41BB-CD3ζ), but CAR expression in T lymphocytes caused fratricide due to the presence of CD7 in the T cells themselves. To downregulate CD7 and control fratricide, we applied a new method (protein expression blocker [PEBL]), based on an anti-CD7 single-chain variable fragment coupled with an intracellular retention domain. Transduction of anti-CD7 PEBL resulted in virtually instantaneous abrogation of surface CD7 expression in all transduced T cells; 2.0% ± 1.7% were CD7+ vs 98.1% ± 1.5% of mock-transduced T cells (n = 5; P < .0001). PEBL expression did not impair T-cell proliferation, interferon-γ and tumor necrosis factor–α secretion, or cytotoxicity, and eliminated CAR-mediated fratricide. PEBL-CAR T cells were highly cytotoxic against CD7+ leukemic cells in vitro and were consistently more potent than CD7+ T cells spared by fratricide. They also showed strong anti-leukemic activity in cell line– and patient-derived T-ALL xenografts. The strategy described in this study fits well with existing clinical-grade cell manufacturing processes and can be rapidly implemented for the treatment of patients with high-risk T-cell malignancies.
Visual Abstract
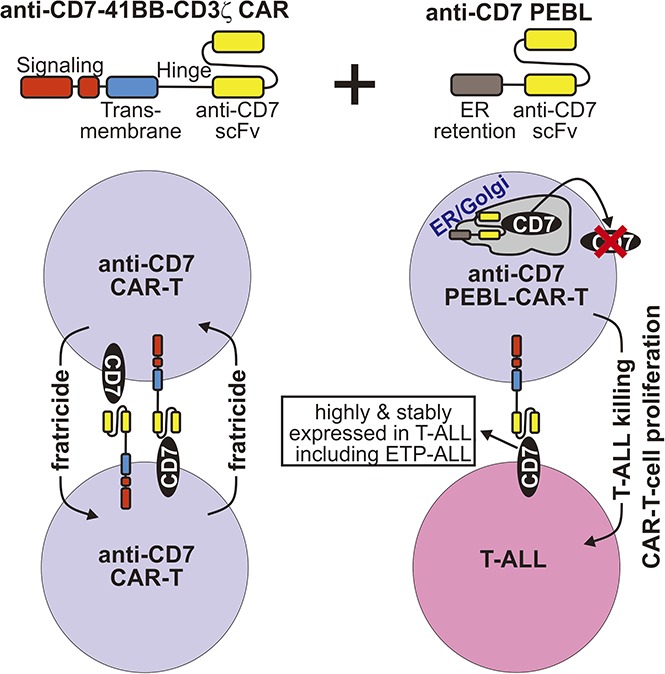
Introduction
T lymphocytes can be induced to specifically recognize and kill tumor cells through the expression of chimeric antigen receptors (CARs).1-5 Central to the effective application of this technology is the identification of a suitable target for the CAR. This must be highly expressed by tumor cells and should be absent in normal cells, or be expressed only by normal cells whose temporary absence is clinically manageable.6 Thus, leukemias and lymphomas of B-cell origin can be targeted with CARs directed against CD195,7 or CD22,8 which are normally expressed only by B-lymphoid cells.9,10 Infusion of autologous T cells expressing anti-CD19 CARs in patients with B-cell refractory leukemia and lymphoma resulted in major clinical responses.11-18 These exciting results have provided indisputable evidence of the power of this technology and suggest the possibility of wider applications in oncology.
The development of CAR T-cell therapies for T-cell malignancies has lagged far behind that of their B-cell counterparts. The need for effective therapies in this area is particularly urgent because of the poor prognosis associated with some T-cell leukemia and lymphoma subtypes. For example, children and adolescents with early T-cell progenitor (ETP) acute lymphoblastic leukemia (ALL) have the poorest response to initial therapy among all patients with ALL.19-21 Intensive chemotherapy and/or allogeneic hematopoietic stem cell transplant often do not prevent treatment-refractory relapse; for these patients, and those with other high-risk features, such as adult age, there is a dearth of treatment options.19,22-25
A major obstacle to the development of effective CAR T cells for T-cell malignancies is that the surface marker profile of malignant T cells (which generally lack CD19 or CD22 expression) largely overlaps that of activated T lymphocytes.19,26 CARs directed against such targets are likely to lead to the self-elimination of the CAR T cells.27,28 In this study, we sought to develop a practical technology for CAR T-cell therapy of ETP-ALL and other T-cell acute lymphoblastic leukemia (T-ALL) subtypes. First, we made a CAR directed against CD7, a 40-kDa type I transmembrane glycoprotein, which is a primary marker for T-cell malignancies,29-32 and is highly expressed in all cases of T-cell ALL, including ETP-ALL.19 Second, we designed a way to rapidly and effectively downregulate CD7 expression in T cells, which averts the fratricide effect, does not involve gene editing, and can be immediately translated into clinical application.
Materials and methods
Cells and culture conditions
The leukemia cell lines Jurkat, CCRF-CEM, Loucy, MOLT4, and KG1a were from the American Type Culture Collection (Rockville, MD). The B-lineage ALL cell line OP-1 was developed in our laboratory.33 We transduced CCRF-CEM cells with a murine stem cell virus (MSCV)–internal ribosome entry site–green fluorescent protein (GFP) retroviral vector (Vector Development and Production Shared Resource Laboratory, St. Jude Children’s Research Hospital, Memphis, TN) containing the firefly luciferase gene. We used the same vector to transduce CCRF-CEM and Jurkat cells with the CD19 gene, which we cloned from the complementary DNA of the RS4;11 B-cell line (American Type Culture Collection). Cell lines were maintained in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin.
Peripheral blood samples were obtained from discarded anonymized byproducts of platelet donations from healthy adult donors at the National University Hospital Blood Bank, Singapore. Bone marrow aspirates from patients with ALL were obtained for diagnostic immunophenotyping and monitoring of treatment response19,26; banked surplus material was used in some experiments, with approval from the Institutional Review Board of the National University of Singapore. Mononucleated cells were separated by centrifugation on a Lymphoprep density step (Axis-Shield, Oslo, Norway) and washed twice in RPMI 1640. T cells were enriched with Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific) and cultured in RPMI 1640, 10% FBS, 1% penicillin-streptomycin, and interleukin-2 (IL-2; 120 IU/mL; Proleukin, Novartis, Basel, Switzerland).
Gene cloning, retroviral transduction, and messenger RNA electroporation
The single-chain variable fragment (scFv) of the anti-CD7 monoclonal antibody TH6934 was joined to the CD8α signal peptide, CD8α hinge and transmembrane domain, and the intracellular domains of 4-1BB and CD3ζ of an anti-CD19–41BB-CD3ζ CAR previously developed in our laboratory.5 The same scFv was also joined to the CD8α signal peptide and sequences encoding endoplasmic reticulum (ER)/Golgi retention peptides EQKLISEEDLKDEL, (GGGGS)4AEKDEL, or CD8α hinge and transmembrane domain followed by LYKYKSRRSFIDEKKMP. These were subcloned into the MSCV vector, with or without GFP or mCherry.
Preparation of the retroviral supernatant and transduction was performed as previously described.5,35 Briefly, pMSCV retroviral vector–conditioned medium was added to RetroNectin-coated (Takara, Otsu, Japan) polypropylene tubes. After centrifugation and removal of the supernatant, T cells were added to the tubes and left at 37°C for 12 hours. Fresh viral supernatant was added on 2 other successive days. T lymphocytes were maintained in RPMI 1640 with FBS, antibiotics, and 200 IU/mL IL-2.
For transient CAR expression, anti-CD7 and anti-CD19 CAR constructs were subcloned into EcoRI and XhoI sites of the pVAXI vector (Thermo Fisher Scientific) and transcribed into messenger RNA (mRNA) using T7 mScript (CellScript, Madison, WI).36 For mRNA electroporation, cells were suspended in electroporation buffer (Amaxa Cell Line Nucleofector Kit V, Lonza, Basel, Switzerland) containing 200 μg of CAR mRNA, and electroporated with an Amaxa Nucleofector 2b (Lonza) using program X-001.36,37 Cells electroporated without mRNA were used as control.
Detection of CAR, protein expression blocker, and surface markers
CARs were detected with a biotin-conjugated goat anti-mouse F(ab′)2 antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) followed by allophycocyanin (APC)-conjugated streptavidin (Jackson ImmunoResearch Laboratories). Phycoerythrin (PE)- or APC-conjugated anti-CD7 (M-T701), CD4 (RPA-T4), CD8 (RPA-T8), CD3 (SK7), and nonreactive isotype-matched antibodies were from BD Biosciences (San Jose, CA). CD19 (LT19) was from Miltenyi Biotech. Cell staining was analyzed using Accuri C6, Fortessa, or LSRII flow cytometers (BD Biosciences) with Diva (BD Biosciences) or FlowJo software (FlowJo, Ashland, OR).
Western blotting was performed as previously described.35 Briefly, cell lysates were extracted using CelLytic M cell lysis reagent (Sigma-Aldrich, Saint Louis, MO) before protein quantification with Pierce BCA protein assay kit (Thermo Fisher Scientific). Cell lysates were diluted with 4× Laemmli sample buffer (Bio-Rad, Hercules, CA) and separated on 10% polyacrylamide gel by electrophoresis under reducing or nonreducing conditions. Blotted membranes were probed with mouse anti-human CD3ζ antibody (8D3; BD Biosciences), goat anti-mouse immunoglobulin G horseradish peroxidase–conjugated (R&D Systems, Minneapolis, MN), and Clarity Western ECL substrate (Bio-Rad). Staining was visualized using ChemiDoc Touch Imager (Bio-Rad).
Cell aggregation, cytotoxicity, cell proliferation, and cytokine production
To measure cell-cell aggregation, Jurkat cells were cocultured with CD7+ or CD7– cells labeled with calcein red-orange AM (Thermo Fisher Scientific) for 30 minutes; cell doublets were counted by flow cytometry. In some experiments, target cells were preincubated for 10 minutes before coculture with a soluble anti-CD7 scFv obtained from the supernatant of Jurkat or 293T cells transduced with a construct consisting of the scFv without transmembrane or signaling sequences.
To test cytotoxicity, target cells were labeled with calcein red-orange AM and placed into a 96-well, round bottom plate (Corning Costar, Corning, NY). T cells were added at different effector-to-target (E:T) ratios with target cells and cultured for 4 hours at 37°C and 5% carbon dioxide. Viable target cells were counted by flow cytometry.38 To measure exocytosis of lytic granules, anti-human CD107a-PE (H4A3; BD Biosciences) was added to the cocultures. After 1 hour, monensin (BD GolgiStop) was added, and cultures were continued for another 3 hours before flow cytometric analysis.
To assess cell proliferation, T cells were cultured alone or in the presence of MOLT-4 cells at a 1:1 E:T ratio in RPMI 1640 with FBS and 120 IU/mL IL-2 at 37°C and 5% carbon dioxide. Target cells, irradiated or treated with Streck cell preservative (Streck Laboratories, Omaha, NE) to inhibit proliferation, were added to the cultures every 7 days. Viable GFP+ or mCherry+ T cells were enumerated by flow cytometry. For interferon-γ (IFN-γ) and tumor necrosis factor–α (TNF-α) production, target and effector cells at a 1:1 E:T ratio were plated as above. After 1 hour, brefeldin A (BD GolgiPlug) was added to the cultures, which continued for another 5 hours. Subsequently, intracellular staining with anti–IFN-γ–PE (clone 25723.11; BD Biosciences) or anti–TNF-α–PE (6401.1111; BD Biosciences) was done before flow cytometric analysis.
Xenograft models
CCRF-CEM cells transduced with luciferase were injected IV in NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NOD/scid IL2RGnull) mice (The Jackson Laboratory, Bar Harbor, ME) at 1 × 106 cells per mouse. Three and/or 7 days later, mice received T cells with downregulated CD7 and anti-CD7 CAR expression at 2 × 107 T cells per mouse. Other mice received T cells transduced with GFP alone or RPMI 1640 with 10% FBS instead of T cells. All mice received 20 000 IU of IL-2 intraperitoneally (IP) every 2 days. Tumor load was determined using the Xenogen IVIS-200 System (Caliper Life Sciences, Waltham, MA) after injecting aqueous d-luciferin potassium salt (Perkin Elmer, Waltham, MA) IP (2 mg per mouse). Luminescence was analyzed with the Living Image 3.0 software. Mice were euthanized when luminescence reached 1 × 1010 photons per second or sooner if physical signs warranting euthanasia appeared.
For the patient-derived xenograft (PDX) model, primary ETP-ALL cells were injected IV in NOD/scid IL2RGnull mice and propagated for 7 to 8 subsequent generations. ETP-ALL cells were then reinjected in NOD/scid IL2RGnull mice, which were either treated with protein expression blocker (PEBL)-CAR T cells or left untreated. Peripheral blood and tissues were monitored for the presence of ALL cells by flow cytometry.19,26 After treatment with red blood cell lysing buffer (Sigma-Aldrich), cells were stained with anti-mouse CD45-PE-cyanine 7 (30-F11, BioLegend), as well as anti-human CD45-APC-H7 (2D1), CD7-PE (M-T701), CD3 APC (SK7), CD34-peridinin chlorophyll protein (8G12) (all from BD Biosciences), and CD33-Brilliant Violet 421 (WM53, BioLegend). Cells were analyzed with a Fortessa flow cytometer using Diva and FlowJo software.
Results
Validation of CD7 as a target for CAR T-cell therapy in leukemia
In leukemic cells from diagnostic bone marrow samples obtained from 49 patients with T-ALL (including 14 patients with ETP-ALL), the median percentage of CD7 expression was >99% (range, 79% to >99%). In only 3 cases (6.1%), CD7 was <99%: 98% in 2 cases and 79% in 1 case (Figure 1A). High CD7 expression was also observed in samples collected from 14 patients with relapsed T-ALL (Figure 1A). Mean fluorescence intensity (MFI) of CD7 in leukemic cells at diagnosis or relapse consistently exceeded that measured in residual normal T cells in the same samples. The median MFI was 20 617 (range, 4105-66 674) in T-ALL cells vs 3032 (range, 1301-9582) in normal T cells (n = 19; P < .0001) (Figure 1B).
Figure 1.

CD7 expression in T-ALL. (A) The percentage of ALL cells expressing CD7 at diagnosis, relapse, and during chemotherapy (MRD); the number of bone marrow samples studied at each stage is shown. (B) CD7 mean fluorescence intensity (MFI) in T-ALL cells and residual normal T cells from the same samples (n = 19; ****P < .0001 by paired Student t test). (C) CD7 MFI in T-ALL cells at diagnosis or relapse (D/R) and in follow-up bone marrow samples with MRD (n = 18). (D) Flow cytometric contour plots illustrate CD7 expression in T-ALL cells (CD3 negative) and normal T cells (CD3 positive) at diagnosis, MRD, and relapse in 1 representative patient.
To determine whether chemotherapy affected CD7 expression, we examined bone marrow samples collected during therapy that contained minimal residual disease (MRD). In all 54 samples (from 21 patients), >99% of residual leukemic cells were CD7+ (Figure 1A). In 18 patients, we monitored CD7 levels during the course of the disease. As shown in Fig. 1C-D, CD7 remained high during therapy. These results validate CD7 as a target for CAR T-cell therapy in T-ALL.
Design and expression of an anti-CD7 CAR
To target CD7, we designed an anti-CD7 CAR composed of the scFv of the anti-CD7 antibody TH69 joined to the signaling domains of 4-1BB (CD137) and CD3ζ via the hinge and transmembrane domain of CD8α (Figure 2A). Retroviral transduction of this construct in Jurkat cells resulted in high expression of anti-CD7 CAR (Figure 2B), which appeared as monomer, dimer, and oligomer by western blotting (Figure 2C).
Figure 2.

Design, expression, and signaling of the anti-CD7 CAR. (A) Schema of the anti-CD7–41BB-CD3ζ construct. (B) Flow cytometric analysis of Jurkat cells transduced with either GFP alone (Mock) or GFP plus anti-CD7 CAR. Dot plots illustrate GFP fluorescence, and CAR expression after staining with biotin-conjugated goat anti-mouse F(ab′)2 antibody and streptavidin-APC (Jackson ImmunoResearch Laboratories). (C) Western blot analysis of CAR expression in Jurkat cells. Cell lysates of mock- and CAR-transduced Jurkat cells were separated on a 10% polyacrylamide gel under reducing or nonreducing conditions. The blotted membrane was probed with mouse anti-human CD3ζ antibody (8D3; BD Biosciences) and goat anti-mouse immunoglobulin G conjugated to horseradish peroxidase (R&D Systems). Antibody binding was revealed with Clarity Western ECL Substrate (Bio-Rad). (D) Anti-CD7 CAR induces expression of activation markers on ligation. Bars show the mean (± SD) of CD25 and CD69 MFI in CAR- and mock-transduced Jurkat cells after 24 hours with or without CD7+ MOLT-4 cells. P values by Student t test are shown for significant differences (*P = .016; ***P < .001). (E) Representative flow cytometric histograms of the experiments shown in panel D.
To confirm that the TH69 scFv could bind CD7, we produced it in soluble form and tested it on CD7+ MOLT-4 and CD7– OP-1 cells; MOLT-4 cells were labeled, whereas OP-1 cells were not (supplemental Figure 1A). In addition, staining with an anti-CD7 monoclonal antibody was significantly reduced when MOLT-4 cells were preincubated with the anti-CD7 scFv supernatant; CD7 MFI ± standard deviation (SD) went from 31 730 ± 1144 to 5987 ± 241 (n = 3). Jurkat cells expressing an anti-CD7 CAR formed aggregates with CD7+ MOLT-4 cells, whereas those transduced with GFP only or an anti-CD19 CAR did not; conversely, the anti-CD19 CAR induced cell aggregation with CD19+ OP-1 cells, whereas the anti-CD7 CAR did not (supplemental Figure 1B). Preincubation of MOLT-4 or CCRF-CEM with the soluble anti-CD7 scFv prevented the formation of aggregates (supplemental Figure 1C).
To determine whether the anti-CD7 CAR was functional, we measured levels of the activation markers CD25 and CD69 in Jurkat cells after 24-hour coculture with MOLT4. There was a clear upregulation of both activation markers in cells expressing the anti-CD7 CAR (Figure 2D-E). In sum, the anti-CD7–41BB-CD3ζ CAR can bind to its cognate antigen and transduces activation signals on ligation.
Expression of anti-CD7 CAR in T cells causes fratricide
To determine the effects of anti-CD7–41BB-CD3ζ CAR in peripheral blood T lymphocytes, we used 2 different methods to express it: retroviral transduction (supplemental Figure 2) and mRNA electroporation. However, it markedly reduced T-cell viability. Mean ± SD T-cell recovery 24 hours after mRNA electroporation was 39.8% ± 13.0% (n = 7) of the recovery after electroporation without mRNA (Figure 3A); if the CAR was introduced by viral transduction, cell recovery was 25.1% ± 16.2% (n = 10) that of mock-transduced T cells (Figure 3B); overall, CAR expression reduced cell recovery to 31.1% ± 16.3% (n = 17) after 24 hours. Prolonging cell culture further increased the difference in numbers between CAR- and mock-transduced cells overall (Figure 3C). In the absence of target cells, CAR expression induced exocytosis of lytic granules revealed by CD107a expression (Figure 3D), suggesting that impaired cell recovery was caused by fratricide.
Figure 3.

Expression of anti-CD7 CAR in human peripheral blood T cells results in fratricide, which is prevented by CD7 downregulation. (A) The percentage of viable T cells recovered 24 hours after electroporation with or without anti-CD7 CAR mRNA (n = 7). Viable cells were counted by flow cytometry. (B) The percentage of viable T cells recovered 24 hours after CAR transduction with a retroviral vector as compared with cells from the same donors transduced with GFP alone (Mock) (n = 10). (C) The percentage of viable CAR- or mock-transduced T cells recovered during the week after transduction. Shown are follow-up results for 5 of the 10 experiments shown in panel B. (D) The percentage of CD107a in T cells after electroporation with or without anti-CD7 CAR mRNA. Mean (± SD) of triplicate measurements are shown. (E) Schematic representation of anti-CD7 PEBL constructs. (F) Representative flow cytometric histograms illustrate CD7 expression in T lymphocytes after retroviral transduction of 3 anti-CD7 PEBLs or mock-transduced GFP alone (Mock). T cells were stained with anti-CD7–PE (M-T701; BD Biosciences). (G) The percentage of CD7 expression in T cells retrovirally transduced with the anti-CD7 PEBL-1 or mock-transduced (n = 5). (H) Flow cytometric dot plots illustrate downregulation of CD7 expression in T cells by PEBL transduction together with expression of anti-CD7–41BB-CD3ζ CAR 12 hours after electroporation with or without CAR mRNA. Cells were stained with biotin-conjugated goat anti-mouse F(ab′)2 antibody and streptavidin-APC (Jackson ImmunoResearch Laboratories). (I) The percentage of viable T cells transduced with anti-CD7 PEBL recovered 24 hours after electroporation of anti-CD7 CAR mRNA as compared with cells electroporated with the anti-CD7 CAR mRNA, but transduced with a vector without anti-CD7 PEBL (n = 6). The number of viable cells was measured by flow cytometry. **P < .01; ***P < .001.
Downregulation of CD7 prevents T-cell fratricide and does not affect T-cell function
If the poor T-cell recovery was caused by fratricide mediated by CAR binding to CD7 expressed by the T cells, then it should improve by downregulating CD7 before CAR expression. To test this prediction, we applied a rapid and practical method recently developed in our laboratory (T.K. and D.C., manuscript submitted September 2017) based on the expression of the anti-CD7 scFv linked to amino acid sequences containing the ER retention domains KDEL or KKMP (anti-CD7 PEBL) (Figure 3E). These fasten the constructs to the ER/Golgi, preventing secretion or membrane expression of the targeted protein.39,40 We tested 3 anti-CD7 PEBL constructs and selected PEBL-1 for the next experiments (Figure 3E-F). CD7 surface expression was essentially abrogated in all T cells transduced with this construct, whereas CD7 mRNA expression was retained (Figure 3F; supplemental Figure 3). In 5 experiments, 98.1% ± 1.5% mock-transduced T cells were CD7+ vs 2.0% ± 1.7% for T cells transduced with the anti-CD7 PEBL (P < .0001) (Figure 3G). When the anti-CD7 CAR was expressed by electroporation in cells with downregulated CD7, it was clearly detectable by flow cytometry (Figure 3H). By expressing the CAR in cells with CD7 knockdown, T-cell viability markedly improved (Figure 3I). In 6 paired experiments, viable cell recovery after CAR mRNA electroporation was consistently superior in T cells that had been previously transduced with the anti-CD7 PEBL (P = .008).
After anti-CD7 PEBL transduction, the proportion of CD4 and CD8 cells was similar to that of mock-transduced cells (Figure A). Absence of CD7 expression on the surface membrane did not affect T-cell survival in culture (Figure 4B). To further probe the functional capacity of T cells transduced with anti-CD7 PEBL, we engineered them to express the anti-CD19–CAR (Figure 4C) and tested their capacity to exert cytotoxicity, release cytotoxic granules, and secrete IFN-γ in the presence of CD19+ ALL cells. As shown in Figure 4D-F, PEBL transduction and lack of surface CD7 did not alter CAR-mediated cell function.
Figure 4.

CD7 downregulation by PEBL does not alter T-cell phenotype, proliferation, and functionality. (A) The percentage of CD4 and CD8 cells 7 to 14 days after retroviral transduction with either anti-CD7 PEBL or GFP alone (Mock). Each symbol corresponds to a different T-cell donor. (B) The growth rate of PEBL- and mock-transduced T cells (from 3 donors) maintained with 200 IU/mL IL-2 for 14 days. Symbols represent the mean (± SD) of triplicate measurements. (C) PEBL- and mock-transduced T cells were electroporated with either anti-CD19–41BB-CD3ζ CAR mRNA or no mRNA. Flow cytometric dot plots illustrate GFP and CAR expression 12 hours after electroporation. CAR was detected with biotin-conjugated goat anti-mouse F(ab′)2 antibody and streptavidin-APC (Jackson ImmunoResearch Laboratories). (D) Cytotoxicity of PEBL- or mock-transduced T cells, electroporated with or without anti-CD19 CAR mRNA, against CD19+ ALL cells (OP-1). Bars show the mean (± SD) of 4-hour cytotoxicity at a 1:1 E:T ratio. (E) CD107a expression in T cells from experiments identical to those described in panel D. (F) IFN-γ production in PEBL- or mock-transduced T cells, electroporated with or without anti-CD19 CAR mRNA, and cocultured with OP-1 for 6 hours at an E:T ratio of 1:1. Bars represent the mean (± SD) of triplicate experiments. ***P < .001; ****P < .0001.
Anti-CD7–41BB-CD3ζ CAR induces powerful cytotoxicity against CD7+ leukemic cells
We prepared CD7-negative T cells using anti-CD7 PEBL and electroporated them with the anti-CD7–41BB-CD3ζ CAR mRNA. We then assessed their anti-leukemic capacity in cocultures with the CD7+ leukemia cell lines MOLT-4, CCRF-CEM, Jurkat, Loucy, or KG1a. As shown in Figure 5A, cytotoxicity was dramatically increased by CAR expression. PEBL-CAR T cells were also highly effective against primary T-ALL cells obtained from patients (Figure 5B).
Figure 5.

T cells with downregulated CD7 by PEBL acquire powerful cytotoxicity against CD7+leukemic cells after expression of anti-CD7 CAR. (A) Cytotoxicity of anti-CD7 PEBL-transduced T cells electroporated with or without anti-CD7 CAR mRNA against CD7+ cell lines. Shown are data for 4-hour assays at a 1:1 E:T ratio. Symbols indicate the mean of 3 measurements each with T cells from 4 donors for MOLT-4, CCRF-CEM, and Jurkat, and 5 donors for Loucy and KG1a (P < .001 for each comparison). (B) Cytotoxicity of anti-CD7 PEBL-transduced T cells electroporated with or without anti-CD7 CAR mRNA against primary leukemic cells from patients with T-ALL. Shown are data for 4-hour assays at the indicated E:T ratio. Symbols refer to the mean (± SD) of 3 measurements. (C) Overall specific cytotoxicity of T cells transduced with either anti-CD7 PEBL or GFP alone (Mock) after electroporation with anti-CD7 CAR mRNA against the 5 CD7+ cell lines. T cells from 3 donors were tested at a 1:1 E:T ratio in 4-hour assays. Each symbol represents the specific percentage of cytotoxicity against the CD7+ cell line after subtraction of the percentage of cytotoxicity obtained with the same T cells electroporated without mRNA. Horizontal bars indicate the median for each group. (D) Anti-CD7 PEBL- or mock-transduced T cells from 3 donors were electroporated with or without anti-CD7 CAR mRNA. Cytotoxicity against MOLT-4 was tested at a 1:1 E:T ratio in 4-hour assays. Shown is the MFI of anti-CD107a–PE (H4A3; BD Biosciences). Bars represent the mean (± SD) of triplicate experiments. (E) Anti-CD7 PEBL-transduced T cells were retrovirally transduced with either anti-CD7 CAR or mock-transduced and tested against primary leukemic cells from patients with T-ALL. Each symbol represents the mean (± SD) of triplicate experiments. (F) Mock- or PEBL-transduced T cells, sequentially transduced with or without anti-CD7 CAR, were cultured alone or in the presence of Streck-treated MOLT-4 cells, added weekly and 120 IU/mL IL-2. Symbols indicate the mean (± SD) percentage of cell recovery relative to the number of input cells in triplicate cultures. **P < .01; ***P < .001; ****P < .0001.
We compared the cytotoxicity of PEBL-CAR T cells to that of the residual T cells recovered after CAR electroporation in cells not transduced with PEBL. In 45 experiments with cells from 3 donors, the cytotoxicity of the PEBL-CAR cells consistently surpassed that of non-PEBL T cells (Figure 5C). The superior activity of the former cells was also observed when we compared the expression of CD107a (Figure 5D), IFN-γ, and TNF-α (supplemental Figure 4). The expression of PEBL and CAR by sequential retroviral transduction also produced powerful cytotoxicity against patient-derived T-ALL cells (Figure 5E) and cell lines (supplemental Figure 5). Proliferation of anti-CD7 PEBL-CAR T cells in the presence of CD7+ target cells was much higher than that of CAR T cells without CD7 downregulation by PEBL (P < .01) (Figure 5F). Finally, we compared the cytotoxicity exerted by anti-CD7 PEBL-CAR T cells to that of T cells expressing an anti-CD19–41BB-CD3ζ CAR5 against the same target cells. To this end, we transduced CCRF-CEM and Jurkat cells with CD19 and expressed either CAR in cells previously transduced with anti-CD7 PEBL (supplemental Figure 6A-B). Anti-CD7 and anti-CD19 CAR T cells had similar short- and long-term cytotoxicity (supplemental Figure 6C-D); long-term proliferative capacity in the presence of CD19+ CD7+ target cells was slightly lower for the anti-CD7 CAR T cells (supplemental Figure 6E), which might be explained by the lower expression of CD7 vs CD19 on target cells (supplemental Figure 6B).
Antileukemic activity of anti-CD7 PEBL-CAR T cells in murine models of T-ALL
To further gauge the anti-tumor capacity of anti-CD7 PEBL-CAR T cells, we engrafted NOD/scid IL2RGnull mice with CCRF-CEM cells. T cells retrovirally transduced with anti-CD7 PEBL and anti-CD7 CAR produced a considerable antileukemic effect, with a marked reduction in leukemia cell burden and a decrease in leukemia cell growth (Figure 6A-C; supplemental Figure 7). Three weeks after leukemic cell injection, the median percentage of CCRF-CEM cells in peripheral blood by flow cytometry was 68% for control mice (n = 5) and 67% for those who receive GFP-alone T cells (n = 5), but they were undetectable in mice treated with anti-CD7 PEBL-CAR T cells (supplemental Figure 8A). Relapse occurring after anti-CD7 PEBL-CAR T-cell treatment was not due to CCRF-CEM cell subsets lacking CD7; leukemic cells continued to express high levels of CD7, and sensitivity to anti-CD7 CAR cytotoxicity remained high regardless of whether CCRF-CEM cells were derived from livers or spleens of relapsing mice or directly from the original cell culture (supplemental Figure 8B).
Figure 6.

PEBL-transduced T cells expressing an anti-CD7–41BB-CD3ζ CAR exert antitumor activity in xenografts. NOD-SCID-IL2RGnull mice were infused IV with 1 × 106 CCRF-CEM cells labeled with luciferase. A total of 2 × 107 PEBL-CAR T cells were administered IV on day 7 (A) or on day 3 and day 7 (B) after leukemic cell infusion to 3 and 5 mice, respectively. The remaining mice received either mock-transduced T cells or RPMI 1640 instead of cells (Control). All mice received 20 000 IU IL-2 once every 2 days IP. Shown is in vivo imaging of leukemia cell growth after d-luciferin IP injection. Ventral images of mice on day 3 in panel B are shown with enhanced sensitivity to demonstrate CCRF-CEM engraftment in all mice. The complete set of luminescence images is shown in supplemental Figure 7. (C) Leukemia cell growth in mice shown in panels A and B is expressed as photons per second. Each symbol corresponds to bioluminescence measurements in each mouse, normalized to the average of ventral plus dorsal signals in all mice before CAR T-cell infusion. (D) Kaplan-Meier curves show overall survival of mice in the different groups (8 in each group). Mice were euthanized when the total bioluminescence signal reached 1 × 1010 photons per second. The P values were calculated by log-rank test. N.S., not significant.
To test PEBL-CAR T cells against primary leukemic cells in vivo, we used a PDX model of ETP-ALL, which allows propagation of leukemic cells derived from a patient with ETP-ALL at diagnosis in NOD/scid IL2RGnull mice. Leukemic cells expanding in mice retained an immunophenotype matching that of the patient's diagnostic sample, with expression of CD7, CD34, and CD33 and absence of surface CD3, CD1a, CD8, and CD5 (supplemental Figure 9). The cells were unable to survive and expand ex vivo and needed to be injected in mice for propagation. All mice had ETP-ALL in peripheral blood at the time of CAR T-cell treatment (Figure 7A). As shown in Figure 7B, ETP-ALL cells represented the majority of leukocytes in the bone marrow, spleens, livers, and lungs. After the administration of PEBL CAR T cells (2 × 107 in 1 mouse, 2 × 106 in the remaining 4 mice), leukemic cell numbers in peripheral blood decrease dramatically, whereas PEBL-CAR T cells became detectable in all mice (Figure 7A). In blood smears, smudge cells were prominent, suggesting leukemia cell lysis (Figure 7C). Leukemia progressed in all 5 control mice, which were euthanized when ETP-ALL constituted ≥80% of peripheral blood CD45+ cells. The mouse treated with 2 × 107 PEBL-CAR T cells died of apparent graft-versus-host disease 23 days after PEBL-CAR T-cell infusion. No ETP-ALL could be detected in blood, bone marrow, livers, spleens, lungs and brains, whereas PEBL-CAR T cells were detectable in all tissues (Figure 7D-E). The 4 mice treated with 2 × 106 PEBL-CAR T cells were alive 25 (n = 1) to 39 (n = 3) days postinfusion with no signs of graft-versus-host disease.
Figure 7.

PEBL-CAR T-cell activity against ETP-ALL in a patient-derived xenograft (PDX) model. (A) Primary ETP-ALL cells, previously propagated in NOD-SCID-IL2RGnull mice, were infused IV in 10 NOD-SCID-IL2RGnull mice at 2 × 106 cells per mouse. Five mice (Control) were left untreated. The remaining 5 mice received a single IV infusion of PEBL-CAR T cells (2 × 107 in PEBL-CAR#1 mouse, 2 × 106 in the remaining 4 mice) at the indicated time point (blue arrow), as well as 20 000 IU IL-2 IP every 2 days; IL-2 was also administered to 2 of the 5 control mice. Red symbols (left y-axes) indicate the number of ETP-ALL cells per milliliter counted in peripheral blood. Blue symbols (right y-axes) show the numbers of PEBL-CAR T cells. The mice were euthanized when the percentage of ETP-ALL cells among blood mononucleated cells reached ≥80%. (B) The percentage of ETP-ALL (denominator, total human plus mouse CD45+ cells) in various organs of the 5 untreated mice. (C) Blood smears of treated (PEBL-CAR#1) and untreated ETP-ALL 7 days after infusion of T cells; smudge cells were prominent in blood after infusion of PEBL-CAR T cells. Original magnification ×40, Wright stain. (D) Flow cytometric dot plots show the presence of CD7+ CD3– ETP-ALL cells in the tissues of an untreated control mouse with ETP-ALL and of CD7– CD3+ PEBL-CAR T cells in the PEBL-CAR#1 mouse treated with PEBL-CAR T cells. No ETP-ALL (<0.01%) was detected in the treated mouse. The events shown were normalized to the events acquired for the corresponding plots shown in the control mouse. (E) Spleens of treated (PEBL-CAR#1) and untreated mice. BM, bone marrow.
Discussion
Durable remissions in patients with B-cell leukemia and lymphoma can be achieved with CAR T cells, but effective options are lacking for patients with T-cell malignancies. To bridge this gap, we sought to develop a CAR T-cell approach that could be rapidly translated into clinical intervention. We targeted CD7, a widely expressed surface T-cell marker that is highly stable even in T-ALL cells exposed to chemotherapy. We designed a second-generation anti-CD7 CAR and found that suppression of CD7 surface expression in T cells was essential; without it, the CAR caused severe T-cell loss, and the full functional potential of CAR T cells could not be achieved. Transduction of anti-CD7 PEBL resulted in virtually instantaneous abrogation of CD7 expression. Expression of anti-CD7 CAR in such cells produced powerful anti-leukemic activity in vitro, as well as in xenograft and PDX models of T-ALL. Thus, by using this strategy, we could rapidly generate large numbers of CAR T cells exerting robust and specific cytotoxicity against T-cell malignancies, including one of the most aggressive forms, ETP-ALL.
The PEBL technology that we used to downregulate endogenous CD7 is based on the use of a scFv directed against the targeted antigen coupled with an ER/Golgi-retention motif. In this way, any newly synthesized CD7 remains anchored in the ER and/or Golgi, and its surface expression is prevented. We found this method to be remarkably effective in downregulating CD7 and suppressing CAR-mediated fratricide. Importantly, intracellular retention of CD7 did not alter T-cell function and allowed normal expansion, cytokine secretion, and cytotoxicity. This is consistent with results of studies with CD7-deficient mice, which showed normal lymphocyte populations in lymphoid tissues.41,42 An alternative approach to downregulate CD7 would be to apply gene editing methods, such as meganucleases, TALEN, or CRISPR/Cas9.43 To this end, a recent study reported an anti-CD7 CAR that was expressed in T cells with CD7 gene deletion by CRISPR/Cas9.44 Besides differences in costimulatory molecules (our CAR has 4-1BB instead of CD28), which may have clinical impact,45,46 the high specificity and practical nature of the PEBL strategy make it particularly attractive for current clinical use. This method requires a simple transduction with the same viral vector carrying the CAR, either as 2 sequential transductions or a single transduction with a bicistronic vector carrying both constructs. It fits well with established, clinical-grade cell manufacturing processes and does not raise possible regulatory concerns associated with off-target activity.47,48
CD7 is a hallmark molecule for early T-cell differentiation; it is nearly universally expressed in T-ALL and, among normal cells, its expression is limited to T cells.19,29-32 In a clinical study with an anti-CD7–ricin-A–chain immunotoxin in patients with T-cell lymphoma, the dose-limiting toxicity was vascular leak syndrome, a side-effect seen with other toxin conjugates; no binding of anti-CD7 was found in the endothelial cells of various tissues.49 Nevertheless, transient expression of the CAR by mRNA electroporation might be considered in early studies assessing the potential for acute toxicities of anti-CD7 PEBL-CAR T cells. A concern of anti-CD7 CAR therapy is the depletion of normal T cells by the infused cells, leading to immunodeficiency. We envisage the initial application of our technology as a means to reduce MRD in patients with high-risk T-ALL, therefore maximizing the success of allogeneic hematopoietic stem cell transplantation.50 In such instances, anti-CD7 CAR T cells would be eliminated by the transplant conditioning and the T-cell compartment reconstituted from donor stem cells. Outside the transplant setting, “suicide genes” could be activated once leukemia eradication has been achieved.51 Ultimately, this may not be an issue, because the infused anti-CD7 T cells (which retain their endogenous CD3/TCR complex) might reconstitute a sufficiently wide T-cell repertoire. To this end, it should be noted that subsets of CD4 memory and CD8 effector T cells in human blood lymphocytes that do not express CD7 have been described52,53 and that T-ALL cells express CD7 at higher levels than normal T cells. Thus, CD7-dim subsets might help to repopulate the T-cell repertoire even after CD7-directed therapy.
The standard treatment of T-ALL mainly relies on intensive chemotherapy plus hematopoietic stem cell transplant for patients with high-risk disease, but results are far from satisfactory.54,55 The findings presented in this study suggest the infusion of anti-CD7 PEBL-CAR T cells could significantly enhance, or perhaps replace, existing chemotherapy- and transplant-based strategies. Conceivably, CAR expression together with downregulation of the targeted antigen in T cells should also be applicable to other T-cell markers, such as CD3, CD2, and CD5, whose expression is prevalent in T-cell lymphoproliferative neoplasms. Because a fraction of high-risk acute myeloid leukemia cases express CD7,19,30,56 testing the potential of anti-CD7 CAR T cells for this leukemia subtype is also warranted.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by a Singapore Translational Research Award from the National Medical Research Council of Singapore, the Children’s Cancer Foundation, the Lee Foundation, and the Viva Foundation for Children with Cancer of Singapore.
Authorship
Contribution: Y.T.P. and N.V. performed the experiments and analyzed the data; T.K. developed and optimized the PEBLs; N.S. generated the PDX model of ETP-ALL; E.C.-S. preformed leukemia immunophenoytping and MRD studies; and D.C. designed the study, analyzed the data, and wrote the manuscript with Y.T.P. and the input of all other authors.
Conflict-of-interest disclosure: Y.T.P., N.V., T.K., E.C.-S., and D.C. are coinventors on patent applications describing some of the technologies used in this study. N.S. declares no competing financial interests.
Correspondence: Dario Campana, Department of Pediatrics, Yong Loo Lin School of Medicine, National University of Singapore, Centre for Translational Medicine, 14 Medical Dr, Level 9 South, Singapore 117599; e-mail: paedc@nus.edu.sg.
References
- 1.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90(2):720-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geiger TL, Leitenberg D, Flavell RA. The TCR zeta-chain immunoreceptor tyrosine-based activation motifs are sufficient for the activation and differentiation of primary T lymphocytes. J Immunol. 1999;162(10):5931-5939. [PubMed] [Google Scholar]
- 3.Brentjens RJ, Latouche JB, Santos E, et al. . Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9(3):279-286. [DOI] [PubMed] [Google Scholar]
- 4.Cooper LJ, Topp MS, Serrano LM, et al. . T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101(4):1637-1644. [DOI] [PubMed] [Google Scholar]
- 5.Imai C, Mihara K, Andreansky M, et al. . Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676-684. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentjens RJ, Santos E, Nikhamin Y, et al. . Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13(18 Pt 1):5426-5435. [DOI] [PubMed] [Google Scholar]
- 8.Haso W, Lee DW, Shah NN, et al. . Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nadler LM, Anderson KC, Marti G, et al. . B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J Immunol. 1983;131(1):244-250. [PubMed] [Google Scholar]
- 10.Campana D, Janossy G, Bofill M, et al. . Human B cell development. I. Phenotypic differences of B lymphocytes in the bone marrow and peripheral lymphoid tissue. J Immunol. 1985;134(3):1524-1530. [PubMed] [Google Scholar]
- 11.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grupp SA, Kalos M, Barrett D, et al. . Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Till BG, Jensen MC, Wang J, et al. . CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119(17):3940-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kochenderfer JN, Dudley ME, Feldman SA, et al. . B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709-2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davila ML, Riviere I, Wang X, et al. . Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maude SL, Frey N, Shaw PA, et al. . Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. . T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turtle CJ, Hanafi LA, Berger C, et al. . CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123-2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coustan-Smith E, Mullighan CG, Onciu M, et al. . Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Ding L, Holmfeldt L, et al. . The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inukai T, Kiyokawa N, Campana D, et al. . Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children’s Cancer Study Group Study L99-15. Br J Haematol. 2012;156(3):358-365. [DOI] [PubMed] [Google Scholar]
- 22.Neumann M, Heesch S, Gökbuget N, et al. . Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. 2012;2(1):e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain N, Lamb AV, O’Brien S, et al. . Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. 2016;127(15):1863-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood. 2017;129(9):1134-1142. [DOI] [PubMed] [Google Scholar]
- 25.Campana D, Pui CH. Minimal residual disease-guided therapy in childhood acute lymphoblastic leukemia. Blood. 2017;129(14):1913-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coustan-Smith E, Sancho J, Hancock ML, et al. . Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood. 2002;100(7):2399-2402. [DOI] [PubMed] [Google Scholar]
- 27.Mihara K, Yanagihara K, Takigahira M, et al. . Activated T-cell-mediated immunotherapy with a chimeric receptor against CD38 in B-cell non-Hodgkin lymphoma. J Immunother. 2009;32(7):737-743. [DOI] [PubMed] [Google Scholar]
- 28.Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126(8):983-992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haynes BF, Eisenbarth GS, Fauci AS. Human lymphocyte antigens: production of a monoclonal antibody that defines functional thymus-derived lymphocyte subsets. Proc Natl Acad Sci USA. 1979;76(11):5829-5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vodinelich L, Tax W, Bai Y, Pegram S, Capel P, Greaves MF. A monoclonal antibody (WT1) for detecting leukemias of T-cell precursors (T-ALL). Blood. 1983;62(5):1108-1113. [PubMed] [Google Scholar]
- 31.Janossy G, Coustan-Smith E, Campana D. The reliability of cytoplasmic CD3 and CD22 antigen expression in the immunodiagnosis of acute leukemia: a study of 500 cases. Leukemia. 1989;3(3):170-181. [PubMed] [Google Scholar]
- 32.Yeoh EJ, Ross ME, Shurtleff SA, et al. . Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1(2):133-143. [DOI] [PubMed] [Google Scholar]
- 33.Manabe A, Coustan-Smith E, Kumagai M, et al. . Interleukin-4 induces programmed cell death (apoptosis) in cases of high-risk acute lymphoblastic leukemia. Blood. 1994;83(7):1731-1737. [PubMed] [Google Scholar]
- 34.Peipp M, Küpers H, Saul D, et al. . A recombinant CD7-specific single-chain immunotoxin is a potent inducer of apoptosis in acute leukemic T cells. Cancer Res. 2002;62(10):2848-2855. [PubMed] [Google Scholar]
- 35.Kudo K, Imai C, Lorenzini P, et al. . T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014;74(1):93-103. [DOI] [PubMed] [Google Scholar]
- 36.Shimasaki N, Fujisaki H, Cho D, et al. . A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14(7):830-840. [DOI] [PubMed] [Google Scholar]
- 37.Shimasaki N, Campana D. Natural killer cell reprogramming with chimeric immune receptors. Methods Mol Biol. 2013;969:203-220. [DOI] [PubMed] [Google Scholar]
- 38.Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73(6):1777-1786. [DOI] [PubMed] [Google Scholar]
- 39.Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48(5):899-907. [DOI] [PubMed] [Google Scholar]
- 40.Jackson MR, Nilsson T, Peterson PA. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J. 1990;9(10):3153-3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonilla FA, Kokron CM, Swinton P, Geha RS. Targeted gene disruption of murine CD7. Int Immunol. 1997;9(12):1875-1883. [DOI] [PubMed] [Google Scholar]
- 42.Lee DM, Staats HF, Sundy JS, et al. . Immunologic characterization of CD7-deficient mice. J Immunol. 1998;160(12):5749-5756. [PubMed] [Google Scholar]
- 43.Boettcher M, McManus MT. Choosing the right tool for the job: RNAi, TALEN, or CRISPR. Mol Cell. 2015;58(4):575-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gomes-Silva D, Srinivasan M, Sharma S, et al. . CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130(3):285-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campana D, Schwarz H, Imai C. 4-1BB chimeric antigen receptors. Cancer J. 2014;20(2):134-140. [DOI] [PubMed] [Google Scholar]
- 46.Zhao Z, Condomines M, van der Stegen SJC, et al. . Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsai SQ, Joung JK. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat Rev Genet. 2016;17(5):300-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cameron P, Fuller CK, Donohoue PD, et al. . Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods. 2017;14(6):600-606. [DOI] [PubMed] [Google Scholar]
- 49.Frankel AE, Laver JH, Willingham MC, Burns LJ, Kersey JH, Vallera DA. Therapy of patients with T-cell lymphomas and leukemias using an anti-CD7 monoclonal antibody-ricin A chain immunotoxin. Leuk Lymphoma. 1997;26(3-4):287-298. [DOI] [PubMed] [Google Scholar]
- 50.Leung W, Pui CH, Coustan-Smith E, et al. . Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood. 2012;120(2):468-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Straathof KC, Spencer DM, Sutton RE, Rooney CM. Suicide genes as safety switches in T lymphocytes. Cytotherapy. 2003;5(3):227-230. [DOI] [PubMed] [Google Scholar]
- 52.Reinhold U, Abken H, Kukel S, et al. . CD7- T cells represent a subset of normal human blood lymphocytes. J Immunol. 1993;150(5):2081-2089. [PubMed] [Google Scholar]
- 53.Aandahl EM, Sandberg JK, Beckerman KP, Taskén K, Moretto WJ, Nixon DF. CD7 is a differentiation marker that identifies multiple CD8 T cell effector subsets. J Immunol. 2003;170(5):2349-2355. [DOI] [PubMed] [Google Scholar]
- 54.Raetz EA, Teachey DT. T-cell acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2016;2016(1):580-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jabbour E, O’Brien S, Konopleva M, Kantarjian H. New insights into the pathophysiology and therapy of adult acute lymphoblastic leukemia. Cancer. 2015;121(15):2517-2528. [DOI] [PubMed] [Google Scholar]
- 56.Kita K, Miwa H, Nakase K, et al. ; The Japan Cooperative Group of Leukemia/Lymphoma. Clinical importance of CD7 expression in acute myelocytic leukemia. Blood. 1993;81(9):2399-2405. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.