

Abstract
Based on promising results in preclinical models, clinical trials have been performed to evaluate the efficacy of the first-in-class proteasome inhibitor bortezomib towards malignant pleural mesothelioma (MPM), an aggressive cancer arising from the mesothelium of the serous cavities following exposure to asbestos. Unexpectedly, only minimal therapeutic benefits were observed, thus implicating that MPM harbors inherent resistance mechanisms. Identifying the molecular bases of this primary resistance is crucial to develop novel pharmacologic strategies aimed at increasing the vulnerability of MPM to bortezomib. Therefore, we assessed a panel of four human MPM lines with different sensitivity to bortezomib, for functional proteasome activity and levels of free and polymerized ubiquitin. We found that highly sensitive MPM lines display lower proteasome activity than more bortezomib-resistant clones, suggesting that reduced proteasomal capacity might contribute to the intrinsic susceptibility of mesothelioma cells to proteasome inhibitors-induced apoptosis. Moreover, MPM equipped with fewer active proteasomes accumulated polyubiquitinated proteins, at the expense of free ubiquitin, a condition known as proteasome stress, which lowers the cellular apoptotic threshold and sensitizes mesothelioma cells to bortezomib-induced toxicity as shown herein. Taken together, our data suggest that an unfavorable load-versus-capacity balance represents a critical determinant of primary apoptotic sensitivity to bortezomib in MPM.
Introduction
The 26S proteasome is an ATP-dependent protease complex abundantly expressed in eukaryotic cells, responsible for the regulated hydrolysis of most cellular proteins1. The 26S is a multi-subunit enzyme consisting of a 20S particle, where polypeptides get hydrolyzed, associated with the 19S regulatory particles, responsible for recognizing, unfolding, and translocating polyubiquitinated proteins into the inner degradative chamber of the 20S2. This is a ~700 kDa cylindrical macromolecular machine formed by four overlapping heptameric rings, consisting of α (the outer rings) or β (the inner rings) subunits3. Normally, the hydrolyzing activity is yielded by subunits β5, β2, and β1 of the 20S proteasome. However, cells of the immune system or exposed to proinflammatory cytokines are induced to express three additional homologous subunits (β5i, β2i, β1i), which replace their constitutive counterparts in newly synthesized and shorter-lived immunoproteasomes4. In principle, proteasomes can hydrolyze the C-terminal amide bond of every amino acid but proline5; however, proteolytic activities assessed by means of short fluorogenic peptides identify three defined hydrolyzing preferences: trypsin-like (i.e. hydrolysis at the C-terminus of basic residues, β2/β2i), chymotrypsin-like (i.e. hydrolysis at the C-terminus of hydrophobic residues, β5/β5i), and caspase-like activity (i.e. at the C-terminus of acidic residues, β1/β1i). A variety of low molecular weight inhibitors of the proteolytic sites of 20S have been identified that can enter cells and block proteasomal protein degradation6. These agents proved essential for investigating the biological role of the ubiquitin-proteasome system and led to the discovery of diverse key regulatory functions of this pathway. Furthermore, since proteasome inhibitors (PIs) also induce adaptive and maladaptive responses (e.g. the unfolded protein and heat-shock responses), showing previously unpredicted specificity against certain tumor cells, they became the paradigm of negative proteostasis regulators in cancer therapy7.
The anti-cancer use of PIs stemmed from the original observation of their remarkable toxicity against a variety of cancer cells at doses that had little or no toxicity against normal, non-transformed cells8. Further studies and clinical trials allowed the rapid approval of the modified boronic dipeptide bortezomib (Btz, PS-341 or Velcade®) for the treatment of multiple myeloma (MM)9 and refractory mantle cell lymphoma10. Encouraging results were also reported on other hematological and solid cancers11,12. Moreover, the anti-tumor activity of five second generation PIs is currently being evaluated in dedicated clinical trials13. In the case of MM, the accumulation of polyubiquitinated proteins is an established mechanism of PI-induced apoptosis14,15. Such a condition, referred to as proteasome stress 16,17, in plasma cells is particularly aggravated by the extremely high load of protein degradation imposed by large-scale immunoglobulin production, which makes the apoptotic threshold particularly low18–20. This load-versus-capacity model offers an explanation for the exquisite sensitivity of malignant plasma cells to PIs21, and also for the variability observed among different MM cell lines and primary tumors, which differ in both proteasome capacity and degradative workload18. Furthermore, demonstrating cause-effect relationships, increasing proteasome expression18 and reducing protein synthesis21 independently increased bortezomib resistance. Whether and to what extent the load-versus-capacity model may also contribute to explain PI responsiveness of other cancers, and particularly solid tumors, is currently unknown.
Malignant pleural mesothelioma (MPM) is a highly deadly cancer arising from the mesothelium of serous cavities following exposure to asbestos22. Circa 3,000 new MPM cases are diagnosed yearly in the United States, with ~250,000 deaths predicted to be caused by MPM in the next 30 years in occidental Europe23,24. Although measures have been implemented to limit further exposure to asbestos, the long latency of MPM generated a strikingly increasing incidence of this malignancy. MPM is normally intractable by to local therapies and typically progresses with a median overall survival of 12–36 months for localized illness and only 8–14 months for advanced disease25. The currently available chemotherapeutic drugs are poorly active against MPM, with usual single-agent response rates of ≤20%26, and first-line radiation therapy is generally ineffective27. Combination therapy with pemetrexed and cisplatin is the current standard first-line treatment28. However, median survival on this regimen is below 1 year, with <50% response rate. Notable, a randomized phase 3 trial recently reported a significant gain in overall survival (median 18.8 vs. 16.1 months) in MPM patients treated with bevacizumab in addition to the standard pemetrexed and cisplatin regimen29. Moreover, no approved standard of care for patients who have relapsed is available, since several trials have failed to show useful activities in a second-line setting30. Hence, identifying and developing new treatments for MPM is an unmet need.
Although some molecular profiling studies of malignant mesothelioma identified the ubiquitin-proteasome system (UPS) as a potential therapeutic target31–33, several others could not establish any significant correlation between clinical and/or anatomo-histological features of MPM and alterations of the proteasome proteolytic pathway34–41. Notwithstanding, the first-in-class PI bortezomib demonstrated promising anti-tumoral activity in both in vitro and in vivo preclinical models of MPM. Specifically, bortezomib was shown to induce cytotoxicity, cell cycle arrest, and apoptosis in several primary patient-derived and immortalized MPM cell lines, while sparing normal mesothelial cells42–44. The effector mechanisms of such toxicity are poorly understood, with an intricate network of pro- and anti-apoptotic factors possibly implicated45–48. These results provide the rationale to clinically evaluate the effect of bortezomib, alone or in combination with cisplatin, against MPM49,50. However, only modest therapeutic benefits were observed. In particular, data from a multicenter Phase II study of bortezomib as monotherapy in an unselected population of MPM patients demonstrated a poor (5%) response rate, implying that inherent mechanisms of resistance protect primary tumors49. Identifying the molecular bases of the differential sensitivity of MPM cells to proteasome inhibition is, therefore, critical to design novel pharmacologic strategies aimed at increasing responsiveness to bortezomib, alone or in therapeutic combinations. To this end, we hereby challenged the load-versus-capacity model in a panel of four human MPM cell lines characterized by differential apoptotic sensitivity to bortezomib.
Results
Human MPM cell lines show differential sensitivity to the pro-apoptotic effects of bortezomib
In an effort aimed at verifying whether different MPM cell lines display differential sensitivity to apoptosis triggered by proteasome inhibition, we initially tested the biological effects of bortezomib on a panel of four human MPM clones. In particular, MSTO-211H, REN, MM98, and MMB cell lines were treated with increasing amounts of bortezomib and apoptosis was measured after 48 hours by FACS assessment of annexin V and propidium iodide positive cells. As shown in Fig. 1, all clones displayed a marked dose-dependent susceptibility to the pro-apoptotic effects of bortezomib in the nanomolar range. However, the sensitivity to the inhibitor of the four cell lines clearly differed. In particular, MM98 and REN cells displayed EC50 values for bortezomib-induced apoptosis of 17 and 22 nM respectively, while the MSTO-211H line was relatively more resistant with an EC50 of 60 nM. The MMB line was characterized by an intermediate sensitivity (EC50 33 nM; Table 1). Thus, we demonstrated that the aforementioned MPM lines are characterized by a well-defined differential sensitivity/resistance to bortezomib and are therefore suitable for investigating the molecular mechanisms responsible for the different susceptibility of MPM cells to apoptosis caused by proteasome inhibition.
Figure 1.
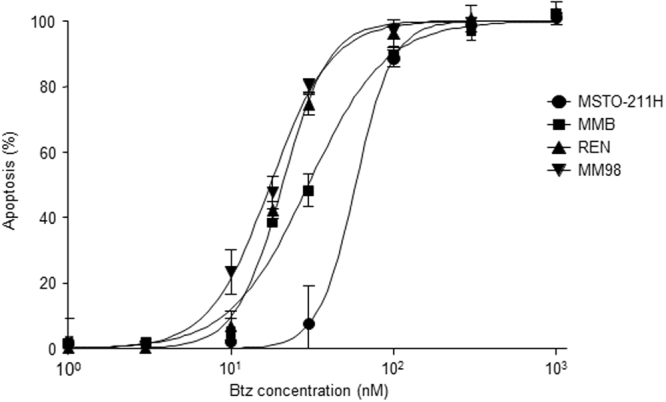
Effects of bortezomib on the viability of malignant pleural mesothelioma cell lines. MM98, REN, MMB, and MSTO-211H cells were treated with increasing concentrations of bortezomib and apoptosis was measured after 48 hours by FACS of annexin V and propidium iodide positive cells, and expressed as variation vs. untreated cells. Values are mean ± SE of three independent experiments. R2 for a sigmoidal dose-response curve ≥0.9 in all cases.
Table 1.
Differential sensitivity of MPM cell lines to bortezomib.
| Cell line | EC50 Btz (nM) |
|---|---|
| MM98 | 16.9 ± 1.7 |
| REN | 22.1 ± 1.4 |
| MMB | 33.4 ± 2.0 |
| MSTO-211H | 60.1 ± 8.6 |
The Malignant Pleural Mesothelioma cell lines MM98, REN, MMB, MSTO-211H were treated for 48 hours with increasing concentrations of bortezomib (Btz) and the proportion of apoptotic (annexin V + propidium iodide-) cells was measured by FACS. The indicated EC50 are an average of 3 independent experiments ± SE.
Different sensitivity of MPM clones to bortezomib correlates with proteasome activity
To assess whether in MPM, as in the paradigmatic PIs-sensitive cancer MM, enhanced susceptibility towards cytotoxic effects of bortezomib correlates with lower overall potential proteasomal proteolytic capacity, we measured the three main peptidase activities of 26S proteasomes in cellular extracts from the four MPM clones by specific fluorogenic substrates in the presence of ATP. With this method proteasome cleavage specificities can be selectively assessed in crude extracts, avoiding time-consuming and laborious chromatographic purifications, since the contribution of non-proteasomal enzymes is determined by highly specific inhibitors of proteasome active sites51. By this approach, we could demonstrate that rates of fluorogenic peptides hydrolysis clearly differ between the MPM lines (Fig. 2A). More importantly, the strongly Btz-sensitive and relatively BTZ-resistant MM98 and MSTO-211H lines displayed, respectively, lower and higher 26S chymotrypsin-like and trypsin-like activities, which are the main proteasomal cleavage specificities and the rate limiting activities for protein turnover52,53. Moreover, for all MPM clones a clear, direct correlation between proteasome chymotrypsin- and trypsin-like activities and resistance to apoptosis induced by bortezomib was seen (Fig. 2B), strongly suggesting that the size of the proteasomal compartment contributes to the sensitivity to PIs in mesothelioma cells. Only for the minor caspase-like activity could a clear positive correlation with Btz-EC50 values of the corresponding MPM lines not be established (Fig. 2B).
Figure 2.

Reduced proteasome activity in MPM cell lines with increased sensitivity to bortezomib. (A) Chymotrypsin-like, trypsin-like, and caspase-like specific activities of proteasomes were assessed in cell extracts as described in the Materials and Methods and expressed on a per-cell basis. Data are the mean of six independent measurements. **P < 0.01. (B) Correlations of 26S proteasome peptidase activities and EC50 values relative to the apoptotic effect of bortezomib in MPM cell lines.
Loss of concerted expression of proteasomal subunits in MPM clones
To investigate the reasons for the difference in proteasome activities of MPM clones in more detail at the molecular level, we measured the expression levels of several proteasome subunits by western blot analysis. In this regard, it is worth noting that the steady-state levels of mature β-subunits, which are subjected to autocatalytic cleavage upon assembly to generate the active form, are directly responsible for proteasome peptidase activities54. Unexpectedly, the expression patterns of the catalytic β-subunits of both constitutive and immuno proteasomes was complex and highly variable between MPM lines (Fig. 3A). In fact, indicative of a loss of the regulatory mechanisms that normally ensure concerted expression of the catalytic subunits of constitutive and immuno proteasomes, each clone showed an individual pattern of content of β-subunits, with some antigens clearly up-regulated and others down-regulated, with no clear delineations between the different lines (Fig. 3B). Similarly, the steady-state levels of the α-subunits also appeared variable and not concertedly regulated in each MPM line, although in this case expression levels seemed concordantly reduced in MM98 cells, which are characterized by lower proteasome activity (Fig. 2). However, α-subunits do not necessarily reflect assembled, functional proteasomes, since they also include the pool of free intracellular subunits54.
Figure 3.
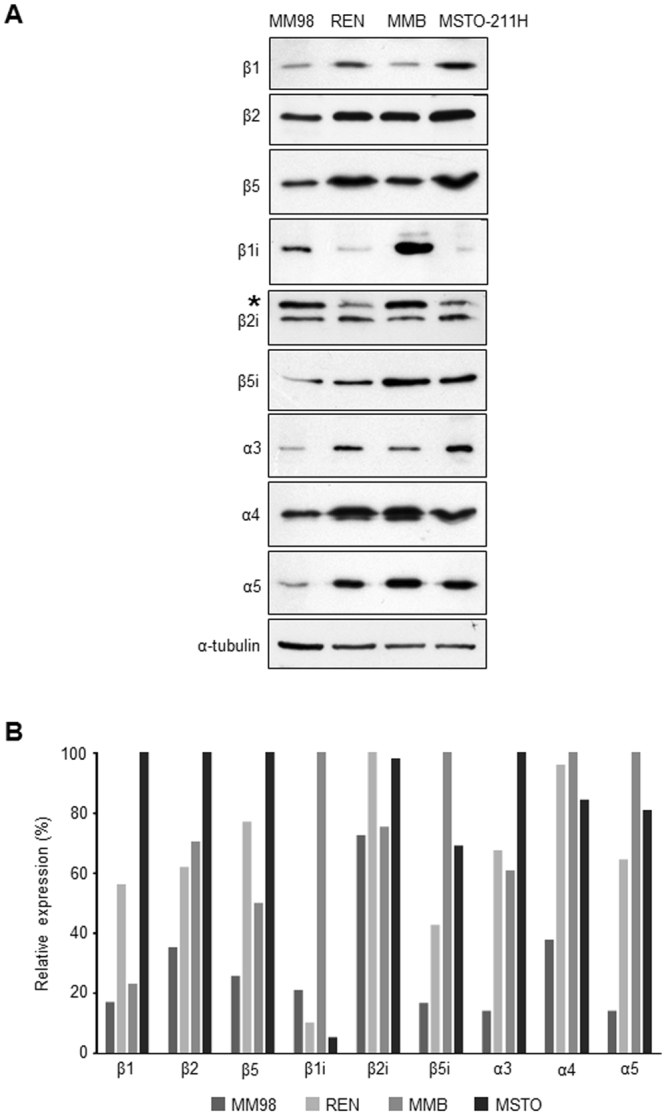
Steady-state levels of catalytic β and non-catalytic α-subunits of constitutive and immunoproteasomes in MPM cell lines. (A) Cellular extracts from the MM98, REN, MMB, and MSTO-211H lines were resolved by SDS-PAGE and analyzed by western blotting with Abs to different proteasome catalytic β and non-catalytic α-subunits. Equal protein amounts (6 µg) were loaded in each lane, with α-tubulin serving as a loading control. *Unspecific band. All the blots were cropped according to their locations in the membrane in order to fit the size, see the original figures in the supplementary information. (B) Relative densitometric quantification of western blot bands shown in panel A normalized by the control loading signal.
More Btz-sensitive MPM lines display higher levels of proteasome stress
A reduced proteasomal compartment may not be effectively suited to cope with the degradative needs of the cell. As a consequence of the resulting unbalance in the load-versus-capacity ratio, cells are expected to experience a higher basal level of proteotoxic stress that, in turn, lowers their intrinsic apoptotic threshold. To verify this scenario, we therefore looked for signs of proteasomal sufferance in the MPM lines by assessing the intracellular levels of free and polymerized ubiquitin. As shown in Fig. 4A, western blotting analysis unambiguously demonstrated that the more Btz-sensitive MM98 and REN clones are characterized by a significantly lower content of unbound ubiquitin compared to the relatively more resistant MMB and MSTO-211H cell lines (Fig. 4B). This deficiency of free ubiquitin reflects its role in building polyubiquitin chains, which, in fact, were greatly accumulated in the more Btz-sensitive clones (Fig. 4C).
Figure 4.
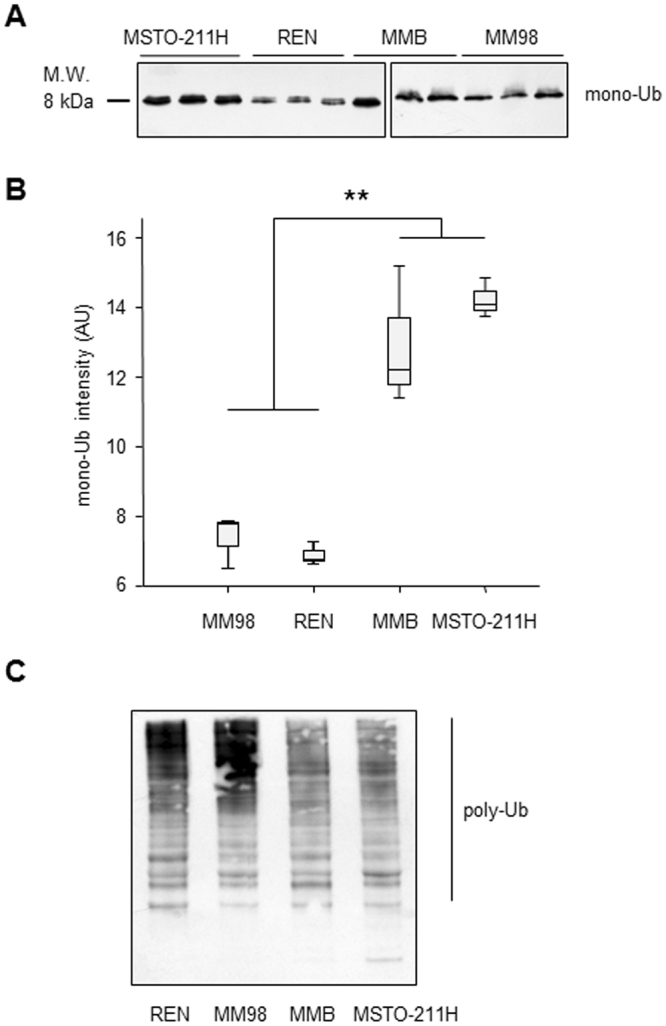
Basal accumulation of polyubiquitinated proteins at the expense of the pool of free ubiquitin in MPM cell lines more sensitive to bortezomib. (A) 15 µg of cellular extracts from the MM98, REN, MMB, and MSTO-211H lines were incubated with anti-ubiquitin (Ub) Ab. For each clone, data from three independent cell extracts are shown. Samples derive from the same experiment and that gels/blots were processed in parallel. The blots were cropped according to their locations in the membrane in order to fit the size, see the original figures in the supplementary information. (B) Densitometric quantification of mono-ubiquitin bands shown in panel A. **P < 0.01. (C) Polyubiquitinated proteins (poly-Ub, 5 µg of cellular extract) were resolved as a smear in an 12% SDS-Page gel, while free Ub was separated in an 18% gel.
To investigate these findings, indicating saturation of proteasome proteolytic capacity with consequent accumulation of undegraded substrates in MPM cells that were more vulnerable to bortezomib, we evaluated polyubiquitinated proteins at the single cell level by confocal immunofluorescence microscopy. To this aim, we used FK2, an antibody that recognizes different types of polyubiquitin chains55 and therefore it is particularly well-suited to detect all potential proteasome substrates. In fact, several recent evidences indicate that not only the canonical lysine-48 linked polyubiquitin chains but also other alternative ubiquitin linked chains support degradation by the 26S proteasome56,57. In line with the western blot data, this analysis revealed strong basal accumulation of polyubiquitinated proteins in REN and MM98 cells with a discrete cytosolic and nuclear pattern, clearly exceeding the signal present in MMB and MSTO-211H cells (Fig. 5). Subsequent quantification and statistical analysis of FK2-dependent fluorescence confirmed that basal accumulation of polyubiquitinated proteins differed substantially in all four MPM clones (Fig. 6A). Most importantly, the fluorescence intensity associated to polyubiquitinated proteins negatively correlated both with the main functional activities of 26S (Fig. 6B) and the EC50 of the corresponding MPM clone for the pro-apoptotic effect of bortezomib (Fig. 6C), clearly indicating a role for overload of proteasomal degradative capacity in determining cellular susceptibility to PIs.
Figure 5.

Immunofluorescence microscopy of polyubiquitinated proteins in MPM cell lines. Cells were stained with FK2 antibody, which specifically recognizes poly-ubiquitin chains, and analyzed by confocal microscopy in three independent experiments. Nuclei labelled red by propidium iodide (PI); size bar: 50 μm lower magnification (left) and 10 μm higher magnification (right). Representative images. Cell area (µ2), mean ± St. dev.: MM98 108 ± 11, REN 117 ± 9, MMB 93 ± 12, MSTO-211H 110 ± 8.
Figure 6.

Basal accumulation of polyubiquitinated proteins inversely correlates with proteasome activity and with intrinsic resistance of MPM clones to the pro-apoptotic effects of bortezomib. (A) Different levels of polyubiquitin conjugates in MPM cell lines. Quantification of the FK2-dependent fluorescence in MPM cells was performed using Image J software (n ≥ 100 cells in ≥ 16 distinct fields). ***P < 0.001. Inverse correlation between levels of polyubiquitinated proteins in MPM clones and 26S proteasome chymotrypsin- and trypsin-like activities (B) and EC50 values relative to apoptotic sensitivity to bortezomib (C).
Discussion
Previous studies have shown that bortezomib and other PIs exhibit significant anti-tumoral activity in preclinical models of MPM both in vitro and in vivo 42–44,58,59. Disappointingly, however, this efficacy was not successfully translated in clinical activity due to either primary and/or acquired resistance49,50. In an effort aimed at identifying the mechanisms of the different intrinsic sensitivities of MPM cells to cytotoxicity induced by proteasomal inhibition, we initially assessed the pro-apoptotic effects of bortezomib in a panel of four human MPM cell lines. In accordance with published data, this analysis revealed that all four MPM clones tested displayed pronounced apoptotic vulnerability to bortezomib, with EC50 values comparable to those determined by the same experimental approach for different cell lines of human multiple myeloma, the paradigmatic proteasome inhibitors responsive cancer21. Crucially, however, the primary sensitivity of these MPM lines to bortezomib varied, with MM98 and REN cells characterized by higher vulnerability to the drug (EC50 values of 17 and 22 nM, respectively), while the MSTO-211H line was relatively more resistant (EC50 of 60 nM) and the MMB clone hallmarked by intermediate susceptibility (EC50 of 33 nM). The finding that these mesothelioma cell lines are not equally responsive to the cytotoxic effects of bortezomib prompted us to investigate the molecular mechanisms of their different intrinsic sensitivity to PIs. In fact, primary resistance to bortezomib represents a crucial therapeutic challenge not only for solid tumors like MPM, but also for prototypical PIs-responsive hematological malignancies. More than 50% of MM patients, for example, fail to respond to bortezomib and the basis of this different intrinsic responsiveness remains elusive. We18,21 and others60 have recently shown that exquisite apoptotic sensitivity of both primary or immortalized MM cells arises from exuberant synthesis of abnormal proteins impinging on a reduced proteasome pool. The resulting saturation of cellular proteolytic capacity (a condition referred to as proteostenosis) has profound consequences for cell viability and tumor chemosensitivity21.
The present study is, to the best of our knowledge, the first documenting that different levels of basal proteasome stress, intended as a situation of precarious equilibrium, characterized by lack of overt signs of cell sufferance (e.g. cell cycle arrest), but deeply affecting the capacity of the cell to successfully cope with additional stress, underlies the intrinsic apoptotic sensitivity to bortezomib for a solid cancer such as pleural mesothelioma. Under normal conditions mammalian cells are capable of adapting their proteasome content to proteolytic needs (through complex feedback loops involving the transcription factor NRF1, and the mTorc1 and ERK5 pathways), thus relieving proteasome stress, as demonstrated in several experimental settings61,62. However, as shown in many studies, cancer cells generally suffer from more stress than their benign counterparts, and are generally more reliant on stress adaptive pathways, making them less efficient at coping with additional stress, hence the model of non-oncogene addiction, which provides a rationale for targeting protein homeostasis or other stress adaptive pathways63. Consistent with this concept, the neoplastic cell lines (both in the case of MPM or MM) hallmarked by an intrinsic, exquisite sensitivity to PI-induced cytotoxic effects, invariably show pathognomonic signs of proteasome stress (e.g. marked accumulation of polyubiquitinated conjugates at the expense of free monomeric ubiquitin), even in the absence of any other condition or stimulus that might negatively impinge on proteasome cellular content or activity. Although other mechanisms such as differential activities of ubiquitin-ligases or deubiquitinating enzymes might contribute to the increase in steady-state levels of ubiquitin-conjugates, the strong correlation between accumulation of polyubiquitinated substrates and low chymotrypsin-like and trypsin-like proteasome activities (that are rate limiting for degradation of proteins in vivo and in vitro 52,53) strongly indicates that reduced proteasome levels and activities play a pivotal role in this process. In fact, in MPM, as already well documented for MM, enhanced levels of proteotoxic stress invariably correspond to a strong reduction of the cellular proteasome compartment. Indeed, the two MPM lines (MM98 and REN) displaying the lower overall 26S activity were found to accumulate higher levels of polyubiquitinated proteins than the two lines (MMB and MSTO-211H) characterized by a higher proteasome content. Interestingly, our results show that proteasome levels may vary greatly among different MPMs, with profound implications for the intrinsic capability of coping with cytotoxic stress, given the key role of the proteasome in integrating signals that control cell-cycle progression, apoptosis, and metabolism. Accordingly, chymotrypsin-like and trypsin-like proteasome activities inversely correlate with the sensitivity to bortezomib of MPM lines, indicating that reduction of the proteasome cellular pool represents a crucial determinant of proteotoxic stress that predisposes mesothelioma cells to the pro-apoptotic effects of PIs. Importantly, bortezomib mainly targets chymotrypsin-like proteasomal activity, while discordant results were reported concerning its effects toward trypsin-like sites11,64. However, it is worth noting that in vivo both bortezomib and other PIs exert their toxic effects by blocking catabolism of full length proteins, whose rates of hydrolysis are determined by the concomitant and coordinated action of all three proteasome active sites, acting through a complex (and not yet completely understood) network of allosteric functional interactions65,66.
Moreover, western blot analysis of the steady-state levels of several proteasome subunits revealed that the MPM cells analyzed in our study have lost the standard, concerted expression of all three catalytic β-subunits of constitutive or immuno proteasomes. On the contrary, each MPM clone shows a specific and individual pattern of catalytic β-subunit expression, indicative of a loss of the regulatory mechanisms that under physiological conditions ensure coordinated expression of active β-subunits, with the purpose to adapt proteasome capacity to cellular needs so as to maintain homeostasis61. Such dysregulated expression patterns may be a consequence of genotoxic stress typical of neoplastic cells, and are likely to account for the difference in the proteasome activity between MPM cell lines that our study unveiled. In fact, reduced expression of individual catalytic subunits and conceivable formation of non-standard mixed proteasomes containing β-subunits of both immuno and constitutive proteasomes may substantially affect overall proteasome enzymatic efficiency67. Of note, loss of the concerted, physiological expression of all proteasome subunits is likely to account for the inability of several DNA microarrays studies to demonstrate a crucial role of the proteasome proteolytic pathway in the biology and pathogenesis of MPM34–41. These results emphasize the need to integrate microarrays expression data with functional measurements of proteasome enzymatic activities.
Of great interest, our study revealed a striking positive correlation between high levels of polyubiquitinated conjugates in MPM cell lines and primary sensitivity to the pro-apoptotic effects of bortezomib, which strongly suggests a cause-effect relationship. In fact, the more Btz-sensitive MM98 and REN lines displayed high levels of basal proteasomal stress indicative of a reduced efficiency of adaptive strategies, including induction of proteasome biogenesis (so-called Proteasome stress response)16,17, which might relieve the proteostatic imbalance experienced by these MPM clones. Of note, the stressful condition revealed by basal accumulation of endogenous ubiquitin conjugates is still compatible with apparently normal cell functions, implying regular turnover of key proteasomal substrates (e.g. cyclins). However, this precarious equilibrium can be easily pushed towards a toxic collapse of proteostasis by further compromising the proteolytic route at doses of PIs that are lower than those required to achieve the same result in cells that, like MSTO-211H (and to a lesser extent MMB), are characterized by a more-favorable load-versus-capacity ratio. Importantly, saturation of the proteasomal proteolytic route may stabilize pro-apoptotic effectors, such as the Bcl-2 homology domain 3 (BH3)-only proteins Bim and Noxa and the mitochondrial outer membrane permeabilizers BAX/BAK, or abrogate activity of survival factors like NF-kB, which are all known to play a pivotal role in modulating the sensitivity of MPM cells to bortezomib44,45,47,49,68. So far, it has been generally assumed that the load-versus-capacity model may account for the exquisite sensitivity to PIs of MM and other professional secretory cells that are intrinsically predisposed to saturation of the proteasomal degradative route due to their overwhelming rates of protein synthesis. Our data, on the contrary, suggest that a compromised proteostatic equilibrium, arising at least in part from a reduced proteasome pool, might be a typical hallmark of PIs-sensitive cancers in general. Although further studies will be required to fully understand the molecular mechanisms of this proteostatic unbalance in MPM and possibly in other PIs-responsive solid cancers, it is possible that the high levels of genotoxic stress typically experienced by tumor cells, with the consequent enhanced generation of mutated/abnormal polypeptides requiring extremely fast degradation69, accompanied by a reduced efficiency of appropriate adaptive strategies counteracting this stressful and potentially harmful condition, might underlie the basal proteotoxic stress seen in our study in MPM clones that are more sensitive to bortezomib.
Our results documenting that an unfavorable load-vs-capacity ratio represents a critical determinant of apoptotic sensitivity and underlies vulnerability to bortezomib not only for the prototypical PIs-responsive hematological cancer multiple myeloma, but also for an extremely recalcitrant solid tumor like MPM, potentially provide a framework for identifying indicators of responsiveness to PIs and for improving their clinical efficacy. Conceivably, our data indicate that assessment of proteasome stress and capacity in MPM patients might represent a potential predictor of individual responsiveness to bortezomib with both prognostic and therapeutic value. Moreover, pharmacological strategies aimed at exacerbating the proteotoxic stress experienced by MPM cells are predicted to lower their intrinsic threshold to apoptosis triggered by PIs and therefore substantially increase their efficacy against this malignancy.
Methods
Cell lines
Human malignant pleural mesothelioma (MPM) cell lines were generously provided by Dr. S. Biffo (University of Milan, Milan, Italy) (MM98) and Dr. G. Gaudino (University of Hawaii, Honolulu, HI, USA) (MMB, MSTO-211H, REN). MSTO-211H were maintained in RPMI1640 containing 10% fetal bovine serum (FBS), L-glutamine, and antibiotics; REN and MM98 in DMEM containing 10% FBS, L-glutamine, and antibiotics; MMB in nutrient mixture F-12 Ham supplemented with 110% FBS, L-glutamine, and antibiotics. All MPM cell lines were cultured at 37 °C in an humidified incubator in an atmosphere of 5% CO2. Under these growth conditions, the doubling time for all MPM clones was ~24 h. All cell culture reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Flow-cytometric analyses of apoptosis
Cells were treated with bortezomib (Millennium Pharmaceuticals, Cambridge, MA, USA) as indicated, harvested, stained with FITC-conjugated Annexin V (1 μg/ml) and propidium iodide (2.5 μg/ml) according to the manufacturer’s instructions, and analyzed by FACScalibur (BD Biosciences, Franklin Lakes, NJ, USA) as described18,19,21.
Proteasome activity assays
Proteasome activity was assessed in MPM crude extracts using fluorogenic peptides according to a method that avoids long and laborious chromatographic purification of 26S particles51. Accordingly, proteasome-specific peptidase activities were assayed by monitoring the production of 7-amino-4-methylcoumarin (amc) from the following fluorogenic peptides (Bachem, Bubendorf, Switzerland): 100 μM Suc-LLVY-amc (for chymotrypsin-like), 500 μM Bz-VGR-amc (for trypsin-like), and 100 μM Ac-YVAD-amc (for caspase-like activity) in 20 mM Tris-HCl (pH 7.5), 1 mM ATP, 2 mM MgCl2, and 0.2% bovine serum albumin (BSA). Two of these substrates (Suc-LLVY-amc and Suc-YVAD-amc) were used at a concentration highly exceeding their Km value for the relative proteasome catalytic β-subunit, and therefore hydrolysis rates were assessed at Vmax. The third substrate (Bz-VGR-amc) has a lower affinity for β2/β2i subunit and was used at a higher concentration to approach its Vmax of hydrolysis (Not shown). Reactions were started by adding an aliquot of cellular extract, and the fluorescence of released amc (excitation, 380 nm; emission, 460 nm) was monitored continuously at 37 °C with a Carry Eclipse spectrofluorometer (Varian, Palo Alto, CA, USA). Background activity (caused by non-proteasomal degradation) was determined by addition of the PI epoxomicin (Sigma-Aldrich, St. Louis, MO, USA) for chymotrypsin-like activity, MG132 (Calbiochem, San Diego, CA, USA) for caspase-like activity and β-lactone (Enzo Life Sciences, Farmingdale, NY, USA) for trypsin-like activity, each used at a final concentration (2 μM, 10 μM, and 20 μM, respectively) known to completely suppress the relative proteasomal peptidase activity without affecting other proteases with the same cleavage specificity6. Assays were calibrated using standard solutions of free fluorophore, and reaction velocities were calculated from the slopes of the initial linear portions of the curves. Substrate consumption at the end of incubation never exceeded 1%.
Immunoblot analyses
Immunoblot analyses of α and β-subunits of constitutive and immuno proteasomes, α-tubulin, free ubiquitin and polyubiquitinated proteins were performed as previously described21,54. Briefly, extracts were resolved by 18% (for free ubiquitin) or 12% (for polyubiquitinated conjugates and all other antigens) SDS-PAGE gel and transferred on Nitocellulose (Sigma-Aldrich, St. Louis, MO, USA, for polyubiquitinated conjugates) or Immobilon®-P (Merck Millipore, Darmstadt, Germany, for all other antigens) transfer membranes. Nitrocellulose membrane was boiled for 5 min to unmask poly-Ub antigens and so make more sensitive and quantitative the immunoblot. The membranes were then incubated in blocking buffer (5% BSA, 0.1%Tween-20 in 1 × PBS), followed by incubation with mAb anti-ubiquitin (P4D1, Santa Cruz Biotechnologies, Santa Cruz, CA, USA), anti α-tubulin (T5168, Sigma-Aldrich, St. Louis, MO, USA), anti-α3, -α4, -α5 (MCP196, MCP79, MCP257, Enzo Life Sciences, Farmingdale, NY, USA), anti-β1, -β2, -β5, -β1i -β2i, -β5i (gift of Prof. S. Ferrone, Harvard Medical School, Boston, MA, USA). Bound antibodies were visualized using the ECL technique and bands were detected with Hyperfilm™ ECL™ (GE Healthcare). Densitometric analysis was performed with a VersaDoc 1000 Imaging System and Quantity One software (Bio-Rad, Hercules, CA, USA).
Immunofluorescence
MPM cell lines were seeded on poly-L-lysine-coated slides, fixed with 4% paraformaldehyde for 10 min at room temperature (RT), and permeabilized with PBS 0.1% Triton X-100 for 10 min at RT. Cells were then preincubated with PBS-1% normal goat serum (NGS) for 30 min at RT, incubated with monoclonal mouse anti-ubiquitin (clone FK2, 1/100; Enzo Life Sciences, Farmingdale NY, USA) diluted in PBS-1% NGS for 1 hour at RT, rinsed in PBS, and then stained with goat anti-mouse IgG Alexa Fluor 488 (1/200, Life Technologies, Paisley, UK) diluted in PBS-1% NGS for 1 hour at RT. After rinsing in PBS, cells were stained with propidium iodide for 5 min at RT (1/1000; Sigma-Aldrich, St. Louis, MO, USA), washed in PBS, and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). Images were acquired as single-stack images (capture images) at 40× water immersion with a confocal laser scanning microscope (TCS SP5, Leica Microsystems). Laser parameters were kept unchanged for all the different cell groups so that the detection of staining was maximal while avoiding pixel saturation. Quantitative analysis was performed by an operator unaware of the experimental group. Images were analyzed with ImageJ software (NIH, Bethesda, MD, USA) measuring cell area, mean gray value, and integrated density. More than 100 cells per group were counted and analyzed.
Statistical analysis
To compare average measurements of peptidase proteasomal activities, western blot densitometries, and immunofluorescence intensities, we adopted a non-parametric Mann-Whitney test, since preliminary Shapiro-Wilk tests have proven that data were not normally distributed. Data were graphically visualized using box plots.
Data Availability
No datasets were generated or analyzed during the current study.
Electronic supplementary material
Acknowledgements
We thank S. Biffo and G. Gaudino for providing human MPM lines, S. Ferrone for antibodies, P. Moore for assistance in preparation of the manuscript, and C. Cocito for valuable assistance in statistical analysis of the data. This work was supported through grants to P.C. from Fondazione della Regione autonoma Valle d’Aosta per la ricerca sul cancro, and Fondazione Cassa di Risparmio di Torino. S.C. is supported by Associazione Italiana per la Ricerca sul Cancro (AIRC, Investigator Grant 18858 and Special Program Molecular Clinical Oncology 5 per mille n.9965).
Author Contributions
P.C. and F.C. conceived and designed the experiments; F.C., G.J., C.S., L.O., L.P., B.A. performed the experiments; F.C., G.J., C.S., L.O., L.P., B.A., S.M., G.D., G.N., S.C., and P.C. analyzed the data; F.C., S.C. and P.C. wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-17977-9.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Voges D, Zwickl P, Baumeister W. The Ubiquitin 26S Proteasome Proteolytic Pathway. Annu. Rev. Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 2.Baumeister W, Walz J, Zühl F, Seemüller E. The proteasome: Paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/S0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 3.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 4.Cascio P. PA28αβ: The Enigmatic Magic Ring of the Proteasome? Biomolecules. 2014;4:566–84. doi: 10.3390/biom4020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raule M, et al. PA28αβ reduces size and increases hydrophilicity of 20S immunoproteasome peptide products. Chem. & Biol. 2014;21:470–480. doi: 10.1016/j.chembiol.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem. & Biol. 2001;8:739–758. doi: 10.1016/S1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 7.Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–421. doi: 10.1016/S1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 8.Orlowski RZ, et al. Tumor growth inhibition induced in a murine model of human Burkitt’s lymphoma by a proteasome inhibitor. Cancer Res. 1998;58:4342–4348. [PubMed] [Google Scholar]
- 9.San Miguel JF, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N. Engl. J. Med. 2008;359:906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 10.O’Connor OA, et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J. Clin. Oncol. 2005;23:676–684. doi: 10.1200/JCO.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 11.Kisselev AF, Van Der Linden WA, Overkleeft HS. Proteasome inhibitors: An expanding army attacking a unique target. Chem. & Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: Lessons from the first decade. Clin. Cancer Res. 2008;14:1649–1657. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 13.Molineaux SM. Molecular pathways: Targeting proteasomal protein degradation in cancer. Clin. Cancer Res. 2012;18:15–20. doi: 10.1158/1078-0432.CCR-11-0853. [DOI] [PubMed] [Google Scholar]
- 14.Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia. 2009;23:1964–1979. doi: 10.1038/leu.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hideshima T, Anderson KC. Biologic Impact of Proteasome Inhibition in Multiple Myeloma Cells-From the Aspects of Preclinical Studies. Semin. Hematol. 2012;49:223–227. doi: 10.1053/j.seminhematol.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CS, et al. A proteasomal stress response: Pre-treatment with proteasome inhibitors increases proteasome activity and reduces neuronal vulnerability to oxidative injury. J. Neurochem. 2004;91:996–1006. doi: 10.1111/j.1471-4159.2004.02813.x. [DOI] [PubMed] [Google Scholar]
- 17.London MK, Keck BI, Ramos PC, Dohmen RJ. Regulatory mechanisms controlling biogenesis of ubiquitin and the proteasome. FEBS Lett. 2004;567:259–264. doi: 10.1016/j.febslet.2004.04.078. [DOI] [PubMed] [Google Scholar]
- 18.Bianchi G, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood. 2009;113:3040–3049. doi: 10.1182/blood-2008-08-172734. [DOI] [PubMed] [Google Scholar]
- 19.Cascio P, et al. Dampening Ab responses using proteasome inhibitors following in vivo B cell activation. Eur. J. Immunol. 2008;38:658–667. doi: 10.1002/eji.200737743. [DOI] [PubMed] [Google Scholar]
- 20.Cenci S, et al. Progressively impaired proteasomal capacity during terminal plasma cell differentiation. EMBO J. 2006;25:1104–1113. doi: 10.1038/sj.emboj.7601009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cenci S, et al. Pivotal Advance: Protein synthesis modulates responsiveness of differentiating and malignant plasma cells to proteasome inhibitors. J. Leukoc. Biol. 2012;92:921–931. doi: 10.1189/jlb.1011497. [DOI] [PubMed] [Google Scholar]
- 22.Kamp DW. Asbestos-induced lung diseases: an update. Translational Research. 2009;153:143–152. doi: 10.1016/j.trsl.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson BWS, Lake RA. Advances in Malignant Mesothelioma. N. Engl. J. Med. 2005;353:1591–1603. doi: 10.1056/NEJMra050152. [DOI] [PubMed] [Google Scholar]
- 24.Tsao AS, Wistuba I, Roth JA, Kindler HL. Malignant pleural mesothelioma. J. Clin. Oncol. 2009;27:2081–2090. doi: 10.1200/JCO.2008.19.8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelvez-Zapata SM, Gaffney D, Scarci M, Coonar AS. What is the survival after surgery for localized malignant pleural mesothelioma? Interact. Cardiovasc. Thorac. Surg. 2013;16:533–537. doi: 10.1093/icvts/ivs542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fennell Da, Gaudino G, O’Byrne KJ, Mutti L, van Meerbeeck J. Advances in the systemic therapy of malignant pleural mesothelioma. Nat. Clin. Pract. Oncol. 2008;5:136–147. doi: 10.1038/ncponc1039. [DOI] [PubMed] [Google Scholar]
- 27.Goudar RK. New therapeutic options for mesothelioma. Curr. Oncol. Rep. 2005;7:260–265. doi: 10.1007/s11912-005-0048-3. [DOI] [PubMed] [Google Scholar]
- 28.Vogelzang NJ, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003;21:2636–2644. doi: 10.1200/JCO.2003.11.136. [DOI] [PubMed] [Google Scholar]
- 29.Zalcman, G., Brosseau, S. & Scherpereel, A. Prophylactic radiotherapy to prevent procedure-tract metastases. Lancet Oncol. 17 (2016). [DOI] [PubMed]
- 30.Krug LM. An overview of chemotherapy for mesothelioma. Hematology/Oncology Clinics of North America. 2005;19:1117–1136. doi: 10.1016/j.hoc.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Røe OD, et al. Malignant pleural mesothelioma: Genome-wide expression patterns reflecting general resistance mechanisms and a proposal of novel targets. Lung Cancer. 2010;67:57–68. doi: 10.1016/j.lungcan.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 32.Borczuk AC, et al. Molecular profiling of malignant peritoneal mesothelioma identifies the ubiquitin-proteasome pathway as a therapeutic target in poor prognosis tumors. Oncogene. 2007;26:610–617. doi: 10.1038/sj.onc.1209809. [DOI] [PubMed] [Google Scholar]
- 33.Bott M, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011;43:668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pass HI, et al. Gene Expression Profiles Predict Survival and Progression of Pleural Mesothelioma. Clin. Cancer Res. 2004;10:849–859. doi: 10.1158/1078-0432.CCR-0607-3. [DOI] [PubMed] [Google Scholar]
- 35.Gordon GJ, et al. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am. J. Pathol. 2005;166:1827–40. doi: 10.1016/S0002-9440(10)62492-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.López-Ríos F, et al. Global gene expression profiling of pleural mesotheliomas: Overexpression of aurora kinases and P16/CDKN2A deletion as prognostic factors and critical evaluation of microarray-based prognostic prediction. Cancer Res. 2006;66:2970–2979. doi: 10.1158/0008-5472.CAN-05-3907. [DOI] [PubMed] [Google Scholar]
- 37.Røe, O. D. et al. Genome-wide profile of pleural mesothelioma versus parietal and visceral pleura: The emerging gene portrait of the mesothelioma phenotype. PLoS One4 (2009). [DOI] [PMC free article] [PubMed]
- 38.de Reyniès A, et al. Molecular classification of malignant pleural mesothelioma: identification of a poor prognosis subgroup linked to the epithelial-to-mesenchymal transition. Clin. Cancer Res. 2014;20:1323–34. doi: 10.1158/1078-0432.CCR-13-2429. [DOI] [PubMed] [Google Scholar]
- 39.Bueno R, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016;48:407–416. doi: 10.1038/ng.3520. [DOI] [PubMed] [Google Scholar]
- 40.Quispel-Janssen, J. M. et al. Comprehensive pharmacogenomic profiling of malignant pleural mesothelioma identifies a subgroup sensitive to FGFR inhibition. Clin. Cancer Res. 23, 10.1158/1078-0432.CCR-17-1172 (2017). [DOI] [PubMed]
- 41.Schunselaar, L. et al. Chemical profiling of primary mesothelioma cultures defines subtypes with different expression profiles and clinical responses. Clin. Cancer Res. 10.1158/1078-0432.CCR-17-1345 (2017). [DOI] [PubMed]
- 42.Wang Y, et al. Targeted proteasome inhibition by Velcade induces apoptosis in human mesothelioma and breast cancer cell lines. Cancer Chemother. Pharmacol. 2010;66:455–466. doi: 10.1007/s00280-009-1181-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gordon GJ, et al. Preclinical studies of the proteasome inhibitor bortezomib in malignant pleural mesothelioma. Cancer Chemother. Pharmacol. 2008;61:549–558. doi: 10.1007/s00280-007-0500-1. [DOI] [PubMed] [Google Scholar]
- 44.Sartore-Bianchi A, et al. Bortezomib inhibits nuclear factor-kappaB-dependent survival and has potent in vivo activity in mesothelioma. Clin. Cancer Res. 2007;13:5942–5951. doi: 10.1158/1078-0432.CCR-07-0536. [DOI] [PubMed] [Google Scholar]
- 45.Busacca, S. et al. BAK and NOXA Are Critical Determinants of Mitochondrial Apoptosis Induced by Bortezomib in Mesothelioma. PLoS One8 (2013). [DOI] [PMC free article] [PubMed]
- 46.Barbone D, Yang TM, Morgan JR, Gaudino G, Broaddus VC. Mammalian target of rapamycin contributes to the acquired apoptotic resistance of human mesothelioma multicellular spheroids. J. Biol. Chem. 2008;283:13021–13030. doi: 10.1074/jbc.M709698200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbone D, et al. The Bcl-2 repertoire of mesothelioma spheroids underlies acquired apoptotic multicellular resistance. Cell Death Dis. 2011;2:e174. doi: 10.1038/cddis.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan B-Z, et al. TRAIL and proteasome inhibitors combination induces a robust apoptosis in human malignant pleural mesothelioma cells through Mcl-1 and Akt protein cleavages. BMC Cancer. 2013;13:140. doi: 10.1186/1471-2407-13-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fennell Da, et al. Phase II clinical trial of first or second-line treatment with bortezomib in patients with malignant pleural mesothelioma. J. Thorac. Oncol. 2012;7:1466–1470. doi: 10.1097/JTO.0b013e318260dfb9. [DOI] [PubMed] [Google Scholar]
- 50.O’Brien, M. et al. Phase II study of bortezomib with cisplatin as first-line treatment of malignant pleural mesothelioma (MPM): EORTC 08052. J. Clin. Oncol. 30 (2012).
- 51.Kisselev AF, Goldberg AL. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol. 2005;398:364–378. doi: 10.1016/S0076-6879(05)98030-0. [DOI] [PubMed] [Google Scholar]
- 52.Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH. The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J. Biol. Chem. 1997;272:25200–25209. doi: 10.1074/jbc.272.40.25200. [DOI] [PubMed] [Google Scholar]
- 53.Raule M, Cerruti F, Cascio P. Enhanced rate of degradation of basic proteins by 26S immunoproteasomes. Biochim. Biophys. Acta - Mol. Cell Res. 2014;1843:1942–1947. doi: 10.1016/j.bbamcr.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt M, Kloetzel PM. Biogenesis of eukaryotic 20S proteasomes: the complex maturation pathway of a complex enzyme. FASEB J. 1997;11:1235–1243. doi: 10.1096/fasebj.11.14.9409542. [DOI] [PubMed] [Google Scholar]
- 55.Schwertman P, et al. An immunoaffinity purification method for the proteomic analysis of ubiquitinated protein complexes. Anal. Biochem. 2013;440:227–236. doi: 10.1016/j.ab.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 56.Grice GL, Nathan JA. The recognition of ubiquitinated proteins by the proteasome. Cellular and Molecular Life Sciences. 2016;73:3497–3506. doi: 10.1007/s00018-016-2255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu P, et al. Quantitative Proteomics Reveals the Function of Unconventional Ubiquitin Chains in Proteasomal Degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun X, Gulyás M, Hjerpe A, Dobra K. Proteasome inhibitor PSI induces apoptosis in human mesothelioma cells. Cancer Lett. 2006;232:161–169. doi: 10.1016/j.canlet.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 59.Yuan BZ, Chapman JA, Reynolds SH. Proteasome Inhibitor MG132 Induces Apoptosis and Inhibits Invasion of Human Malignant Pleural Mesothelioma Cells. Transl. Oncol. 2008;1:129–140. doi: 10.1593/tlo.08133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shabaneh, T. B. et al. Molecular Basis of Differential Sensitivity of Myeloma Cells to Clinically Relevant Bolus Treatment with Bortezomib. PLoS One8 (2013). [DOI] [PMC free article] [PubMed]
- 61.Rousseau A, Bertolotti A. An evolutionarily conserved pathway controls proteasome homeostasis. Nature. 2016;536:184–189. doi: 10.1038/nature18943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Radhakrishnan SK, et al. Transcription Factor Nrf1 Mediates the Proteasome Recovery Pathway after Proteasome Inhibition in Mammalian Cells. Mol. Cell. 2010;38:17–28. doi: 10.1016/j.molcel.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blackburn C, et al. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S β5-subunit. Biochem. J. 2010;430:461–476. doi: 10.1042/BJ20100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ruschak AM, Kay LE. Proteasome allostery as a population shift between interchanging conformers. Proc. Natl. Acad. Sci. 2012;109:E3454–E3462. doi: 10.1073/pnas.1213640109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kisselev AF, et al. The caspase-like sites of proteasomes, their substrate specificity, new inhibitors and substrates, and allosteric interactions with the trypsin-like sites. J. Biol. Chem. 2003;278:35869–35877. doi: 10.1074/jbc.M303725200. [DOI] [PubMed] [Google Scholar]
- 67.Guillaume B, et al. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class Imolecules. Proc. Natl. Acad. Sci. 2010;107:18599–18604. doi: 10.1073/pnas.1009778107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barbone, D. et al. Vorinostat Eliminates Multicellular Resistance of Mesothelioma 3D Spheroids via Restoration of Noxa Expression. PLoS One7 (2012). [DOI] [PMC free article] [PubMed]
- 69.Schubert U, et al. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analyzed during the current study.