

Abstract
S. aureus and A. baumannii are among the ESKAPE pathogens that are increasingly difficult to treat due to the rise in the number of drug resistant strains. Novel therapeutics targeting these pathogens are much needed. The bacterial enoyl reductase (FabI) is as potentially significant drug target for developing pathogen-specific antibiotics due to the presence of alternate FabI isoforms in many other bacterial species. We report the identification and development of a novel N-carboxy pyrrolidine scaffold targeting FabI in S. aureus and A. baumannii, two pathogens for which FabI essentiality has been established. This scaffold is unrelated to other known antibiotic families, and FabI is not targeted by any currently approved antibiotic. Our data shows that this scaffold displays promising enzyme inhibitory activity against FabI from both S. aureus and A. baumannii, as well as encouraging antibacterial activity in S. aureus. Compounds also display excellent synergy when combined with colistin and tested against A. baumannii. In this combination the MIC of colistin is reduced by 10 fold. Our first generation compound displays promising enzyme inhibition, targets FabI in S. aureus with a favorable selectivity index (ratio of cytotoxicity to MIC), and has excellent synergy with colistin against A. baumannii, including a multidrug resistant strain.
Graphical abstract
1. Introduction
The need for, and role of antibiotics in treatment of bacterial infections, and their prophylactic use prior to surgeries, or in immunocompromised patients is undisputed and well understood.1 While this widespread, and often indispensable, use of antibiotics has led to an improved quality of life for many patients, extensive use has also led to an unprecedented rise in the emergence of multidrug resistant bacterial strains.2 The World Health Organization has recognized antimicrobial resistance as a major threat to global health. Furthermore, the economic burden caused by antibiotic resistance in the United States is estimated to be $20 billion per year with an additional $35 billion per year in lost productivity.3 There is a large gap between the burden of infections due to multidrug resistant bacteria and the development of new antibiotics to tackle the problem due in part to the steep decrease in the number of pharmaceutical companies investing in antibiotic research.2, 4 Additionally, many antibacterials, including telithromycin, linezolid, and bedaquiline among others, have limited use due to low safety profiles. Thus, there is a need for novel antibiotics to address the rising problem of antibacterial resistance.
Staphylococcus aureus and Acinetobacter baumannii, both members of the ESKAPE pathogens group,5 are major threats to public health due to the emergence of drug resistant strains. Methicillin-resistant S. aureus (MRSA) and A. baumannii have been identified by the CDC as serious healthcare threats. MRSA, a major cause of community and hospital associated infections, causes serious and life threatening infections including bacteremia, endocarditis, and osteomyelitis.6 A. baumannii is a causative pathogen for serious infections in hospital settings, such as pneumonia, meningitis and bacteremia.7 Newer molecules within existing drug classes have been introduced to the market or are in clinical drug development to address MRSA or A. baumannii to overcome the resistance seen with older antibiotics,8 but it is only a matter of time before we see a surge in development of resistance to these drugs. More alarmingly, resistance to recent novel antibiotics such as daptomycin,9 and the last line antibiotic colistin,10 which target S. aureus and A. baumannii infections respectively, have already been reported, underscoring the urgent need for developing newer antibiotics with novel targets and mechanisms of action.
The bacterial fatty acid biosynthesis (FASII) pathway has recently emerged as a promising novel target for antibacterial drug discovery, because of the differences between the human FASI and bacterial FASII pathways11 and the essentiality of this pathway in Gram-negative pathogens such as E. coli,12 F. tularensis13 and such Gram-positive ones as S. aureus.14 The enoyl-ACP reductase enzyme, FabI, catalyzes the rate-limiting step in the bacterial FASII pathway, and reduces the enoyl-ACP substrate in the presence of the cofactor NADH or NADPH, depending on the bacterial species.15 FabI is aa promising narrow-spectrum antimicrobial target due to the presence of diverse isoforms of FabI (FabV, FabK, FabL)16 in many bacterial species, along with the fact that some Gram-positive bacteria can circumvent FabI inhibition by uptake of exogenous fatty acids.14b As FabI essentiality has been established in S. aureus 14 and A. baumannii,7, 17 it is a promising target for developing pathogen specific therapeutics.
We report here the identification and development of a novel series of FabI inhibitors based on the N-carboxy pyrrolidine scaffold targeting both the FabI enzyme from S. aureus (SaFabI) and the FabI enzyme from A. baumannii (AbFabI). This scaffold was initially discovered through a high throughput screen conducted on an antibacterial focused library18 with the FabI enzyme from F. tularensis (FtFabI). The initial hit, compound 1 (Table1), displayed promising enzyme inhibitory activity against both SaFabI and AbFabI as well as FtFabI. Due to the high unmet need in healthcare facilities worldwide for a novel antibiotic to combat the emerging resistant strains of S. aureus and A. baumannii, our study focuses on developing this scaffold as an antibiotic for these two pathogens. We tested ∼70 analogs of the initial hit and discuss the structure-activity relationship against both enzyme targets, SaFabI and AbFabI. These enzymes share a high sequence identity (45% identity) and structural similarity (RMSD for all residues < 1Å when the various crystal structures of SaFabI are overlaid with the crystal structure of AbFabI). As a result the SAR of the analogs are largely similar for both enzymes, although differences are observed that correlate with the differences in the active site residues between the two enzymes. We also report the on-target vs off-target effects of our inhibitors using a S. aureus strain that overexpresses SaFabI.15 Compounds that effectively inhibit SaFabI and AbFabI, with promising antibacterial activity show excellent synergy in A. baumannii when combined with colistin. These active compounds were tested with clinical isolates including methicillin resistant S. aureus (MRSA) and multidrug resistant A. baumannii (MDR-Ab) and found to maintain similar activities. Thus, this work lays a solid foundation for development of N-carboxy pyrrolidine based FabI inhibitors targeting S. aureus as a monotherapy and A. baumannii as a combination therapy.
Table 1. Inhibitory activities of N-carboxy pyrrolidine analogs against SaFabI and AbFabI.

|
2. Materials and Methods
2.1. HTS Library
The compound library screened against FtFabI consisted of 25,000 lead-like compounds purchased from Life Chemicals, and has been previously described.18
2.2. Compounds
All compounds, including the hit compound 1, were custom synthesized by Life Chemicals through their CRO services with their identity and purity confirmed through mass spectrometry and NMR methods. All compounds tested herein have purity >95%. Compounds 1-29 are racemic mixtures. Crotonyl-CoA was purchased from Sigma Aldrich and Life Sciences Inc., while NADH and NADPH were from Fisher Scientific. Polymixin B nonapeptide was purchased from Sigma Aldrich. AbFabI was a generous gift from the Seattle Structural Genomics Center for Infectious Disease (SSGCID).
2.3. Bacterial strains
Methicillin resistant S. aureus (subsp. aureus Rosenbach) is a clinical isolate from ATCC (ATCC 43300). Acinetobacter baumannii strain 2208 is also a clinical isolate from ATCC (ATCC19606). Multidrug resistant A. baumannii is a clinical isolate from ATCC (BAA-1605) that is resistant to ceftazidime, gentamicin, ticarcillin, piperacillin, aztreonam, cefepime, ciprofloxacin, imipenem, and meropemem.
2.4. IC50 determinations
Individual compound IC50 experiments were carried out by measuring fluorescence of NADPH (for SaFabI) or NADH (for AbFabI) at 340 nm/460 nm wavelength, as previously described.15 Briefly, the assay was started by the addition of 400 μM, and 300 μM crotonyl-CoA for SaFabI and AbFabI, respectively, to the assay buffer containing 200 μM NADPH / NADH, 0.1 mg/mL BSA and 0.01% triton in 50 mM MES/100 mM NaCl buffer, with 300 nM SaFabI or 200 nM AbFabI respectively. Compounds were tested at concentrations ranging from 0.4 nM to 200 μM. Slopes from the first 10 minutes of the enzyme reaction were used to calculate the percent enzyme inhibition relative to the control (no compound, with DMSO). All compounds were tested in at least two independent experiments (with each experiment run in duplicate) to ensure reproducibility of the results. The reported IC50 has a two-fold experimental uncertainty. Representative compounds were shown to bind to the enzyme by SPR methods, indicating that activity is specific, and not artifactual. Additionally, compounds were confirmed not to interfere with the fluorescence assay by monitoring the fluorescence signal of the assay solution prior to adding the substrate to begin the catalysis reaction.
2.5. MIC determinations
Individual compound MICs were measured using the microbroth dilution method, with the plate set-up as described previously.15 The reported MICs are the mean of at least two runs and are within a two-fold experimental uncertainty.
2.6. Checkerboard MIC experiments
In the checkerboard assay, concentrations of colistin and each N-carboxy pyrrolidine analog were simultaneously varied - one across columns, the other down the rows. In effect, each well had a different combination of concentrations of both compounds. The range of each compound chosen for the .checkerboard assay was such that the individual compound MIC is in the middle of the range. The concentration of colistin was varied from 4 μg/mL to 0.03 μg/mL, while that of the N-carboxy pyrrolidine analogs was varied from 100 μg/mL to 0.19 μg/mL.
A. baumannii was grown to mid log-phase and then diluted to an OD600 of 0.004 with fresh media (tryptic soy broth). 50 μL of this freshly diluted culture was added to all wells on the plate. The checkerboard MIC plates were incubated overnight at 37 oC without shaking.
For each clear well observed on the checkerboard MIC plate the total fractional inhibitory concentration (ΣFIC) was calculated as follows:
Where, C = Concentration of the compound in the combination, MIC = Minimum Inhibitory Concentration of compound alone, FIC = Fractional Inhibitory Concentration
A ΣFIC value of < 0.5 indicates synergy, while a value between 0.5 and 2 indicates additivity or indifference and a value > 2 indicates antagonism.19
2.7. Cytotoxicity experiments
We used HepG2 cells (human liver cancer cell line; source ATCC: HB8065) to determine cytotoxicity. HepG2 cells were grown in EMEM media (Eagle's minimal essential media) supplemented with either 10% FBS (fetal bovine serum) or with 0.5% FBS + 10% Cell-Ess media. Cell-Ess media is a serum free, chemically defined serum replacement from Essential Pharmaceuticals. HepG2 cells were seeded in 96-well plates at 25,000 cells/well, respectively, in 100 μl. Following overnight incubation at 37 oC in 5% CO2 these cells were treated with compounds at concentrations varying from 100 μg/mL to 3 μg/mL. Varying concentrations of DMSO only (matching the volume used for compounds) were used as controls to monitor the effect of DMSO on cells at every stage. After a 24hr incubation, drug induced cytotoxicity was measured by the standard MTT assay.20 In brief 10 μL of 5 mg/mL MTT solution was added to all wells and incubated at 37 oC for 2 hrs. The MTT media was then removed and 100 μL of DMSO added to all wells. After mixing to extract formazan the metabolic activity of the cells were measured by quantifying the conversion of the yellow MTT to purple MTT-formazan. Absorbance was read at 570nm in a plate reader (Biotek Synergy HTX). Each compound was run in duplicate and the average absorbance noted. From a plot of cell viability (% reduction in absorbance for compound compared to DMSO control) the IC50 value (concentration of the drug that reduced cell viability by 50%) was calculated. The reported IC50s are mean of at least two runs and are within a two-fold experimental uncertainty.
2.8. Docking analysis
Before docking, the Protein Preparation Wizard in Schrödinger Suite (release 2016-1) was utilized to optimize the crystal structures of the apo AbFabI (PDB code 4ZJU with resolution of 1.2 Å) and SaFabI in complex with AFN-1252 (PDB code 4FS3 with resolution of 1.8 Å). After adding all hydrogens and deleting waters, restrained minimization was performed on hydrogens first, and then the RMSD of the heavy atoms was converged to less than 0.30 Å in the OPLS3 force field. The 3D structures of compounds 8, 9, 24, and 25 were created by LigPrep (Schrödinger Release 2016-1: Ligprep, version 3.7). Meanwhile, the OPLS3 force field was applied for ligand geometric optimization, with all possible ionization and tautomeric forms created at pH 7.4 by EPIK.21 Default values were used for other parameters for protein and ligand preparations. GOLD v5.2.222 was used for docking with the above prepared proteins and ligands. AFN-1252 was extracted from the optimized SaFabI complex before docking and selected as the reference ligand during docking. The active sites for AbFabI and SaFabI were set as a 10 Å sphere around residues Tyr159 and Tyr157, respectively. The proteins were kept rigid during docking. The ligand was flexible during docking with “flip amide bonds”, “detect internal H bonds” and “flip ring corners” on during ligand conformation searching. The best scoring poses for these ligands were chosen for further analysis. Figures were prepared with PyMol (Open-Source PyMOL 1.2r1).
3. Results
The N-carboxy pyrrolidine scaffold was identified through a high-throughput screen conducted with a previously described customized 25,000-compound drug-like chemical library18 from which the initial hit, compound 1, was identified. Compound 1 displayed a promising IC50 of 10.7 μM against SaFabI and 3.1 μM against AbFabI (Table 1). To determine if this is a potential new antibiotic scaffold we pursued a detailed analysis by designing analogs and monitoring enzyme inhibitory activity with the SaFabI and AbFabI enzymes. Data for the most relevant compounds is presented in Table 1. We also monitored the antibacterial activities, evaluated the possibility of antibacterial activity arising from FabI inhibition and determined cytotoxicity of these compounds. The results are discussed below.
3.1. Structure-Activity Relationship of the N-carboxy pyrrolidine scaffold with SaFabI
Close analogs of the initial hit (compound 1) were synthesized by modifying groups at positions R1 and R2, as shown in Table 1. Co-crystallization of compound 1 in the FabI active site provided a preliminary binding mode that helped guide this structure-based design process. Consequently, the SAR table may seem incomplete due to the absence of obvious compounds. The co-crystal structure is not of high enough quality for PDB deposition hence is not discussed in detail here. However, the compounds presented here were rationally designed based in part on this binding mode.
The parent compound of the initial hit 1 (without –Cl on R1) was tested but displayed poor enzyme inhibition and had solubility issues, hence is not shown in Table 1. Replacement of the 2-chlorobenzyl at the R2 position with a 2-fluorobenzyl as in compound 2 did not significantly affect activity (2-fold improvement in IC50). Compounds 3 (2-methoxyphenyl at R1 with 2-fluorobenzyl at R2), 4 (2,3-difluorobenzyl at R2 with an unsubstituted phenyl ring at R1), and 5 (2,3-difluorobenzyl at R2 with 2-methoxyphenyl at R1) also did not show much improvement in inhibitory activity when compared to compound 1 (IC50s of 4.6 μM, 7.3 μM and 3 μM respectively, compared to 10.7 μM for 1). Compound 6 with a 2,6-difluorobenzyl at R2 showed a 5-fold improvement in enzyme inhibitory activity when compared to the initial hit compound 1 (IC50 of 1.9 μM compared to 10.7 μM) while a 2-methoxyphenyl at R1 in compound 7 did not significantly impact inhibitory activity with a ∼3-fold poorer inhibitory activity when compared to compound 6.
Addition of a 2-methoxy group on the phenyl ring at R1 significantly improved enzyme inhibitory activity in compound 8 (0.5 μM) when compared to compound 1 (10.7 μM). The inhibitory potency of a 2-methoxy group on the phenyl ring at R1 is likely dictated by the type of halogen present on the benzyl group at R2, as compound 8, with a 2-chlorobenzyl group at R2, was 9-fold more potent than compound 3 (2-fluorobenzyl at R2), 6-fold more potent than compound 5 (2,3-difluorobenzyl at R2) and 11-fold more potent than 7 (2,6-difluorobenzyl group at R2). A 4-methoxy substitution on the phenyl ring of R1 in compound 9, on the other hand, did not impact enzyme inhibition when compared to compound 1 (12.5 μM compared to 10.7 μM respectively), but 9 was significantly less potent than compound 8 (12.5 μM compared to 0.5 μM respectively). A pyridine ring (compound 10) or a thiazole ring at R1 (compound 11) were well tolerated, as they did not significantly impact enzyme inhibitory activity when compared to the initial hit compound 1 (18.4 μM and 6 μM compared to 10.7 μM respectively). A methylenedioxy-substituted phenyl ring at R1 (compound 12) improved enzyme inhibitory activity compared to the initial hit (2.5 μM compared to 10.7 μM). However, replacing the 2-chloro substitution on the R2 benzyl in compound 12 with 2-fluoro (compound 13), 2,6-difluoro (compound 14), 2,3-difluoro (compound 15), or 2-chloro-6-fluoro (compound 16) did not have a significant impact on enzyme inhibitory activity when compared to compound 12. The SAR observed with compounds 13, 14 and 15 is similar to that observed with compounds 2, 4 and 6.
A bulky 4-acetamide substitution on the R2 benzyl ring resulted in a complete loss of enzyme inhibitory activity irrespective of the substitution on the phenyl ring at R1 as seen in compounds 17-21. Similarly a 4-isopropyl sulfonyl group on the benzyl ring at R2 was not tolerated as seen with compound 22.
To improve both our understanding of the SAR, and the SaFabI inhibitory properties of the N-carboxy pyrrolidine series, we expanded the scaffold by introducing an ethylene linker at R2, and incorporating an acetamide-substituted pyridine ring, somewhat analogous to the central linker in AFN-125223 (or Debio145224). Our co-crystal structure revealed the binding mode of compound 1 in the active site of FtFabI and we designed a series of analogs with polar atoms at positions appropriate for interaction with polar moieties in the binding site. In this series, compound 23 with a phenyl ring at R1 displayed a promising IC50 of 1.2 μM with SaFabI. 2-methoxyphenyl (compound 24) or 4-methoxyphenyl (compound 25) groups at R1 displayed a slight but not significant reduction in enzyme inhibitory activity compared to compound 23 (3.5 μM and 3.6 μM respectively compared to 1.2 μM), while a methylenedioxy-substituted phenyl ring at R1 (compound 26) did not significantly change enzyme inhibitory activity (1.1 μM compared to 1.2 μM for compound 26 and 23, respectively). Thiazole (compound 27) at R1 showed a slight reduction in enzyme inhibitory activity (4.5 μM compared to 1.2 μM), while a pyridine ring at R1 (compound 28) showed a significant loss in inhibitory activity (28.4 μM compared to 1.2 μM). We also note the importance of the pyridine ring at the R2 position as replacing it with a phenyl, as in compound 29, produced a complete loss in enzyme inhibitory activity. Figure 1A presents the overview of the SAR with the SaFabI enzyme.
Figure 1.
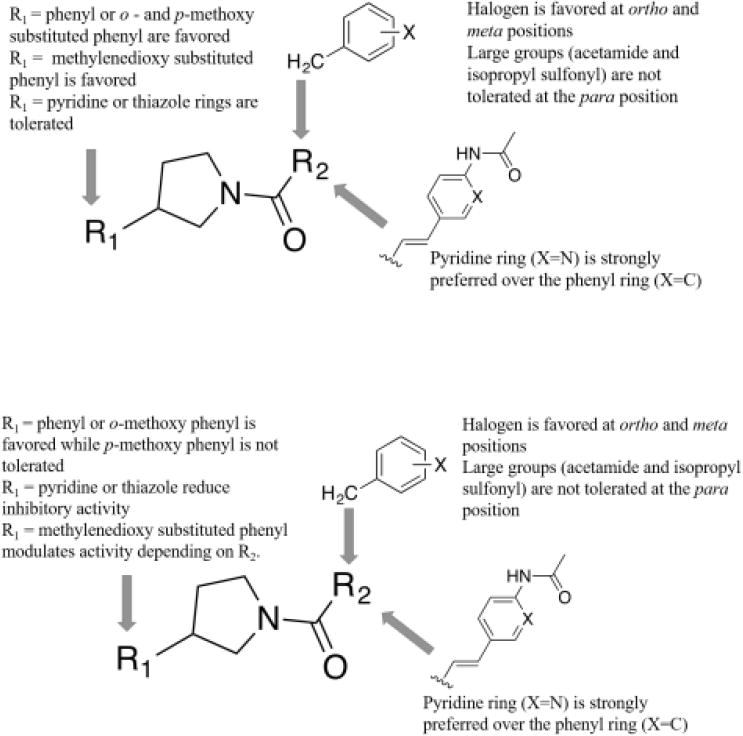
(A) Overview of SAR with the SaFabI enzyme.
(B) Overview of SAR with the AbFabI enzyme
3.2. SAR of N-carboxy pyrrolidine scaffold with AbFabI
Given the structural similarity between AbFabI and SaFabI, we expected that the pyrrolidine inhibitors would bind to and inhibit AbFabI. Testing with our initial hit compound 1 confirmed this hypothesis, producing an IC50 of 3.1 μM with AbFabI. All analogs tested with SaFabI were also subsequently tested with AbFabI.
With the AbFabI enzyme we observe that the 2-fluorobenzyl group in compound 2 (3.9 μM IC50) did not impact inhibitory activity when compared to the 2-chlorobenzyl group in compound 1, similar to that observed with the SaFabI enzyme. A 2-methoxy substitution on the phenyl ring at R1 (compound 3) also did not impact enzyme inhibition when compared to compound 2 (IC50 2.1 μM compared to 3.9 μM). Addition of a di-fluoro substitution at R2 and an unsubstituted or 2-methoxy-substituted phenyl ring at R1 as in compounds 4-7 resulted in a 2-3 fold variation in IC50 when compared to the initial hit compound 1. We observed a significant change in enzyme inhibition upon comparison of compounds 8 and 9 where the 2-methoxy group on the phenyl ring at R1 was substituted by a 4-methoxy group. This change in the methoxy position from ortho to para caused the compound to lose inhibitory activity for AbFabI (IC50 changing from 3.6 μM to >200 μM). This drastic change in enzyme inhibition was not observed with SaFabI.
Introduction of a pyridine ring (compound 10) or a thiazole ring (compound 11) at the R1 position led to a reduction in enzyme inhibitory activity with almost a 10-fold reduction seen in the case of the pyridine ring (compound 10), when compared to compound 1 (IC50 of 30.7 μM vs. 3.1 μM). A 3,4-methylenedioxy ring on the phenyl ring of R1 (compound 12) was relatively well tolerated with a 2 to 3-fold increase in IC50 compared to compound 1 (7.8 μM compared to 3.1 μM). However, replacing the 2-Cl group on the benzyl ring at the R2 position in compound 12 with 2-F as in compound 13 or 2,6-difluoro substitution (compound 14) reduced enzyme inhibitory activity (32 μM and 54 μM IC50 for compounds 13, and 14, respectively, compared to 7.8 μM for compound 12) while 2,3-difluoro substitution (compound 15) did not have much of an impact (13.7 μM compared to 7.8 μM in compound 12). Comparing compound 12 with compound 16 we see that the addition of a 6-fluoro group to the 2-chlorobenzyl ring at position R2 led to a significant loss in enzyme inhibitory activity from 7.8 μM to > 200 μM.
Similar to observations with SaFabI we see that 4-acetamide substitution on the benzyl ring at R2 resulted in a complete loss of enzyme inhibitory activity irrespective of the substitution on the phenyl ring at R1 (compounds 17-21). Similarly a 4-isopropyl sulfonyl group on the benzyl ring at R2 (compound 22) also caused loss in activity.
Our most promising compounds in this series were compounds 23 and 24 which exhibited sub-micromolar IC50s against AbFabI. Moving the 2-methoxy group on the phenyl ring in compound 24 to the 4-methoxy position in compound 25 caused a very significant loss in enzyme inhibitory activity from an IC50 of 0.42 μM to >200 μM. A similar pattern was observed with compounds 8 and 9 where the IC50 increased from 3.6 μM to >200 μM. A methylenedioxy substituted phenyl ring at position R1 (compound 26) was better tolerated than the 4-methoxyphenyl as in compound 25 (IC50 of 8.5 μM compared to >200 μM). Introduction of a thiazole ring (compound 27) or pyridine ring (compound 28) at the R1 position restored enzyme inhibitory activity when compared to compound 25. However, the IC50s for these compounds were still 15 to 40-fold higher than for compound 23. Finally, as observed with SaFabI, replacing the pyridine ring with a phenyl in compound 29 caused complete loss in enzyme inhibitory activity (from 0.42 μM for compound 24 to >100 μM for compound 29), demonstrating the importance of this pyridine ring. An overview of the SAR with the AbFabI enzyme is presented in Figure 1B.
3.3. Antibacterial Activity
Our initial hit, compound 1, displayed a promising MIC of 12.5-25 μg/mL against the three S. aureus strains tested including MRSA (Table 2). We also tested antibacterial activity with the S. aureus strain that overexpresses SaFabI. FabI overexpression in S. aureus has been previously used to confirm the mechanism of antibacterial activity of FabI inhibitors by our group and others.15, 25 Additionally, a 4-fold increase in FabI concentration in S. aureus has been shown to have minimal impact on bacterial fitness.25 Although we have not determined the precise FabI concentration in the SaFabI overexpressing strain, the plasmid copy number suggests it to be ≥ 4-fold. Hence a 4-fold or higher increase in MIC with the SaFabI overexpression strain is indicative of on-target FabI inhibition. With triclosan we observed an 8-fold increase in MIC with the S. aureus strain overexpressing FabI, which serves as a good control.15 In order to confirm that the increase in MIC was indeed due to the increased concentration of SaFabI and not due to a general mechanism of resistance such as efflux, we tested MICs of antibiotics such as streptomycin, chloramphenicol, tetracycline, and erythromycin, with cellular targets other than FabI, against both native and SaFabI overexpressing strains and found that the MICs for these antibiotics remained unchanged (data not shown).
Table 2. Antibacterial activities of N-carboxy pyrrolidine analogs.
| Compound | S. aureus strain Newman (μg/mL)a | Methicillin Resistant S. aureus (MRSA) (μg/mL) | S. aureus strain RN4220 (μg/mL) | S. aureus strain RN4220 overexpressing SaFabI (μg/mL) | E. coli strain BW25113 ΔTolC (μg/mL) |
|---|---|---|---|---|---|
| 1 | 12.5 (41.7) | 25 | 12.5 | 50 * | 50 |
| 2 | 50 (176.5) | 100 | 50 | 200 | >200 |
| 3 | 50 (159.6) | 100 | 50 | 100 | >200 |
| 4 | 50 (165.9) | 100 | 50 | 200 | >200 |
| 5 | 100 | 100 | 100 | >200 | >200 |
| 6 | 50 (165.9) | >200 | 50 | >200 | >200 |
| 7 | 50 (150.9) | >200 | 50 | 50 | >200 |
| 8 | 25 (75.8) | 25 | 12.5 | 50 * | >200 |
| 10 | 50 (166.3) | 200 | 100 | 200 | >200 |
| 11 | 50 (163) | 100 | 50 | >200 | >100 |
| 12 | 6.25 18.2) | 25 | 12.5 | >200 * | 25 |
| 16 | 25 (69.1) | >200 | 25 | >200 * | >200 |
| 23 | >200 | >200 | >200 | >200 | 12.5 |
| 24 | 12.5 (34.2) | 25 | 12.5 | 50 * | 0.75 |
MIC values in μM concentrations are reported in brackets for compounds with MIC <50 μM
Compounds showing on-target activity against S. aureus
Compounds 9, 13-15, 17-22, 25-29 display MICs >200 μg/mL with all strains tested
All compounds were tested with native A. baumannii strain 2208 and E. coli strain BW25113 and found to display MICs > 200 μg/mL
With compound 1 we observed a 4-fold increase in MIC when tested with the S. aureus strain overexpressing SaFabI. Thus, compound 1 displays on-target FabI inhibition in S. aureus. However, this does not rule out the possibility that the compounds could also exhibit activity against a secondary target.
Modification at the R2 position by introducing -F atoms on the benzyl ring and a 2-methoxy group on the phenyl ring at the R1 position as in compounds 2-7 did not significantly impact enzyme inhibitory activity, but reduced antibacterial activity compared to compound 1. This suggests that the -F substitution impairs membrane penetration. With compounds 2-7 the on vs. off-target activity is inconclusive at this point as they possess relatively higher MICs (≥ 50 μg/mL) and the change is not significant. However, a 2-methoxyphenyl group at R1, as in compound 8, significantly improves enzyme inhibitory activity against SaFabI, but did not change antibacterial activity when compared to compound 1. Compound 8 also continues to maintain on-target specificity, with a 4-fold increase in MIC with the SaFabI overexpressing strain. Moving the position of the methoxy group to the para position as in compound 9 led to a significant increase in MIC, corresponding to the 25-fold increase in IC50 when compared to compound 8 (with SaFabI). Compounds 10 and 11 have poor MICs of 50-200 μg/mL due to the replacement of the phenyl ring at the R1 position in compound 1 with a pyridine ring or a thiazole ring. Compound 12 displays a promising MIC of 6.25-25 μg/mL in S. aureus, while compounds 13-15 have poor antibacterial activity. Similar to that observed with compounds 2-7, poor MICs with compounds 13-15 correlate with the introduction of the -F substituent on the R2 benzyl ring. Compound 12 displays on-target antibacterial activity, as there is a > 4-fold increase in MIC with the SaFabI overexpressing strain. Compound 16 is similar to compound 12 with an additional 6-fluoro atom on the 2-chlorobenzyl group at the R2 position. It is interesting to note that compound 16 displayed an MIC of 25 μg/mL in S. aureus strains Newman and RN4220 but is >200ug/mL with MRSA. On comparing the MIC with S. aureus strain RN4220 and strain RN4220 overexpressing SaFabI we believe this MIC arises from on-target antibacterial activity due to the > 4 fold increase in MIC with the SaFabI overexpressing strain. The lack of antibacterial activity with MRSA needs to be further investigated. Compounds 17-22 have MICs > 200 μg/mL which is not surprising since they have very poor enzyme inhibitory activity. Compounds 23, 25, and 26-29 show no antibacterial activity at concentrations tested (MIC >200 μg/mL) while compound 24, a close analog to compound 25, displays a promising MIC of 12.5-25 μg/mL with the antibacterial activity likely arising from on-target inhibition (4-fold increase in MIC with the SaFabI overexpressing S. aureus strain).
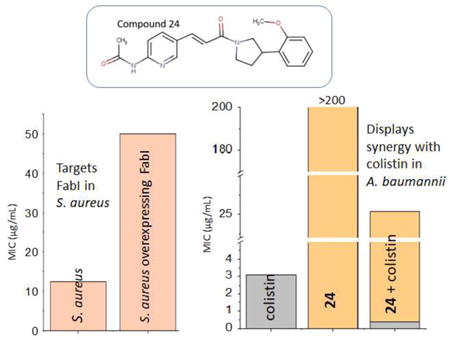
These compounds displayed poor antibacterial activity against Gram-negative A. baumannii with MIC >200 μg/mL. A similar trend was observed in Gram-negative E. coli. However, when the TolC-(efflux pump mutant) strain of E. coli was tested, some compounds displayed MICs of 50 μg/mL (compound 1), 25 μg/mL (compound 12), 12.5 μg/mL (compound 23) and 0.75 μg/mL (compound 24) indicating that efflux contributed to their inactivity in the native strains. It is interesting to note that compound 24 displays an excellent MIC of 0.75 μg/mL (or 2 μM) with the TolC- strain.
3.4. Compound synergy with colistin against A. baumannii
Compounds with promising enzyme inhibitory activity against AbFabI display poor antibacterial activity in A. baumannii, which we hypothesized to be due to poor cell penetration properties. To evaluate this hypothesis we tested several antibiotics that are known to disrupt the outer bacterial membrane, thereby potentially increasing cell permeation in combination with our compounds. Compounds with promising enzyme inhibitory activity against AbFabI were tested in these experiments. We found excellent synergy in A. baumannii strain 2208 when the compounds were combined with colistin (polymixin E, a last line antibiotic) in a checkerboard MIC assay. The MIC of colistin against A. baumannii ranges from 1.5 – 3.1 μg/mL. However, when colistin is combined with our compounds, the MIC of colistin in the combination was ∼10-fold lower when synergy was observed (Table 3). Compounds 1, 2, 4, 6, 7, 8, 12 and 24 showed excellent synergy in combination with colistin (Table 3). The IC50s of these compounds are <10 μM. Compounds 10, 11, 23, 26, 27 and 28 did not show synergistic behavior in combination with colistin, despite having promising IC50s against AbFabI. Compounds 23 and 24, with the most promising IC50 values of 0.47 μM and 0.42 μM, respectively, with AbFabI, displayed differing effects, with compound 23 showing no synergy in A. baumannii, while compound 24, a close analog, showed good synergy. This also correlates well with the difference in observed individual MICs and on-target vs off-target activities of compounds 23 and 24 in S. aureus (Table 2). Additionally, as expected compounds with poor MIC in S. aureus did not show synergy with colistin in A. baumannii. These include compounds 11, 23, 26, 27, and 28. However, it is to be noted that not all compounds that lack MIC in S. aureus were tested in these synergy experiments. These include compounds 3, 5, 9, 13, 14-22, 25 and 29.
Table 3.
Synergistic activities of N-carboxy pyrrolidine analogs in combination with colistin against A. baumannii strain 2208. MIC of compound and colistin is the concentration in the clear well (well with lowest concentrations and no growth). MIC of colistin alone is 1.5-3.1 μg/mL
| Compound in combination with colistin | MIC of colistin in combination (μg/mL) | MIC of compound in combination (μg/mL) | ΣFIC | Effect |
|---|---|---|---|---|
| 1 | 0.39 | 12.5 | 0.31 | Synergy |
| 2 | 0.19 | 25 | 0.25 | Synergy |
| 4 | 0.19 | 25 | 0.25 | Synergy |
| 6 | 0.19 | 25 | 0.25 | Synergy |
| 7 | 0.19 | 50 | 0.38 | Synergy |
| 8 | 0.39 | 12.5 | 0.19 | Synergy |
| 10 | 0.39 | 50 | 0.5 | No synergy/Synergy |
| 11 | 0.39 | 50 | 0.75 | No synergy |
| 12 | 0.78 | 6.25 | 0.28 | Synergy |
| 23 | 3.1 | n/a | n/a | No synergy |
| 24 | 0.39 | 25 | 0.37 | Synergy |
| 26 | 1.56 | n/a | n/a | No synergy |
| 27 | 0.39 | 100 | 1 | No synergy |
| 28 | 0.39 | 100 | 1 | No synergy |
To determine whether the observed synergy with colistin is due to improved cell permeability of our compounds we also conducted a checkerboard MIC assay with polymixin B nonapeptide. Polymixin B nonapeptide has an MIC >120 μg/mL against A. baumannii strain 2208 but is known to increase cell membrane permeability.26 When combined with two of our compounds we find synergy similar to that with colistin (Table 4) thus confirming that the lack of direct antibacterial activity of our FabI inhibitors is due to poor cell permeation.
Table 4.
Synergistic activities of N-carboxy pyrrolidine analogs in combination with polymixin B nonapeptide against A. baumannii strain 2208. MIC of compound and polymixin B nonapeptide is the concentration in the clear well (well with lowest concentrations and no growth).
| Compound in combination with polymixin B | MIC of polymixin B in combination (μg/mL) | MIC of compound in combination (μg/mL) | ΣFIC | Effect |
|---|---|---|---|---|
| 1 | 3.75 | 50 | 0.28 | Synergy |
| 24 | 15 | 50 | 0.37 | Synergy |
To better understand the potential clinical relevance of this work we tested four of our most active compounds (compounds with the lowest MIC values in the combination with colistin in Table 3) with a multidrug resistant clinical isolate of A. baumannii (MDR-Ab). We find that these compounds display similar synergy with colistin against MDR-Ab (Table 5).
Table 5.
Synergistic activities of N-carboxy pyrrolidine analogs in combination with colistin against MDR A. baumannii. MIC of compound and colistin in the clear well (well with lowest concentrations and no growth) is reported.
| Compound in combination with colistin | MIC of colistin in combination (μg/mL) | MIC of compound in combination (μg/mL) | ΣFIC | Effect |
|---|---|---|---|---|
| 1 | 0.78 | 25 | 0.25 | Synergy |
| 8 | 0.39 | 25 | 0.25 | Synergy |
| 12 | 0.78 | 12.5 | 0.31 | Synergy |
| 24 | 0.78 | 12.5 | 0.31 | Synergy |
3.5. Cytotoxicity
Promising compounds were tested for cytotoxicity with HepG2 cells. HepG2 cells were incubated for 24 hours with different concentrations of each compound and the cell viability determined. We employed two methods for this testing. The conventional method utilizes 10% FBS (fetal bovine serum) while a slightly modified method utilizes 0.5% FBS (20-fold less) with the media supplemented with 10% Cell-Ess media. Cell-Ess is a serum free, chemically defined serum replacement. This replacement was done to understand the contribution of serum binding to toxicity. The presence of FBS has differing effects on the toxicity towards HepG2 cells. We find that compounds 1, 2, 4, 7, 8, and 12 have 2 to 4-fold lower IC50s in the presence of 0.5% of serum indicating the potential for these compounds to bind to serum (Table 6). Compounds 6 and 24 have minimal toxicity compared to the rest of the compounds in both media. Compound 24 is especially promising due to the fact that it exhibits the best selectivity index (ratio of cytotoxicity to MIC) of >10 in two S. aureus strains highlighting the therapeutic potential of this compound.
Table 6.
Cytotoxicity of the N-carboxy pyrrolidine analogs at standard serum concentration of 10% FBS and a low serum concentration of 0.5% FBS. Also included are the MICs against S. aureus strain Newman for comparison purposes.
| Compound | IC50 in HepG2 cells (μg/mL) in 10% FBS | IC50 in HepG2 cells (μg/mL) in 0.5% FBS | MIC against S. aureus strain Newman (μg/mL) |
|---|---|---|---|
| 1 | 79 | 55 | 12.5 |
| 2 | 160 | 76 | 50 |
| 4 | 125 | 68 | 50 |
| 6 | 237 | >200 | 50 |
| 7 | 279 | 65 | 50 |
| 8 | 75 | 39 | 25 |
| 12 | 64 | 46 | 6.25 |
| 24 | 219 | 138 | 12.5 |
4. Discussion and Conclusions
Antibacterial resistance is a major clinical and public health problem plaguing society today. With the rapid rise in pathogenic strains resistant to most known antibiotics, society is on the brink of entering the post-antibiotic era where infections can kill, and most routine hospital procedures (chemotherapy, organ transplantation and all surgeries) could turn deadly. If no steps are taken to address this emerging threat, death from antibiotic resistant strains could surpass 10 million by the year 2050, a number higher than that due to cancer.27
Gram-positive S. aureus and Gram-negative A. baumannii are two nosocomial pathogens that are difficult to treat due to the rising number of drug resistant strains. We have identified a novel scaffold that displays antibacterial activities targeting these two pathogens. We were able to co-crystallize compound 1 with FtFabI (FabI from F. tularensis) and this binding mode helped guide the rational structure based drug design process although this structure was not of sufficient quality for PDB deposition. Figure 2 presents an overlay of the crystal structure of compound 1 with the crystal structures of two known FabI inhibitors – a benzimidazole-based FabI inhibitor (PDB ID 4NZ9), and the well-known FabI inhibitor AFN1252 (PDB ID 4FS3). Compound 1 occupies the same region in the active site as known FabI inhibitors and also maintains H-bond with the –OH group of NADH and Tyr 156 in FtFabI that are seen with most known FabI inhibitors.
Figure 2.
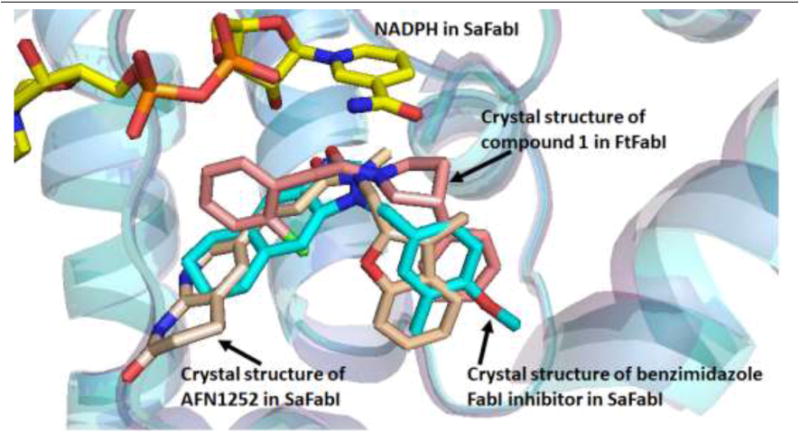
Overlay of the crystal structures of compound 1 in the active site of FtFabI (salmon) with two known FabI inhibitors - benzimidazole based FabI inhibitor in SaFabI (PDB ID 4NZ9) (cyan), and AFN1252 (PDB ID 4FS3) (beige). Compound 1 occupies the same region in the active site as known FabI inhibitors.
We conducted a detailed SAR analysis by designing analogs based on this binding mode and tested with both SaFabI and AbFabI and found that although the SAR are similar between these two enzymes, there are differences that help us better understand the binding modes in the two isoforms. Generally a compound with promising IC50 in SaFabI (IC50 <15 μM) tends to also be promising in AbFabI, although with exceptions that include compounds 9, 13, 14, 16 and 25. These exceptions are all related in that they all carry a 4-methoxy or methylenedioxy substituted phenyl ring at the R1 position that is not tolerated in AbFabI. One of the most significant differences is the substitution of a 2-methoxyphenyl in compound 24 by a 4-methoxyphenyl in compound 25 that causes a complete loss in enzyme inhibitory activity in AbFabI compared to that in SaFabI. A similar comparison of compounds 8 and 9 reveals the same pattern in AbFabI. To better understand the reason for this we performed a computational docking analysis and selected the binding poses that had features in common with most known FabI inhibitors11 including the co-crystal structure of compound 1. Figure 3 displays an overlay of the docked pose of compound 1 with that of the crystal structure. The features commonly seen with FabI inhibitors include a delocalized planar moiety or an aromatic ring that can participate in the π-stacking interaction with the aromatic nicotinamide ring of NAD+ and a hydrogen bond accepting group that can interact with an active site tyrosine residue and is within H-bonding distance of the -OH group
Figure 3.
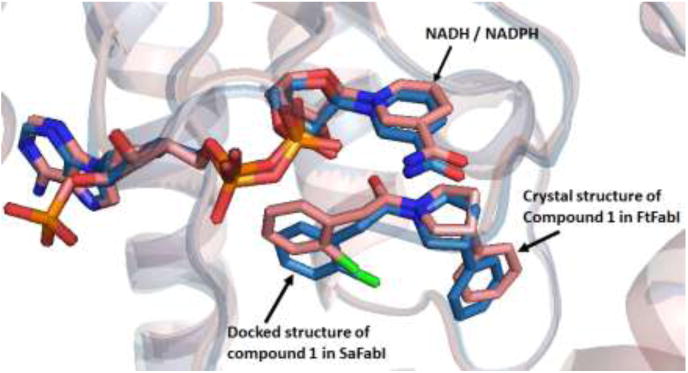
Overlay of the co-crystal structure of compound 1 in the active site of FtFabI (salmon) and the docked pose of compound 1 (blue) in the active site of SaFabI. The docked pose of compound 1 maintains a binding mode similar to the crystal structure.
These docked poses can explain the lack of enzyme inhibitory activity in compound 9 compared to compound 8 in AbFabI. The R1 groups in compounds 8 and 9 occupy different positions in the active site of AbFabI, as seen in Figure 5A. The 2-methoxyphenyl ring in compound 8 is well accommodated in the active site, while the 4-methoxyphenyl is shifted away, as the 4-methoxy group clashes with Asn156. This shift positions the 4-methoxy ring in an unfavorable hydrophobic region surrounded by Leu100, and Ile201 (Figure 5B). It is possible that compound 9 does not really have a stable binding pose due to clash with a side chain and unfavorable hydrophobic interactions arising from the re-positioning of the 4-methoxy group, resulting in poor enzyme inhibition.
Figure 5.
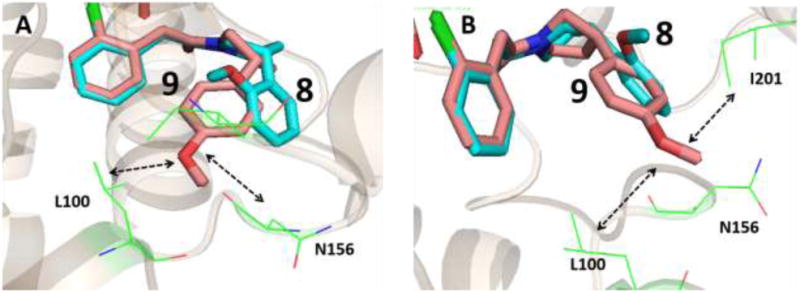
Docking pose of compound 8 (cyan backbone) and compound 9 (salmon backbone) in the active site of AbFabI (PDB ID 4ZJU). (A) The 4-methoxy phenyl in compound 9 clashes with N156 if it maintains a binding pose similar to compound 8. As a result we observe a shift in the binding mode. (B) The shift in binding mode of compound 9 positions it in the unfavorable hydrophobic environment of L100 and I201 which likely explains the weak inhibitory activity of compound 9 in AbFabI.
This same explanation can be applied to compounds 24 and 25 in AbFabI. On comparing the activities of compounds 9 and 25 in AbFabI and SaFabI, we find that they show significant differences in inhibition of the two enzymes. We believe this to be due to differences in the positioning of the loop with the catalytic Tyr (Tyr 157) (Figure 6). In SaFabI, this loop is shifted away from the cofactor when compared to the same loop in AbFabI. As a result of this shift, the inhibitors with the 4-methoxyphenyl group at the R1 position are better accommodated in SaFabI than in AbFabI.
Figure 6.
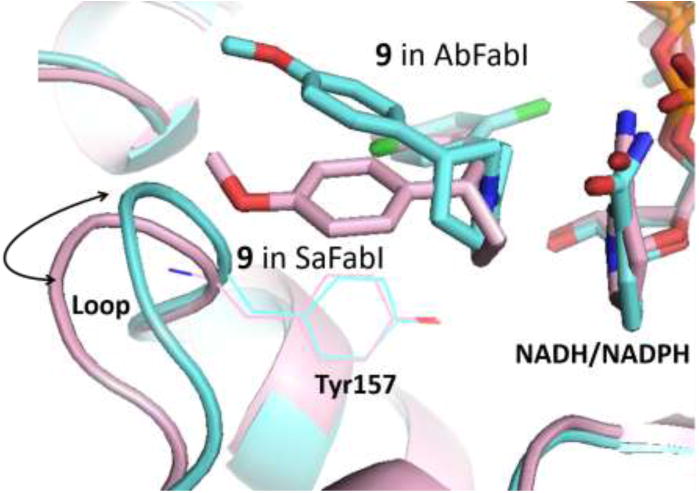
Overlay of SaFabI (pink) and AbFabI (blue) with compound 9 docked in the active site. The loop with the catalytic Tyr 157 residue is shifted in toward the active site in the case of AbFabI, leading to compounds with 4-methoxy phenyl groups at the R1 position being better accommodated in SaFabI than in AbFabI.
In SaFabI, compounds 24 and 25 do not show a change in IC50 between the 2-methoxyphenyl and the 4-methoxyphenyl at the R1 position, while compounds 8 and 9 show a 25-fold increase in IC50 when going from 2-methoxyphenyl and the 4-methoxyphenyl at the R1 position. We believe this to be due to differences in the substituents at the R2 position.
We do not observe a correlation between IC50 and MIC values in S. aureus, which is most likely due to different cell membrane penetration properties of these compounds. In S. aureus our initial hit compound 1 has on-target antibacterial activity, and the more promising MIC of 12.5-25 μg/mL (42 -84 μM), in the IC50 range of 10.7 μM. Introduction of -F atoms on the benzyl ring as in compounds 2-7 seems to lead to cell penetration issues as these compounds show poor antibacterial activity. It is interesting to note that compounds 8 and 9, which differ in the positioning of the methoxy group on the phenyl ring at the R1 position, show a > 8-fold difference in MIC values (with compound 8 having a MIC between 12.5-25 μg/mL, while compound 9 has a MIC >200 μg/mL) which corresponds with the difference in IC50 values (with compound 8 having an IC50 of 0.5 μM vs. 12.5 μM for compound 9).
When combined with colistin we find that the compounds with promising enzyme inhibitory activities against AbFabI also display excellent synergy with colistin in A. baumannii. Colistin was discovered in the 1940s and is active mainly against Gram-negative pathogens. Clinical use decreased in the 1970s due to issues with nephrotoxicity and neurotoxicity.28 The rapid rise in multidrug resistant strains has led to the use of colistin as a last line antibiotic. The mechanism of action of colistin is not well understood, but it is known to bind to the lipid A component of the cell membrane lipopolysaccharides (LPS) which are found in the outer layer of the Gram-negative cell membrane and to destabilize the cell membrane.28 We find that the MIC of colistin varies from 1.5-3.1 μg/mL in our lab conditions. In our synergy experiments the minimum inhibitory concentration of colistin drops by ∼10-fold which could significantly reduce its toxic side effects if the administered dose can be similarly reduced. We believe that the possible increased permeability of the A baumannii membrane rendered by colistin provides our compounds an access to the cell interior where they inhibit FabI leading to cell death. We confirmed this hypothesis by testing with polymixin B nonapeptide. Polymixin B nonapeptide has no antibacterial activity on its own but is known to disrupt the cell membrane of A baumannii thus increasing permeation.26, 29 When combined with the FabI inhibitors we observe synergy similar to that observed with colistin. This observation confirms that the lack of FabI inhibitor antibacterial activity in A. baumannii is mostly due to poor cell permeation properties.
We observe that most compounds that are good inhibitors of SaFabI and AbFabI (IC50 <15 μM), with a MIC ≤ 50 μg/mL in S. aureus, and that are on-target in S. aureus, display synergy with colistin in A. baumannii. These compounds include compounds 1, 2, 4, 6, 8, 12 and 24. The significance of this work is highlighted by the fact that similar synergy is observed when compounds 1, 8, 12, and 24 were combined with colistin and tested against a clinical isolate of A. baumannii that is multidrug resistant.
The cytotoxic potential of these compounds was evaluated in a HepG2 cell line in the presence of 10% FBS and 0.5% FBS to understand the impact of serum binding on toxicity. Several compounds exhibit lower IC50s in low serum media compared to the higher serum media with these IC50s being in the range of the MICs observed. This indicates that these compounds have the potential to be cytotoxic. However, binding to serum is not viewed to be a drawback as many pharmaceutically active compounds are known to be >90% bound to serum. This includes the FabI inhibitor AFN-1252 (currently Debio1452) which is known to be 95% bound to serum proteins of mouse, rat, dog, and humans.30 Compounds 6 and 24 are two compounds whose IC50s do not change significantly in the presence of different serum concentrations. These compounds have promising selectivity indices when the IC50 against the HepG2 cell line is compared with the MIC observed in S. aureus. Our most promising compound is compound 24. This compound efficiently inhibits the FabI enzyme (IC50 of 3.5 μM and 0.42 μM against SaFabI and AbFabI respectively), and exhibits a promising MIC of 12.5 μg/mL in S. aureus. It has minimal off-target activity in S. aureus, and displays good synergy with colistin against A. baumannii, with a promising selectively index. With this first generation of compounds we have not studied the potential for resistance development via active site mutations. Design of the next generation of inhibitors will involve making additional interactions in the active site with either the protein backbone or key catalytic residues to minimize resistance through mutations. The potential for resistance will then be explored.
In conclusion, we report a novel antibiotic scaffold that differs significantly from that of any currently marketed antibiotics. FabI is a narrow spectrum drug target and therapeutics targeting FabI could potentially minimally disrupt the gut microbiome. Our promising preliminary data indicates that this scaffold has the potential to address the need for novel antimicrobials to treat drug resistant strains of both S. aureus and A. baumannii. This inhibitor series shows promise for developing antibiotics treating S. aureus as a monotherapy and A. baumannii as a combination therapy with colistin.
Supplementary Material
Figure 4.
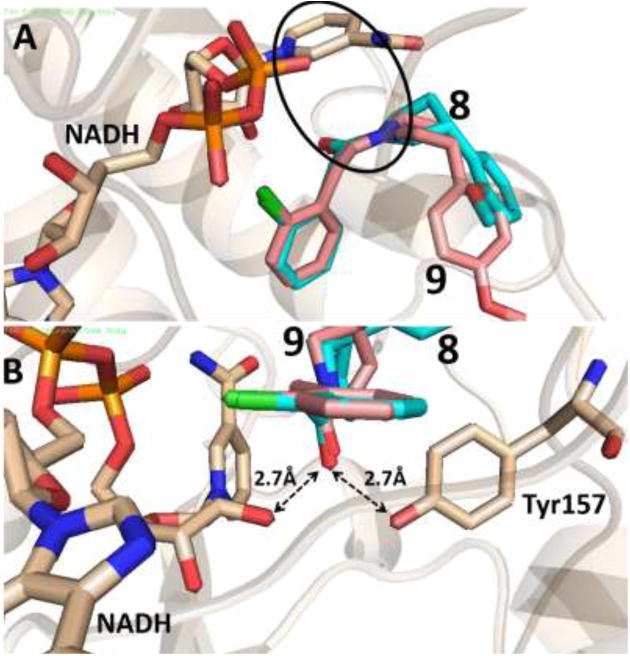
Docked pose of compound 8 (cyan backbone) and 9 (salmon backbone) in the active site of AbFabI (PDB ID 4ZJU). This docking pose maintains the important π-stacking interaction with the aromatic ring of NADH as shown by the circle in (A) while also being within H-bonding distance of the ribose ring of NADH and Tyr157, as seen in (B).
Acknowledgments
We thank Dr. Adam Elhofy and Dr. Hyun-Young Jeong for their guidance with the cytotoxicity experiments. We also thank Essential Pharmaceuticals for providing us with the Cell-Ess media and the Seattle Structural Genomics Center for Infectious Disease (SSGCID) for providing us with the AbFabI enzyme. This work was funded, in part, through the National Institutes of Health Grant AI110090 (Phase I STTR).
Abbreviations Used
- ESKAPE
Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species
- FabI
enoyl-ACP reductase
- MRSA
Methicillin-resistant Staphylococcus aureus
- FASI
human fatty acid biosynthesis
- FASII
bacterial fatty acid biosynthesis
- SaFabI
S. aureus FabI enzyme
- AbFabI
Acenitobacter baumannii FabI enzyme
- FtFabI
Francisella tularensis FabI enzyme
- MDR-Ab
multidrug resistant A. baumannii
- MIC
Minimum Inhibitory Concentration
- LPS
lipopolysaccharides
- FIC
Fractional Inhibitory Concentration
- FBS
fetal bovine serum
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide
- EMEM media
Eagle's Minimal Essential Media
- BSA
Bovine Serum Albumin
Footnotes
Appendix: LC-MS and NMR spectra supporting compound characterization.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ. Molecular mechanisms of antibiotic resistance. Nature reviews Microbiology. 2015;13(1):42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett JG, Gilbert DN, Spellberg B. Seven ways to preserve the miracle of antibiotics. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2013;56(10):1445–50. doi: 10.1093/cid/cit070. [DOI] [PubMed] [Google Scholar]
- 3.Golkar Z, Bagasra O, Pace DG. Bacteriophage therapy: a potential solution for the antibiotic resistance crisis. Journal of infection in developing countries. 2014;8(2):129–36. doi: 10.3855/jidc.3573. [DOI] [PubMed] [Google Scholar]
- 4.Bax R, Green S. Antibiotics: the changing regulatory and pharmaceutical industry paradigm. The Journal of antimicrobial chemotherapy. 2015;70(5):1281–4. doi: 10.1093/jac/dku572. [DOI] [PubMed] [Google Scholar]
- 5.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 6.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, MJR, Talan DA Chambers, H. F. Infectious Diseases Society of America. Clinical practice guidelines by the infectious diseases society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18–55. doi: 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- 7.Wang N, Ozer EA, Mandel MJ, Hauser AR. Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. MBio. 2014;5(3):e01163–14. doi: 10.1128/mBio.01163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bassetti M, Righi E. Development of novel antibacterial drugs to combat multiple resistant organisms. Langenbecks Arch Surg. 2015;400(2):153–65. doi: 10.1007/s00423-015-1280-4. [DOI] [PubMed] [Google Scholar]
- 9.Cameron DR, Mortin LI, Rubio A, Mylonakis E, Moellering RC, Jr, Eliopoulos GM, Peleg AY. Impact of daptomycin resistance on Staphylococcus aureus virulence. Virulence. 2015;6(2):127–31. doi: 10.1080/21505594.2015.1011532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai Y, Chai D, Wang R, Liang B, Bai N. Colistin resistance of Acinetobacter baumannii: clinical reports, mechanisms and antimicrobial strategies. The Journal of antimicrobial chemotherapy. 2012;67(7):1607–15. doi: 10.1093/jac/dks084. [DOI] [PubMed] [Google Scholar]
- 11.Mehboob S, Hevener KE, Truong K, Boci T, Santarsiero BD, Johnson ME. Structural and enzymatic analyses reveal the binding mode of a novel series of Francisella tularensis enoyl reductase (FabI) inhibitors. Journal of medicinal chemistry. 2012;55(12):5933–41. doi: 10.1021/jm300489v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heath RJ, Rock CO. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem. 1995;270(44):26538–42. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 13.Kingry LC, Cummings JE, Brookman KW, Bommineni GR, Tonge PJ, Slayden RA. The Francisella tularensis FabI enoyl-acyl carrier protein reductase gene is essential to bacterial viability and is expressed during infection. J Bacteriol. 2013;195(2):351–8. doi: 10.1128/JB.01957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Balemans W, Lounis N, Gilissen R, Guillemont J, Simmen K, Andries K, Koul A. Essentiality of FASII pathway for Staphylococcus aureus. Nature. 2010;463(7279):E3–E4. doi: 10.1038/nature08667. [DOI] [PubMed] [Google Scholar]; (b) Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(37):15378–15383. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mistry TL, Truong L, Ghosh AK, Johnson ME, Mehboob S. Benzimidazole-Based FabI Inhibitors: A Promising Novel Scaffold for Anti-staphylococcal Drug Development. ACS infectious diseases. 2017;3(1):54–61. doi: 10.1021/acsinfecdis.6b00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Heath RJ, Su N, Murphy CK, Rock CO. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J Biol Chem. 2000;275(51):40128–33. doi: 10.1074/jbc.M005611200. [DOI] [PubMed] [Google Scholar]; (b) Zhu L, Lin J, Ma J, Cronan JE, Wang H. Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob Agents Chemother. 2010;54(2):689–98. doi: 10.1128/AAC.01152-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu H, Tonge PJ. Mechanism and inhibition of the FabV enoyl-ACP reductase from Burkholderia mallei. Biochemistry. 2010;49(6):1281–9. doi: 10.1021/bi902001a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Heath RJ, Rock CO. Microbiology - A triclosan-resistant bacterial enzyme. Nature. 2000;406(6792):145–146. doi: 10.1038/35018162. [DOI] [PubMed] [Google Scholar]
- 17.(a) Gerusz V. Recent Advances in the Inhibition of Bacterial Fatty Acid Biosynthesis. 2010;45:295–311. [Google Scholar]; (b) Chen Y, Pi B, Zhou H, Yu Y, Li L. Triclosan resistance in clinical isolates of Acinetobacter baumannii. Journal of medical microbiology. 2009;58(Pt 8):1086–91. doi: 10.1099/jmm.0.008524-0. [DOI] [PubMed] [Google Scholar]
- 18.Lee H, Zhu T, Patel K, Zhang YY, Truong L, Hevener KE, Gatuz JL, Subramanya G, Jeong HY, Uprichard SL, Johnson ME. High-throughput screening (HTS) and hit validation to identify small molecule inhibitors with activity against NS3/4A proteases from multiple hepatitis C virus genotypes. PLoS One. 2013;8(10):e75144. doi: 10.1371/journal.pone.0075144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh R, Ramachandran V, Shandil R, Sharma S, Khandelwal S, Karmarkar M, Kumar N, Solapure S, Saralaya R, Nanduri R, Panduga V, Reddy J, Prabhakar KR, Rajagopalan S, Rao N, Narayanan S, Anandkumar A, Balasubramanian V, Datta S. In Silico-Based High-Throughput Screen for Discovery of Novel Combinations for Tuberculosis Treatment. Antimicrobial Agents and Chemotherapy. 2015;59(9):5664–5674. doi: 10.1128/AAC.05148-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of immunological methods. 1983;65(1-2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 21.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. Journal of computer-aided molecular design. 2007;21(12):681–91. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 22.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52(4):609–23. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan N, Albert M, Awrey D, Bardouniotis E, Berman J, Clarke T, Dorsey M, Hafkin B, Ramnauth J, Romanov V, Schmid MB, Thalakada R, Yethon J, Pauls HW. Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrobial agents and chemotherapy. 2012;56(11):5865–74. doi: 10.1128/AAC.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flamm RK, Rhomberg PR, Kaplan N, Jones RN, Farrell DJ. Activity of Debio1452, a FabI inhibitor with potent activity against Staphylococcus aureus and coagulase-negative Staphylococcus spp., including multidrug-resistant strains. Antimicrobial agents and chemotherapy. 2015;59(5):2583–7. doi: 10.1128/AAC.05119-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slater-Radosti C, Van Aller G, Greenwood R, Nicholas R, Keller PM, DeWolf WE, Fan F, Payne DJ, Jaworski DD. Biochemical and genetic characterization of the action of triclosan on Staphylococcus aureus. Journal of Antimicrobial Chemotherapy. 2001;48(1):1–6. doi: 10.1093/jac/48.1.1. [DOI] [PubMed] [Google Scholar]
- 26.Tsubery H, Ofek I, Cohen S, Fridkin M. Structure-Function Studies of Polymyxin B Nonapeptide: Implications to Sensitization of Gram-Negative Bacteria. Journal of medicinal chemistry. 2000;43(16):3085–3092. doi: 10.1021/jm0000057. [DOI] [PubMed] [Google Scholar]
- 27.O'Neill J. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. Review on Antimicrobial Resistance. 2014 [Google Scholar]
- 28.Velkov T, Roberts KD, Nation RL, Thompson PE, Li J. Pharmacology of polymyxins: new insights into an ‘old’ class of antibiotics. Future Microbiol. 2013;8(6):711–24. doi: 10.2217/fmb.13.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsubery H, Ofek I, Cohen S, Eisenstein M, Fridkin M. Modulation of the hydrophobic domain of polymyxin B nonapeptide: effect on outer-membrane permeabilization and lipopolysaccharide neutralization. Molecular pharmacology. 2002;62(5):1036–42. doi: 10.1124/mol.62.5.1036. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan N, Awrey D, Bardouniotis E, Berman J, Yethon J, Pauls HW, Hafkin B. In vitro activity (MICs and rate of kill) of AFN-1252, a novel FabI inhibitor, in the presence of serum and in combination with other antibiotics. Journal of chemotherapy. 2013;25(1):18–25. doi: 10.1179/1973947812Y.0000000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.