

ABSTRACT
MicroRNAs (miRNAs) are small noncoding RNAs that regulate gene expression posttranscriptionally through silencing or degrading their targets, thus playing important roles in the immune response. However, the role of miRNAs in the host response against infectious bursal disease virus (IBDV) infection is not clear. In this study, we show that the expression of a series of miRNAs was significantly altered in DF-1 cells after IBDV infection. We found that the miRNA gga-miR-130b inhibited IBDV replication via targeting the specific sequence of IBDV segment A and enhanced the expression of beta interferon (IFN-β) by targeting suppressors of cytokine signaling 5 (SOCS5) in host cells. These findings indicate that gga-miR-130b-3p plays a crucial role in host defense against IBDV infection.
IMPORTANCE This work shows that gga-miR-130b suppresses IBDV replication via directly targeting the viral genome and cellular SOCS5, the negative regulator for type I interferon expression, revealing the mechanism underlying gga-miR-130-induced inhibition of IBDV replication. This information will be helpful for the understanding of how host cells combat pathogenic infection by self-encoded small RNA and furthers our knowledge of the role of microRNAs in the cell response to viral infection.
KEYWORDS: microRNA, type I IFN, IBDV, SOCS
INTRODUCTION
Infectious bursal disease (IBD), also called Gumboro disease, is an acute, highly contagious disease in young chickens across the world (1). Its causative agent, IBD virus (IBDV), causes a severe immunosuppression by destroying its target cells—the B-lymphocyte precursors in diseased chickens—which leads to an increased susceptibility to other pathogens (2). IBDV is an avibirnavirus belonging to the Birnaviridae family, containing two segments of double-stranded RNA (dsRNA) (A and B) (3). The short RNA, segment B (2.8 kb), encodes VP1, an RNA-dependent RNA polymerase (RdRp) (4, 5), while segment A, the large molecule (3.17 kb), contains two partially overlapping open reading frames (ORFs) (6). The first ORF encodes the nonstructural viral protein 5 (VP5), and the second one encodes a 110-kDa pVP2-VP4-VP3 precursor that can be cleaved by the proteolytic activity of VP4 to form viral proteins VP2, VP3, and VP4 (7, 8). The immune system senses virus infection by recognizing pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs) and initiates antiviral responses by producing type I interferons (IFNs). IBDV infection triggers expression of genes involved in Toll-like receptor (TLR)- and IFN-mediated antiviral responses (9). It has been reported that IFN-α has strong antiviral activities in IBDV-infected cells (10), suggesting that type I interferon of host cells may play a critical role in combating IBDV. Interestingly, cellular microRNAs (miRNAs) acted against viral infection by targeting the genomes of viruses (11–13), and meanwhile, some miRNAs were reported to regulate IFN production (14–16) or IFN downstream signals (17). These findings indicate that miRNAs play important roles in host defense against viral infection (18).
MicroRNAs are small noncoding RNAs of 20 to 24 nucleotides that regulate eukaryotic gene expression posttranscriptionally by affecting degradation and translation of target mRNAs (19–21). Some studies have proposed that miRNAs protect and activate gene expression in certain cells (22). As the research on miRNAs goes on, the roles of miRNAs in various biological processes have gradually been deciphered, including roles in the development and differentiation of cancer (23), cell proliferation and differentiation (24), cell cycle and apoptosis (25), and immunoregulation and viral infection (26, 27). The miRNA miR-130b-3p, belonging to the miR-130/301 family, has been found to be involved in different human physiological activities and cancers, such as pancreatic cancer (28), hepatocellular carcinoma (29), colorectal cancer (30), and bladder cancer (31). Although recent evidence shows that miR-130b takes part in the regulation of cytokine expression (32–34), its role in the cell response to viral infection remains elusive.
In this study, we demonstrate that miR-130b acts as an antagonist against IBDV infection via suppressing virus replication and upregulating type I interferon expression. We identified IBDV segment A and suppressors of cytokine signaling 5 (SOCS5), a negative regulator of the JAK-STAT signaling pathway, as bona fide targets of miR-130b. Ectopic expression of miR-130b effectively suppressed IBDV replication by directly targeting viral RNAs and enhanced IFN-β expression via inhibiting the expression of SOCS5, indicating that miR-130b plays a key role in the host response to IBDV infection.
RESULTS
The expression of miR-130b-3p increases in DF-1 cells with IBDV infection.
To identify the miRNAs involved in the host response to IBDV infection, we performed a high-throughput sequencing assay to obtain miRNA profiles of DF-1 cells infected with IBDV strain Lx at a multiplicity of infection (MOI) of 0.1 for 24 h. Using the KEGG and GO pathway analysis database, we analyzed four major antiviral pathways that were targeted by miRNAs that were differentially expressed upon IBDV infection (Fig. 1A). The results showed that 296 miRNAs were involved. Among them, 214 miRNAs were engaged in a JAK-STAT signaling pathway, 207 in a Toll-like receptor-mediated signaling pathway, 164 in a RIG-I-like receptor (RLR)-mediated signaling pathway, and 244 in a cytokine-cytokine receptor signaling pathway. Several miRNAs, such as miR-27a, miR-30, miR-130b, and miR-146, attracted our attention because their expression changed significantly upon IBDV infection and they had been reported to participate in the immune response (16, 34–37). We focused on the role of miR-130b in the cell response to IBDV infection because this miRNA participated in the antiviral process (38–40).
FIG 1.

Infection of DF-1 cells with IBDV strain Lx enhances gga-miR-130b expression. (A) KEGG pathway enrichment analysis of miRNAs that were differentially expressed in DF-1 cells upon IBDV infection. The major antiviral pathways in which these miRNAs participated were noted and analyzed. The percentage was calculated as follows: number of miRNAs involved in the cytokine-cytokine receptor, TLR, RLR, or JAK-STAT pathway/total number of miRNAs that participated in these four antiviral pathways. (B) Expression of gga-miR-130b in DF-1 cells with IBDV infection at different time points. DF-1 cells were infected with IBDV at an MOI of 0.1 or mock infected. At different time points (12, 24, 36, and 48 h) after IBDV infection, total RNA was extracted and qRT-PCR was performed to detect gga-miR-130b transcripts. The relative level of gga-miR-130b expression was calculated as follows: expression of miR-130b in IBDV-infected or normal cells/expression of miR-130b in normal cells at 12 h. (C) Expression of gga-miR-130b in DF-1 cells infected with IBDV at different doses. DF-1 cells were infected with IBDV (at an MOI of 0.01, 0.1, 1, or 10) or mock infected. Twenty-four hours after infection, total RNA was extracted and qRT-PCR was performed to detect gga-miR-130b transcripts. The relative level of gga-miR-130b expression was calculated as follows: expression of miR-130b in IBDV-infected cells/expression of miR-130b in normal cells. The expression of U6 snRNA was used as an internal control. Data are representative of three independent experiments and are presented as means and SD. ***, P < 0.001; **, P < 0.01.
The results from high-throughput sequencing assay of IBDV-infected cells exhibited a decrease of miR-130b expression, suggesting that cellular miR-130b might be involved in IBDV infection. To further determine the effect of IBDV infection on miRNA expression, we performed quantitative reverse transcription-PCR (qRT-PCR) to examine the expression of mature miRNAs in cells with IBDV infection at an MOI of 0.1 at different time points or at different doses (Fig. 1B and C). Unexpectedly, we found that the expression of miR-130b in cells markedly increased after IBDV infection compared to that in mock-infected cell controls and that the expression of miR-130b was upregulated with increasing doses, reaching the peak level in cells with IBDV infection at an MOI of 1 but declining with a higher dose. These results indicate that expression of miR-130b in cells with IBDV infection increased in a time- and dose-dependent manner.
gga-miR-130b is involved in type I interferon expression.
Host cells combat viral infection by producing type I interferon upon recognition of viral dsRNA by TLR3 or RIG-I molecules (41). To explore the effect of miRNAs on antiviral signaling pathways, we transfected DF-1 cells with miR-130b and examined the expression of type I IFNs and the related transcriptional regulators NF-κB and IRF3 in these cells in response to IBDV infection or poly(I·C) treatment. As a result, overexpression of miR-130b markedly enhanced the expression of IFN-β in cells with IBDV infection or poly(I·C) treatment, while IFN-α expression was not affected (Fig. 2A and B). Further, we tested the expression of two important transcriptional regulators of type I IFN expression: IRF3, also known as IRF7 in chickens (42, 43), and p65, a member of the NF-κB family with transcription activity (44, 45). Consistently, the expression of NF-κB and IRF3 was also upregulated in cells with miR-130b overexpression (Fig. 2C and D). These data suggest that miR-130b is involved in IFN-β expression in cells in response to IBDV infection. To consolidate these findings, we transfected cells with miR-130b inhibitors and examined the expression of type I IFNs, NF-κB, and IRF3 in cells treated as described above. As shown in Fig. 3A and B, the expression of IFN-β but not IFN-α was significantly downregulated in DF-1 cells treated with miR-130b inhibitors compared to that in miRNA inhibitor control cells. Likewise, the expression of NF-κB and IRF3 was also downregulated in cells with miR-130b inhibitors (Fig. 3C and D). These data strongly suggest that miR-130b affects the upstream pathway of NF-κB- and IRF3-regulated type I interferon expression.
FIG 2.

gga-miR-130b enhances poly(I·C)/IBDV-induced expression of type I interferon. (A to D) DF-1 cells were transfected with miRNA controls or miR-130b mimics at 80 nM. Eighteen hours after transfection, cells were infected with IBDV at an MOI of 0.1 or treated with poly(I·C) at a final concentration of 2 μg/ml. Twelve hours after poly(I·C) treatment or IBDV infection, mRNA expression of IFN-α (A), IFN-β (B), IRF3 (C), and NF-κB-p65 (D) was measured by qRT-PCR, using specific primers. The relative levels of gene expression were calculated as follows: mRNA expression of IFN-α, IFN-β, p65, or IRF3 in miR-130b-transfected or normal cells treated with poly(I·C) or IBDV/mRNA expression of miRNA control-transfected cells in control medium. The expression of GAPDH was used as an internal control. Data are representative of three independent experiments and are presented as means and SD. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
FIG 3.

Knockdown of endogenous gga-miR-130b by inhibitors reduces poly(I·C)/IBDV-induced expression of type I interferon. (A to D) DF-1 cells were transfected with miR-130b inhibitors or miRNA inhibitor controls at 80 nM. Eighteen hours after transfection, cells were infected with IBDV at an MOI of 0.1 or treated with poly(I·C) at a final concentration of 2 μg/ml. Twelve hours after poly(I·C) treatment or IBDV infection, cells were harvested for quantification of the expression of IFN-α (A), IFN-β (B), IRF3 (C), and NF-κB-p65 (D). The relative levels of gene expression were calculated as follows: mRNA expression of IFN-α, IFN-β, p65, or IRF3 in miR-130b inhibitor-transfected cells or normal controls treated with poly(I·C) or IBDV/mRNA expression of miRNA inhibitor control-transfected cells in control medium. GAPDH was used as an internal control. Data are representative of three independent experiments and are presented as means and SD. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
gga-miR-130b inhibits IBDV replication.
Through miRNA target site prediction using RNA22.v2, we found a putative target site of miR-130b in the genome of IBDV Lx. As shown in Fig. 4A and B, overexpression of miR-130b inhibited the expression of IBDV VP3 but not VP5, and this inhibition by miR-130b transfection occurred in a dose-dependent manner. In contrast, transfection of cells with miR-130b inhibitors significantly enhanced viral VP3 expression (Fig. 4C and D). These data indicate that miR-130b specifically inhibits viral VP3 expression. To confirm these results, we performed an indirect immunofluorescent-antibody assay (IFA) to examine the effect of miR-130b on IBDV growth in DF-1 cells at 24 h post-IBDV infection, and we measured IBDV growth in cell cultures at different time points by using 50% tissue culture infective dose (TCID50) assays. As shown in Fig. 5A to F, transfection of DF-1 cells with miR-130b significantly suppressed IBDV replication compared to that in control cells. Similarly, miR-130b markedly slowed IBDV growth as determined by TCID50 assays (Fig. 5G). These results revealed the suppressive effect of miR-130b on IBDV replication, including production of progeny, release, spread, and infection of neighboring cells.
FIG 4.
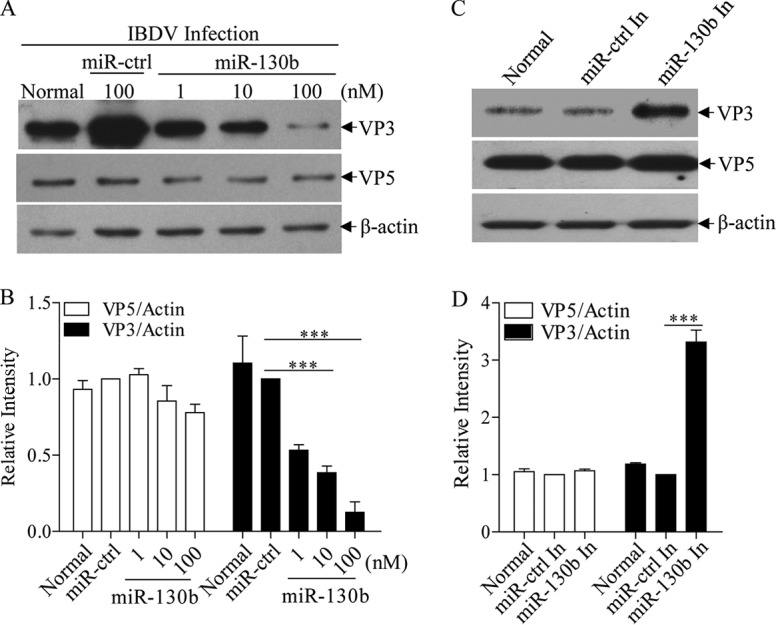
gga-miR-130b inhibits IBDV protein expression. (A and B) Transfection of miR-130b in DF-1 cells reduced expression of VP3 but not VP5. DF-1 cells were transfected with miRNA controls or miR-130b mimics at different doses, followed by infection with IBDV at an MOI of 0.1. Twenty-four hours after infection, cell lysates were prepared and examined by Western blotting using anti-VP3 or anti-VP5 antibodies. Endogenous β-actin expression was used as an internal control. The band densities for VP3 and VP5 in panel A were quantitated by densitometry as shown in panel B. The relative levels of VP3 or VP5 were calculated as follows: band density of VP3 or VP5/band density of β-actin. (C and D) Inhibition of endogenous gga-miR-130b enhanced IBDV VP3 protein expression. DF-1 cells were transfected with miR-130b inhibitors or miRNA control inhibitors before being infected with IBDV at an MOI of 0.1. Twenty-four hours after infection, cell lysates were prepared and examined by Western blotting using anti-VP3 or anti-VP5 antibody. Endogenous β-actin expression was used as an internal control. The band densities of VP3 and VP5 in panel C were quantitated by densitometry as shown in panel D. The relative levels of VP3 and VP5 were calculated as described above. Data are representative of three independent experiments and are presented as means and SD. ***, P < 0.001.
FIG 5.

gga-miR-130b inhibits IBDV replication. (A to F) Examination of IBDV replication by IFA. DF-1 cells were transfected with miRNA controls or miR-130b mimics at 80 nM for 18 h, followed by infection with IBDV Lx at an MOI of 0.1. Twenty-four hours after infection, cells were fixed and examined for the expression of IBDV VP3 protein by immunofluorescent-antibody assays. The pictures in panels A to C were taken under a fluorescence microscope, and those in panels D to F were taken under a light microscope. Magnification, ×100. (G) Analysis of the effect of miR-130b on IBDV replication by TCID50 assay. DF-1 cells were transfected with miRNA controls, miR-130b mimics, or medium only. Eighteen hours after transfection, cells were infected with IBDV Lx at an MOI of 0.1. At different time points (12, 24, 48, and 72 h) after IBDV infection, the viral loads in the cell cultures were determined by TCID50 assays in 96-well plates. The significance of the differences between miR-130b-transfected cells and controls was determined by ANOVA (P < 0.05). The graph shows the average viral loads in DF-1 cells for three individual experiments.
gga-miR-130b directly targets the IBDV genome.
The observation that miR-130b suppressed IBDV replication prompted us to investigate the molecular mechanism underlying miR-130b-mediated suppression of IBDV. To investigate whether the inhibition of viral replication was due to the direct targeting of the IBDV genome by miR-130b, we used the RNA22.v2 database to screen for miR-130b target sites on IBDV genomic RNAs. One target site for miR-130b was noted in IBDV segment A (bp 882 to 901), which was located in the second ORF of segment A and encoded viral proteins VP2, VP3, and VP4. To confirm the interaction of miRNAs with the target site, we constructed a firefly luciferase reporter plasmid (pGL3-target-WT) containing the predicted target site and another construct (pGL3-target-Mut) with a mutation in the seed region (Fig. 6A). DF-1 cells were cotransfected with miRNAs and luciferase reporter gene plasmids plus pRL-TK, and then luciferase reporter gene assays were performed. As shown in Fig. 6B, transfection of cells with miR-130b plus pGL3-target-WT inhibited luciferase activity compared to that of controls, and this inhibitory effect of miR-130b occurred in a dose-dependent manner. However, this inhibitory effect could be overcome by mutation of the inserted target site of miR-130b in the luciferase reporter vector (Fig. 6C). These results indicate that miR-130b directly inhibits IBDV replication via targeting of the viral RNA genome.
FIG 6.
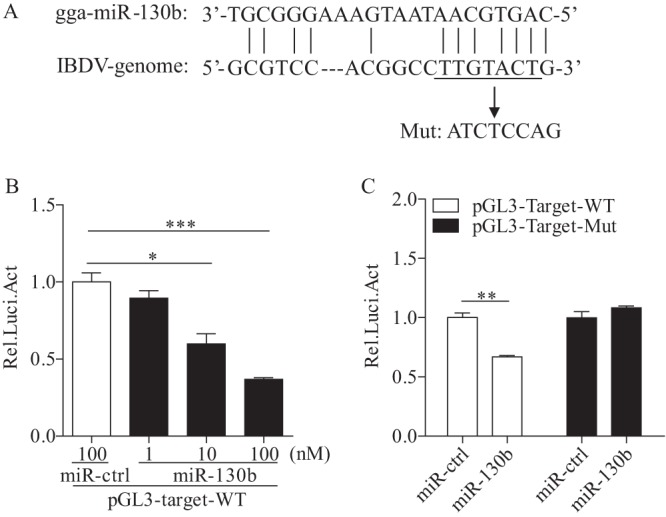
gga-miR-130b directly targets the IBDV genome. (A) Diagram of predicted target sites for miR-130b in IBDV genomic RNA. The seed sequence of miR-130b is underlined and was mutated as indicated by the arrow. (B) miR-130b inhibited target gene expression in a dose-dependent manner. DF-1 cells were cotransfected with luciferase reporter vectors containing wild-type (WT) target sites, pRL-TK, and miR-130b at different concentrations. At 48 h posttransfection, cells were lysed and luciferase reporter gene assays were performed to measure luciferase activities. The relative level of luciferase activity was calculated as follows: luciferase activity of cells cotransfected with the reporter plasmid and miR-130b mimics/luciferase activity of cells cotransfected with the reporter plasmid and miRNA controls. (C) A point mutation in the target gene abolished miR-130b-induced suppression of the target gene. DF-1 cells were cotransfected with miR-130b and the WT or mutant luciferase reporter vector. At 48 h posttransfection, a luciferase reporter gene assay was performed to measure luciferase activity. The relative level of luciferase activity was calculated as follows: luciferase activity of cells cotransfected with the reporter plasmid and miRNA mimics/luciferase activity of cells cotransfected with the WT reporter plasmid and miRNA controls. Data are representative of three independent experiments and are presented as means and SD. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
SOCS5 is a target of gga-miR-130b in host cells.
Since transfection of miR-130b enhances expression of IFN-β, we set out to explore the underlying mechanism. Using Targetscan and the PicTar database, we identified SOCS5 as the target of miR-130b in host cells. The region of the SOCS5 3′ untranslated region (3′UTR) at bp 961 contains the target site of miR-130b. We cloned the target gene into the luciferase reporter gene vector pGL3-control to examine the effect of miR-130b on target gene expression by luciferase reporter gene assay. Thus, we constructed a firefly luciferase reporter gene plasmid (pGL3-socs5-WT) containing the predicted target site in SOCS5 and another construct (pGL3-socs5-Mut) with mutations in the seed region (Fig. 7A). As shown in Fig. 7B, transfection of DF-1 cells with miR-130b markedly suppressed the luciferase activity of cells transfected with pGL3-socs5-WT, indicating that miR-130b inhibited the mRNA expression of SOCS5. In contrast, transfection with pGL3-socs5-Mut abolished miR-130b-mediated inhibition of its target sequence, and inhibition of miR-130b by miR-130b inhibitors increased the expression of the luciferase reporter vector, to some extent (Fig. 7C). These data clearly demonstrate that miR-130b suppresses the activation of the luciferase reporter plasmid carrying the SOCS5 3′UTR by targeting its specific sequence.
FIG 7.
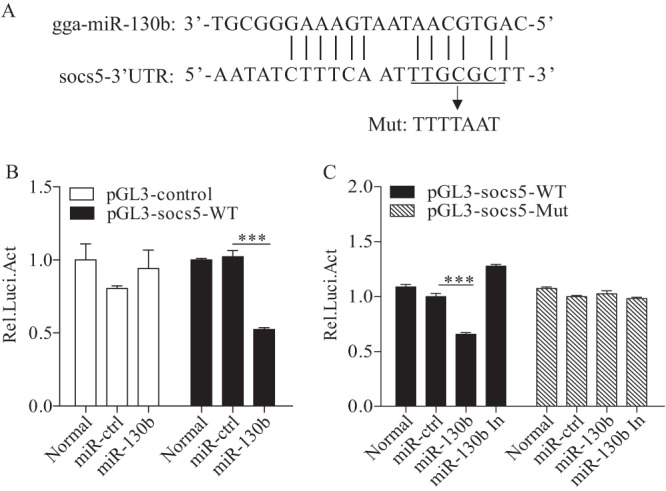
The SOCS5 gene is a cellular target of gga-miR-130b. (A) Diagram of predicted target sites for miR-130b in the SOCS5 gene. The seed sequence of miR-130b is underlined and was mutated as indicated by the arrow. (B) Transfection of gga-miR-130b reduced expression of SOCS5. DF-1 cells were cotransfected with miRNAs and luciferase reporter vectors. At 48 h posttransfection, cells were lysed, and a luciferase reporter gene assay was performed to measure SOCS5 expression. The relative level of luciferase activity was calculated as follows: luciferase activity of reporter plasmid-transfected cells or cells cotransfected with the reporter plasmid and miRNA mimics/luciferase activity of cells cotransfected with the WT reporter plasmid and miRNA controls. (C) Mutation of the target site abolished the inhibition of SOCS5 by miR-130b. DF-1 cells were cotransfected with miRNA controls or miR-130b mimics or inhibitors and luciferase reporter vectors. At 48 h posttransfection, the assay was performed to measure the luciferase activity. The relative level of luciferase activity was calculated as described above. Data are representative of three independent experiments and are presented as means and SD. ***, P < 0.001.
To consolidate these findings, we examined the effect of miR-130b on SOCS5 expression at the protein level by performing Western blotting. Consistently, miR-130b markedly inhibited SOCS5 expression (Fig. 8A and B). As observed above, this inhibition was abolished by the presence of miR-130b inhibitors (Fig. 8C and D). These data strongly suggest that miR-130b enhances the expression of IFN-β via inhibition of SOCS5, the negative regulator of type I IFN expression.
FIG 8.
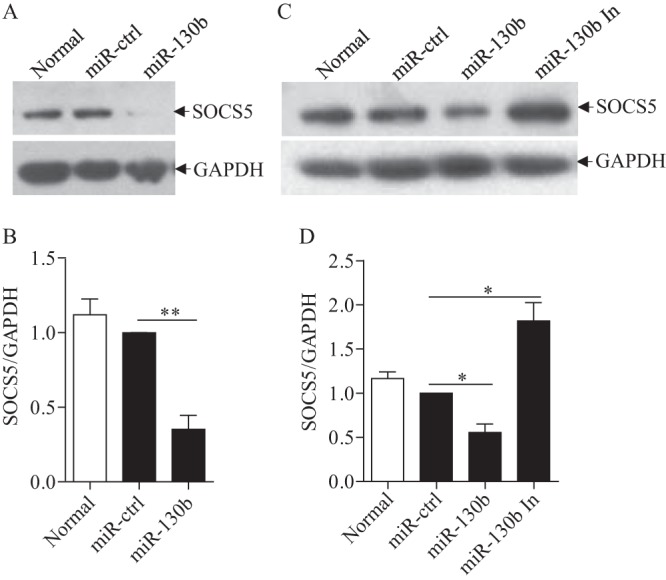
gga-miR-130b inhibits the expression of SOCS5 in DF-1 cells. (A and B) Transfection of miR-130b into DF-1 cells reduced expression of SOCS5 at the protein level. DF-1 cells were transfected with miRNA controls or miR-130b mimics at 80 nM. (A) Forty-eight hours after transfection, cells were harvested, and the cytosolic proteins were examined by Western blotting using anti-SOCS5 polyclonal antibodies. GAPDH expression was used as an internal control. (B) The band densities of SOCS5 in panel A were quantitated by densitometry. The relative level of SOCS5 expression was calculated as follows: band density of SOCS5 in each sample/band density of GAPDH in the same sample. (C and D) miR-130b inhibitors enhanced the expression of SOCS5. DF-1 cells were transfected with miRNA controls or miR-130b mimics or inhibitors. (C) Forty-eight hours after transfection, the expression of SOCS5 was examined by Western blotting. (D) The band densities of SOCS5 in DF-1 cells in panel C were quantitated by densitometry, and the relative levels of SOCS5 expression were calculated as described above. Data are representative of three independent experiments and are presented as means and SD. **, P < 0.01; *, P < 0.05.
gga-miR-130b enhances expression of the STAT family.
Because SOCS5, a member of the SOCS family, is known to be a negative regulator of the JAK-STAT pathway, it was logical to hypothesize that inhibition of SOCS5 by miR-130b would enhance the expression of STATs. To test this hypothesis, we examined the mRNA expression of STAT family members in cells with miR-130b transfection. As shown in Fig. 9A to F, transfection of DF-1 cells with miR-130b mimics enhanced the expression of STATs in general, especially that of STAT1, -3, and -6, regardless of whether cells were infected or not, and inhibition of miR-130 decreased the relative expression level. These data suggest that enhancement of STAT expression via reduction of SOCS5 by miR-130b contributes to the increase of IFN-β expression and, further, partly suppresses the replication of IBDV.
FIG 9.

Inhibition of SOCS5 by gga-miR-130b enhances the expression of STATs. (A to F) DF-1 cells were transfected with miRNA controls or miR-130b mimics or inhibitors at 80 nM. Eighteen hours after transfection, cells were infected with IBDV at an MOI of 0.1 or left uninfected. Twelve hours after infection, mRNA expression levels of STAT1 (A), STAT2 (B), STAT3 (C), STAT4 (D), STAT5 (E), and STAT6 (F) were measured by qRT-PCRs using specific primers, and GAPDH was used as an internal control. The relative levels of gene expression were calculated as follows: mRNA expression of the STAT gene in miR-130b-transfected cells or normal cells/mRNA expression of the STAT gene in miRNA control-transfected cells. Data are representative of three independent experiments and are presented as means and SD. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
gga-miR-130b enhances STAT1 phosphorylation on Tyr701 in DF-1 cells with chIFN-γ stimulation.
Activation of the JAK-STAT pathway is based on the phosphorylation of JAKs and STATs on tyrosine residues (46). As a member of the STAT family, STAT1 has been reported to play a vital role in antiviral responses of host cells (47–49). To determine whether miR-130b affects phosphorylation of STAT1, we transfected DF-1 cells with miR-130b mimics, miR-130b inhibitors, or miRNA controls and performed Western blotting to examine the phosphorylation of STAT1 in DF-1 cells after treatment with chicken IFN-γ (chIFN-γ), an activator of STAT1 (50). As shown in Fig. 10A and B, phosphorylation of STAT1 on Tyr701 was markedly enhanced in miR-130b-transfected cells post-chIFN-γ treatment. In contrast, inhibition of miR-130b reduced phosphorylation of STAT1 on Tyr701 (Fig. 10C and D). These results indicate that downregulation of SOCS5 by miR-130b leads to enhanced phosphorylation of STAT1.
FIG 10.

gga-miR-130b enhances STAT1 phosphorylation on Tyr701 residues. (A and B) miR-130b transfection enhanced phosphorylation of STAT1. DF-1 cells were transfected with miR-130b mimics or miRNA controls at 80 nM. Twenty-four hours after transfection, cells were stimulated with chicken IFN-γ (200 ng/ml) and harvested at the indicated time points. (A) The cell lysates were examined by Western blotting using anti-pSTAT1 (Tyr701) and anti-STAT1 antibodies. GAPDH expression was used as an internal control. (B) The band densities of p-STAT1 in panel A were quantitated by densitometry. The relative level of p-STAT1 was calculated as follows: band density of p-STAT1 in each sample/band density of GAPDH in the same sample. (C and D) miR-130b inhibitors reduced the phosphorylation of STAT1. DF-1 cells were transfected with miR-130b inhibitors or miRNA inhibitor controls and stimulated with chicken IFN-γ (200 ng/ml) for the indicated periods. (C) Phosphorylated STAT1 was examined by Western blotting. (D) The band densities of p-STAT1 in panel C were quantitated by densitometry. The relative level of p-STAT1 expression was calculated as described above. Data are representative of three independent experiments and are presented as means and SD. **, P < 0.01.
Taken together, our data indicate that miR-130b suppresses IBDV replication via direct targeting of the IBDV genome and SOCS5.
DISCUSSION
IBD is a highly contagious viral disease that damages lymphoid organs in birds (51). Because this disease causes immunosuppression in poultry (52, 53), it remains a big threat to the poultry industry all over the world. Recently, miRNAs, as new players of regulatory control over gene expression programs, have been well studied, especially in human cancers. The role of miRNAs in host defense against viral infections has also been well illustrated (12, 54, 55). It was reported that host miRNAs could inhibit virus replication via direct targeting of the viral genome in the process of viral infection, suggesting a new kind of host antiviral mechanism (11, 39, 56). Furthermore, multiple miRNAs triggered innate immune response or modulated host factors to provide a less permissive environment for virus replication (15, 57–60). In this study, we observed a miRNA expression profile for IBDV-infected cells. Among these cellular miRNAs, miR-130b attracted our attention because it had been reported to regulate the innate immune system by targeting tumor necrosis factor alpha (TNF-α) in cervical cancer cells (61) and sustaining NF-κB activation in TCC cell lines (33). In the host response to viral infections, cellular miR-130b inhibited replication of porcine reproductive and respiratory syndrome virus (PRRSV) by directly targeting the PRRSV 5′UTR (39). Similarly, our data show that miR-130b plays an important role in host defense against IBDV infection.
First, our data showed that the expression of miR-130b was upregulated in cells during IBDV infection. Second, we found that miR-130b enhanced type I interferon expression and suppressed IBDV replication and that miR-130b enhanced the mRNA expression of IRF3 and p65. Third, and also importantly, we identified the specific sequences in IBDV segment A and the cellular socs5 gene that were targeted by miR-130b, which led to the upregulation of STATs 1, 3, and 6 and enhanced phosphorylation of STAT1. As a consequence, the expression of type I interferon was upregulated and IBDV replication was inhibited. These results demonstrate that miR-130b plays an important role in host defense against IBDV infection.
It is known that miRNAs exert their functions through complementing their target mRNAs in seed regions (62–64). It has been reported that miR-130b suppresses PRRSV replication via targeting of the PRRSV 5′UTR (39). Using bioinformatics analysis, we found one target of miR-130b in the IBDV genome. In addition, our data also show that the region of the SOCS5 3′UTR at 961 bp contains the target site of miR-130b. These data unveil the mechanism underlying miR-130b-mediated suppression of IBDV replication and support the role of miR-130b in host defense against viral infections.
SOCS5 is a member of the SOCS (suppressor of cytokine signaling) family. This family, also known as the STAT-induced STAT inhibitor (SSI) protein family, contains eight members, including SOCS1 to -7 and the CIS protein (65, 66). The SOCS proteins are key negative regulators of cytokine and growth factor signaling (67), and the upregulated expression of SOCS proteins triggers a negative-feedback process for overactivated cytokine signaling via several inhibitory mechanisms (68, 69). SOCS5 can directly bind to the JAK kinase domain via a region in the N terminus, inhibiting the autophosphorylation of JAK1 and JAK2 and hence negatively regulating JAK-STAT signaling (70, 71). It has been reported that JAK-STAT signaling plays a critical role in the host response to viral infections (72, 73). In this study, we found targeting of miR-130b to SOCS5. Inhibition of SOCS5 by miR-130b enhanced the mRNA expression of STATs 1, 3, and 6 and the phosphorylation of STAT1 on Tyr701 residues. As STATs are involved in type I interferon expression, which plays a major role in the host antiviral response (74–76), the enhancement of STAT expression by miR-130b-mediated suppression of SOCS5 contributes at least in part to the inhibition of IBDV replication.
RNA viruses are characterized by rapid mutation due to the lack of proofreading activity of RNA polymerase (77). With regard to miRNA target and virus evolution, some researchers have suggested that miRNA-binding sites without a positive influence on virus replication can rapidly be deleted in vivo, while a positive selective pressure will result in retention of the sites (78). Some researchers have also claimed that miRNA suppression is one way for viruses to evade innate elimination and result in persistent infection (18, 79). As it is a double-stranded RNA virus, there is the possibility for IBDV to partly escape from miR-130b inhibition if the target site of miR-130b on the IBDV genome is mutated. Interestingly, through analyzing the binding sites of miR-130b in the genomes of different strains of IBDV, we found that the binding site of miR-130b had mutations in vvIBDV, while it was relatively conserved in most classical and low-virulence IBDV strains (data not shown). This information indicates that some vvIBDV strains could evolve to avoid the inhibitory effect of miR-130b on replication by mutating its binding site. But besides direct targeting of the viral genome, miR-130b in host cells can also inhibit IBDV replication by enhancing the expression of IFN-β, a vital cytokine in antiviral responses. Thus, miR-130b plays an important role in the cell response to IBDV infection.
miR-130b inhibited the replication of IBDV via direct targeting of the IBDV genome and suppression of SOCS5, an inhibitor of IFN-β signaling in host cells. For both of these mechanisms, direct targeting of miR-130b to the IBDV genome may play a more important role in the inhibition of IBDV replication. Compared with the direct inhibition of viral RNA by miR-130b, the increase of miR-130b on IFN-β was achieved by inhibition of SOCS5 indirectly, and many other factors in host cells may interfere in this process. Of course, many tests were needed to verify this hypothesis, such as testing the inhibition of IBDV replication after mutating the binding site of miR-130b in the genome.
The mechanism underlying miRNA-mediated suppression of viral replication is intricate. Host cells employ miRNAs to inhibit viral replication, while some pathogens use miRNAs to benefit their own replication (80). Several questions need to be addressed. For example, besides miR-130b, are there any other miRNAs involved in the host response to IBDV infection? Does miR-130b also act as an antivirus component in the host defense against other viruses? And are there more targets in signaling pathways involved in host defense? More efforts will be required to elucidate the molecular mechanisms underlying the pathogenesis of IBDV infection.
In summary, the present study reveals that miR-130b plays an important role in inhibiting IBDV replication. It directly targets the IBDV genome, suppressing VP3 expression and viral replication. Furthermore, miR-130b also targets socs5, decreasing the expression of SOCS5, a negative regulator of the JAK-STAT signaling pathway and thus upregulating the expression of type I interferon. These findings may provide important insight into the roles of miRNAs in host defense against viral infections.
MATERIALS AND METHODS
Cells and virus.
DF-1 cells were obtained from the ATCC and were cultured in Dulbecco modified Eagle medium (DMEM) (Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS) in a 5% CO2 and 37°C incubator. Lx, a cell culture-adapted IBDV strain, was kindly provided by Jue Liu (Beijing Academy of Agriculture and Forestry, Beijing, China).
Reagents.
All miRNA mimics and inhibitors were synthesized by Gene Pharma (Shanghai, China). The pGL3-control plasmid was kindly provided by Wenghai Feng (China Agriculture University, Beijing, China). All restriction enzymes were purchased from TaKaRa (Japan). Endotoxin-free plasmid preparation kits were purchased from Aidlab (Beijing, China). Monoclonal antibodies against IBDV VP3 (EU0206), VP4 (EU0207), and VP5 (EU0208) and chicken IFN-γ (EU0216) were obtained from CAEU Biological Company (Beijing, China). Polyclonal antibodies against chicken SOCS5 were prepared by immunizing BALB/c mice with prokaryotically expressed chicken SOCS5 protein in our laboratory. Anti-actin (sc-1616-R) antibody was obtained from Santa Cruz Biotechnology (USA). Anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) mouse monoclonal antibodies were obtained from GBC Lifetech (China). Anti-STAT1 antibodies were obtained from Cell Signal Technology (USA), and anti-pSTAT1 (Tyr701) antibodies were obtained from Proteintech (USA). Poly(I·C) was purchased from Sigma (USA). Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibodies were purchased from DingGuo (China). OPTI-MEM I, jetPRIME transfection reagents, and Lipofectamine 2000 were purchased from Invitrogen (USA). An enhanced chemiluminescence (ECL) kit was purchased from Kangwei Biological Company (China). A dual-luciferase assay kit was purchased from Promega (USA).
miRNA mimics and miRNA target prediction.
DF-1 cells were mock infected or infected with IBDV at an MOI of 0.1 for 24 h, and then the total RNA was extracted and purified. Deep sequencing was performed by LC Sciences (Hangzhou, China). Mimics/inhibitors of miRNAs were synthesized by GenePharma Company. The sense sequences were as follows: for miR-130b-3p mimics, 5′-CAGUGCAAUAAUGAAAGGGCGU-3′; for miR-130b-3p inhibitors, 5′-ACGCCCUUUCAUUAUUGCACUG-3′; for the negative control, 5′-UUCUCCGAACGUGUCACGUTT-3′; and for negative-control inhibitors, 5′-CAGUACUUUUGUGUAGUACAA-3′. miRNA targets in IBDV RNA were predicted by use of RNA22.v2 (https://cm.jefferson.edu/rna22/Interactive/). miRNA targets in host cells were predicted by use of Targetscan7.0 (http://www.targetscan.org/vert_70/), Microcosm Targets (http://www.ebi.ac.uk/enright-srv/microcosm/cgi-bin/targets/v5/genome.pl), and PicTar (http://www.pictar.org/).
Construction of plasmids.
Target sequences of miR-130b were cloned using the following primers: for the miR-130b target sequence in IBDV segment A, sense primer 5′-GGGGTACCGCTTACCCACATCATA-3′ and antisense primer 5′-GAAGATCTTACCACCATGGATCGTC-3′ (GenBank accession number 171906501); for the miR-130b target sequence in SOCS5, sense primer 5′-GCTCTAGAAAGCTTATGGGAATC-3′ and antisense primer 5′-CGTCTAGATGGATTAAGATTGCATG-3′ (GenBank accession number 971386812). Primers for the mutated target sequence were as follows: for the mutated target sequence of miR-130b in segment A, sense primer 5′-AAGCGTCCACGGCCATCTCCAGGGCGCCACCATCTA-3′ and antisense primer 5′-TAGATGGTGGCGCCCTGGAGATGGCCGTGGACGCTT-3′; and for the mutated target sequence of miR-130b in SOCS5, sense primer 5′-AAACATTTTAAAATTAACAAATTGAAAGATATTTACA-3′ and antisense primer 5′-TGTAAATATCTTTCAATTTGTTAATTTTAAAATGTTTT-3′. All the primers were synthesized by Sangon Company (China). All the fragments were amplified by PCR using LA Taq DNA polymerase under the following conditions: 94°C for 5 min, 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, and 72°C for 10 min. The products were stored at 4°C.
Immunofluorescent-antibody assay (IFA).
DF-1 cells were infected with IBDV strain Lx at an MOI of 0.1 and incubated at 37°C for 2 h. Uninfected DF-1 cells were used as negative controls. Two hours after incubation, the medium was changed to fresh DMEM with 2% FBS and continuously incubated for 24 h. The cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, blocked with 1% bovine serum albumin (BSA), and incubated with anti-IBDV VP3 monoclonal antibody followed by FITC-conjugated goat anti-mouse IgG antibodies. The cells were visualized by use of a fluorescence microscope.
qRT-PCR analysis.
Total RNA was prepared from DF-1 cells by use of an Aidlab RNA extraction and purification kit or miRNA extraction kit per the manufacturer's instructions. One microgram of total RNA was used for cDNA synthesis by reverse transcription, using an RT-PCR kit (TaKaRa). Primers used for qRT-PCR were as follows: for IBVD VP3, sense primer 5′-ATGTGGCTGGAAGAGAATGG-3′ and antisense primer 5′-GCCCTTTGAGACTTGCTACCT-3′; for chicken GAPDH (chGAPDH), sense primer 5′-CAACTACATGGTTTACATGTTCC-3′ and antisense primer 5′-GGACTGTGGTCATGAGTCCT-3′; for chIFN-α, sense primer 5′-CCAGCACCTCGAGCAAT-3′ and antisense primer 5′-GGCGCTGTAATCGTTGTCT-3′; for chIFN-β, sense primer 5′-GCCTCCAGCTCCTTCAGAATACG-3′ and antisense primer 5′-CTGGATCTGGTTGAGGAGGCTGT-3′; for chp65, sense primer 5′-CCACAACACAATGCGCTCTG-3′ and antisense primer 5′-AACTCAGCGGCGTCGATG-3′; for chIRF3, sense primer 5′-GCTCTCTGACTCTTTCAACCTCTT-3′ and antisense primer 5′-AATGCTGCTCTTTTCTCCTCTG-3′; for chSOCS5, sense primer 5′-CGCTTCCTGTTTGATTTGGT-3′ and antisense primer 5′-ACTTCGCTCATCGTGCTTTT-3′; for chSTAT1, sense primer 5′-CTTGATGCTGGGAGAGGAGT-3′ and antisense primer 5′-TGAGGGAGAGAGAGCGAAAG-3′; for chSTAT2, sense primer 5′-AGCAAAACTGTCCTGGTTGG-3′ and antisense primer 5′-GACCCTCATTGGTGCCTCT-3′; for chSTAT3, sense primer 5′-TGTTGGCAGATGAGGAGTTG-3′ and antisense primer 5′-GTGATTCGGCAAGAGAGGTTA-3′; for chSTAT4, sense primer 5′-TGAAAGCAATCTGGGTGGA-3′ and antisense primer 5′-TCGCAGTATGTCAGCAAAGG-3′; for chSTAT5, sense primer 5′-TTGACCTGGACGACACCAT-3′ and antisense primer 5′-GACACAAACACGGCACAGTC-3′; and for chSTAT6, sense primer 5′-GGAAAGCGAAAGGGACAAAC-3′ and antisense primer 5′-GATGGGGTTCAGCAGGTTCT-3′. All primers were designed with reference to previous publications (41) and synthesized by Sangon Company. The analysis of real-time PCR was carried out with a Light Cycler 480 machine (Roche, USA). PCR was performed in a 20-μl volume containing 2 μl of cDNA, 10 μl of 2× SYBR green Ex Taq premix (TaKaRa), and 0.4 M (each) gene-specific primers. Thermal cycling parameters were as follows: 94°C for 2 min; 40 cycles of 94°C for 20 s, 55°C for 20 s, and 72°C for 20 s; and 1 cycle of 95°C for 30 s, 60°C for 30 s, and 95°C for 30 s. qRT-PCR analysis of gga-miR-130b was performed with an RT-PCR quantitation kit (GenePharma, China). Thermal cycling parameters for miRNA were as follows: 95°C for 3 min; 40 cycles of 95°C for 12 s and 62°C for 40 s; and 1 cycle of 95°C for 30 s, 60°C for 30 s, and 95°C for 30 s. The final step was to obtain a melting curve for the PCR products to determine the specificity of the amplification. All samples were used in triplicate on the same plate, and the GAPDH gene or U6 snRNA was utilized as the reference gene. The expression levels of genes were calculated relative to that of the GAPDH gene or U6 snRNA and are presented as fold increases or decreases relative to the control sample level.
Preparation of polyclonal antibodies and Western blotting.
To generate polyclonal antibodies against chSOCS5, 6-week-old BALB/c mice were immunized with prokaryotically expressed chSOCS5 protein three times at 3-week intervals. Blood was taken from the orbital sinuses of immunized mice for measurement of anti-chSOCS5 antibodies by indirect enzyme-linked immunosorbent assay (ELISA). To determine the effect of miR-130b on its target, DF-1 cells (6 × 105) were seeded in 6-well plates and cultured for 24 h before transfection with miR-130b mimics or inhibitors or a negative control by use of Lipofectamine 2000. To examine the effect of miRNAs on IBDV replication, DF-1 cells were transfected with miRNAs. Eighteen hours after transfection, cells were infected with or without IBDV at an MOI of 0.1. Cell lysates were prepared using a lysis buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 1% NP-40; 5 mM EDTA; 10% glycerol; 1× complete protease inhibitor cocktail) before being boiled with 6× SDS loading buffer for 10 min. The samples were fractionated by electrophoresis on 10% SDS-PAGE gels, and resolved proteins were transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with 5% skim milk, the membranes were incubated with anti-VP3, anti-VP5, anti-actin, or anti-GAPDH antibody, followed by appropriate HRP-conjugated secondary antibodies. To examine the effect of miRNAs on cellular targets, DF-1 cells were transfected with miRNA mimics or inhibitors or a negative control. Forty-eight hours after transfection, the cell lysates were subjected to Western blotting using anti-GAPDH or anti-SOCS5 antibody followed by HRP-conjugated goat anti-mouse secondary antibodies. Blots were developed using an ECL kit.
Dual-luciferase reporter gene assays.
DF-1 cells were seeded in 24-well plates and transfected with luciferase reporter gene plasmids (pGL3-target-wt or pGL3-target-mutant) and miRNA mimics, inhibitors, or controls by use of Lipofectamine 2000. To normalize for transfection efficiency, 0.01 μg of the pRL-TK Renilla luciferase reporter gene plasmid was added to each transfection mixture. Forty-eight hours after transfection, luciferase reporter gene assays were performed with a dual-luciferase assay kit (Promega, USA). Firefly luciferase activities were normalized on the basis of Renilla luciferase activities. Each reporter gene assay was repeated at least three times. Data are shown as averages ± standard deviations (SD).
Measurement of IBDV growth in DF-1 cells.
DF-1 cells transfected with miRNA mimics or miRNA controls were infected with IBDV at an MOI of 0.1, and cell cultures were collected at different time points (12, 24, 48, and 72 h) after infection. After being freeze-thawed three times, the cell culture samples were centrifuged at 2,000 × g for 10 min, and the supernatants were saved at −80°C until use. The viral contents in the total cell lysates were titrated using a TCID50 assay with DF-1 cells. Briefly, the viral solution was diluted 10-fold in DMEM. A 100-μl aliquot of each diluted sample was added to the well of a 96-well plate, followed by addition of 100 μl of DF-1 cells at a density of 5 × 105 cells/ml. Cells were cultured at 37°C in 5% CO2 for 5 days. Tissue culture wells with cytopathic effect (CPE) were determined to be positive. The titer was calculated based on a previously described method (81).
Examination of STAT1 phosphorylation.
DF-1 cells were seeded in 24-well plates before transfection with miRNA controls or miR-130b mimics or inhibitors. Twenty-four hours after transfection, the cells were stimulated with chicken IFN-γ (200 ng/ml) for the indicated times. The cells were then lysed, and cell lysates were examined by Western blotting using anti-pSTAT1 (Tyr701), anti-STAT1, and anti-GAPDH antibodies, followed by proper HRP-conjugated secondary antibodies. Blots were developed using an ECL kit.
Study approval.
All procedures for animal experiments were approved by the Institutional Animal Care and Use Committee of China Agricultural University (approval no. SKLAB-2016-01-06) and used in accordance with regulations and guidelines of this committee.
Statistical analysis.
The significance of differences in gene expression, luciferase activities, and viral growth between miRNA mimics or inhibitors and controls was determined by the Mann-Whitney test and analysis of variance (ANOVA), as appropriate.
Accession number(s).
The results of the deep sequencing assay were uploaded to the GEO database under accession number GSE90095.
ACKNOWLEDGMENTS
We thank Jue Liu and Wenhai Feng for their kind assistance.
This work was supported by grants from the National Natural Science Foundation of China (grant 31430085) and the Earmarked Fund for Modern Agro-Industry Technology Research System (grant NYCYTX-41).
REFERENCES
- 1.Pitcovski J, Gutter B, Gallili G, Goldway M, Perelman B, Gross G, Krispel S, Barbakov M, Michael A. 2003. Development and large-scale use of recombinant VP2 vaccine for the prevention of infectious bursal disease of chickens. Vaccine 21:4736–4743. doi: 10.1016/S0264-410X(03)00525-5. [DOI] [PubMed] [Google Scholar]
- 2.Stricker RL, Behrens SE, Mundt E. 2010. Nuclear factor NF45 interacts with viral proteins of infectious bursal disease virus and inhibits viral replication. J Virol 84:10592–10605. doi: 10.1128/JVI.02506-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azad AA, Barrett SA, Fahey KJ. 1985. The characterization and molecular cloning of the double-stranded RNA genome of an Australian strain of infectious bursal disease virus. Virology 143:35–44. doi: 10.1016/0042-6822(85)90094-7. [DOI] [PubMed] [Google Scholar]
- 4.Pan J, Lin L, Tao YJ. 2009. Self-guanylylation of birnavirus VP1 does not require an intact polymerase activity site. Virology 395:87–96. doi: 10.1016/j.virol.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Einem UI, Gorbalenya AE, Schirrmeier H, Behrens SE, Letzel T, Mundt E. 2004. VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J Gen Virol 85:2221–2229. doi: 10.1099/vir.0.19772-0. [DOI] [PubMed] [Google Scholar]
- 6.Kibenge FS, McKenna PK, Dybing JK. 1991. Genome cloning and analysis of the large RNA segment (segment A) of a naturally avirulent serotype 2 infectious bursal disease virus. Virology 184:437–440. doi: 10.1016/0042-6822(91)90865-9. [DOI] [PubMed] [Google Scholar]
- 7.Jagadish MN, Staton VJ, Hudson PJ, Azad AA. 1988. Birnavirus precursor polyprotein is processed in Escherichia coli by its own virus-encoded polypeptide. J Virol 62:1084–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hudson PJ, McKern NM, Power BE, Azad AA. 1986. Genomic structure of the large RNA segment of infectious bursal disease virus. Nucleic Acids Res 14:5001–5012. doi: 10.1093/nar/14.12.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong RT, Hon CC, Zeng F, Leung FC. 2007. Screening of differentially expressed transcripts in infectious bursal disease virus-induced apoptotic chicken embryonic fibroblasts by using cDNA microarrays. J Gen Virol 88:1785–1796. doi: 10.1099/vir.0.82619-0. [DOI] [PubMed] [Google Scholar]
- 10.O'Neill AM, Livant EJ, Ewald SJ. 2010. Interferon alpha-induced inhibition of infectious bursal disease virus in chicken embryo fibroblast cultures differing in Mx genotype. Avian Dis 54:802–806. doi: 10.1637/9001-072309-Reg.1. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Q, Guo XK, Gao L, Huang C, Li N, Jia X, Liu W, Feng WH. 2014. MicroRNA-23 inhibits PRRSV replication by directly targeting PRRSV RNA and possibly by upregulating type I interferons. Virology 450–451:182–195. doi: 10.1016/j.virol.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 12.Song L, Liu H, Gao S, Jiang W, Huang W. 2010. Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J Virol 84:8849–8860. doi: 10.1128/JVI.00456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe Y, Kishi A, Yachie N, Kanai A, Tomita M. 2007. Computational analysis of microRNA-mediated antiviral defense in humans. FEBS Lett 581:4603–4610. doi: 10.1016/j.febslet.2007.08.049. [DOI] [PubMed] [Google Scholar]
- 14.Contreras J, Rao DS. 2012. MicroRNAs in inflammation and immune responses. Leukemia 26:404–413. doi: 10.1038/leu.2011.356. [DOI] [PubMed] [Google Scholar]
- 15.Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X. 2009. MicroRNA-146a feedback inhibits RIG-I-dependent type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol 183:2150–2158. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- 16.Taganov KD, Boldin MP, Chang KJ, Baltimore D. 2006. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papadopoulou AS, Dooley J, Linterman MA, Pierson W, Ucar O, Kyewski B, Zuklys S, Hollander GA, Matthys P, Gray DH, De Strooper B, Liston A. 2011. The thymic epithelial microRNA network elevates the threshold for infection-associated thymic involution via miR-29a mediated suppression of the IFN-alpha receptor. Nat Immunol 13:181–187. doi: 10.1038/ni.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahajan VS, Drake A, Chen J. 2009. Virus-specific host miRNAs: antiviral defenses or promoters of persistent infection? Trends Immunol 30:1–7. doi: 10.1016/j.it.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bushati N, Cohen SM. 2007. microRNA functions. Annu Rev Cell Dev Biol 23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 20.Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 21.Bartel DP, Chen CZ. 2004. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet 5:396–400. [DOI] [PubMed] [Google Scholar]
- 22.Song K, Han C, Dash S, Balart LA, Wu T. 2015. miR-122 in hepatitis B virus and hepatitis C virus dual infection. World J Hepatol 7:498–506. doi: 10.4254/wjh.v7.i3.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. 2005. MicroRNA expression profiles classify human cancers. Nature 435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 24.Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. 2007. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci U S A 104:7080–7085. doi: 10.1073/pnas.0702409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo CJ, Pan Q, Li DG, Sun H, Liu BW. 2009. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: an essential role for apoptosis. J Hepatol 50:766–778. doi: 10.1016/j.jhep.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 26.Zhu B, Ye J, Nie Y, Ashraf U, Zohaib A, Duan X, Fu ZF, Song Y, Chen H, Cao S. 2015. MicroRNA-15b modulates Japanese encephalitis virus-mediated inflammation via targeting RNF125. J Immunol 195:2251–2262. doi: 10.4049/jimmunol.1500370. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. 2007. Requirement of bic/microRNA-155 for normal immune function. Science 316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao G, Zhang JG, Shi Y, Qin Q, Liu Y, Wang B, Tian K, Deng SC, Li X, Zhu S, Gong Q, Niu Y, Wang CY. 2013. miR-130b is a prognostic marker and inhibits cell proliferation and invasion in pancreatic cancer through targeting STAT3. PLoS One 8:e73803. doi: 10.1371/journal.pone.0073803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu AM, Yao TJ, Wang W, Wong KF, Lee NP, Fan ST, Poon RT, Gao C, Luk JM. 2012. Circulating miR-15b and miR-130b in serum as potential markers for detecting hepatocellular carcinoma: a retrospective cohort study. BMJ Open 2:e825. doi: 10.1136/bmjopen-2012-000825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colangelo T, Fucci A, Votino C, Sabatino L, Pancione M, Laudanna C, Binaschi M, Bigioni M, Maggi CA, Parente D, Forte N, Colantuoni V. 2013. MicroRNA-130b promotes tumor development and is associated with poor prognosis in colorectal cancer. Neoplasia 15:1086–1099. doi: 10.1593/neo.13998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bertero T, Cottrill K, Krauszman A, Lu Y, Annis S, Hale A, Bhat B, Waxman AB, Chau BN, Kuebler WM, Chan SY. 2015. The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J Biol Chem 290:2069–2085. doi: 10.1074/jbc.M114.617845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miao Y, Zheng W, Li N, Su Z, Zhao L, Zhou H, Jia L. 2017. MicroRNA-130b targets PTEN to mediate drug resistance and proliferation of breast cancer cells via the PI3K/Akt signaling pathway. Sci Rep 7:41942. doi: 10.1038/srep41942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cui X, Kong C, Zhu Y, Zeng Y, Zhang Z, Liu X, Zhan B, Piao C, Jiang Z. 2016. miR-130b, an onco-miRNA in bladder cancer, is directly regulated by NF-kappaB and sustains NF-kappaB activation by decreasing cylindromatosis expression. Oncotarget 7:48547–48561. doi: 10.18632/oncotarget.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han X, Wang Y, Zhang X, Qin Y, Qu B, Wu L, Ma J, Zhou Z, Qian J, Dai M, Tang Y, Chan EK, Harley JB, Zhou S, Shen N. 2016. MicroRNA-130b ameliorates murine lupus nephritis through targeting the type I interferon pathway on renal mesangial cells. Arthritis Rheumatol 68:2232–2243. doi: 10.1002/art.39725. [DOI] [PubMed] [Google Scholar]
- 35.Cruz LO, Hashemifar SS, Wu CJ, Cho S, Nguyen DT, Lin LL, Khan AA, Lu LF. 2017. Excessive expression of miR-27 impairs Treg-mediated immunological tolerance. J Clin Invest 127:530–542. doi: 10.1172/JCI88415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao M, Sun D, Guan Y, Wang Z, Sang D, Liu M, Pu Y, Fang X, Wang D, Huang A, Bi X, Cao L, He C. 2016. Disulfiram and diphenhydramine hydrochloride upregulate miR-30a to suppress IL-17-associated autoimmune inflammation. J Neurosci 36:9253–9266. doi: 10.1523/JNEUROSCI.4587-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Wu H, Li P, Zhao Y, Liu M, Tang H. 2014. NF-kappaB-modulated miR-130a targets TNF-alpha in cervical cancer cells. J Transl Med 12:155. doi: 10.1186/1479-5876-12-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han B, Lian L, Li X, Zhao C, Qu L, Liu C, Song J, Yang N. 2016. Chicken gga-miR-130a targets HOXA3 and MDFIC and inhibits Marek's disease lymphoma cell proliferation and migration. Mol Biol Rep 43:667–676. doi: 10.1007/s11033-016-4002-2. [DOI] [PubMed] [Google Scholar]
- 39.Li L, Gao F, Jiang Y, Yu L, Zhou Y, Zheng H, Tong W, Yang S, Xia T, Qu Z, Tong G. 2015. Cellular miR-130b inhibits replication of porcine reproductive and respiratory syndrome virus in vitro and in vivo. Sci Rep 5:17010. doi: 10.1038/srep17010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S, Duan X, Li Y, Liu B, McGilvray I, Chen L. 2014. MicroRNA-130a inhibits HCV replication by restoring the innate immune response. J Viral Hepat 21:121–128. doi: 10.1111/jvh.12131. [DOI] [PubMed] [Google Scholar]
- 41.Li Z, Wang Y, Li X, Li X, Cao H, Zheng SJ. 2013. Critical roles of glucocorticoid-induced leucine zipper in infectious bursal disease virus (IBDV)-induced suppression of type I interferon expression and enhancement of IBDV growth in host cells via interaction with VP4. J Virol 87:1221–1231. doi: 10.1128/JVI.02421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim TH, Zhou H. 2015. Functional analysis of chicken IRF7 in response to dsRNA analog poly(I:C) by integrating overexpression and knockdown. PLoS One 10:e133450. doi: 10.1371/journal.pone.0133450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cormican P, Lloyd AT, Downing T, Connell SJ, Bradley D, O'Farrelly C. 2009. The avian Toll-like receptor pathway—subtle differences amidst general conformity. Dev Comp Immunol 33:967–973. doi: 10.1016/j.dci.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 44.Schmitz ML, Mattioli I, Buss H, Kracht M. 2004. NF-kappaB: a multifaceted transcription factor regulated at several levels. Chembiochem 5:1348–1358. doi: 10.1002/cbic.200400144. [DOI] [PubMed] [Google Scholar]
- 45.Vermeulen L, De Wilde G, Notebaert S, Vanden BW, Haegeman G. 2002. Regulation of the transcriptional activity of the nuclear factor-kappaB p65 subunit. Biochem Pharmacol 64:963–970. doi: 10.1016/S0006-2952(02)01161-9. [DOI] [PubMed] [Google Scholar]
- 46.Liu KD, Gaffen SL, Goldsmith MA. 1998. JAK/STAT signaling by cytokine receptors. Curr Opin Immunol 10:271–278. doi: 10.1016/S0952-7915(98)80165-9. [DOI] [PubMed] [Google Scholar]
- 47.Au-Yeung N, Mandhana R, Horvath CM. 2013. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. JAKSTAT 2:e23931. doi: 10.4161/jkst.23931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Najjar I, Fagard R. 2010. STAT1 and pathogens, not a friendly relationship. Biochimie 92:425–444. doi: 10.1016/j.biochi.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ihle JN. 2001. The Stat family in cytokine signaling. Curr Opin Cell Biol 13:211–217. doi: 10.1016/S0955-0674(00)00199-X. [DOI] [PubMed] [Google Scholar]
- 50.Rauch I, Muller M, Decker T. 2013. The regulation of inflammation by interferons and their STATs. JAKSTAT 2:e23820. doi: 10.4161/jkst.23820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muller H, Islam MR, Raue R. 2003. Research on infectious bursal disease—the past, the present and the future. Vet Microbiol 97:153–165. doi: 10.1016/j.vetmic.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 52.Sharma JM, Kim IJ, Rautenschlein S, Yeh HY. 2000. Infectious bursal disease virus of chickens: pathogenesis and immunosuppression. Dev Comp Immunol 24:223–235. doi: 10.1016/S0145-305X(99)00074-9. [DOI] [PubMed] [Google Scholar]
- 53.van den Berg TP, Eterradossi N, Toquin D, Meulemans G. 2000. Infectious bursal disease (Gumboro disease). Rev Sci Tech 19:509–543. doi: 10.20506/rst.19.2.1227. [DOI] [PubMed] [Google Scholar]
- 54.Wang YS, Ouyang W, Pan QX, Wang XL, Xia XX, Bi ZW, Wang YQ, Wang XM. 2013. Overexpression of microRNA gga-miR-21 in chicken fibroblasts suppresses replication of infectious bursal disease virus through inhibiting VP1 translation. Antiviral Res 100:196–201. doi: 10.1016/j.antiviral.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 55.Wang P, Hou J, Lin L, Wang C, Liu X, Li D, Ma F, Wang Z, Cao X. 2010. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol 185:6226–6233. doi: 10.4049/jimmunol.1000491. [DOI] [PubMed] [Google Scholar]
- 56.Ingle H, Kumar S, Raut AA, Mishra A, Kulkarni DD, Kameyama T, Takaoka A, Akira S, Kumar H. 2015. The microRNA miR-485 targets host and influenza virus transcripts to regulate antiviral immunity and restrict viral replication. Sci Signal 8:a126. doi: 10.1126/scisignal.aab3183. [DOI] [PubMed] [Google Scholar]
- 57.Li Z, Chen B, Feng M, Ouyang H, Zheng M, Ye Q, Nie Q, Zhang X. 2015. MicroRNA-23b promotes avian leukosis virus subgroup J (ALV-J) replication by targeting IRF1. Sci Rep 5:10294. doi: 10.1038/srep10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lian L, Li X, Zhao C, Han B, Qu L, Song J, Liu C, Yang N. 2015. Chicken gga-miR-181a targets MYBL1 and shows an inhibitory effect on proliferation of Marek's disease virus-transformed lymphoid cell line. Poult Sci 94:2616–2621. doi: 10.3382/ps/pev289. [DOI] [PubMed] [Google Scholar]
- 59.Ren Z, Ambros VR. 2015. Caenorhabditis elegans microRNAs of the let-7 family act in innate immune response circuits and confer robust developmental timing against pathogen stress. Proc Natl Acad Sci U S A 112:E2366–E2375. doi: 10.1073/pnas.1422858112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martinez-Nunez RT, Louafi F, Friedmann PS, Sanchez-Elsner T. 2009. MicroRNA-155 modulates the pathogen binding ability of dendritic cells (DCs) by down-regulation of DC-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN). J Biol Chem 284:16334–16342. doi: 10.1074/jbc.M109.011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim C, Lee H, Cho YM, Kwon OJ, Kim W, Lee EK. 2013. TNFalpha-induced miR-130 resulted in adipocyte dysfunction during obesity-related inflammation. FEBS Lett 587:3853–3858. doi: 10.1016/j.febslet.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 62.Moore MJ, Scheel TK, Luna JM, Park CY, Fak JJ, Nishiuchi E, Rice CM, Darnell RB. 2015. miRNA-target chimeras reveal miRNA 3′-end pairing as a major determinant of Argonaute target specificity. Nat Commun 6:8864. doi: 10.1038/ncomms9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hausser J, Zavolan M. 2014. Identification and consequences of miRNA-target interactions—beyond repression of gene expression. Nat Rev Genet 15:599–612. doi: 10.1038/nrg3765. [DOI] [PubMed] [Google Scholar]
- 64.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. 2003. Prediction of mammalian microRNA targets. Cell 115:787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 65.Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, Kile BT, Kent SB, Alexander WS, Metcalf D, Hilton DJ, Nicola NA, Baca M. 1999. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci U S A 96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, Sprigg NS, Starr R, Nicholson SE, Metcalf D, Nicola NA. 1998. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A 95:114–119. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Linossi EM, Nicholson SE. 2015. Kinase inhibition, competitive binding and proteasomal degradation: resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol Rev 266:123–133. doi: 10.1111/imr.12305. [DOI] [PubMed] [Google Scholar]
- 68.Liang Y, Xu WD, Peng H, Pan HF, Ye DQ. 2014. SOCS signaling in autoimmune diseases: molecular mechanisms and therapeutic implications. Eur J Immunol 44:1265–1275. doi: 10.1002/eji.201344369. [DOI] [PubMed] [Google Scholar]
- 69.Rico-Bautista E, Flores-Morales A, Fernandez-Perez L. 2006. Suppressor of cytokine signaling (SOCS) 2, a protein with multiple functions. Cytokine Growth Factor Rev 17:431–439. doi: 10.1016/j.cytogfr.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 70.Sharma N, Kumawat KL, Rastogi M, Basu A, Singh SK. 2016. Japanese encephalitis virus exploits the microRNA-432 to regulate the expression of suppressor of cytokine signaling (SOCS) 5. Sci Rep 6:27685. doi: 10.1038/srep27685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Linossi EM, Chandrashekaran IR, Kolesnik TB, Murphy JM, Webb AI, Willson TA, Kedzierski L, Bullock AN, Babon JJ, Norton RS, Nicola NA, Nicholson SE. 2013. Suppressor of cytokine signaling (SOCS) 5 utilises distinct domains for regulation of JAK1 and interaction with the adaptor protein Shc-1. PLoS One 8:e70536. doi: 10.1371/journal.pone.0070536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin RJ, Chang BL, Yu HP, Liao CL, Lin YL. 2006. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J Virol 80:5908–5918. doi: 10.1128/JVI.02714-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heim MH, Moradpour D, Blum HE. 1999. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J Virol 73:8469–8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao L, He S, Liu Y, Zhao P, Bian Z, Qi Z. 2016. Inhibition of STAT pathway impairs anti-hepatitis C virus effect of interferon alpha. Cell Physiol Biochem 40:77–90. doi: 10.1159/000452526. [DOI] [PubMed] [Google Scholar]
- 75.Wesoly J, Szweykowska-Kulinska Z, Bluyssen HA. 2007. STAT activation and differential complex formation dictate selectivity of interferon responses. Acta Biochim Pol 54:27–38. [PubMed] [Google Scholar]
- 76.Darnell JJ, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 77.Steinhauer DA, Domingo E, Holland JJ. 1992. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 122:281–288. doi: 10.1016/0378-1119(92)90216-C. [DOI] [PubMed] [Google Scholar]
- 78.Trobaugh DW, Klimstra WB. 2017. MicroRNA regulation of RNA virus replication and pathogenesis. Trends Mol Med 23:80–93. doi: 10.1016/j.molmed.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Umbach JL, Cullen BR. 2009. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev 23:1151–1164. doi: 10.1101/gad.1793309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sullivan CS, Ganem D. 2005. MicroRNAs and viral infection. Mol Cell 20:3–7. doi: 10.1016/j.molcel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 81.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497. [Google Scholar]