

Abstract
Cryogenic electron microscopy (cryo‐EM) and single‐particle analysis now enables the determination of high‐resolution structures of macromolecular assemblies that have resisted X‐ray crystallography and other approaches. We developed the SIMPLE open‐source image‐processing suite for analysing cryo‐EM images of single‐particles. A core component of SIMPLE is the probabilistic PRIME algorithm for identifying clusters of images in 2D and determine relative orientations of single‐particle projections in 3D. Here, we extend our previous work on PRIME and introduce new stochastic optimization algorithms that improve the robustness of the approach. Our refined method for identification of homogeneous subsets of images in accurate register substantially improves the resolution of the cluster centers and of the ab initio 3D reconstructions derived from them. We now obtain maps with a resolution better than 10 Å by exclusively processing cluster centers. Excellent parallel code performance on over‐the‐counter laptops and CPU workstations is demonstrated.
Keywords: Cryo‐EM, single‐particle analysis, ab initio 3D reconstruction, clustering
Abbreviations
- 2D
2‐dimensional
- 3D
3‐dimensional
- A
Ångström unit (10−10 m)
- Ca
calcium
- Cn
rotational point‐group symmetry (order n)
- CPU
central processing unit
- Cryo‐EM
cryogenic electron microscopy
- CTF
contrast transfer function (of the electron microscope)
- EM
electron microscopy
- EO
extremal optimization
- Fglobal
fraction of search space scanned for the entire particle ensemble
- Fparticle
fraction of search space scanned per‐particle
- GPU
graphics processing unit
- MPI
message passing interface
- MRA
multi reference alignment
- MRC
Medical Research Council (EM file format)
- PBS
portable batch system
- PRIME
probabilistic initial model generation for electron microscopy
- PRIME2D
PRIME for simultaneous 2D alignment and clustering
- PRIME3D
PRIME for ab initio 3D reconstruction
- Rrate
Randomization rate
- SAC
Simultaneous 2D alignment and clustering
- SGE
Sun grid engine (now Oracle Grid Engine)
- SHC
Stochastic hill‐climbing
- SIMPLE
single‐particle image processing Linux engine
- SLURM
simple Linux utility for resource management
- SNHC
stochastic neighbourhood hill‐climbing
- SNR
signal to noise ratio
- SPIDER
system for processing image data from electron microscopy and related fields (EM file format)
- Textremal
temperature (used in annealing)
- TRPA1
transient receptor Potential cation channel, subfamily A, member 1
- TRPV1
transient receptor potential cation channel subfamily V member 1
Introduction
High‐resolution imaging of biological macromolecules with an electron microscope requires embedding of the single‐particles in vitreous ice, keeping the particles hydrated in a near‐native state.1 Generation of an accurate 3D density map from large sets of single‐particle cryo‐EM projection images is challenging due to the low SNR, the many parameters that need to be determined in an unsupervised manner and the large risk of getting trapped in local optima. Despite the mathematical elegance of single‐algorithm solutions to ab initio single‐particle 3D reconstruction,2, 3, 4 real world data sets are generally too challenging for one‐step approaches to be successful. Multi‐protein complexes often partially unfold when they interact with the air‐water interface during specimen preparation, they form micro‐aggregations because of insufficient solubility, or they are mistaken for ice contaminations by automatic particle identification procedures. These factors, and many more,5 contribute to single‐particle data sets being intrinsically heterogeneous. Hence, the first task in single‐particle image processing is to identify the particle images we are interested in analysing further in terms of structure, dynamics and composition. Clustering algorithms therefore play a pivotal role in initial data quality assessment. However, referring to the problem of identifying particle subsets that are homogeneous in projection direction and structure as a bona fide clustering problem is misleading, since it is non‐trivial to define a function that computes a distance between two images. Prior 2D registration6 or generation of rotation and shift invariant representations would be required,7 since direct distance measures depend on the random in‐plane rotation and random rotational origin offsets of the single‐particle projections. Accurate registration of image pairs or generation of good invariant representations cannot readily be accomplished because of the high level of noise. A common alternative approach is to view the problem of simultaneous 2D alignment and clustering (SAC) as a statistical parameter optimization problem.8, 9, 10, 11 Developing fast and robust algorithms for solving SAC is important because:
Large data sets need to be rapidly processed in 2D to assess their amenability to high‐resolution 3D reconstruction.
Contaminating images of, for example, ice or particle micro‐aggregates often constitute a significant portion of a data set because of errors in particle identification. These images need to be identified and removed.
Identifying highly homogeneous subsets of images in accurate register allows averaging to improve the SNR, which is important for the subsequent steps of ab initio 3D reconstruction10 and structural heterogeneity analysis.12, 13
The dimensionality reduction provided by solving SAC is ∼100‐fold, which enables rapid downstream analysis of the resulting cluster centers (often referred to as 2D class averages) on lightweight computer architectures, such as workstations or laptops.
One of the earliest proposed general approaches for solving SAC was the multi‐reference alignment (MRA) algorithm.14, 15 Most cryo‐EM image‐processing packages13, 16, 17, 18, 19, 20, 21 implement their own variant of MRA, deploying a k‐means‐like strategy that involves randomized initialization of cluster centers, followed by iterative greedy local‐search‐based optimization of cluster assignments and in‐plane rotations.
We recently developed a new SAC solver based on formulation of the multi‐reference alignment problem as a combinatorial optimization problem.10 We applied stochastic hill‐climbing (SHC)22 to estimate the parameters subject to optimization: cluster assignments, in‐plane rotations and rotational origin shifts. In the previous study, we showed that our SHC‐based SAC solver overcomes inherent limitations of other commonly used center‐based clustering approaches. However, it remains an open question of how far SAC solvers can be improved in terms of the resolution of the cluster centers obtained and the ability to resolve structural heterogeneity arising from conformational or compositional variations.
We introduce a probabilistic single‐particle projection alignment framework that includes CTF‐dependent Wiener restoration23 and implements new stochastic optimization methods with improved orientation search diversity. We invested significant efforts into optimizing the parallel CPU code and provide benchmarks on several experimental data sets on different computer architectures. Data sets of realistic size can now be processed on lightweight workstations or laptops. Our implementation is simple, efficiently parallelized, and improves the resolution of the cluster centers and the ab initio 3D reconstructions derived from them to subnanometer resolution.
Algorithms
Improved simultaneous 2D alignment and clustering with PRIME2D
The PRIME2D algorithm has three components: (1) an initialization step that selects a random subset of the analysed images as initial references, (2) a stochastic search step that updates the parameters subject to optimization (cluster assignments, in‐plane rotations and rotational origin shifts) and (3) a cluster center update step (a mathematical description of the cluster center is provided in Supporting Information Section 1). Steps (2) and (3) are iterated until the parameter assignments are consistent between successive iterations. Our improvements to PRIME2D include a new stochastic search approach described below, an accelerated algorithm for matching particle images with cluster centers (see Supporting Information Section 2) and re‐implementation of a standard approach for CTF correction (briefly described in Supporting Information Section 1).
A problem with iterative center‐based approaches for solving SAC is that decisions made early in the search will strongly influence later decisions. The effects of poor initial choices may be propagated all the way to the final solution. This causes the final result to strongly depend on the initialization condition and only convergence to a local optimum is guaranteed. In attempt to overcome these issues, we designed a new stochastic optimization approach for solving SAC. Consider optimization problems where each solution element can be assigned an individual score. In our case, a correlation‐based score is associated with each particle image (Supporting Information Section 2, Equations 8 and 9). It has been shown that random update of the worst scoring elements, without any improvement through search, cause all individuals to reach fitness values above a certain threshold.24 This so‐called “Extremal Optimization” (EO) approach provides means for wide exploration of a configuration space and has been applied to solve difficult combinatorial problems.25, 26, 27 However, direct application of EO to SAC would be computationally inefficient. Therefore, we designed a hybrid approach that combines EO with our previous stochastic local search method (SHC, refs 4, 10 & Supporting Information Section 2, Equation 16), leveraging the diversity of EO and its capability to continue the search beyond a local optimum in a computationally efficient manner. We define a temperature (T extremal) and a randomization rate (R rate) to anneal the temperature throughout the search via T(t)extremal = T(t – 1)*R rate where t denotes iteration number [see Fig. 1(B)], analogous to simulated annealing.28 T extremal = 0.5 causes half of the least fit images to be subjected to extremal rather than SHC‐based update, where the extremal update is defined as
Figure 1.
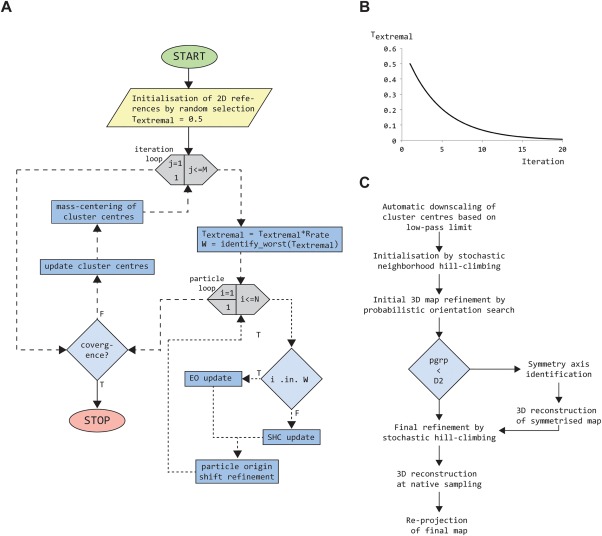
Flowcharts for PRIME2D/PRIME3D. (A) Flowchart for the PRIME2D hybrid EO/SHC SAC solver. Loop constructs are gray hexagons, computations blue rectangles and decisions light blue diamonds. (B) Extremal temperature (Textremal), controlling the rate of EO vs. SHC update, plotted as a function of iteration number. (C) PRIME3D workflow for analysis of cluster centers
Assign the particle image a random cluster label so that the label must change.
Optimize in‐plane parameters by exhaustive search.
Accept the new parameter assignment unconditionally.
The annealing causes the EO update rate to be high initially (50%), when the need to overcome bias is high, and low toward the end of a run when the SAC solution needs refinement rather than re‐shaping. Balancing EO and SHC in this way allows rapid identification of a near‐optimal SAC solution. The SHC‐based update is done as previously described by us10 (see also Supporting Information Section 2). An initial temperature of T extremal = 0.5 and randomization rate of R rate = 0.8 worked well for all data sets we have processed so far. Figure 1(A) provides a schematic representation of the PRIME2D algorithm.
We implemented an automated PRIME2D workflow that takes care of the initialization and automatically downscales the images based on the input low‐pass limit range. The search is divided into two stages. The first stage uses only low‐resolution information (typically 20 Å) and highly down‐sampled images. This substantially accelerates performance, since the computations grow with the square of the image size. In the second stage, the low‐pass limit and degree of downscaling are updated. The final cluster centers are calculated at the original sampling and ranked according to decreasing population.
Improved analysis of cluster centers of cn symmetric molecules with PRIME3D
We have previously demonstrated that PRIME3D provides a robust solution to the ab initio 3D reconstruction problem4, 10 and the algorithm has been used in the hands of others to generate starting models that were subsequently refined to near‐atomic resolution.29, 30, 31, 32, 33, 34 PRIME3D can be used to obtain 3D reconstructions directly from the noisy individual images. However, a faster route that provides better success/failure statistics is to first cluster the data with PRIME2D to generate cluster centers that are subjected to PRIME3D analysis. Asymmetric molecules (ribosomes, spliceosomes and so forth) and molecules with point‐group symmetries equal to or higher than D2 (β‐galactosidase, proteasome, GroEL, and so forth) are easy to reconstruct. We have found Cn‐symmetric particles to be the most difficult targets. Although it is possible to constraint the orientation search from the start according to the input point‐group symmetry, we typically start off assuming no symmetry and then analyse the converged asymmetric map to identify the correct point‐group and principal symmetry axis orientation.35 However, this approach sometimes fails for Cn symmetric single‐particles due to incorrect identification of the symmetry axis. A brute force method to overcome this problem would be to restart the procedure, which is a practical but costly solution to the problem. We hypothesized that Cn‐symmetries do not constraint the orientation search enough to provide any useful regularization of the inverse problem. In support of this notion, asymmetric reconstructions of dihedral, tetrahedral, octahedral and icosahedral molecules come out almost perfectly symmetrical, whereas asymmetric maps of Cn‐symmetric molecules can be distorted due to the orientations tending to cluster within a single asymmetric unit rather than spreading evenly over the point‐group. We therefore designed a new stochastic optimiser—stochastic neighborhood hill climbing (SNHC)—to overcome this bias, resulting in a success rate of 97% over 60 test runs (see Results). Briefly, SNHC defines a small stochastic neighborhood per cluster center, consisting of five projection directions randomly sampled from the entire discrete search space of 1000 directions. The stochastic neighborhood is evaluated exhaustively and the best orientation found is assigned to the cluster center, whether or not it improves the correlation vs. the previous orientation. This approach is more than an order of magnitude faster than the original PRIME3D approach. Large fluctuations ensure that many local optima are being explored. The extreme diversity of SNHC ensures rapid identification of a low‐resolution molecular shape. We use SNHC in replacement of random initialization of PRIME3D to improve the success rate for Cn‐symmetric single‐particles. We implemented an automated PRIME3D workflow for analysing cluster centers obtained with PRIME2D (executed via simple_distr_exec prg = ini3D_from_cavgs), outlined in Figure 1(C).
Resolution estimation
The parameters determined for the individual particle images by PRIME2D (cluster assignments, in‐plane rotations, origin shifts) are combined with those obtained with PRIME3D for the cluster centers (projection directions, in‐plane rotations, origin shifts) to yield per‐particle 3D orientation estimates. This step is implemented in the SIMPLE application map2ptcls. A resolution estimate is obtained by calculating even/odd 3D reconstructions and applying the FSC = 0.143 threshold criterion.36 None of the maps presented below were obtained by 3D refinement of the individual particle images—the analysis was done exclusively using cluster centers and the resolution was estimated following mapping of the orientation parameters back to the particle images.
Shared‐memory parallelization
One PRIME2D iteration consists of (1) update of the parameters subject to optimization followed by (2) update of the cluster centers. Step (1) represents the majority of the computations but can be done for every particle image independently (“embarrassingly parallel” problem). We encapsulate the entire stochastic parameter update procedure in an abstract data type and declare an array with one search object per particle image. Shared‐memory parallelization is applied to the loop over particle indices to allow the reference and particle images to be shared across CPUs. Since the time taken to process one particle image is stochastic, we use a scheduling scheme where large batches are assigned to each CPU initially and, as the loop progresses, the batches decrease in size to allow efficient load balancing. The cluster center update (Step 2) also involves significant computations (rotation of the images with convolution interpolation,37 CTF multiplication, origin shifting and summation). For each cluster in sequence, we identify the indices of the particle members and create load‐balanced batches of images (one batch per available CPU) so that the update of the cluster centers can be executed efficiently in parallel.
Multi‐user distributed computing environments
We developed our own tool for distributed‐memory parallelization, supporting common cluster queuing systems (PBS, SLURM and SGE) in addition to local execution on multi‐socket workstations. In multi‐user environments, this tool has distinct advantages over codes based on the message‐passing interface (MPI). Instead of requesting a large number of CPU cores for the entire duration of the program execution, the computations associated with individual PRIME2D/PRIME3D iterations are divided into balanced partitions and submitted as individual jobs to the queuing system. If there are multiple SIMPLE users on the same cluster, there is no risk that any single user uptakes more time than it takes to complete a single PRIME2D/PRIME3D iteration, before a user competing for the same resources is given chance to access. There is a slight overhead (matter of seconds) with this mode of execution, since the assembly of cluster centers from partitions (PRIME2D) and the assembly of a 3D reconstruction from partial volumes (PRIME3D) are done serially. However, this short delay has the advantage of freeing the computer resources so that other users may gain access. The best resource utilization when using SIMPLE on distributed computer systems is accomplished in multi‐user environments where many users compete for the same machines. The PRIME2D and PRIME3D cluster center analyses are executed as follows in a distributed computing environment:
where prg is the program name, stk is the input image stack (MRC or SPIDER format), smpd is the sampling distance of the images (in Å), msk is the Gaussian mask radius in pixels, ncls is the number of clusters, ctf is the CTF status flag; yes meaning images have CTF, flip meaning images have been pre‐corrected with phase‐flipping, no meaning images have no CTF, deftab is a text‐file with CTF parameters, nparts is the number of partitions to divide the job into and pgrp is the point‐group symmetry.
Results
Benchmarking on over‐the‐counter computers
We tested the PRIME2D/PRIME3D approach using data sets available at the Electron Microscopy Public Image ARchive (EMPIAR). All data characteristics are described online.38 The first two benchmarks were executed on a laptop (MacBook Pro mid 2015, 2.8 GHz Intel i7, four physical cores) using 5000 images randomly selected from larger data sets (Figs. 2 and 3). Already after ∼10 PRIME2D iterations (wall‐clock time of ∼40 minutes for the proteasome vs. ∼20 minutes for β‐galactosidase), we saw evidence of projected secondary structure elements in the cluster centers (100 centers in total). The proteasome reconstruction obtained from cluster centers (Fig. 2) had better resolution than the β‐galactosidase map (Fig. 3) because the effective number of images is 50,000 (D7 point‐group symmetry) vs. 20,000 for β‐galactosidase (D2 point‐group symmetry). PRIME2D achieves a 50‐fold dimensionality reduction (from 25,000 to 500 free parameters) in these tests. Therefore, the following PRIM3D step can be executed rapidly (∼10 minutes) to generate an initial 3D volume. PRIME3D automatically updates the low‐pass limit based on search statistics and the final limit is set to 10 Å by default. That we obtain resolutions (following the FSC = 0.143 criterion36) that extend beyond the hard low‐pass limit is reassuring and reflects the quality of cluster centers obtained. We further benchmarked the approach on additional data sets; the results are summarized in Figures 4 and 5. In all tests, we obtained maps with a resolution better than 10 Å (8.0–9.6 Å) in less than two hours wall‐clock time on over‐the‐counter machines that cost less than 2000 USD. Cluster centers produced without the use of a 3D reference volume in other widely used program packages17, 18, 19, 20, 21, 39, 40 have not been demonstrated to yield maps with a resolution better than 10 Å.
Figure 2.
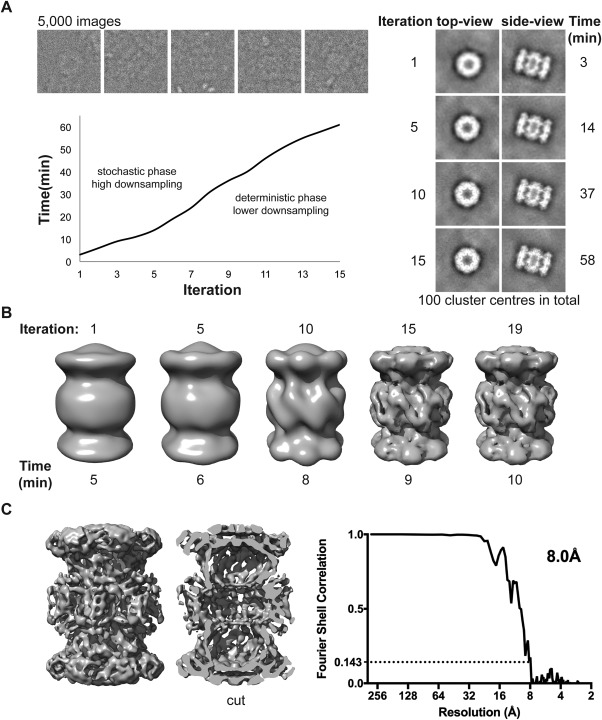
Benchmark on a laptop (MacBook Pro mid 2015, 2.8 GHz Intel i7, four physical cores) using 5000 images of the proteasome extracted from a larger data set (EMPIAR‐10025): (A) Grouping of the 5000 images (box = 224) into 100 clusters with PRIME2D. The left panel shows the evolution of two cluster centers throughout the stochastic search. The right panel shows examples of the experimental images (top) and the time per iteration (bottom). (B) Ab initio 3D reconstruction from cluster centers obtained in 10 minutes on a two‐year‐old laptop. (C) Map obtained by mapping the orientations obtained for cluster centers back to the particle images (left) and Fourier Shell Correlation plot (FSC) (right), demonstrating a resolution of 8.0 Å
Figure 3.
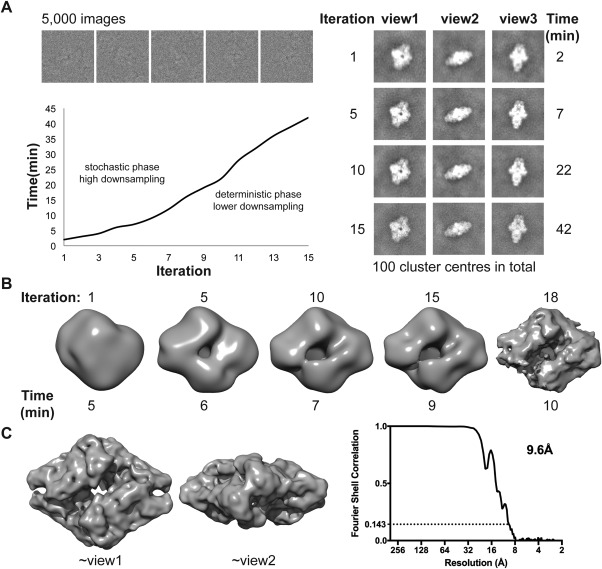
Benchmark on a laptop (MacBook Pro mid 2015, 2.8 GHz Intel i7, four physical cores) using 5000 images of beta‐galactosidase extracted from a larger data set (EMPIAR‐10013): (A) Grouping of the 5000 images (box = 256) into 100 clusters. The left panel shows the evolution of three cluster centers throughout the stochastic search. The right panel shows examples of the experimental images (top) and the time per iteration (bottom). (B) Asymmetric ab initio 3D reconstruction from cluster centers obtained in 10 minutes on a two‐year‐old laptop. (C) Symmetrized map obtained by mapping the orientations obtained for cluster centers back to the particle images (left) and Fourier Shell Correlation (FSC) plot (right), demonstrating a resolution of 9.6 Å
Figure 4.
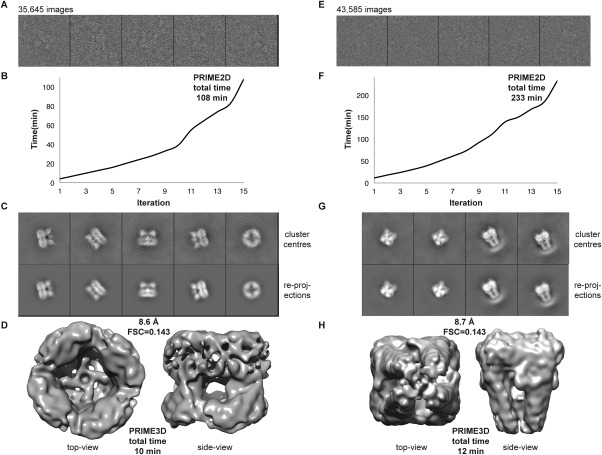
Benchmark on a workstation (3.0 Ghz Intel i7, eight physical cores) using, 35,645 images of TRPV1 (EMPIAR‐10005; left panel) and 43,585 images of TRPA1 (EMPIAR‐10024; right panel): (A, E) Sample images from the data set. (B, F) Wall‐clock time as a function of PRIME2D iteration number. (C, G) Cluster centers (top) and corresponding re‐projections of the ab initio maps (bottom). (D, H) Top‐ (left) and side‐view (right) of the density maps obtained with PRIME3D from cluster centers. The resolutions obtained are 8.6 Å (TRPV1) and 8.7 Å (TRPA1)
Figure 5.
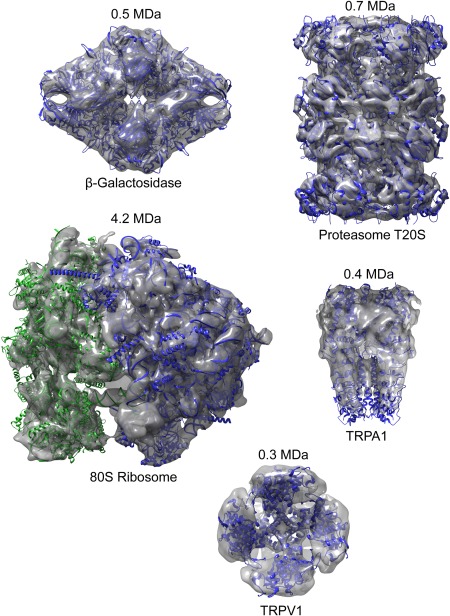
A gallery of ab initio maps obtained from cluster centers with the improved PRIME2D/PRIME3D approach
Robustness analysis of PRIME3D when applied to cn symmetric molecules
We repeated our improved PRIME3D analysis 20 times on cluster centers derived from three data sets that were difficult targets for our previous approach.4 The three data sets analysed were all of C4 symmetric membrane receptors: (1) the calcium release channel IP3R41 (kindly shared by Steven Ludtke), (2) TRPA1 (EMPIAR‐10024), and (3) TRPV1 (EMPIAR‐10005). We obtained success rates of (1) 100%, (2) 100%, and (3) 90%. Figure 6 shows the spatial median map and the map furthest from the spatial median map for each of the tests, illustrating the variance of the map distribution for the set of successful runs. In conclusion, the PRIME3D approach is robust (failure rate of 3% over 60 tests) but when it fails, it fails dramatically [see example in Fig. 6(C)]. The 2/20 TRPV1 failures are caused by the rotational symmetry axis being incorrectly identified because of a pseudo 4‐fold axis orthogonal to the true axis. We therefore recommend users to restart the procedure a few times and compare the results obtained, which is not a huge effort as it takes roughly ten minutes.
Figure 6.
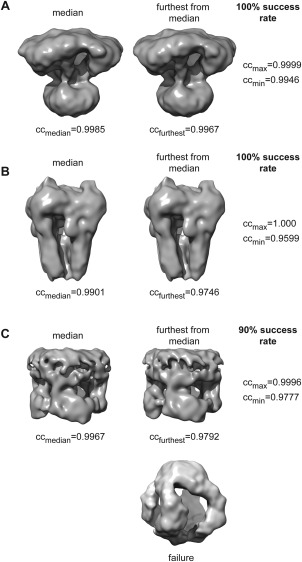
Stress‐testing PRIME3D. The three panels (A–C) show (from left to right) the median map of the successful tests, the map furthest from the median, and the success rate for data sets: (A) the calcium release channel IP3R, (B) TRPA1 (EMPIAR‐10024), and (C) TRPV1 (EMPIAR‐10005). ccmedian indicates the average correlation between the median map and all others in the set of successful runs, ccfurthest indicates the average correlation between the map furthest from the median and all other maps in the set, ccmax is the maximum pairwise volume‐to‐volume correlation identified and ccmin is the minimum pairwise volume‐to‐volume correlation identified
Discussion
Because of the rapidly increasing popularity of cryo‐EM techniques and the many newcomers to the field, it is important that we address the outstanding problems in data analysis. We have identified five areas that need further investigation/development:
Reproducibility and comparative analysis of existing algorithms.
Development of new robust image‐processing methods.
Reducing execution speed and computational requirements.
Automation and creation of high‐level workflows that are less prone to user mistakes and can be stitched together to create protocols.
Development of hybrid orientation search techniques and intelligent algorithms.
Reproducibility and comparative testing is strengthened by developments like Scipion42 that integrate many different packages under a unified graphical user‐interface, allowing users to mix and match algorithms while the complete workflow is being stored so that it can be automatically re‐executed at any later point. We have integrated the developments described here within the Scipion framework (planned release August 2017) and they are available for download at http://simplecryoem.com/as part of SIMPLE 2.5.
With “robustness” we mean the ability of the algorithm to cope with erroneous input. Single‐particle cryo‐EM data sets have large errors because of the high‐level of noise of the images, the structurally heterogeneous nature of macromolecules and errors in particle identification. We previously demonstrated the robustness of the PRIME3D approach toward initialization by erroneous starting models.4 Here, we demonstrate increased convergence radius for Cn‐symmetric molecules when using SNHC to initialize PRIME3D. Restarting the algorithm many times and analysing the success/failure statistics is important during development, to identify and eliminate bottlenecks. It is also important for the user to understand what to expect, how many times the algorithm should be restarted and how much the obtained solutions will differ. We do anticipate larger map variations upon restart analysis of more challenging (i.e., conformationally heterogeneous) data sets than those analysed here. This will be a subject of future studies.
Reducing execution speed and computational requirements is essential for making the technique available to as many as possible. Computer clusters are expensive to buy and maintain and not available to all end‐users. GPU acceleration will play a central role.2, 43 However, not all algorithms are well suited for GPUs and not all computers have GPU capabilities. Therefore, it is equally important to engineer high‐performance CPU code. In this release (SIMPLE 2.5), we have focused exclusively on accelerating the CPU code via code optimization techniques such as data reorganization, pipelining and load balancing of the parallel implementation.
As the number of cryo‐EM users increase, the “synchrotron model” for data acquisition will become more common, as evidenced by the number of centralized cryo‐EM resources being established world‐wide. Enabling users to analyse a substantial subset of the images while the data is still being acquired, using their own laptops or a nearby workstation, will increase insight into the nature of the data and potentially change the way the images are being collected.
Automation is another key area that we address here by creating highly automated workflows for generating cluster centers with PRIME2D and for calculating ab initio 3D reconstructions from cluster centers with PRIME3D. A web‐based graphical user‐interface is planned for the next SIMPLE release, but even novice computer users will be able to execute the two command lines controlling PRIME2D and PRIME3D, respectively.
We believe that development of hybrid orientation search techniques and the use of intelligent algorithms will accelerate progress in the area of cryo‐EM image processing. A few mature tools implement tuneable 3D reconstruction algorithms. For example, EMAN217 implements a large number of different scoring functions; some work better for certain molecular shapes and their suitability may vary with the quality of the map during the refinement cycle. Rather than experimenting with different scoring functions, we have focused on implementing different variants of stochastic orientation search that we now try to combine in intelligent ways to accelerate the execution speed and improve the robustness of the PRIME approach. We anticipate that hybrid methods and algorithms capable of making on the fly decisions about what search procedure or scoring function to use will make single‐particle 3D reconstruction more automated, accessible and reproducible.
Conclusions
We introduced improved methods for analysis of single‐particle cryo‐EM projection images: PRIME2D for simultaneous 2D alignment and clustering and PRIME3D for ab initio 3D reconstruction. These algorithms are part of the open‐source SIMPLE image‐processing suite, version 2.5, available for download at http://simplecryoem.com/.
Supporting information
Supporting Information
Acknowledgments
Calculations were done at the Multi‐modal Australian Sciences Imaging and Visualisation Environment (MASSIVE) (www.massive.org.au). We thank Nikolaus Grigorieff and Susan Lea for stimulating discussions, Joseph Caesar for identifying critical bugs, Matthew Beloussoff for careful testing and Jose Miguel de la Rosa Trevin for integrating SIMPLE 2.5 within the Scipion integrative package.
Contributor Information
Dominika Elmlund, Email: dominika.elmlund@monash.edu.
Hans Elmlund, Email: hans.elmlund@monash.edu.
References
- 1. Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P (1988) Cryo‐electron microscopy of vitrified specimens. Q Rev Biophys 21:129–228. [DOI] [PubMed] [Google Scholar]
- 2. Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA (2017) cryoSPARC: algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods 14:290–296. [DOI] [PubMed] [Google Scholar]
- 3. Singer A, Shkolnisky Y (2011) Three‐dimensional structure determination from common lines in cryo‐EM by Eigenvectors and semidefinite programming. Siam J Imag Sci 4:543–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elmlund H, Elmlund D, Bengio S (2013) PRIME: probabilistic initial 3D model generation for single‐particle cryo‐electron microscopy. Structure 21:1299–1306. [DOI] [PubMed] [Google Scholar]
- 5. Elmlund D, Le SN, Elmlund H (2017) High‐resolution cryo‐EM: the nuts and bolts. Curr Opin Struct Biol 46:1–6. [DOI] [PubMed] [Google Scholar]
- 6. Penczek PA, Grassucci RA, Frank J (1994) The ribosome at improved resolution ‐ new techniques for merging and orientation refinement in 3D cryoelectron microscopy of biological particles. Ultramicroscopy 53:251–270. [DOI] [PubMed] [Google Scholar]
- 7. Zhao Z, Singer A (2014) Rotationally invariant image representation for viewing direction classification in cryo‐EM. J Struct Biol 186:153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scheres SHW, Valle M, Nunez R, Sorzano COS, Marabini R, Herman GT, Carazo JM (2005) Maximum‐likelihood multi‐reference refinement for electron microscopy images. J Mol Biol 348:139–149. [DOI] [PubMed] [Google Scholar]
- 9. Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA (2012) Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure 20:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reboul CF, Bonnet F, Elmlund D, Elmlund H (2016) A stochastic hill climbing approach for simultaneous 2D alignment and clustering of cryogenic electron microscopy images. Structure 24:988–996. [DOI] [PubMed] [Google Scholar]
- 11. Sorzano COS, Bilbao‐Castro JR, Shkolnisky Y, Alcorlo M, Melero R, Caffarena‐Fernandez G, Li M, Xu G, Marabini R, Carazo JM (2010) A clustering approach to multireference alignment of single‐particle projections in electron microscopy. J Struct Biol 171:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elmlund D, Davis R, Elmlund H (2010) ab initio structure determination from electron microscopic images of single molecules coexisting in different functional states. Structure 18:777–786. [DOI] [PubMed] [Google Scholar]
- 13. Elmlund D, Elmlund H (2012) SIMPLE: Software for ab initio reconstruction of heterogeneous single‐particles. J Struct Biol 180:420–427. [DOI] [PubMed] [Google Scholar]
- 14. van Heel M (1984) Multivariate statistical classification of noisy images (randomly oriented biological macromolecules). Ultramicroscopy 13:165–183. [DOI] [PubMed] [Google Scholar]
- 15. van Heel M, Stoffler‐Meilicke M (1985) Characteristic views of Escherichia‐coli and Balantidium‐coli Stearothermophilis‐30S ribosomal‐subunits in the electron‐microscope. EMBO J 4:2389–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shaikh TR, Gao H, Baxter WT, Asturias FJ, Boisset N, Leith A, Frank J (2008) SPIDER image processing for single‐particle reconstruction of biological macromolecules from electron micrographs. Nat Protoc 3:1941–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ (2007) EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157:38–46. [DOI] [PubMed] [Google Scholar]
- 18. Scheres SH (2012) RELION: implementation of a Bayesian approach to cryo‐EM structure determination. J Struct Biol 180:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de la Rosa‐Trevin JM, Oton J, Marabini R, Zaldivar A, Vargas J, Carazo JM, Sorzano CO (2013) Xmipp 3.0: an improved software suite for image processing in electron microscopy. J Struct Biol 184:321–328. [DOI] [PubMed] [Google Scholar]
- 20. Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, Ludtke SJ (2007) SPARX, a new environment for Cryo‐EM image processing. J Struct Biol 157:47–55. [DOI] [PubMed] [Google Scholar]
- 21. van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M (1996) A new generation of the IMAGIC image processing system. J Struct Biol 116:17–24. [DOI] [PubMed] [Google Scholar]
- 22. Russel SJ, Norvig P. 2003. Artificial intelligence: A modern approach. Upper Saddle River, New Jersey: Prentice Hall. [Google Scholar]
- 23. Penczek PA (2010) Image restoration in cryo‐electron microscopy. Methods Enzymol 482:35–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boettcher S, Percus AG (2001) Optimization with extremal dynamics. Phys Rev Lett 86:5211–5214. [DOI] [PubMed] [Google Scholar]
- 25. Boettcher S (2005) Extremal optimization for Sherrington‐Kirkpatrick spin glasses. Eur Phys J B 46:501–505. [Google Scholar]
- 26. Boettcher S, Percus AG (2004) Extremal optimization at the phase transition of the three‐coloring problem. Phys Rev E 69:PMID: ISI:000222502800131 {Medline} [DOI] [PubMed] [Google Scholar]
- 27. Chen YW, Lu YZ, Chen P (2007) Optimization with extremal dynamics for the traveling salesman problem. Phys Statist Mech Appl 385:115–123. [Google Scholar]
- 28. Kirkpatrick S, Gelatt CD, Vecchi MP (1983) Optimization by simulated annealing. Science 220:671–680. [DOI] [PubMed] [Google Scholar]
- 29. Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, Shi Y (2014) Three‐dimensional structure of human gamma‐secretase. Nature 512:166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park J, Elmlund H, Ercius P, Yuk JM, Limmer DT, Chen Q, Kim K, Han SH, Weitz DA, Zettl A, Alivisatos AP (2015) Nanoparticle imaging. 3D structure of individual nanocrystals in solution by electron microscopy. Science 349:290–295. [DOI] [PubMed] [Google Scholar]
- 31. Paulsen CE, Armache JP, Gao Y, Cheng Y, Julius D (2015) Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520:511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nguyen TH, Galej WP, Bai XC, Savva CG, Newman AJ, Scheres SH, Nagai K (2015) The architecture of the spliceosomal U4/U6.U5 tri‐snRNP. Nature 523:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boland A, Martin TG, Zhang Z, Yang J, Bai XC, Chang L, Scheres SH, Barford D (2017) Cryo‐EM structure of a metazoan separase‐securin complex at near‐atomic resolution. Nat Struct Mol Biol 24:414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ekiert DC, Bhabha G, Isom GL, Greenan G, Ovchinnikov S, Henderson IR, Cox JS, Vale RD (2017) Architectures of lipid transport systems for the bacterial outer membrane. Cell 169:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reboul CF, Elmlund H (2017) Single‐particle cryo‐EM—Statistical Test for Point‐group Symmetry. In preparation.
- 36. Rosenthal PB, Henderson R (2003) Optimal determination of particle orientation, absolute hand, and contrast loss in single‐particle electron cryomicroscopy. J Mol Biol 333:721–745. [DOI] [PubMed] [Google Scholar]
- 37. Yang Z, Penczek PA (2008) Cryo‐EM image alignment based on nonuniform fast Fourier transform. Ultramicroscopy 108:959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. https://www.ebi.ac.uk/pdbe/emdb/empiar/.
- 39. Ludtke SJ, Baldwin PR, Chiu W (1999) EMAN: Semiautomated software for high‐resolution single‐particle reconstructions. J Struct Biol 128:82–97. [DOI] [PubMed] [Google Scholar]
- 40. Frank J, Radermacher M, Penczek P, Zhu J, Li YH, Ladjadj M, Leith A (1996) SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol 116:190–199. [DOI] [PubMed] [Google Scholar]
- 41. Ludtke SJ, Tran TP, Ngo QT, Moiseenkova‐Bell VY, Chiu W, Serysheva II (2011) Flexible architecture of IP3R1 by Cryo‐EM. Structure 19:1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de la Rosa‐Trevin JM, Quintana A, Del Cano L, Zaldivar A, Foche I, Gutierrez J, Gomez‐Blanco J, Burguet‐Castell J, Cuenca‐Alba J, Abrishami V, Vargas J, Oton J, Sharov G, Vilas JL, Navas J, Conesa P, Kazemi M, Marabini R, Sorzano CO, Carazo JM (2016) Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J Struct Biol 195:93–99. [DOI] [PubMed] [Google Scholar]
- 43. Kimanius D, Forsberg BO, Scheres SH, Lindahl E (2016) Accelerated cryo‐EM structure determination with parallelisation using GPUs in RELION‐2. Elife 5. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information