

ABSTRACT
Protective symbiosis has been reported in many organisms, but the molecular mechanisms of the mutualistic interactions between the symbionts and their hosts are unclear. Here, we sequenced the 424-kbp genome of “Candidatus Spiroplasma holothuricola,” which dominated the hindgut microbiome of a sea cucumber, a major scavenger captured in the Mariana Trench (6,140 m depth). Phylogenetic relationships indicated that the dominant bacterium in the hindgut was derived from a basal group of Spiroplasma species. In this organism, the genes responsible for the biosynthesis of amino acids, glycolysis, and sugar transporters were lost, strongly suggesting endosymbiosis. The highly decayed genome consists of two chromosomes and harbors genes coding for proteolysis, microbial toxin, restriction-methylation systems, and clustered regularly interspaced short palindromic repeats (CRISPRs), composed of three cas genes and 76 CRISPR spacers. The holothurian host is probably protected against invading viruses from sediments by the CRISPRs/Cas and restriction systems of the endosymbiotic spiroplasma. The protective endosymbiosis indicates the important ecological role of the ancient Spiroplasma symbiont in the maintenance of hadal ecosystems.
IMPORTANCE Sea cucumbers are major inhabitants in hadal trenches. They collect microbes in surface sediment and remain tolerant against potential pathogenic bacteria and viruses. This study presents the genome of endosymbiotic spiroplasmas in the gut of a sea cucumber captured in the Mariana Trench. The extreme reduction of the genome and loss of essential metabolic pathways strongly support its endosymbiotic lifestyle. Moreover, a considerable part of the genome was occupied by a CRISPR/Cas system to provide immunity against viruses and antimicrobial toxin-encoding genes for the degradation of microbes. This novel species of Spiroplasma is probably an important protective symbiont for the sea cucumbers in the hadal zone.
KEYWORDS: symbiosis, hadal, CRISPR, genome reduction, Spiroplasma
INTRODUCTION
Deep-sea scavengers and other benthic biota are major players in deep-sea ecosystems. They recycle organic materials and filter microbes as food sources. The deep-sea surface sediments are replete with viruses, which are often much more abundant than bacteria (1, 2). Owing to the low nutrient content of this environment, the microbial biomass becomes key to scavengers' survival. These organisms evolved so as to filter through their digestive system as much surface sediment material as possible. As a trade-off, the intimate contact with bacteria and viruses that this process implies posed lethal threats. Marine invertebrates have developed an efficient immune system to cope with this situation (3). As cases in point, the echinoderms of coastal waters are hardly vulnerable to pathogenic viruses and bacteria (4). This raised an interesting issue regarding the strategy employed by deep-sea invertebrates to efficiently cope with the massive influx of potential pathogens that transit through their gut.
The gut microbiota is essential to harvest nutrients and promote health in humans and other organisms (5, 6). Symbiotic microbes, particularly those establishing long-term mutualism, carry out pivotal functions in the gut of their host. For example, symbionts can help the host digest wood debris in the termite gut (7), degrade recalcitrant organics (8), and evade pathogens (9). Previous observations established that the microbial community in the gut of deep-sea animals was not as rich as that of mammals, ending up with a limited collection of major symbionts apparently dominating the gut microbiome (8, 10). Among those, Mollicutes (Tenericutes) were frequently detected in metazoan guts and in plants (11–13). In the gut of a deep-sea giant isopod, symbiotic mycoplasmas located in the stomach were found to provide the host with sugars and amino acids using enzymes secreted at the cell membrane of the bacteria (8). Phylogenetically related spiroplasmas were observed in the gut and gill of deep-sea chitons, but their hypothetical role in wood degradation lacked strong supporting evidence (10). To our knowledge, there are not studies investigating whether Tenericutes might protect the deep-sea hosts against viruses. In insects, protective symbiosis has been demonstrated by the manipulation of endosymbiotic Wolbachia bacteria in Drosophila strains to delay virus accumulation and enhance the viral immunity of the insect host (9). Spiroplasma spp. have been frequently found to coexist with Wolbachia while constituting a mutualistic relationship with the Drosophila host (14). It was also reported that the spiroplasmas in Drosophila could hinder invasion of the insect by parasitic wasps or pathogenic worms rather than viruses (13). The molecular mechanisms of the protective effects remained largely speculative, with a possible metabolic interference with the phospholipid metabolism of the parasites (15).
Genome sequencing has fundamentally improved our understanding of the mutualistic relationship between symbionts and hosts. Deciphering the Wolbachia genome allowed the identification of ankyrin-encoding genes that enable the bacterium to evade recognition and attack by the host (16). Of particular interest, eukaryote-derived genes encoding leucine-rich regions and fibronectin essential for the host-symbiont interaction were identified in the symbiotic genomes (17, 18). Models of mutualistic symbiosis were then proposed on the basis of genome features (18).
Echinoderms represented by sea cucumbers (Echinodermata: Holothuroidea) are dominant invertebrates in hadal trenches (>6,000 m depth) (19). As primary consumers, they may profoundly influence microbial populations in the trench ecosystem. In the food chain, sea cucumbers are preys of fish and sea stars, serving as a key player in the ecosystem. Understanding how deep-sea Holothuroidea survive in the low-nutrient sediment is a question of considerable interest. Furthermore, their patchy distribution in deep-sea and hadal regions may cause geographic isolation, leading to niche-specific biological features. Here, we worked on a sea cucumber obtained from the Mariana Trench at a depth of 6,140 m. DNA from the gut sample was sequenced and assembled into the genome of an endosymbiotic Spiroplasma. The apparent considerable streamlining of this genome with a loss of numerous essential genes was indicative of long-term mutualistic interactions and niche-specific evolution. The maintenance of pathways for viral defense in an otherwise-streamlined genome suggests a protective symbiosis of the Spiroplasma as an adaptive function for the sea cucumber in the trench.
RESULTS
Gut microbiota of the deep-sea sea cucumber.
A transparent sea cucumber was collected in the southern slope of the Mariana Trench (see Fig. S1 in the supplemental material). Its body length was about 8 cm. Taxonomic classification using the cytochrome c oxidase subunit I (CoxI) gene and 18S rRNA gene indicates that the sea cucumber is a species close to Zygothuria oxysclera (98% similar between 18S rRNA genes). Sequencing of full-length 16S rRNA genes from the gut microbiota of the sea cucumber revealed the major inhabitants of the different gut sections. Classification of the 16S rRNA genes showed that unclassified Gammaproteobacteria (mostly belonging to incertae sedis groups originating from deep-sea surface sediments) were dominant in the foregut and the midgut (Fig. 1). In contrast, the hindgut was overwhelmingly occupied by a single unknown bacterial species, which was not detected in the surface sediment of the sampling site. The holothurian foregut and hindgut are developed from different cells and therefore probably carry out different physiological functions (20).
FIG 1.
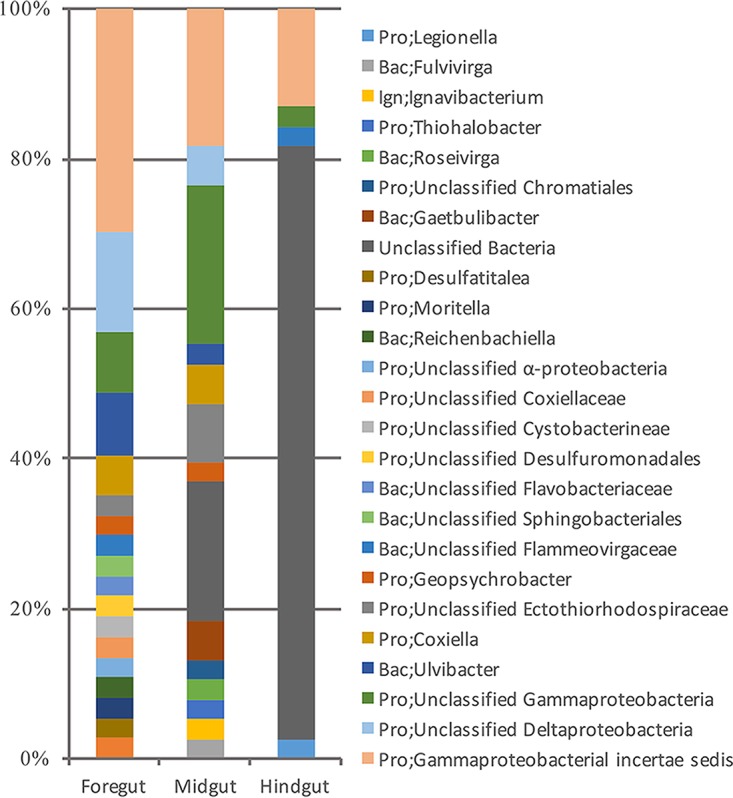
Bacterial community in the gut of the sea cucumber. The 16S rRNA genes in the foregut, midgut, and hindgut of the sea cucumber were amplified and cloned into vectors. A total of 114 cloning sequences were sequenced and classified into genera in the RDP database at a confidence level of 80% (60). The phyla in the classification are abbreviated: Pro, Proteobacteria; Bac, Bacteroidetes; Ign, Ignavibacteria.
Genomic binning.
Illumina sequencing data (7.8 Gbp) were obtained for the hindgut sample. After quality control, 6.1 Gbp of data was used for assembly and genome binning. Optimized pipelines for genomic binning allowed us to achieve a genome consisting of two chromosomes of 280,592 bp and 143,947 bp in size (Fig. S2 and 2). The 424,539-bp genome is characterized by a low G+C content of 29.6%.
FIG 2.

Circular representation of “Ca. Spiroplasma holothuricola” chromosomes. The “Ca. Spiroplasma holothuricola” 424-kbp genome was assembled into two chromosomes. CDSs in categories of Clusters of Orthologous Groups (COG) were depicted with different colors. G+C content and GC skew are displayed on the inner two cycles. Clustered regularly interspaced short palindromic repeats (CRISPRs) and three cas genes are located at the 3′ end of the chromosome 2.
There were two rRNA genes (16S plus 23S rRNA) and 32 tRNA genes for 20 amino acids in the genome. A total of 98 conserved single-copy genes (CSCGs) were identified in the genome (Table S1). Most of the apparently missing CSCGs correspond to functions that were the result of orthologous replacement (21), in particular, glycyl-tRNA synthetase, preprotein translocation factor, and the degradosome (22). The 16S rRNA gene was extracted for taxonomic assignment and phylogenetic analysis. The closest known relative was Spiroplasma sp. strain Bratislava (identity 79%) in the NCBI database. This prompted a further phylogeny-based placement of the bacterial species in the whole kingdom. In the phylogenetic tree with reference sequences from 27 phyla, the 16S rRNA gene from the genome was within the clade for Tenericutes (Fig. S3). Within a phylogenetic tree for Tenericutes, the bacterium from the sea cucumber was located in Spiroplasma group, including a known species of Spiroplasma ixodetis (Fig. 3). However, the large distance from S. ixodetis was indicative of a long-term symbiotic life history or environmental selection under deep-sea conditions for the Spiroplasma-like bacterium. Indeed, the closest species was a symbiotic Spiroplasma bacterium found in a deep-sea chiton and jellyfish (Fig. 3) (10). For the holothurian gut symbiont, its relationship with Spiroplasma species was further supported by a phylogenetic tree constructed by using concatenated conserved proteins (Fig. S4). Therefore, the bacterium in the sea cucumber hindgut represents a novel species of Spiroplasma. The species name of the symbiotic bacterium should be temporarily designated “Candidatus Spiroplasma holothuricola” (holo, thuri'cola. N.L. gen. pl. holothuri of sea cucumbers belonging to the taxon Holothuroidea; L. suff. -cola, inhabitant, dweller; N.L. n. holothuricola, sea cucumber-dweller, referring to the host of the microorganism). A strain name, MT37, was allocated to the organism.
FIG 3.

Phylogenetic positions of “Ca. Spiroplasma holothuricola” in Tenericutes. 16S rRNA genes from different Tenericutes groups were collected to determine the phylogenetic position of “Ca. Spiroplasma holothuricola.” A maximum likelihood phylogenetic tree was reconstructed, and bootstrap values are indicated on the branches of the tree. If a sequence is not associated with a species name, the host and source were present. Accession numbers are presented in parentheses next to the organism names.
Comparison of symbiotic genomes.
The genome of “Ca. Spiroplasma holothuricola” was compared with six genomes of symbionts (Table 1). The chosen genomes were characterized by a low G+C content, except for that of “Candidatus Moranella endobia.” Although the size of the “Ca. Moranella endobia” genome is only 538 kbp, it contains 101 conserved single-copy genes (CSCGs), which is close to the number of the full set of CSCGs in the genomes of free-living species (23). The other genomes larger than 500 kbp also had more than 100 CSCGs. The smallest genome, that of “Candidatus Nasuia deltocephalinicola,” contains only 27 CSCGs.
TABLE 1.
Genomic features of symbiotic species
| Species | Genome size (bp) | No. of contigs | No. of CDSs | G+C content (%) | No. of rRNAs | No. of tRNAs | No. of CSCGs | Coding density (%) |
|---|---|---|---|---|---|---|---|---|
| “Ca. Spiroplasma holothuricola” | 424,539 | 2 | 347 | 29.6 | 2 | 32 | 98 | 90.2 |
| Buchnera aphidicola | 641,454 | 1 | 589 | 25.3 | 3 | 32 | 105 | 86 |
| “Ca. Moranella endobia” | 538,294 | 1 | 431 | 43.5 | 5 | 41 | 101 | 77.4 |
| Mycoplasma sp. Bg1 | 785,028 | 5 | 674 | 26.5 | 3 | 32 | 101 | 91.8 |
| “Ca. Nasuia deltocephalinicola” | 112,091 | 1 | 154 | 17.1 | 2 | 30 | 27 | 66.3 |
| Spiroplasma diminutum | 945,296 | 1 | 858 | 25.4 | 3 | 29 | 106 | 92.8 |
| “Ca. Sulcia muelleri” | 190,657 | 1 | 180 | 24 | 3 | 30 | 60 | 93.5 |
The KEGG pathways were compared between the genomes (Table S2). Most pathways essential to most of bacteria were eliminated from these genomes (Table S2 and Fig. 4). Purine metabolism, pyrimidine metabolism, and ribosome and aminoacyl-tRNA biosynthesis were relatively complete, but, as with the genomes of “Ca. Nasuia deltocephalinicola” and “Candidatus Sulcia muelleri,” some genes of these ubiquitous pathways are still lacking. Most of the Tenericutes seem to have an abundance of sugar phosphotransferase systems (PTSs) (24), as exemplified with Mycoplasma sp. strain Bg1 and Spiroplasma diminutum (Fig. 4). However, this is not always the case, e.g., the “Ca. Spiroplasma holothuricola” genome does not have any PTSs in its genome. The lack of sugar uptake is consistent with the absence of the genes involved in glycolysis and citrate cycle (tricarboxylic acid [TCA] cycle) in the genome. In addition, the “Ca. Spiroplasma holothuricola” genome harbored the smallest number of genes responsible for amino acid biosynthesis, strongly indicating an endosymbiotic lifestyle.
FIG 4.

Heatmap for metabolic pathways of symbiotic bacteria. The color depth was set according to the gene numbers of the KEGG pathways in the genomes.
To examine what essential genes were absent from the “Ca. Spiroplasma holothuricola” genome, 149 core genes for minimal metabolism (25) were probed against the Mycoplasma mycoides synthetic genome JCVI-syn3.0 and the Buchnera aphidicola genome. Synthetic M. mycoides could conduct an autonomous life cycle with only 474 genes (26). With a 618-kbp genome, the B. aphidicola genome is also highly reduced (27). A total of 24 core genes were absent from the “Ca. Spiroplasma holothuricola” genome, in contrast to 9 core genes in the M. mycoides genome and 8 core genes in the B. aphidicola genome (Table S3). In particular, the genes involved in the formylation of the first methionine of initiator tRNA and peptide bond synthesis of triple consecutive proline residues (28) were lost in the “Ca. Spiroplasma holothuricola” genome. However, some of the missing genes, such as those for GroEL and glutaminyl-tRNA synthetase, could be at least partially replaced by other genes. On the other hand, 6 of the core genes were proven to be dispensable to minimal metabolism, as they were not identified in the synthetic M. mycoides genome or in the “Ca. Spiroplasma holothuricola” genome (Table S3).
Major metabolic activities inferred from the genome.
Autonomous gene annotation revealed that 75 out of 347 genes did not have any homolog in the NCBI database. This may be expected, bearing in mind that selection for symbiosis accelerated evolution. On the other hand, high pressure has a considerable impact on protein structure, demanding significant changes in order to maintain protein function (29). Despite the lack of predicted function for a certain proportion of the genome, the metabolic network and symbiotic mode of life could be predicted with the gene content in the genome. The “Ca. Spiroplasma holothuricola” genome did not possess the genes involved in carbohydrate utilization pathways, such as glycolysis, the TCA cycle, and phosphorylation/respiration (Fig. 5). It is notable, however, that citrate might be present in the organism, possibly as a metal chelator and a regulator, as the four conserved motifs that have been shown to bind citrate in the highly conserved polynucleotide phosphorylase (30) are present in its sequence (Fig. S5). Alternatively, this could mean that these motifs are necessary for the activity of the protein and are accidentally involved in citrate binding. The endosymbiotic spiroplasma is probably an acetogen, because it has the genes responsible for acetyl-coenzyme A (acetyl-CoA) fermentation to produce acetate. In this process, an ATP is generated. However, in the predicted metabolic network, the source of acetyl-CoA is not evident. Alcohol is possibly used to produce acetyl-CoA, but an obvious gene encoding acetaldehyde dehydrogenase is missing (Fig. 5).
FIG 5.

Schematic symbiotic model between the Spiroplasma and holothurian host. The gene prediction for the “Ca. Spiroplasma holothuricola” genome was used to illustrate the metabolic network and virus resistance strategies. Viruses invading the host cell were supposed to be destroyed by the “Ca. Spiroplasma holothuricola” with CRISPRs/Cas and restriction-methylation (RM) systems. SDF, sodium dicarboxylate symporter family; Ktr, potassium uptake protein; TK, thymidine kinase; ComEC, late competence protein; FHY, bifunctional riboflavin kinase; DPCK, dephospho-CoA kinase; Pi, inorganic phosphate; FMN, flavin mononucleotide.
In bacteria, the fermentation of arginine (arginolysis) is an alternative to ATP generation when sugar sources are not available (31). The endosymbiotic spiroplasma in the sea cucumber likely employed arginolysis to supply ATP rather than acetyl-CoA fermentation, because all the genes encoding the enzymes that take part in arginolysis via the arginine deiminase system (ADI pathway) were identified in the genome. The ADI pathway is widely used to cope with unusually low pH in a variety of lactic acid bacteria, as well as in Tenericutes (32). The arginine deiminase gene (arcA) has homologs identified in Entomoplasma, Spiroplasma, and Mesoplasma (identity between 44% and 46%). Carbamoyl-P, as a product of citrulline degradation, is likely catabolized by carbamate kinase (CKase) to generate ATP, ammonia, and CO2 (Fig. 5). The CKase of our organism is most similar (52%) to the homolog from Staphylococcus hominis. ATP generation via this pathway will lead to the accumulation of ornithine and shortage of arginine. This imbalance can presumably be solved by the ArcD permease that carries out cross-membrane exchange of arginine and ornithine (Fig. 5) (33). In the genome of “Ca. Spiroplasma holothuricola,” an arcD gene (KEGG gene K03758) with the highest identity (35%) to the homolog of Mycoplasma yeatsii was identified.
Cross-membrane transport.
“Ca. Spiroplasma holothuricola” probably depended on some sodium-driven symporters to carry C4 carbon sources, amino acids, and likely, choline, into the cell (Fig. 5). A Ktr channel that acts on exchange of Na+ and K+ may pump extra Na+ out of cytoplasm to maintain homeostatic balance (34). The identified ABC transporters might be involved in the uptake of phosphate, ribose, manganese, and magnesium. Possible secretion of bacteriocins and toxic waste also relies on ABC transporters. Both types of exporters are encoded by other Spiroplasma genomes, but the identities between the homologs were low (<40%). The presence of a mere four genes for amino acid biosynthesis indicates that “Ca. Spiroplasma holothuricola” seems to obtain most amino acids from the host. Cardiolipin is essential for bacterial membrane curvature (35). A cardiolipin synthase gene was identified, but the other related genes for the upstream synthesis of cardiolipin were not found. This indicates that the precursors of cardiolipin might be imported from the host.
We used correspondence analysis (CA) to obtain insight into the proteins likely to be located in the membrane. CA separated all of the predicted proteins into two clusters (Fig. 6). The most prominent factor on axis 1 was the amino acid composition driven by the G+C content of their codons (0.93), and the factors driving axis 2 were hydrophilicity (0.86) and hydrophobicity (−0.87). A total of 27 proteins were separated into the second group with a probability higher than 0.95. Fifteen of them were known transmembrane proteins, including sodium-dependent transporters, preprotein translocase, and ABC transporter permeases, whereas 10 of the proteins could not be annotated. Glutamic acid and phenylalanine were characteristic amino acids in cluster 2 proteins, perhaps corresponding to transmembrane properties (Fig. 6). As in other instances of novel species, many cytoplasmic membrane proteins could not yet be associated with a known function in the Spiroplasma genome.
FIG 6.
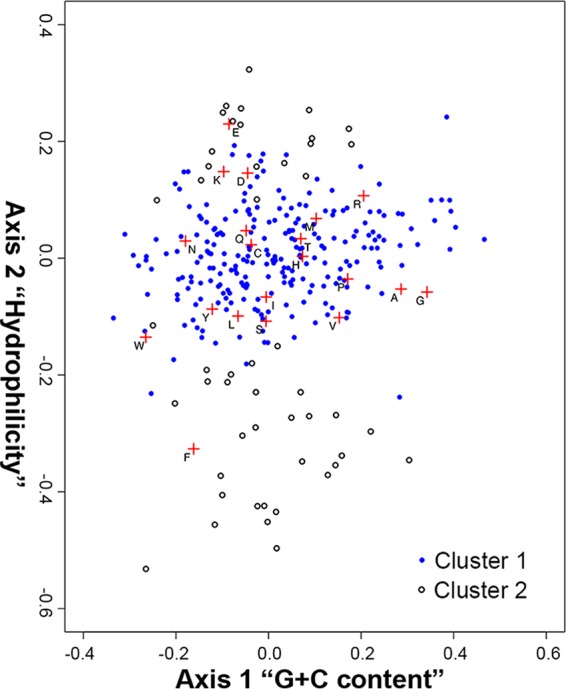
Correspondence analysis of “Ca. Spiroplasma holothuricola” proteins. The factors on the two most informative axes of CA analysis separated the proteins of “Ca. Spiroplasma holothuricola” into two clusters, denoted by blue solid dots and blank dots. The red crosses denote representative amino acids for the proteins nearby.
Strategies of host protection.
Remarkably, despite the small genome size, there was a very long clustered regularly interspaced short palindromic repeat (CRISPR) system in the “Ca. Spiroplasma holothuricola,” genome consisting of three cas genes (cas1-cas2-cas9) and 76 spacers (total of 12 kbp) (Fig. 2). The homologous cas genes with the highest identity were found in Spiroplasma syrphidicola genome (46% for Cas1, 52% for Cas2, and 32% for Cas9). Compared with its relatives in Tenericutes in hosts of fungi and insects, “Ca. Spiroplasma holothuricola” had the highest number of CRISPR spacers (Table 2). In contrast, the S. diminutum genome contains one CRISPR system but with only two spacers. The spacers are DNA tags left during the process of immunity generation against invading phages (36). The large number of CRISPR spacers in the genome indicates that “Ca. Spiroplasma holothuricola” is likely very efficient in protection of the host against viruses (Fig. 5). Remarkably, a search against the NCBI database identified one archaeal and three eukaryotic viral complements of the CRISPR spacers: Acanthamoeba castellanii mimivirus (identity, 93%), the envelope gene of feline immunodeficiency virus (identity, 86%), the gag gene of human immunodeficiency virus (identity, 92%), and hyperthermophilic archaeal virus 1 (identity, 99%).
TABLE 2.
Statistics of CRISPR/Cas in genomes of Tenericutes
| Species | Accession no. | cas type | No. of spacers | Host |
|---|---|---|---|---|
| Spiroplasma syrphidicola | NC_021284 | cas1-cas2-cas9 | 35 | Fly |
| Mycoplasmataceae bacterium | JQIB01000000 | cas1-cas2 | 3 | Fungus |
| Spiroplasma turonicum | CP013860.1 | cas2-cas9 | 46 | Horsefly |
| Spiroplasma helicoides | NZ_CP017015 | cas2-cas9 | 45 | Horsefly |
| Spiroplasma apis | NC_022998 | cas1-cas2-cas9 | 55 | Honeybee |
| “Candidatus Hepatoplasma crinochetorum” | CP006932 | cas1-cas9 | 35 | Arthropod |
In the genome, a 2,742-amino acid (aa) predicted protein (encoded by gene 10005) was characterized with 8 transmembrane domains and seven YD-repeat units. We speculate that it could be a nucleic acid binding membrane toxin originating from phages and allowing resistance of the host against parasites, as reported in insect symbionts (37). In this protein, the identification of two SalY domains (COG0577, a permease component of an ABC-type antimicrobial peptide transport system) also supports its role in defense. In Streptococcus pyogenes, the expression of SalY was critical for intracellular survival in murine macrophage (38). The genome of Acholeplasma multilocale, as a remote relative of Spiroplasma in Tenericutes, also has the salY gene. Altogether, this transmembrane protein might be produced to kill other microbes, which is possibly beneficial to the host. A search against the MEROPS database (39) resulted in at least 12 peptidase genes in the genome. Antimicrobial streptococcin, lacticin, and mersacidin are potentially encoded by the genome and function with these associated peptidases. The “Ca. Spiroplasma holothuricola” might also rely on the peptidases for the acquisition of leucine, serine, methionine, and proline.
There were three restriction-methylation (RM) systems likely obtained through horizontal gene transfer (HGT). An RM system consists of restriction, modification, and specificity domains (40). Bacteria depend on the RMs to prevent invasion of plasmids and phages (41). The cytosine-5 methyltransferase genes were not derived from Tenericutes and perhaps from Staphylococcus, based on sequential similarity (61%). Furthermore, the presence of a CRISPR/Cas system in the genome will strengthen the protection provided by RMs against phages (42). In the genome, the comEC gene involved in the transmembrane uptake of single-stranded naked DNA was identified. This transport process could not only provide the spiroplasma with nutrient sources (43, 44) but also be a weapon to degrade bacterial and viral DNA that spread in the cytoplasm of the host.
DISCUSSION
As dominant inhabitants in the hadal zone, sea cucumbers confront pathogen infection, starvation, and extreme hydrostatic pressure. In addition to low-nutrient detrital materials, all sorts of microbes are overlooked major food sources of the hadal scavengers. In this study, the biodiversity of the bacteria located in the front, middle, and hindgut of the sea cucumber dropped gradually, suggesting a filtration process of the microbes in the digestive tract (45). The hindgut bacterial population was dominated by “Ca. Spiroplasma holothuricola,” suggesting prior clearance of microbes flowing from the foregut and midgut. The digestion of the microbes was possibly helped by antimicrobial toxins secreted by the spiroplasma symbiont. Besides prokaryotic microbes, a wealth of viruses was caught into the gut, since they are major inhabitants of the benthic prokaryotic biomass (46). How could the sea cucumber retain immunity to the potential pathogens that keep coming through its gut? To date, innate cellular immune mechanisms of holothurians are rarely reported. In this study, we propose that the symbiont “Ca. Spiroplasma holothuricola” probably carries out the protective role for sea cucumbers in the Mariana Trench.
We have recently shown the importance of symbiotic mycoplasmas in the digestion of detrital nutrients for deep-sea isopod host (8). Together with the current study, it appears that Tenericutes bacteria tend to evolve into symbionts critical for the survival of scavengers in deep-sea and hadal areas. The reasons for this protective symbiosis had not been deciphered. Our observations may bring about a novel view for protective actions in other deep-sea scavengers. Such a mutual beneficial relationship must have been established for a long period of time, resulting in a drastic reduction of the “Ca. Spiroplasma holothuricola” genome. Subsequently, despite limited studies on holothurian symbionts, we speculate that vertical transmission between generations resulted in a stable relationship between the holothurian host and the endosymbiotic Spiroplasma. However, considering the patchy distribution of the sea cucumber populations in the abyssal and hadal zones (47), it is likely that sea cucumber individuals of different species or habitats did not have the spiroplasma symbionts. Therefore, the transmission between generations and populations could not be answered at present. Since “Ca. Spiroplasma holothuricola” was not present in the surface sediments (our unpublished data), vertical transmission of the endosymbiotic spiroplasma between sea cucumber generations is reasonable. For the sea cucumbers in shallow waters, “Ca. Spiroplasma holothuricola” was not observed in their gut microbiomes (48). Moreover, the composition of the gut bacterial communities also much differed between the sea cucumbers from the coastal (48) and the hadal zones in the current study.
Here, we constructed the binned genome of a representative for all “Ca. Spiroplasma holothuricola” sequences in the sea cucumber sample. Taking into account the coverage levels of different genomic regions, the genomic variations of the spiroplasmas could be examined (Fig. S6). Some genomic regions were associated with an abnormally high coverage, while others were covered by the reads to a limited extent. Among genes associated with a high coverage were those encoding RM proteins and Cas proteins, suggesting that the “Ca. Spiroplasma holothuricola” genome is very dynamic at these regions. It can be expected that the genomic variants also reflected to some extent the reorganization of the genomic contents under pressure of various irregular attacks from different pathogens. Novel genes might be recruited by the uptake of foreign DNAs (44), while the overall trend seems to be in favor of gene loss.
Long-term symbiosis and high hydrostatic pressure yield an accumulation of mutations in the 16S rRNA gene of “Ca. Spiroplasma holothuricola,” blurring the phylogenetic position of the organism among Tenericutes. Perhaps this is also the reason why we could not locate a 5S rRNA gene in the genome. The phylogeny-based placement of the symbiont was somewhere between the Spiroplasma and Mycoplasma clades. The closest known species was S. ixodetis, a basal phylogenetic clade among the six clades in Spiroplasma (49). The appearance of S. ixodetis in a neighboring branch of the phylogenetic tree indicates that “Ca. Spiroplasma holothuricola” was derived from an ancient lineage of Spiroplasma. The hadal condition of high hydrostatic pressure imposes considerable constraints on the protein structures (50), which certainly affects the corresponding gene sequences. The present study highlights the low representation of 16S rRNA sequences for environmental Tenericutes, particularly in deep-sea zones in the public databases. The small number of reference sequences prohibits studies on their origin and evolution. An exploration of the two-dimensional (2D) structural feature of stems and loops could perhaps help trace the deep-sea spiroplasma back to its origin. Although we could not provide microscopic evidence in the present study, the taxonomy of the symbiont was strongly supported by the spiral morphology of its close relative in a deep-sea chiton (10).
Genomic features offer insights about the coevolution between hosts and symbiotic bacteria. Despite the small size of the genome, about 22% of the predicted genes of the “Ca. Spiroplasma holothuricola” remained completely unknown. It can be surmised that some of these genes are responsible for substance exchange between the host and the symbiont. As cases in point, unknown transmembrane proteins show great potential in host-symbiont communications. In some symbiosis systems, the host genomes harbored numerous genes essential to the symbiotic microbes, and the encoded proteins were synthesized in their cytoplasm before being transferred into the symbionts (51). Most tiny genomes (<400 kbp) were isolated from insect symbiotic bacteria (49, 52). The provision of essential amino acids and vitamins was the major contribution to the insect host by the endosymbionts (49, 52). In contrast, the holothurian host probably provided amino acids to the symbiotic spiroplasmas.
Here, we provide evidence for a protective effect of symbiotic Spiroplasma on its deep-sea animal host. Similar symbiotic effects have yet only been demonstrated in insects (13). The specific features of the genome of “Ca. Spiroplasma holothuricola,” missing all major metabolic requirements except for nucleic acid scavenging, are highly suggestive of specialization for a defensive symbiotic role. In this organism, genes serving as defensive mechanisms were enriched and recruited through HGT. Due to a low representation of deep-sea viral proteins in the public database, we could not identify the exact targets of the CRISPR/Cas system, but we note that it must correspond to a considerable panel of viruses (76 repeats). In a complementary work, we obtained the sequence of an unknown viral genome (approximately 57 kbp; our unpublished data). Similarity with predicted viral proteins showed affinity to ostreid and abalone herpesvirus, a cause of worldwide outbreaks of fatal diseases of abalones and mollusks (53). Although the identification of a herpesvirus was not confirmed in the current study, frequent contacts with viruses were obvious for “Ca. Spiroplasma holothuricola.” The large number of CRISPR spacers was rarely found in other bacteria, mostly <50 (54). The T+A-rich CRISPR cas9 gene is a potential useful tool for the modification at T+A-rich genomic regions. Any specific properties of the cas9 gene in “Ca. Spiroplasma holothuricola” were of great interest for further experimental examination.
Finally, the “Ca. Spiroplasma holothuricola” genome is one of the smallest known symbiotic genomes (49). The formation of two chromosomes in the genome has never been reported in Spiroplasma species. Because there are not homologous regions between the chromosomes, it is unlikely they will be further integrated into one circular genome at present. Although Spiroplasma species are well-known symbionts, genomic reduction to about 400 kbp with evolution towards protective symbiosis has not yet been reported. Here, some 24 core genes were depleted from the “Ca. Spiroplasma holothuricola” genome, which might substantially compromise the molecular machines for basic life processes. However, if one takes into account conserved functions rather than conserved genes, the number of missing activities is less than a handful (Table S1). This provides strong evidence that the genome sequence is virtually complete. The niche specification evolution of the genes dramatically affected the efficiency of automatic gene annotation by the hmmsearch and BLAST programs, so we had to resort to manual annotation. Among the missing CSCGs (Table S1), the presence of a specific Ile-tRNA with rare UAU anticodon accounts for the loss of the tilS gene responsible for the anticodon U34 modification (55). The glycine-tRNA ligase is probably encoded by glyR, which is a streamlined convergent version of widespread glyS and glyQ (56). Several elements (ftsY, ffh, and secE) for the preprotein translocation were missing and could be replaced by proteins involved in division (57). Due to the absence of glycolysis, phosphoglycerate kinase is reasonably one of the kinases without identification. The RNA degradosome and DNA management in T+A-rich Firmicutes are structurally different from what happens in other bacteria. This was also the case in “Ca. Spiroplasma holothuricola,” where the absent RNase III gene might be replaced by rnr, rnrY, and rny (22); the missing uvrB gene might be replaced by the novel nuclease genes that encode a structurally different class of proteins. For the missing core proteins, the loss of the EF-P gene should be compensated by introducing new factors for the translation of triple prolines (58). Adjacent to the proline-tRNA ligase gene, there were three novel genes with unknown functions. Altogether, the lack of these CSCGs and core genes in the “Ca. Spiroplasma holothuricola” genome was required by the depletion of genomic redundancy under endosymbiosis.
MATERIALS AND METHODS
Sampling.
A sea cucumber was captured with help of the Jiaolong submersible in the South Mariana Trench (10°89′N, 142°23′E) at a depth of 6,140 m during DY37-II Dive122 on 1 July 2016 (Fig. S1). It was preserved in ethanol before laboratory treatment. DNA was extracted from the foregut, midgut, and hindgut, using the PowerSoil DNA isolation kit (Mo Bio, Carlsbad, CA, USA). It was subsequently purified, according to the manufacturer's instructions. The quality and quantity of the DNA sample were checked with a Qubit 2.0 fluorometer (Life Technologies, USA).
Cloning and classification of 16S rRNA gene sequences.
The bacterial 16S rRNA genes were obtained by PCR amplification using a pair of universal primers, (27F, 5′-AGAGTTTGATC[C/A]TGGCTCAG-3′; 1492R, 5′-TACGG[C/T]TACCTTGTTACGAC-3′) (59) and PrimeSTAR HS DNA polymerase (TaKaRa, Dalian, China). The PCR products were separated by gel electrophoresis, and the bands with expected size (approximately 1,500 bp) were purified with a QIAquick purification kit (Qiagen, Germany).
Bacterial 16S rRNA full-length fragments were cloned into the pMD-18T vector (TaKaRa, Dalian, China) and then transformed into JM109 competent cells (TaKaRa). Thirty-eight cloning sequences were obtained for each of the samples using a Sanger sequencer (ABI 3730) and were then classified by the RDP Classifier (60), with a confidence threshold of 80%.
Genome binning of gut metagenome.
Approximately 100 ng of DNA sample from the hindgut was used for library preparation and sequencing on an Illumina MiSeq platform. The quality of the reads was checked with the NGS QC toolkit (61). Assembly of the Illumina paired-end reads was performed using the SPAdes 3.5 (62). Binning of the draft genomes was carried out by separating the contigs in reference to their different coverage levels, G+C contents, and tetranucleotide frequencies (23). The R scripts for the binning process were obtained from https://github.com/MadsAlbertsen/multi-metagenome. In a binned genome with low G+C content, coding sequences (CDSs) were predicted using Prodigal (version 2.60) and then translated into proteins (63). CheckM was applied to check the quality of the draft genome (64). A manual check was performed to link up the three contigs, which resulted in two circular chromosomes. The replication origin of the chromosomes was predicted using Ori-Finder (65). The coxI and 18S rRNA genes of the sea cucumber host were extracted from the assembled metagenome using a BLAST search.
Genome annotation and metabolic prediction.
CSCGs were identified by searching the CDSs of the genome against a revised HMM database containing 108 CSCG genes (three original genes for Archaea and Cyanobacteria were excluded) (23) using hmmsearch 3.0, with default settings (66). The tRNA genes in the genome were searched using tRNAscan (67).
In the binned genome, coverage of the contigs by the reads was calculated using SAMtools with a setting of no mismatch in “seed alignment” (68). Using a sliding window of 500 bp, the coverage levels of the genome in different regions were calculated. The distance of each movement of the sliding window was 250 bp on the contigs. Coverage of the CDSs was estimated using the average of the values in the sliding windows that moved across the region where the CDSs resided.
The genome was compared with a collection of endosymbiotic genomes listed in the review by Lo et al. (49), along with Spiroplasma diminutum and Mycoplasma sp. Bg1 (8). Initial annotation of the deduced proteins in these genomes was carried out using BLASTP against the NCBI_nr, KEGG, MEROPS (39), and COG databases (69), with a maximum E value cutoff of 1e-04. The KEGG genes were grouped in pathways for comparison in a heatmap using Cluster3 (70). Final annotation benefited from the reannotation of the synthetic M. mycoides genome Syn3.0 (26, 71). CRISPRs and their targets were searched by CRISPRFinder (72). Transmembrane domains in the predicted proteins were predicted by TMHMM2.0 (73). Prior to clustering, CA was employed to order proteins along multiple factors (74, 75). The CDSs in the genome were analyzed for CA clustering by most significant factors using the FactoMineR package (http://factominer.free.fr/), as described elsewhere (76).
Reconstruction of phylogenetic trees.
To determine the taxonomic positions of the unknown bacteria, rRNA genes for a total of 27 phyla were downloaded from the SILVA database (77). From each of the phyla, five representative sequences from different classes were picked out to reconstruct a phylogenetic tree. The sequences were aligned with MUSCLE3.5, with manual adjustments (78). Alignment positions with gaps present in more than 50% of the sequences were deleted. Using a model of GTRGAMMA, a maximum likelihood (ML) phylogenic tree with 1,000 bootstraps was constructed with raxmlGUI (79). A second ML tree that consisted of the 16S rRNA gene and Tenericutes with more references from the NCBI was built with the same method. Furthermore, 49 Tenericutes genomes that belonged to different taxonomic groups were pooled (80). A phylogenomic analysis was conducted using a total of 20 CSCGs that were shared among all the genomes. The proteins for the CSCGs were concatenated for further alignment with MUSCLE3.5, followed by manual adjustments, as described above. The third ML phylogenetic tree based on 1,000 replicates was constructed using raxmlGUI with the PROTGAMMALG model (79).
Accession number(s).
The genome of the sea cucumber host was deposited in the NCBI database under accession numbers CP022928 and CP022929. It was further manually annotated using the MicroScope platform (81) under accession number 9445.
Supplementary Material
ACKNOWLEDGMENTS
We thank Y. Zhang and the team members of the Jiaolong manned submersible for their efforts in the sampling cruise.
This study was supported by the National Science Foundation of China (grants 31460092, 41476104, and 31460001), the Strategic Priority Research Program B of the Chinese Academy of Sciences (grants XDB06010103 and XDB06010201), and the National Key Research and Development Program of China (grant 2016YFC0302500).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01965-17.
REFERENCES
- 1.Hara S, Koike I, Terauchi K, Kamiya H, Tanoue E. 1996. Abundance of viruses in deep oceanic waters. Mar Ecol Prog Ser 145:269–277. doi: 10.3354/meps145269. [DOI] [Google Scholar]
- 2.Yoshida M, Takaki Y, Eitoku M, Nunoura T, Takai K. 2013. Metagenomic analysis of viral communities in (hado)pelagic sediments. PLoS One 8:e57271. doi: 10.1371/journal.pone.0057271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiaramonte M, Russo R. 2015. The echinoderm innate humoral immune response. Ital J Zool 82:300–308. doi: 10.1080/11250003.2015.1061615. [DOI] [Google Scholar]
- 4.Sweet MJ, Bateman KS. 2015. Diseases in marine invertebrates associated with mariculture and commercial fisheries. J Sea Res 104:16–32. doi: 10.1016/j.seares.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clemente JC, Ursell LK, Parfrey LW, Knight R. 2012. The impact of the gut microbiota on human health: an integrative view. Cell 148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sommer F, Backhed F. 2013. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol 11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 7.Brune A. 2014. Symbiotic digestion of lignocellulose in termite guts. Nat Rev Microbiol 12:168–180. doi: 10.1038/nrmicro3182. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Huang JM, Wang SL, Gao ZM, Zhang AQ, Danchin A, He LS. 2016. Genomic characterization of symbiotic mycoplasmas from the stomach of deep-sea isopod Bathynomus sp. Environ Microbiol 18:2646–2659. doi: 10.1111/1462-2920.13411. [DOI] [PubMed] [Google Scholar]
- 9.Hedges LM, Brownlie JC, O'Neill SL, Johnson KN. 2008. Wolbachia and virus protection in insects. Science 322:702–702. doi: 10.1126/science.1162418. [DOI] [PubMed] [Google Scholar]
- 10.Duperron S, Pottier MA, Leger N, Gaudron SM, Puillandre N, Le Prieur S, Sigwart JD, Ravaux J, Zbinden M. 2013. A tale of two chitons: is habitat specialisation linked to distinct associated bacterial communities? FEMS Microbiol Eco 83:552–567. doi: 10.1111/1574-6941.12014. [DOI] [PubMed] [Google Scholar]
- 11.Clark TB. 1982. Spiroplasmas: diversity of Arthropod reservoirs and host-parasite relationships. Science 217:57–59. doi: 10.1126/science.217.4554.57. [DOI] [PubMed] [Google Scholar]
- 12.Gasparich GE. 2010. Spiroplasmas and phytoplasmas: microbes associated with plant hosts. Biologicals 38:193–203. doi: 10.1016/j.biologicals.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Anbutsu H, Fukatsu T. 2011. Spiroplasma as a model insect endosymbiont. Environ Microbiol Rep 3:144–153. doi: 10.1111/j.1758-2229.2010.00240.x. [DOI] [PubMed] [Google Scholar]
- 14.Jaenike J, Stahlhut JK, Boelio LM, Unckless RL. 2010. Association between Wolbachia and Spiroplasma within Drosophila neotestacea: an emerging symbiotic mutualism. Mol Ecol 19:414–425. doi: 10.1111/j.1365-294X.2009.04448.x. [DOI] [PubMed] [Google Scholar]
- 15.Paredes JC, Herren JK, Schüpfer F, Lemaitre B. 2016. The role of lipid competition for endosymbiont-mediated protection against parasitoid wasps in Drosophila. mBio 7:e01006-16. doi: 10.1128/mBio.01006-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N, Bhattacharyya A, Kapatral V, Kumar S, Posfai J, Vincze T, Ingram J, Moran L, Lapidus A, Omelchenko M, Kyrpides N, Ghedin E, Wang S, Goltsman E, Joukov V, Ostrovskaya O, Tsukerman K, Mazur M, Comb D, Koonin E, Slatko B. 2005. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol 3:599–614. doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naito M, Morton JB, Pawlowska TE. 2015. Minimal genomes of mycoplasma-related endobacteria are plastic and contain host-derived genes for sustained life within Glomeromycota. Proc Natl Acad Sci U S A 112:7791–7796. doi: 10.1073/pnas.1501676112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao ZM, Wang Y, Tian RM, Wong YH, Batang ZB, Al-Suwailem AM, Bajic VB, Qian PY. 2014. Symbiotic adaptation drives genome streamlining of the cyanobacterial sponge symbiont “Candidatus Synechococcus spongiarum”. mBio 5:e00079-14. doi: 10.1128/mBio.00079-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jamieson AJ, Gebruk A, Fujii T, Solan M. 2011. Functional effects of the hadal sea cucumber Elpidia atakama (Echinodermata: Holothuroidea, Elasipodida) reflect small-scale patterns of resource availability. Mar Biol 158:2695–2703. doi: 10.1007/s00227-011-1767-7. [DOI] [Google Scholar]
- 20.Mashanov VS, Dolmatov IY, Heinzeller T. 2005. Transdifferentiation in holothurian gut regeneration. Biol Bull 209:184–193. doi: 10.2307/3593108. [DOI] [PubMed] [Google Scholar]
- 21.Poptsova MS, Gogarten JP. 2007. The power of phylogenetic approaches to detect horizontally transferred genes. BMC Evol Biol 7:45. doi: 10.1186/1471-2148-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danchin A. 2009. A phylogenetic view of bacterial ribonucleases. Prog Mol Biol Trans Sci 85:1–41. doi: 10.1016/S0079-6603(08)00801-5. [DOI] [PubMed] [Google Scholar]
- 23.Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 24.Siebold C, Flukiger K, Beutler R, Erni B. 2001. Carbohydrate transporters of the bacterial phosphoenolpyruvate: sugar phosphotransferase system (PTS). FEBS Lett 504:104–111. doi: 10.1016/S0014-5793(01)02705-3. [DOI] [PubMed] [Google Scholar]
- 25.Gil R, Silva FJ, Pereto J, Moya A. 2004. Determination of the core of a minimal bacterial gene set. Microbiol Mol Biol Rev 68:518–537. doi: 10.1128/MMBR.68.3.518-537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutchison CA III, Chuang RY, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, Gill J, Kannan K, Karas BJ, Ma L, Pelletier JF, Qi ZQ, Richter RA, Strychalski EA, Sun L, Suzuki Y, Tsvetanova B, Wise KS, Smith HO, Glass JI, Merryman C, Gibson DG, Venter JC. 2016. Design and synthesis of a minimal bacterial genome. Science 351:aad6253. doi: 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- 27.Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H. 2000. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- 28.Katoh T, Wohlgemuth I, Nagano M, Rodnina MV, Suga H. 2016. Essential structural elements in tRNAPro for EF-P-mediated alleviation of translation stalling. Nat Commun 7:11657. doi: 10.1038/ncomms11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohmae E, Miyashita Y, Kato C. 2013. Thermodynamic and functional characteristics of deep-sea enzymes revealed by pressure effects. Extremophiles 17:701–709. doi: 10.1007/s00792-013-0556-2. [DOI] [PubMed] [Google Scholar]
- 30.Stone CM, Butt LE, Bufton JC, Lourenco DC, Gowers DM, Pickford AR, Cox PA, Vincent HA, Callaghan AJ. 2017. Inhibition of homologous phosphorolytic ribonucleases by citrate may represent an evolutionarily conserved communicative link between RNA degradation and central metabolism. Nucleic Acids Res 45:4655–4666. doi: 10.1093/nar/gkx114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vander Wauven C, Piérard A, Kley-Raymann M, Haas D. 1984. Pseudomonas aeruginosa mutants affected in anaerobic growth on arginine: evidence for a four-gene cluster encoding the arginine deiminase pathway. J Bacteriol 160:928–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casiano-Colón A, Marquis RE. 1988. Role of the arginine deiminase system in protecting oral bacteria and an enzymatic basis for acid tolerance. Appl Environ Microbiol 54:1318–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bourdineaud JP, Heierli D, Gamper M, Verhoogt HJ, Driessen AJ, Konings WN, Lazdunski C, Haas D. 1993. Characterization of the arcD arginine:ornithine exchanger of Pseudomonas aeruginosa. Localization in the cytoplasmic membrane and a topological model. J Biol Chem 268:5417–5424. [PubMed] [Google Scholar]
- 34.Corratgé-Faillie C, Jabnoune M, Zimmermann S, Very AA, Fizames C, Sentenac H. 2010. Potassium and sodium transport in non-animal cells: the Trk/Ktr/HKT transporter family. Cell Mol Life Sci 67:2511–2532. doi: 10.1007/s00018-010-0317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wingreen NS, Huang KC. 2015. Physics of intracellular organization in bacteria. Annu Rev Microbiol 69:361–379. doi: 10.1146/annurev-micro-091014-104313. [DOI] [PubMed] [Google Scholar]
- 36.Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Degnan PH, Moran NA. 2008. Diverse phage-encoded toxins in a protective insect endosymbiont. Appl Environ Microbiol 74:6782–6791. doi: 10.1128/AEM.01285-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phelps HA, Neely MN. 2007. SalY of the Streptococcus pyogenes lantibiotic locus is required for full virulence and intracellular survival in macrophages. Infect Immun 75:4541–4551. doi: 10.1128/IAI.00518-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rawlings ND, Barrett AJ, Finn R. 2016. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 44:D343–D350. doi: 10.1093/nar/gkv1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi I. 2001. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res 29:3742–3756. doi: 10.1093/nar/29.18.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson GG, Murray NE. 1991. Restriction and modification systems. Annu Rev Genet 25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
- 42.Dupuis MÈ, Villion M, Magadan AH, Moineau S. 2013. CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat Commun 4:2087. doi: 10.1038/ncomms3087. [DOI] [PubMed] [Google Scholar]
- 43.Kaufenstein M, van der Laan M, Graumann PL. 2011. The three-layered DNA uptake machinery at the cell pole in competent Bacillus subtilis cells is a stable complex. J Bacteriol 193:1633–1642. doi: 10.1128/JB.01128-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen I, Dubnau D. 2004. DNA uptake during bacterial transformation. Nat Rev Microbiol 2:241–249. doi: 10.1038/nrmicro844. [DOI] [PubMed] [Google Scholar]
- 45.Plotieau T, Lavitra T, Gillan DC, Eeckhaut I. 2013. Bacterial diversity of the sediments transiting through the gut of Holothuria scabra (Holothuroidea; Echinodermata). Mar Biol 160:3087–3101. doi: 10.1007/s00227-013-2297-2. [DOI] [Google Scholar]
- 46.Danovaro R, Dell'Anno A, Corinaldesi C, Magagnini M, Noble R, Tamburini C, Weinbauer M. 2008. Major viral impact on the functioning of benthic deep-sea ecosystems. Nature 454:1084–1087. doi: 10.1038/nature07268. [DOI] [PubMed] [Google Scholar]
- 47.Roberts D, Gebruk A, Levin V, Manship BAD. 2000. Feeding and digestive strategies in deposit-feeding holothurians. Oceanogr Mar Biol 38:257–310. [Google Scholar]
- 48.Gao F, Li F, Tan J, Yan J, Sun H. 2014. Bacterial community composition in the gut content and ambient sediment of sea cucumber Apostichopus japonicus revealed by 16S rRNA gene pyrosequencing. PLoS One 9:e100092. doi: 10.1371/journal.pone.0100092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lo WS, Huang YY, Kuo CH. 2016. Winding paths to simplicity: genome evolution in facultative insect symbionts. FEMS Microbiol Rev 40:855–874. doi: 10.1093/femsre/fuw028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Somero GN. 1992. Adaptations to high hydrostatic pressure. Annu Rev Physiol 52:557–577. doi: 10.1146/annurev.ph.54.030192.003013. [DOI] [PubMed] [Google Scholar]
- 51.Woolfit M, Iturbe-Ormaetxe I, McGraw EA, O'Neill SL. 2009. An ancient horizontal gene transfer between mosquito and the endosymbiotic bacterium Wolbachia pipientis. Mol Biol Evol 26:367–374. doi: 10.1093/molbev/msn253. [DOI] [PubMed] [Google Scholar]
- 52.Nakabachi A, Yamashita A, Toh H, Ishikawa H, Dunbar HE, Moran NA, Hattori M. 2006. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science 314:267. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- 53.Savin KW, Cocks BG, Wong F, Sawbridge T, Cogan N, Savage D, Warner S. 2010. A neurotropic herpesvirus infecting the gastropod, abalone, shares ancestry with oyster herpesvirus and a herpesvirus associated with the amphioxus genome. Virol J 7:308. doi: 10.1186/1743-422X-7-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Horvath P, Barrangou R. 2010. CRISPR/Cas, the immune system of Bacteria and Archaea. Science 327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 55.Taniguchi T, Miyauchi K, Nakane D, Miyata M, Muto A, Nishimura S, Suzuki T. 2013. Decoding system for the AUA codon by tRNAIle with the UAU anticodon in Mycoplasma mobile. Nucleic Acids Res 41:2621–2631. doi: 10.1093/nar/gks1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valencia-Sánchez MI, Rodríguez-Hernández A, Ferreira R, Santamaría-Suárez HA, Arciniega M, Dock-Bregeon A-C, Moras D, Beinsteiner B, Mertens H, Svergun D, Brieba LG, Grøtli M, Torres-Larios A. 2016. Structural insights into the polyphyletic origins of glycyl tRNA synthetases. J Chem Biol 291:14430–14446. doi: 10.1074/jbc.M116.730382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harwood CR, Cranenburgh R. 2008. Bacillus protein secretion: an unfolding story. Trends Microbiol 16:73–79. doi: 10.1016/j.tim.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 58.Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, Jung K. 2013. Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 339:82–85. doi: 10.1126/science.1228985. [DOI] [PubMed] [Google Scholar]
- 59.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175. In Stackebrandt E, Goodfellow M (ed), Nucleic acid techniques in bacterial systematics. John Wiley and Sons, New York, NY. [Google Scholar]
- 60.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:141–145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patel RK, Jain M. 2012. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao F, Zhang CT. 2008. Ori-Finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinformatics 9:79. doi: 10.1186/1471-2105-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krogh A, Brown M, Mian IS, Sjölander K, Haussler D. 1994. Hidden Markov models in computational biology. J Mol Biol 235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 67.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/MAP format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eisen MB, Spellman PT, Brown PO, Botstein D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Danchin A, Fang G. 2016. Unknown unknowns: essential genes in quest for function. Microb Biotechnol 9:530–540. doi: 10.1111/1751-7915.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 74.Hill MO. 1974. Correspondence analysis: a neglected multivariate method. J R Stat Soc Ser C Appl Stat 23:340–354. doi: 10.2307/2347127. [DOI] [Google Scholar]
- 75.Pascal G, Medigue C, Danchin A. 2006. Persistent biases in the amino acid composition of prokaryotic proteins. Bioessays 28:726–738. doi: 10.1002/bies.20431. [DOI] [PubMed] [Google Scholar]
- 76.Riley M, Staley JT, Danchin A, Wang TZ, Brettin TS, Hauser LJ, Land ML, Thompson LS. 2008. Genomics of an extreme psychrophile, Psychromonas ingrahamii. BMC Genomics 9:210. doi: 10.1186/1471-2164-9-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Silvestro D, Michalak I. 2012. raxmlGUI: a graphical front-end for RAxML. Org Div Evol 12:335–337. doi: 10.1007/s13127-011-0056-0. [DOI] [Google Scholar]
- 80.Leclercq S, Dittmer J, Bouchon D, Cordaux R. 2014. Phylogenomics of “Candidatus Hepatoplasma crinochetorum,” a lineage of Mollicutes associated with noninsect arthropods. Genome Biol Evol 6:407–415. doi: 10.1093/gbe/evu020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, Le Fèvre F, Longin C, Mornico D, Roche D, Rouy Z, Salvignol G, Scarpelli C, Thil Smith AA, Weiman M, Médigue C. 2013. MicroScope—an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res 41:D636–D647. doi: 10.1093/nar/gks1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.