

Abstract
Transfer RNAs acquire a variety of naturally occurring chemical modifications during their maturation; these fine-tune their structure and decoding properties in a manner critical for protein synthesis. We recently reported that in the eukaryotic parasite, Trypanosoma brucei, a methylation and deamination event are unexpectedly interconnected, whereby the tRNA adenosine deaminase (TbADAT2/3) and the 3-methylcytosine methyltransferase (TbTrm140) strictly rely on each other for activity, leading to formation of m3C and m3U at position 32 in several tRNAs. Still however, it is not clear why these two enzymes, which work independently in other systems, are strictly codependent in T. brucei. Here, we show that these enzymes exhibit binding synergism, or a mutual increase in binding affinity, that is more than the sum of the parts, when added together in a reaction. Although these enzymes interact directly with each other, tRNA binding assays using enzyme variants mutated in critical binding and catalytic sites indicate that the observed binding synergy stems from contributions from tRNA-binding domains distal to their active sites. These results provide a rationale for the known interactions of these proteins, while also speaking to the modulation of substrate specificity between seemingly unrelated enzymes. This information should be of value in furthering our understanding of how tRNA modification enzymes act together to regulate gene expression at the post-transcriptional level and provide a basis for the interdependence of such activities.
Keywords: tRNA, modification, RNA editing, T. brucei
INTRODUCTION
All nucleic acids in cells undergo some type of post-transcriptional chemical modification; these may involve chemically simple modifications such as methylations and thiolations or more complex ones requiring several chemical building blocks. Regardless, the importance of modifications is highlighted by their prevalence and degree of evolutionary conservation, and further made obvious by the fact that organisms dedicate more than 1% of their genome to encode modification enzymes (Björk and Kohli 1990; Hopper and Phizicky 2003; de Crécy-Lagard 2007). Functionally, some modifications act as structural modulators increasing flexibility or rigidity as needed, in turn affecting nucleic acid stability; others can directly impact gene function by altering the genetic information of the nucleic acids they target.
By far, tRNAs are the recipients of the largest diversity of modifications. In general, modifications at the anticodon loop play critical roles in translational efficiency and/or fidelity, whereas modifications that are more distal from the anticodon arm ensure proper folding of the tRNA and thus affect tRNA function indirectly. To date over 100 different modifications have been identified in tRNA and in some organisms, for example, Saccharomyces cerevisiae, almost a complete set of modification enzymes have been described. In most cases, however, it is less clear how each enzyme at the molecular level recognizes and targets specific substrates in a pool of very similar nonsubstrate tRNAs. Predictably, their chemical diversity and the variety of tRNA positions targeted dictate that modification enzymes may have evolved numerous ways of substrate recognition; some binding to local secondary structural features, some recognizing the global L-shaped tertiary fold, and yet others surveying specific sequences within the tRNA molecule. Notably, modification enzymes may act on a tRNA molecule at any point in its folding pathway, with some modifications playing critical roles early, ensuring the formation of certain structures while avoiding formation of unwanted conformers. These enzymes lay the structural foundation for subsequent modification enzymes that rely on tRNA architecture for activity (Grosjean et al. 1996b; Helm 2006; Swinehart and Jackman 2015; McKenney et al. 2017). For instance, introduction of m1A at position 9 in human mitochondrial tRNALys favors formation of its canonical L-shape structure (Helm et al. 1998; Helm 2006; Kobitski et al. 2011). Further modification of m2G10 and pseudouridine (ψ) at positions 27 and 28 strongly depends on proper formation of tertiary structure, and by extension, upon prior synthesis of m1A9 (Helm 2006). Many modification enzymes require a fully folded tRNA for activity; among these, S. cerevisiae tRNA adenosine deaminase, ScADAT2/3, strictly relies on the global structure of the tRNA to form inosine at position 34 (I34) (Haumont et al. 1984; Auxilien et al. 1996; Grosjean et al. 1996b). However, its bacterial counterpart, ADATa or TadA, in vitro can efficiently deaminate a minimal substrate composed only of the anticodon arm (Auxilien et al. 1996; Wolf et al. 2002). Likewise, some tRNA methyltransferases, including bacterial TrmJ and eukaryotic Trm5 methyltransferases, require full-length tRNA, whereas others are less stringent and can modify shorter substrates in vitro (Liu et al. 2015).
A growing theme in the RNA modification field is that many modifications do not occur in isolation and may be part of well-orchestrated cascades, whereby one modification is essential or may influence the synthesis of another; in such cases, modifications are predicted to follow a strictly ordered set of reactions. A model of modification interdependence, where modifications may rely on prior modifications, was first proposed after the observation of potential modification and editing cascades in mitochondrial tRNAs (Mörl et al. 1995; Crain et al. 2002; Rubio and Alfonzo 2005; Rubio et al. 2006a, 2017). Since then, more examples of modification cascades have surfaced (Rubio and Alfonzo 2005; Shigi et al. 2006; Rider et al. 2009; Tomikawa et al. 2010; Ishida et al. 2011; Guy et al. 2012; Zhou et al. 2015; Arimbasseri et al. 2016). In terms of tRNA methylation, there are several cases in which methyltransferases specifically act on a previously modified substrate, or where a methylation serves as a prerequisite for further modification (McKenney et al. 2017). In E. coli, for instance, the TrmL methyltransferase requires formation of i6A at position 37 (Zhou et al. 2015). The sequence A36–A37–A38 is important for EcTrmL methylation, but whether this sequence is necessary for recognition by the enzyme itself or for the i6A37 modification is yet unclear; nonetheless, incorporation of i6A37 is sufficient to recruit EcTrmL to the tRNA in vitro (Zhou et al. 2015). Likewise, in yeast N6-isopentenyl adenosine (i6A) at position 37 is required for m3C32 formation on tRNASer (Arimbasseri et al. 2016). Indeed, yeast Trm140 recognizes the sequence identity element G35–U36–t6A37 in tRNAThr substrates; however, the tRNASer substrate lacks this sequence (Han et al. 2016). Instead, tRNASer recognition was stimulated by seryl-tRNA synthetase (Ses1) and relies on its unique variable loop in addition to t6A37 and i6A37 (Han et al. 2016). In yet another example, deamination of adenosine to inosine (A-to-I) is required for further methylation at position 37 of tRNAAla in eukaryotic tRNAs (Grosjean et al. 1995, 1996a; Gerber et al. 1998). In marsupials, deamination editing from cytidine to uridine (C-to-U) at position 35 acts as a determinant for queuosine formation at position 34 in the anticodon loop of tRNAAsp (Mörl et al. 1995; Arimbasseri et al. 2016). There is also direct precedence for methylation as a requirement for deamination; in Archaea, position 58 and/or 57 can be modified with m1A depending on the organism (Grosjean et al. 1995; Roovers et al. 2004). If m1A occurs at position 57, as in the majority of Archaea, it is subsequently converted to m1I57 via adenosine deamination (Grosjean et al. 1995, 1996a; Roovers et al. 2004).
We recently described an unexpected and uniquely extreme instance of modification interdependence involving a tRNA deaminase (TbADAT2/3) and a methyltransferase (TbTrm140) from T. brucei. TbADAT2/3 and TbTrm140 act together to edit and modify a single nucleotide position (cytosine 32, C32) in the anticodon loop of several tRNAs. This position is first methylated to form 3-methylcytosine (m3C) and then deaminated to generate 3-methyluridine (m3U). Remarkably, to form these products both enzymes must be present in the reaction and consequently they form a stable protein complex in vitro and in vivo (Rubio et al. 2017). This example of strict interdependence by both enzymes then raises questions as to what each enzyme contributes to each other in targeting C32. In the present report, we have taken advantage of the robustness of m3C formation in vitro and performed a series of binding and kinetic studies that show that interdependence may be driven by the synergistic effect that both enzymes have on substrate binding when added together in the same reaction. We also show that active site residues in each enzyme contribute minimally to binding synergy and that the observed mutual enhancement of substrate binding depends on domains that are more distal to the active sites. These findings have implications for multisubstrate recognition and are discussed in the context of how binding synergy may be exploited to ensure high specificity.
RESULTS
TbADAT2/3 and TbTrm140 bind synergistically to tRNAThr
To explore the basis for the co-requirement of TbTrm140 and TbADAT2/3 for methylation and deamination activity at position 32 of tRNAThr, an electrophoretic mobility shift assay (EMSA) was established. In these experiments, a slower migrating band was observed when either TbADAT2/3 or TbTrm140 was incubated with the tRNA, indicating formation of a stable protein–RNA complex when compared to a tRNA alone control (Fig. 1A,B). The resulting data were fitted to a binding isotherm with a single exponential and an apparent dissociation constant (Kdapp) was calculated. TbADAT2/3 yielded a Kdapp of 0.21 ± 0.03 µM, comparable to that shown by our laboratory with a different tRNA (Fig. 1A,C; Ragone et al. 2011). There are three different isoacceptors of tRNAThr (anticodons AGU, UGU, and CGU); all are substrates for m3C/m3U formation at position 32 (Gaston et al. 2007), and all showed similar Kdapp in these experiments. However, given that position 32 methylation occurs even in tRNAs that lack an adenosine at position 34 (which is converted to inosine by the same TbADAT2/3 deaminase), we focused on tRNAThrCGU for further binding studies, thus uncoupling contributions, however negligible, to binding from A34 (Arluison et al. 1999; Ragone et al. 2011; Sibert and Patton 2012). No binding parameters have been previously determined for TbTrm140. Thus, before exploring what the interaction between TbADAT2/3 deaminase and TbTrm140 contributes to binding, we performed similar binding experiments as above with recombinant TbTrm140 and tRNAThrCGU (Fig. 1B,D). Constant concentrations of substrate tRNA were incubated with increasing concentration of enzymes and, as above, subjected to EMSA. This yielded a binding isotherm with a nominal Kdapp of 0.21 ± 0.05 µM (Fig. 1D), which is similar for the binding of TbADAT2/3 to the same substrate.
FIGURE 1.
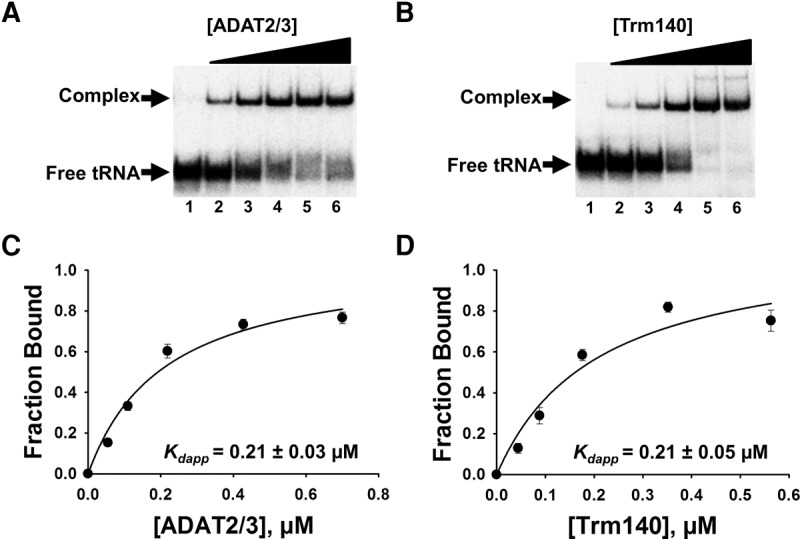
TbADAT2/3 and TbTrm140 stably bind tRNA in vitro. Analysis of protein–tRNA interactions using EMSA where radioactively labeled tRNAThrCGU (2.5 nM) was incubated with increasing concentrations of enzyme and separated on a native acrylamide gel. (A) Representative EMSA of TbADAT2/3 incubated with tRNAThrCGU. Lane 1 is a no-enzyme control; lanes 2–6 show tRNA with an increasing concentration of TbADAT2/3 (0.06, 0.12, 0.24, 0.48, and 0.7 µM, respectively). (B) EMSA of TbTrm140 incubated with tRNAThrCGU. Lane 1 is a no-enzyme control; lanes 2–6 show tRNA with an increasing concentration of TbTrm140 (0.04, 0.08, 0.16, 0.32, and 0.56 µM, respectively). The fraction of total bound tRNA from the EMSA gels was quantified and plotted as a function of protein concentration. The data were fit to a single-ligand binding isotherm and the apparent dissociation constant (Kdapp) was determined as described in Materials and Methods. These graphs are shown in (C) TbADAT2/3 with tRNAThrCGU and (D) TbTrm140 with tRNAThrCGU. Each figure represents at least five independent replicates.
Given that TbADAT2/3 and TbTrm140 form stable complexes in vivo and in vitro and both enzymes are required for methylation (Rubio et al. 2017), we determined the impact of each protein on substrate binding when incubated together with the tRNA substrate. Similar EMSAs were performed, but this time, radiolabeled tRNAThrCGU was incubated with one enzyme at a constant concentration while adding an increasing concentration of the other. First, TbTrm140 was held constant at 0.210 µM and different increasing concentrations of TbADAT2/3 were added, resulting in a stable protein–RNA complex (Fig. 2A) and yielding a Kdapp of <0.03 µM (Fig. 2C), representing over a sevenfold increase in affinity from what is seen with TbADAT2/3 alone (compare to Fig. 1B,D). The Kdapp value determined from the binding isotherm is below the detectable limits of this assay, therefore it is reported here as below 0.03 µM. An analogous increase in binding affinity was determined when the reciprocal experiment was performed. In this case, holding TbADAT2/3 constant while increasing TbTrm140 again resulted in a Kdapp below 0.03 µM (Fig. 2B,D). These results show that, when added together, TbTrm140 and TbADAT2/3 bind synergistically, exhibiting an improvement in binding that is more than the sum of the individual binding affinities.
FIGURE 2.
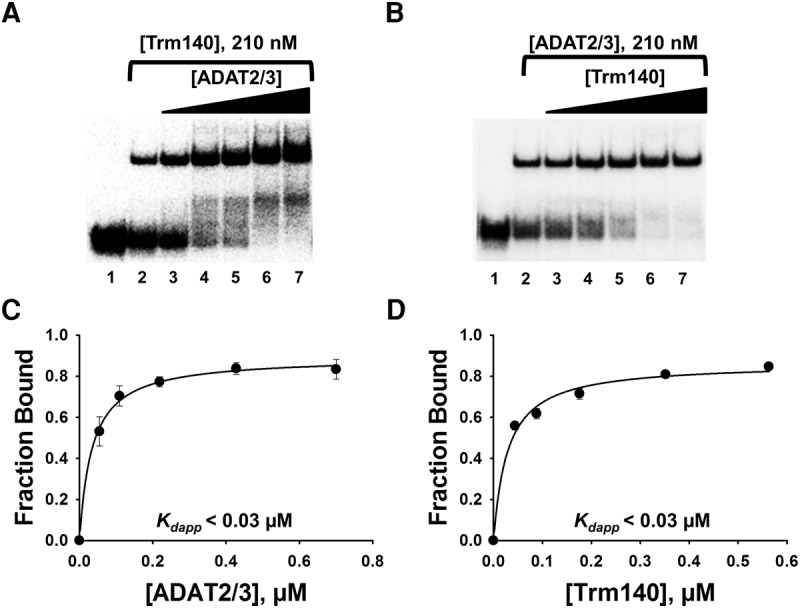
TbADAT2/3 and TbTrm140 bind tRNA synergistically. (A) EMSA of TbADAT2/3 incubated with tRNAThrCGU in the presence of a constant concentration (210 nM) of TbTrm140. Lanes 1 and 2 show a no-enzyme control reaction and a control reaction with no TbTrm140 added, respectively. Lanes 3–7 show an increasing concentration of TbADAT2/3 (0.06, 0.12, 0.24, 0.48, and 0.7 µM, respectively). (B) EMSA of TbTrm140 incubated with tRNAThrCGU in the presence of a constant concentration (210 nM) of TbADAT2/3. Lanes 1 and 2 show a no-enzyme control reaction and control reaction with no TbADAT2/3 added, respectively. Lanes 3–7 show an increasing concentration of TbTrm140 (0.04, 0.08, 0.16, 0.32, and 0.56 µM, respectively). The data were fit to a single-ligand binding isotherm and the apparent dissociation constant (Kdapp) calculated as in Figure 1. These graphs are shown in (C) and (D) for TbADAT2/3 and TbTrm140, respectively. Each figure represents at least five independent replicates.
To rule out the possibility that the observed increase in binding is simply the result of potential molecular crowding, we performed similar experiments with TbADAT2 alone without its partner TbADAT3 (Supplemental Fig. S1A). This protein can still form a stable homodimer, but by itself is unable to either bind tRNA or catalyze the deamination reaction. To test this, a threefold excess of TbADAT2 (600 nM) when compared to the levels of TbADAT2/3 in the previous reactions were added in the presence of increasing concentrations of TbTrm140 as before. This yielded a Kdapp of 0.26 ± 0.06 μM, which represents binding by TbTrm140 alone. Similar experiments were performed with bovine serum albumin still at 600 nM constant concentration while increasing either TbADAT2/3 or TbTrm140 (Supplemental Fig. S1B,C). This yielded a Kdapp of 0.180 ± 0.03 μM and 0.150 ± 0.04 μM, respectively. Therefore, no sign of synergy was observed, ruling out the possibility of nonspecific crowding effects as the root cause of the synergy seen with TbADAT2/3 and TbTrm140. Importantly, in these experiments the order of addition of the proteins to the tRNA had no impact on binding behavior. Taken together, these experiments suggest that part of the co-requirements of both enzymes for activity may rest on their mutual contribution to binding affinity.
Synergistic binding is independent of enzyme activity
To further understand how the two enzymes work in complex, we explored whether the synergy observed was simply due to two different active sites converging on a single position. This being the case, it is expected that mutations at the active site of either enzyme that impair activity should abrogate binding synergy. To test this, previously described active-site and binding mutants of TbADAT2/3 were tested in the presence of TbTrm140. Catalytic mutants were previously generated by alanine substitutions of the proton-shuttling glutamate (E92A) of TbADAT2 and a critical zinc binding residue (C291A) of TbADAT3. These catalytically dead mutants can still form heterodimers and bind tRNA with a similar affinity as wild-type TbADAT2/3 alone (Ragone et al. 2011). As a control, a tRNA binding-deficient mutant created previously by deletion of the last 10 amino acids of the C-terminal end of TbADAT2 was also tested (Ragone et al. 2011). Constant concentrations of each mutant were incubated with radiolabeled tRNA in the presence of increasing concentrations of wild-type TbTrm140 as before, and the resulting dissociation constant was estimated. Neither catalytic mutant (ADAT2/3 E92A and ADAT2/3 C291A) had any effect on binding synergy with Kdapps comparable to wild-type TbADAT2/3 in the presence of TbTrm140 (Fig. 3A,B), whereas the TbADAT2/3 binding-deficient mutant (ADAT2/3 C-terΔ10) exhibited a Kdapp of 0.12 ± 0.03, reflective of the binding by TbTrm140 with no contribution by TbADAT2/3 (Fig. 3C). This last observation is expected given that the 10-amino acid C-terminal deletion of TbADAT2 completely abrogates tRNA binding. Taken together this suggests that binding domains, which are not directly involved in catalysis, are the major contributors to the observed synergy.
FIGURE 3.

TbADAT2/3 catalytic residues do not contribute to increased affinity of TbTrm140 for tRNA. (A) EMSA of TbTrm140 to assess binding to tRNAThrCGU in the presence of two TbADAT2/3 catalytic mutants (E92A) and (B) (C291A). (C) EMSA of TbTrm140 to tRNAThrCGU in the presence of TbADAT2/3 C-terminal deletion binding mutant. In each panel, lanes 1 and 2 show a no-enzyme control reaction and a control reaction with no mutant TbADAT2/3 added, respectively. Lanes 3–7 show an increasing concentration of TbTrm140 (0.04, 0.08, 0.16, 0.32, and 0.56 µM, respectively). The bottom panels show the single-ligand binding isotherms used to calculate the individual Kdapp. Each graph represents at least five independent replicates.
We then generated mutants at conserved SAM-binding residues of TbTrm140. Methyltransferases that utilize S-adenosylmethionine (SAM) as a methyl donor are categorized into at least five classes (I–V) based on their structural folds. Most SAM methyltransferases, including TbTrm140, are class I, which have a conserved and predictable Rossmann-like fold. Class I methyltransferases are made up of a parallel β-sheet surrounded by helices, but unlike the Rossmann-fold, they contain an additional antiparallel β-strand. The class I methyltransferases often vary in sequence, but generally contain a characteristic nucleotide binding motif, GXGXG, important for SAM binding. Analogous conserved residues are found in TbTrm140 and a potential TbTrm140 catalytic mutant was produced by alanine substitution of two of these residues (G124A and G126A). This mutant (Trm140 G124/G126A) had no detectable m3C formation activity in vitro (data not shown), but despite this, no effect on binding synergy was observed in the presence of wild-type TbADAT2/3 (Fig. 4B), once again reinforcing the view that analogous to the TbADAT2/3, active-site residues contribute little to binding synergy.
FIGURE 4.
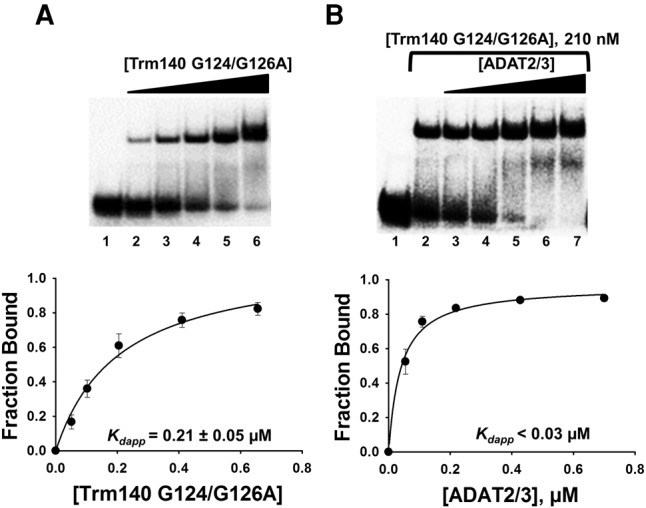
TbTrm140 catalytic residues do not contribute to increased affinity of TbADAT2/3 for tRNA. (A) EMSA of TbTrm140 catalytic mutant (G124/G126A) to tRNAThrCGU. Lane 1 is a no-enzyme control reaction. Lanes 2–6 show an increasing concentration of TbTrm140 catalytic mutant (0.04, 0.08, 0.16, 0.32, and 0.56 µM, respectively). (B) EMSA of TbADAT2/3 to tRNAThrCGU in the presence of the TbTrm140 catalytic mutant. Lanes 1 and 2 show a no-enzyme control reaction and control reaction with no mutant TbTrm140 added, respectively. Lanes 3–7 show an increasing concentration of TbADAT2/3 (0.06, 0.12, 0.24, 0.48, and 0.7 µM, respectively). The bottom panels show the single-ligand binding isotherms used to calculate the individual Kdapp. Each graph represents at least 5 independent replicates.
Some methyltransferases use common specific RNA-binding domains to bind tRNA while others use motifs that are not easily recognizable or that have not been described yet. No obvious RNA binding motif(s) is present in Trm140 from either S. cerevisiae (NP_014882.4) or T. brucei. However, the activity of ScTrm140 was abolished upon deletion of a string of residues at the C terminus (D602-Q621); notably this domain is distal to the conserved SAM-binding residues and presumably does not form part of the active site, suggesting that this region may be important for tRNA binding (Noma et al. 2011). The protein sequence of TbTrm140 was analyzed by the DNA- and RNA-binding prediction tool, BindN (Supplemental Fig. S2), revealing a region of positively charged residues found in the N-terminal portion of the protein (Wang and Brown 2006). A string of positively charged residues equivalent to those deleted in ScTrm140 was also found at the C terminus of TbTrm140. The N-terminal and C-terminal ends of TbTrm140 were, respectively, deleted, and of the resulting mutants analyzed by EMSA, only the former impaired binding (Supplemental Fig. S3). Therefore, the N-terminal deletion mutant (TbTrm140 ΔS2-G17) was chosen for subsequent studies. Comparable to wild-type TbTrm140, the N-terminal deletion mutant can still form homodimers (Supplemental Fig. S4). The C-terminal deletion mutant (TbTrm140 ΔI320-S340) showed similar binding behavior as the wild type and was therefore not pursued further (Supplemental Fig. S5). Incubation of the binding-defective mutant, TbTrm140 ΔS2-G17, with increasing concentrations of TbADAT2/3 abrogated synergy and a dissociation constant reflective of TbADAT2/3 binding alone was observed (Supplemental Fig. S3B). This analysis has thus identified an important tRNA-binding domain in TbTrm140 that is distinct from the analogous domain in yeast. These results also emphasize how tRNA binding domains more distal to the active sites of TbTrm140 and TbTrm140 are important for binding synergy; these must have a direct bearing on the co-requirement for both enzymes in the reaction.
Validation of binding constants by single turnover kinetics
A limitation of EMSA in our situation is that a precise measurement of dissociation constants cannot be achieved. This is due to the fact that titration of one enzyme while the other is held constant limits the linear range of the assay; eventually the dissociation constant calculation becomes reflective of only the enzyme that was held constant. Thus, single turnover kinetic assays were performed to corroborate and more precisely define the binding affinities established by EMSA. First, to compare affinities between TbADAT2/3 alone and TbADAT2/3 with TbTrm140, we performed A-to-I deamination assays. Here it is expected that since A-to-I formation is predictably independent of m3C formation, no synergy will be observed when using inosine formation as a reporter. In this experiment a saturating amount of enzyme, to ensure single-turnover conditions, was incubated with tRNAThrAGU substrate radiolabeled at every adenosine and product formation assessed over time by 1D-TLC (Supplemental Fig. S6A). The resulting data were fit to the equation [f = a(1 − e−kt)] as previously described to obtain an observed rate constant (kobs), which was then used to derive an apparent dissociation constant (Kdapp). TbADAT2/3 alone exhibited a Kdapp of 0.18 ± 0.06 µM, which is in agreement with the results from EMSA (0.21 ± 0.03 µM) (Fig. 5A,C; Ragone et al. 2011). As expected, this Kdapp was virtually unchanged when A-to-I activity was tested in the presence of TbTrm140 (Fig. 5B,D). Similar experiments were performed while monitoring m3C formation (Supplemental Fig. S6B) to calculate kobs and derive a Kdapp for methylation, but this time tRNAThrCGU radiolabeled at every cytosine was used as a substrate. Here, however, because both enzymes are required for m3C production, the assay was performed with both present in the reaction. From this experiment the kinetically determined Kdapp for m3C was 0.02 ± 0.01 µM, which is in agreement with our results from the EMSAs (<0.03 µM) and supports the view that when together both enzymes act synergistically. Importantly, these experiments highlight the fact that while synergy is important for m3C formation, it plays no role on A-to-I despite both reactions requiring the same deaminase (Fig. 6A,B).
FIGURE 5.

Kinetic determination of the dissociation constant of TbADAT2/3 for tRNAThr in the presence of TbTrm140. Single turnover assays of (A) TbADAT2/3 with tRNAThrCGU alone and (B) in the presence of TbTrm140 were performed as described in Materials and Methods. The fraction of inosine formed for each TbADAT2/3 protein concentration ranging from 10 to 2500 nM was measured over time as indicated in the graph. Determination of dissociation constants for (C) TbADAT2/3 with tRNAThrCGU alone and (D) in the presence of TbTrm104. The fraction of inosine produced was plotted as a function of time and fit to a single exponential curve [f = a(1 − e−kt)], where f represents inosine formed, a denotes inosine produced at the end point of the reaction, k signifies kobs, and t is time. The resulting kobs values were plotted against the concentration of TbADAT2/3 and fit to a single ligand binding isotherm. The Kdapp was then determined by nonlinear regression using Sigmaplot. Each graph represents at least five independent replicates.
FIGURE 6.
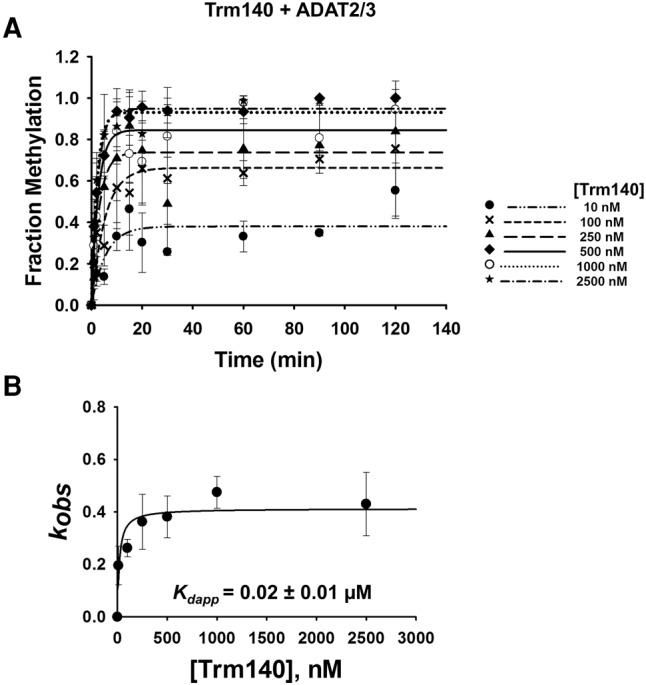
Kinetic determination of dissociation constant of TbTrm140 to tRNAThr in the presence of TbADAT2/3. (A) Single turnover assays of TbTrm140 to tRNAThrCGU in the presence of TbADAT2/3 were performed as described in Materials and Methods. The fraction of methylated cytosine 32 was measured for each TbTrm140 protein concentration ranging from 10 to 2500 nM as shown in the graph. The methylated fraction was plotted as a function of time and fit to a single exponential curve [f = a(1 − e−kt)], where f represents methylated cytosine formed, a denotes methylated cytosine produced at the end point of the reaction, k signifies kobs, and t is time. (B) The resulting kobs values were plotted against the concentration of TbTrm140 and fit to a single ligand binding isotherm. The Kdapp was then determined by nonlinear regression using Sigmaplot. Each graph represents at least five independent replicates.
To validate the observation that the active site residues are not involved in binding synergy, we performed methylation assays with different combinations of catalytic and binding mutants of TbTrm140 and TbADAT2/3. TbTrm140 was tested with the catalytically dead mutant TbADAT2/3 E92A; coincubation of TbTrm140 with this mutant still yielded an active methylase with a kinetically determined dissociation constant of 0.02 ± 0.01 µM. This value is comparable to the wild-type enzymes and again in line with the EMSA results (Supplemental Fig. S7). No detectable methylation activity was observed with the TbADAT2/3 C-terminal binding-impaired deletion mutant, even at an upper concentration range for an extended incubation time (Supplemental Fig. S8A). Similarly, no methylation activity was detected with the TbTrm140 binding mutant, ΔS2-G17, in the presence of the wild-type TbADAT2/3 (Supplemental Fig. S8B). This is expected since the Trm140 tRNA-binding mutant is unable to bind the tRNA; therefore, it cannot methylate it. These results support our findings by EMSA and underscore the importance of tRNA binding domains as a driving force for synergy, and consequently, for m3C formation.
DISCUSSION
Enzymes interact with partner proteins that may alter their specificity, activity, and affinity for a given substrate; tRNA modification enzymes are no exception, with methyltransferases being particularly prone to subunit recruitment (Guy and Phizicky 2014; Hori 2014; Swinehart and Jackman 2015; McKenney et al. 2017). The yeast 2′-O-methyltransferase, Trm7 for example, interacts with two different proteins, Trm732 and Trm734, to modulate its specificity for positions C32 and G34 of the same tRNA anticodon (Pintard et al. 2002; Purta et al. 2006; Benítez-Páez et al. 2010; Guy et al. 2012; Guy and Phizicky 2014; Hori 2014; Swinehart and Jackman 2015; McKenney et al. 2017). Similarly, the m2G10 methyltransferase, Trm11, and the mcm5U34 methyltransferase, Trm9, both utilize the protein Trm112 to direct substrate specificity (Kalhor and Clarke 2003; Purushothaman et al. 2005; Studte et al. 2008; Mazauric et al. 2010; Chen et al. 2011; Liger et al. 2011; Guy and Phizicky 2014; Hori 2014; Swinehart and Jackman 2015; McKenney et al. 2017). Other enzymes require both subunits for tRNA binding, as is the case with Trm6 and Trm61, although Trm6 is the catalytic component (Anderson et al. 1998, 2000; Ozanick et al. 2007; Guy and Phizicky 2014; Hori 2014; Swinehart and Jackman 2015; McKenney et al. 2017). In yet another instance, the enzymes Trm8 and Trm82, which methylate tRNA to form m7G at position 46, are both required to form an active enzyme complex (Alexandrov et al. 2002; Matsumoto et al. 2007; Leulliot et al. 2008; Guy and Phizicky 2014; Hori 2014; Swinehart and Jackman 2015; McKenney et al. 2017)
We previously demonstrated that two different enzymes, a methyltransferase and a deaminase, converge on a single anticodon loop nucleotide position to catalyze formation of m3C and m3U at position 32 of tRNAThr. Unlike many of the aforementioned multiprotein enzymes, we show here that TbADAT2/3 and TbTrm140 are both capable of binding tRNA directly; however, binding individually is nonproductive, leading to neither methylation nor deamination of position 32 of tRNAs; their intended target. The question then remains as to why these enzymes require each other for function. One possibility could be the increase in binding affinity observed upon addition of both binding partners; this can be attributed to several potential factors. First, it could be a result of coactivation upon protein complex formation prior to substrate binding, whereby complex formation leads to a conformational change that then makes the enzymes poised for activity. However, were this the case, it is expected that complex formation alone would be sufficient for activity, even if one of the partners in the complex is unable to bind tRNA, which goes against the observations presented here. The answer instead may lie in how they bind tRNA, where a rearrangement of the enzymes and/or the tRNA could possibly facilitate binding synergy and stimulate enzyme activity. Most enzymes and enzyme complexes do not bind their substrate as rigid entities, but instead display flexibility upon binding. Binding between protein and RNA is often achieved by an induced-fit mechanism (Williamson 2000; Leulliot and Varani 2001; Uter and Perona 2004). The idea of induced-fit was first used to explain how enzymes in an inactive state become catalytically active upon substrate binding (Jones and Peterlin 1994; Leulliot and Varani 2001). This mode of binding generally utilizes the favorable binding energy to drive entropically unfavorable conformational changes made by either the protein, the RNA, or both (Williamson 2000; Leulliot and Varani 2001; Uter and Perona 2004). These conformational changes can in turn govern specificity and affinity which often go hand in hand (Eaton et al. 1995; Leulliot and Varani 2001; Uter and Perona 2004).
There are many cases where recognition of substrate tRNA, particularly by tRNA methyltransferases, involves multiple steps, including initial binding and induced-fit processes (Hori 2014). The bacterial 2′-O-methyltransferase enzyme, TrmH for instance, methylates G18 on several substrates depending on the organism. In Thermus thermophilus, the enzyme acts on all tRNAs with G18 whereas it methylates only a subset of tRNAs in E. coli (Liu et al. 2015). It was shown that the regions in the N- and C-terminal ends of TrmH support initial tRNA binding while the active-site residues participate in substrate discrimination (Ochi et al. 2013). Therefore, the enzyme undergoes two steps, the initial tRNA binding, facilitated by the ends, followed by induced fit, enabled by the active site (Ochi et al. 2010, 2013). Similar to TrmH, the eukaryotic ADAT2/3 deaminase contains a binding domain away from the active site, which permits accommodation of multiple different substrates, while the bacterial deaminase edits a single A34-containing tRNAArg substrate and recognizes a specific sequence in the anticodon loop with high affinity. The evolution of substrate specificity was proposed whereby the ADAT2/3 enzyme acquired a “general” tRNA-binding domain away from its active site, which coupled with active site structural relaxation over time, may have facilitated the accommodation of multiple different substrates. Thus, together, TbTrm140 and TbADAT2/3 may undergo multiple binding steps to bind and accommodate their tRNA substrates.
The mode of enzymatic activation described here differs from m3C formation at position 32 in other systems. The yeast, Schizosaccharomyces pombe, relies on two Trm140-related homologs for m3C methylation, SpTrm140 specific for tRNAThr and SpTrm141 specific for tRNASer (D'Silva et al. 2011; Noma et al. 2011; Arimbasseri et al. 2016). It is unclear whether SpTrm140 and SpTrm141 function as a multisubunit complex; however, upon deletion of either gene individually, m3C levels decreased in all tRNA substrates, suggesting a connection between these methylation events (Arimbasseri et al. 2016). Interestingly, T. brucei also harbors two homologs of TbTrm140 as well. As of yet, only TbTrm140 mentioned here methylates tRNA whereas the other homolog, TbMTase37, performs an unrelated function in ribosomal RNA biogenesis (Fleming et al. 2016). Other metazoans and fungi contain multiple homologs as well, some with m3C found in an additional substrate, tRNAArg, and present at novel sites of tRNASer (Arimbasseri et al. 2016; Xu et al. 2017). Instead of using two methyltransferase homologs, the ScTrm140 enzyme exploits differences in sequence elements, anticodon loop modifications, and utilizes seryl-tRNA synthetase, Ses1, to modulate specificity (Han et al. 2016). These observations are in line with our data showing that TbADAT2/3 is required for methylation by TbTrm140 and that tRNA-binding plays a major role in T. brucei as an activity determinant (Rubio et al. 2017).
In addition to the functional necessity for the binding strategies described here, binding synergy could also provide a competitive advantage over other tRNA modification enzymes in the nucleus. This would be particularly important for TbADAT2/3, as the bulk of the enzyme localizes to the cytoplasm and is in low abundance in the nucleus where C-to-U editing takes place. One could also envision the use of binding synergy for regulatory purposes. For instance, we previously showed that complex formation between these two enzymes prevents TbADAT2/3 from mutagenizing the genome (Rubio et al. 2017). The increase in binding affinity for tRNA offers a possible explanation for how the complex may be sequestered away on the tRNA, precluding interaction and potentially rampant deamination of the genome by TbADAT2/3.
In the current manuscript, we have presented a rationale for the observed codependence of TbADAT2/3 and TbTrm140 based on the observed synergistic increase in substrate binding affinity when combined. Thus, binding synergy is an important component of enzyme coactivation. Interestingly, the presence of Trm140 in vitro does not affect the A-to-I activity of TbADAT2/3 at position 34. This is not entirely unexpected as the localization of these activities are restricted to separate compartments where A-to-I occurs in the cytoplasm and methylation in the nucleus. The delicate interplay between these seemingly separate editing and methylation events represents an example of the intricacy of tRNA modification pathways and provides a potentially new layer to how modification cascades could be enacted and likely regulated.
MATERIALS AND METHODS
Recombinant protein expression and purification from E. coli
The coding sequences of wild-type TbADAT2/3 and TbTrm140 were introduced into expression vectors and mutants generated as previously described (Rubio et al. 2007, 2017; Ragone et al. 2011). The TbTrm140 catalytic mutant was produced by quick change mutagenesis (Stratagene). For expression, a 10 mL overnight culture was added to 1.5 L of prewarmed 2XYT media and grown at 37°C to an OD600 of 0.6–0.8. Recombinant protein expression was then induced with a final concentration of 0.5 mM IPTG overnight at 25°C. Cells were pelleted, suspended in lysis buffer (100 mM Tris pH 8.0, 100 mM NaCl, protease inhibitor cocktail and 0.1% NP40), and broken by sonication with a Sonifier 450. The resulting extract was spun at 100,000g for 30 min to remove cellular debris. The supernatant was collected and bound to 1 mL bed-volume Ni2+-nitrilotriacetic acid (NTA) agarose beads for 1 h. The NTA–agarose beads were washed in buffer containing 100 mM Tris pH 8.0, 100 mM NaCl, 25–50 mM imidazole; the bound protein was eluted with 400 mM imidazole. The resulting protein samples were dialyzed overnight in 100 mM Tris pH 8.0, 0.5 mM MgCl2, and 2 mM 1,4-dithiothreitol. Protein samples were further purified through a Q-sepharose FPLC column and stored at 80°C in buffer containing 100 mM Tris pH 8.0, 100 mM NaCl, and 20% glycerol.
Size-exclusion chromatography
TbTrm140 N-terminal deletion mutant (ΔS2-G17) and wild-type proteins were recombinantly expressed and purified as described in Materials and Methods. Five hundred microliters of the protein samples were injected into a Superdex 200 column (GE Healthcare) and separated at a flow rate of 0.5 mL/min in a buffer composed of 100 mM Tris pH 8 and 100 mM NaCl. The elution was monitored at a UV absorption at 280 nm. The column was calibrated using known standards: dextran blue (2000 kDa), thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.35 kDa) (Bio-Rad).
Synthesis of full-length tRNA substrates
Full-length substrate tRNAs were in vitro transcribed using a T7 promoter and either internally labeled with [α-32P]-ATP for activity assays or end-labeled with [γ-32P]-ATP for binding assays as previously described (Rubio et al. 2006b; Ragone et al. 2011).
Electrophoretic mobility shift assays
Radiolabeled tRNA (2.5 nM) was denatured by heating to 70°C for 3 min and folded at 37°C for 15 min. The tRNA was incubated on ice with various concentrations of the enzyme(s) in a reaction buffer containing a final concentration of 50 mM HEPES pH 7.5, 1 mM MgCl2, 5 mM KCl for 30 min. The products were separated on a 4% nondenaturing polyacrylamide gel at 80 V for 3 h at room temperature. The gel was dried and exposed to a PhosphoImager screen. The results were analyzed using a Storm Phosphoimager and quantified with the ImageQuant software. The percent of enzyme-bound tRNA was calculated using the equation, tRNAcomplex/(tRNAfree + tRNAcomplex). The percent bound was plotted as a function of protein concentration and fit to a single-ligand binding curve, and the apparent dissociation constant (Kdapp) was calculated using SigmaPlot software. A no protein control was used for background subtraction.
A-to-I and m3C single turnover kinetic activity assays
Adenosine deaminase and methyltransferase assays were performed in reaction buffers containing 40 mM Tris, pH 7.9, 5 mM MgCl2, 10 mM DTT for the A-to-I assays and 25 mM Tris pH 7, 2.5 mM MgCl2, 25 mM NaCl, 50 µM EDTA, and 1 mM SAM (S-adenosylmethionine) for the methylation assays. The enzymes were provided in excess (10 nM to 2.5 µM) to the labeled tRNA substrate (2.5 nM), which was folded as described above, and the reactions were incubated at 27°C. Aliquots were taken at various timepoints, reactions quenched by phenol extraction, and the tRNA was ethanol precipitated. Nuclease P1 was added to the tRNA pellets and incubated overnight at 37°C. The pellet was dried, suspended in 3 µL of water, spotted on a cellulose thin-layer chromatography (TLC) plate (SelectoScientific, cat. no. 10089), and products were resolved for 1 to 2 h in one dimension using solvent C (0.1 M sodium phosphate [pH 6.8]:ammonium sulfate:n-propylalcohol [100:60:2, v/w/v]). The TLC plate was dried and exposed to a PhosphoImager screen. The results were analyzed using Storm imaging and quantified with the ImageQuant software. The fraction of inosine or methylated cytosine produced was calculated using the equation, pI/(pA + pI) for deamination or pm3C/(pC + pm3C) for methylation. The fraction of product was plotted as a function of time and fit to a single exponential curve [f = a(1 − e−kt)], where f represents product formed, a denotes product formed at the end point of the reaction, k signifies kobs and t is time. The resulting kobs values were plotted against the protein concentration and fit to a single ligand binding isotherm. The Kdapp was then determined by nonlinear regression using Sigmaplot.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Jane Jackman for the useful discussions and helpful advice. We are appreciative of all members of the Alfonzo laboratory for their support and suggestions. This work was partially supported by National Institutes of Health grant GM084065 to J.D.A. and a Graduate Fellowship from The Ohio State University Center for RNA Biology to K.M.M.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062893.117.
REFERENCES
- Alexandrov A, Martzen MR, Phizicky EM. 2002. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA 8: 1253–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Phan L, Cuesta R, Carlson BA, Pak M, Asano K, Björk GR, Tamame M, Hinnebusch AG. 1998. The essential Gcd10p–Gcd14p nuclear complex 15 required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev 12: 3650–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Phan L, Hinnebusch AG. 2000. The Gcd10p/Gcd14p complex is the essential two-subunit tRNA(1-methyladenosine) methyltransferase of Saccharomyces cerevisiae. Proc Natl Acad Sci 97: 5173–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimbasseri AG, Iben J, Wei F, Rijal K, Tomizawa K, Hafner M, Maraia RJ. 2016. Evolving specificity of tRNA 3-methyl-cytidine-32 (m3C32) modification: a subset of tRNAsSer requires N6-isopentenylation of A37. RNA 22: 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arluison V, Buckle M, Grosjean H. 1999. Pseudouridine synthetase Pus1 of Saccharomyces cerevisiae: kinetic characterisation, tRNA structural requirement and real-time analysis of its complex with tRNA. J Mol Biol 289: 491–502. [DOI] [PubMed] [Google Scholar]
- Auxilien S, Crain PF, Trewyn RW, Grosjean H. 1996. Mechanism, specificity and general properties of the yeast enzyme catalysing the formation of inosine 34 in the anticodon of transfer RNA. J Mol Biol 262: 437–458. [DOI] [PubMed] [Google Scholar]
- Benítez-Páez A, Villarroya M, Douthwaite S, Gabaldón T, Armengod ME, Hori H, Farabaugh PJ, Kramer E, Vallabhaneni H, Raman A, et al. 2010. YibK is the 2′-O-methyltransferase TrmL that modifies the wobble nucleotide in Escherichia coli tRNALeu isoacceptors. RNA 16: 2131–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk GR, Kohli J. 1990. Synthesis and function of modified nucleosides in tRNA. In Chromatography and modification of nucleosides. Part B. Biological roles and function of modification (ed. Gehrke C, Kuo K), pp. B13–B67. http://linkinghub.elsevier.com/retrieve/pii/S0301477008614888. [Google Scholar]
- Chen C, Huang B, Anderson JT, Byström AS. 2011. Unexpected accumulation of ncm5U and ncm5s2U in a trm9 mutant suggests an additional step in the synthesis of mcm5U and mcm5s2U. PLoS One 6: e20783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain PF, Alfonzo JD, Rozenski J, Kapushoc ST, McCloskey JA, Simpson L. 2002. Modification of the universally unmodified uridine-33 in a mitochondria-imported edited tRNA and the role of the anticodon arm structure on editing efficiency. RNA 8: 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crécy-Lagard V. 2007. Identification of genes encoding tRNA modification enzymes by comparative genomics. Methods Enzymol 425: 153–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Silva S, Haider SJ, Phizicky EM. 2011. A domain of the actin binding protein Abp140 is the yeast methyltransferase responsible for 3-methylcytidine modification in the tRNA anti-codon loop. RNA 17: 1100–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton BE, Gold L, Zichi DA. 1995. Let's get specific: the relationship between specificity and affinity. Chem Biol 2: 633–638. [DOI] [PubMed] [Google Scholar]
- Fleming IMC, Paris Z, Gaston KW, Balakrishnan R, Fredrick K, Rubio MAT, Alfonzo JD. 2016. A tRNA methyltransferase paralog is important for ribosome stability and cell division in Trypanosoma brucei. Sci Rep 6: 21438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston KW, Rubio MAT, Spears JL, Pastar I, Papavasiliou NF, Alfonzo JD. 2007. C to U editing at position 32 of the anticodon loop precedes tRNA 5′ leader removal in trypanosomatids. Nucleic Acids Res 35: 6740–6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber A, Grosjean H, Melcher T, Keller W. 1998. Tad1p, a yeast tRNA-specific adenosine deaminase, is related to the mammalian pre-mRNA editing enzymes ADAR1 and ADAR2. EMBO J 17: 4780–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H, Constantinesco F, Foiret D, Benachenhou N. 1995. A novel enzymatic pathway leading to 1-methylinosine modification in Haloferax volcanii tRNA. Nucleic Acids Res 23: 4312–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H, Auxilien S, Constantinesco F, Simon C, Corda Y, Becker HF, Foiret D, Morin A, Jin YX, Fournier M, et al. 1996a. Enzymatic conversion of adenosine to inosine and to N1-methylinosine in transfer RNAs: a review. Biochimie 78: 488–501. [DOI] [PubMed] [Google Scholar]
- Grosjean H, Edqvist J, Stråby KB, Giegé R. 1996b. Enzymatic formation of modified nucleosides in tRNA: dependence on tRNA architecture. J Mol Biol 255: 67–85. [DOI] [PubMed] [Google Scholar]
- Guy MP, Phizicky EM. 2014. Two-subunit enzymes involved in eukaryotic post-transcriptional tRNA modification. RNA Biol 11: 1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Podyma BM, Preston MA, Shaheen HH, Krivos KL, Limbach PA, Hopper AK, Phizicky EM. 2012. Yeast Trm7 interacts with distinct proteins for critical modifications of the tRNAPhe anticodon loop. RNA 18: 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Marcus E, D'Silva S, Phizicky EM. 2016. S. cerevisiae Trm140 has two recognition modes for 3-methylcytidine modification of the anticodon loop of tRNA substrates. RNA 23: 406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumont E, Fournier M, De Henau S, Grosjean H. 1984. Enzymatic conversion of adenosine to inosine in the wobble position of yeast tRNAAsp: the dependence on the anticodon sequence. Nucleic Acids Res 12: 2705–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M. 2006. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res 34: 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M, Brulé H, Degoul F, Cepanec C, Leroux JP, Giegé R, Florentz C. 1998. The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA. Nucleic Acids Res 26: 1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper AK, Phizicky EM. 2003. tRNA transfers to the limelight. Genes Dev 17: 162–180. [DOI] [PubMed] [Google Scholar]
- Hori H. 2014. Methylated nucleosides in tRNA and tRNA methyltransferases. Front Genet 5: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida K, Kunibayashi T, Tomikawa C, Ochi A, Kanai T, Hirata A, Iwashita C, Hori H. 2011. Pseudouridine at position 55 in tRNA controls the contents of other modified nucleotides for low-temperature adaptation in the extreme-thermophilic eubacterium Thermus thermophilus. Nucleic Acids Res 39: 2304–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Peterlin BM. 1994. Control of RNA initiation and elongation at the HIV-1 promoter. Annu Rev Biochem 63: 717–743. [DOI] [PubMed] [Google Scholar]
- Kalhor HR, Clarke S. 2003. Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol Cell Biol 23: 9283–9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobitski AY, Hengesbach M, Seidu-Larry S, Dammertz K, Chow CS, van Aerschot A, Nienhaus GU, Helm M. 2011. Single-molecule FRET reveals a cooperative effect of two methyl group modifications in the folding of human mitochondrial tRNALys. Chem Biol 18: 928–936. [DOI] [PubMed] [Google Scholar]
- Leulliot N, Varani G. 2001. Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry 40: 7947–7956. [DOI] [PubMed] [Google Scholar]
- Leulliot N, Chaillet M, Durand D, Ulryck N, Blondeau K, van Tilbeurgh H. 2008. Structure of the yeast tRNA m7G methylation complex. Structure 16: 52–61. [DOI] [PubMed] [Google Scholar]
- Liger D, Mora L, Lazar N, Figaro S, Henri J, Scrima N, Buckingham RH, Van Tilbeurgh H, Heurgué-Hamard V, Graille M. 2011. Mechanism of activation of methyltransferases involved in translation by the Trm112 “hub” protein. Nucleic Acids Res 39: 6249–6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RJ, Long T, Zhou M, Zhou XL, Wang ED. 2015. tRNA recognition by a bacterial tRNA Xm32 modification enzyme from the SPOUT methyltransferase superfamily. Nucleic Acids Res 43: 7489–7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Toyooka T, Tomikawa C, Ochi A, Takano Y, Takayanagi N, Endo Y, Hori H. 2007. RNA recognition mechanism of eukaryote tRNA (m7G46) methyltransferase (Trm8-Trm82 complex). FEBS Lett 581: 1599–1604. [DOI] [PubMed] [Google Scholar]
- Mazauric MH, Dirick L, Purushothaman SK, Bjö Rk GR, Lapeyre B. 2010. Trm112p is a 15-kDa zinc finger protein essential for the activity of two tRNA and one protein methyltransferases in yeast. J Biol Chem 285: 18505–18515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenney KM, Rubio MAT, Alfonzo JD. 2017. The evolution of substrate specificity by tRNA modification enzymes. Enzymes 41: 51–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mörl M, Dörner M, Pääbo S. 1995. C to U editing and modifications during the maturation of the mitochondrial tRNAAsp in marsupials. Nucleic Acids Res 23: 3380–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Yi S, Katoh T, Takai Y, Suzuki T, Suzuki T. 2011. Actin-binding protein ABP140 is a methyltransferase for 3-methylcytidine at position 32 of tRNAs in Saccharomyces cerevisiae. RNA 17: 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi A, Makabe K, Kuwajima K, Hori H. 2010. Flexible recognition of the tRNA g18 methylation target site by trmh methyltransferase through first binding and induced fit processes. J Biol Chem 285: 9018–9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi A, Makabe K, Yamagami R, Hirata A, Sakaguchi R, Hou YM, Watanabe K, Nureki O, Kuwajima K, Hori H. 2013. The catalytic domain of topological knot tRNA methyltransferase (TrmH) discriminates between substrate tRNA and nonsubstrate tRNA via an induced-fit process. J Biol Chem 288: 25562–25574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozanick SG, Bujnicki JM, Sem DS, Anderson JT. 2007. Conserved amino acids in each subunit of the heteroligomeric tRNA m1A58 Mtase from Saccharomyces cerevisiae contribute to tRNA binding. Nucleic Acids Res 35: 6808–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintard L, Lecointe F, Bujnicki JM, Bonnerot C, Grosjean H, Lapeyre B. 2002. Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J 21: 1811–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purta E, van Vliet F, Tkaczuk KL, Dunin-Horkawicz S, Mori H, Droogmans L, Bujnicki JM. 2006. The yfhQ gene of Escherichia coli encodes a tRNA:Cm32/Um32 methyltransferase. BMC Mol Biol 7: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman SK, Bujnicki JM, Grosjean H, Lapeyre B. 2005. Trm11p and Trm112p are both required for the formation of 2-methylguanosine at position 10 in yeast tRNA. Mol Cell Biol 25: 4359–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragone FL, Spears JL, Wohlgamuth-Benedum JM, Kreel N, Papavasiliou FN, Alfonzo JD. 2011. The C-terminal end of the Trypanosoma brucei editing deaminase plays a critical role in tRNA binding. RNA 17: 1296–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider LW, Ottosen MB, Gattis SG, Palfey BA. 2009. Mechanism of dihydrouridine synthase 2 from yeast and the importance of modifications for efficient tRNA reduction. J Biol Chem 284: 10324–10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers M, Wouters J, Bujnicki JM, Tricot C, Stalon V, Grosjean H, Droogmans L. 2004. A primordial RNA modification enzyme: the case of tRNA (m1A) methyltransferase. Nucleic Acids Res 32: 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio M, Alfonzo JD. 2005. Editing and modification in trypanosomatids: the reshaping of non-coding RNAs. Top Curr Genet 12: 71–86. [Google Scholar]
- Rubio MAT, Ragone FL, Gaston KW, Ibba M, Alfonzo JD. 2006a. C to U editing stimulates A to I editing in the anticodon loop of a cytoplasmic threonyl tRNA in Trypanosoma brucei. J Biol Chem 281: 115–120. [DOI] [PubMed] [Google Scholar]
- Rubio MAT, Ragone FL, Gaston KW, Ibba M, Alfonzo JD. 2006b. C to U editing stimulates A to I editing in the anticodon loop of a cytoplasmic threonyl tRNA in Trypanosoma brucei. J Biol Chem 281: 115–120. [DOI] [PubMed] [Google Scholar]
- Rubio MAT, Pastar I, Gaston KW, Ragone FL, Janzen CJ, Cross GAM, Papavasiliou FN, Alfonzo JD. 2007. An adenosine-to-inosine tRNA-editing enzyme that can perform C-to-U deamination of DNA. Proc Natl Acad Sci 104: 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio MAT, Gaston KW, McKenney KM, Fleming IMC, Paris Z, Limbach PA, Alfonzo JD. 2017. Editing and methylation at a single site by functionally interdependent activities. Nature 542: 494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigi N, Suzuki T, Terada T, Shirouzu M, Yokoyama S, Watanabe K. 2006. Temperature-dependent biosynthesis of 2-thioribothymidine of Thermus thermophilus tRNA. J Biol Chem 281: 2104–2113. [DOI] [PubMed] [Google Scholar]
- Sibert BS, Patton JR. 2012. Pseudouridine synthase 1: a site-specific synthase without strict sequence recognition requirements. Nucleic Acids Res 40: 2107–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studte P, Zink S, Jablonowski D, Bär C, von der Haar T, Tuite MF, Schaffrath R. 2008. tRNA and protein methylase complexes mediate zymocin toxicity in yeast. Mol Microbiol 69: 1266–1277. [DOI] [PubMed] [Google Scholar]
- Swinehart WE, Jackman JE. 2015. Diversity in mechanism and function of tRNA methyltransferases. RNA Biol 12: 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomikawa C, Yokogawa T, Kanai T, Hori H. 2010. N7-Methylguanine at position 46 (m7G46) in tRNA from Thermus thermophilus is required for cell viability at high temperatures through a tRNA modification network. Nucleic Acids Res 38: 942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uter NT, Perona JJ. 2004. Long-range intramolecular signaling in a tRNA synthetase complex revealed by pre-steady-state kinetics. Proc Natl Acad Sci 101: 14396–14401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brown SJ. 2006. BindN: a web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res 34: W243–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson JR. 2000. Induced fit in RNA-protein recognition. Nat Struct Biol 7: 834–837. [DOI] [PubMed] [Google Scholar]
- Wolf J, Gerber AP, Keller W. 2002. tadA, an essential tRNA-specific adenosine deaminase from Escherichia coli. EMBO J 21: 3841–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Liu X, Sheng N, Oo KS, Liang J, Chionh YH, Xu J, Ye F, Gao YG, Dedon PC, et al. 2017. Three distinct 3-methylcytidine (m3C) methyltransferases modify tRNA and mRNA in mice and humans. J Biol Chem 292: 14695–14703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Long T, Fang ZP, Zhou XL, Liu RJ, Wang ED. 2015. Identification of determinants for tRNA substrate recognition by Escherichia coli C/U34 2′-O-methyltransferase. RNA Biol 12: 900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.