

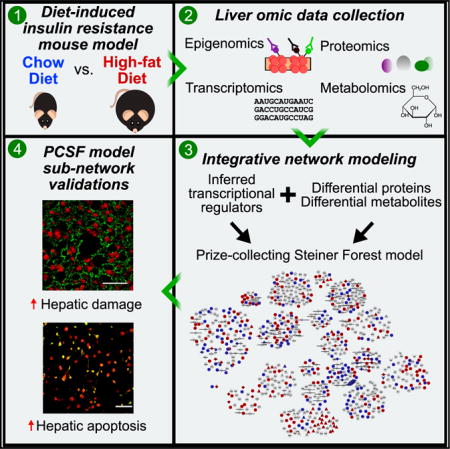
SUMMARY
Obesity is a major human health crisis that promotes insulin resistance and, ultimately, type 2 diabetes. The molecular mechanisms that mediate this response occur across many highly complex biological regulatory levels that are incompletely understood. Here, we present a comprehensive molecular systems biology study of hepatic responses to high-fat feeding in mice. We interrogated diet-induced epigenomic, transcriptomic, proteomic, and metabolomic alterations using high-throughput omic methods and used a network modeling approach to integrate these diverse molecular signals. Our model indicated that disruption of hepatic architecture and enhanced hepatocyte apoptosis are among the numerous biological processes that contribute to early liver dysfunction and low-grade inflammation during the development of diet-induced metabolic syndrome. We validated these model findings with additional experiments on mouse liver sections. In total, we present an integrative systems biology study of diet-induced hepatic insulin resistance that uncovered molecular features promoting the development and maintenance of metabolic disease.
Graphical abstract
INTRODUCTION
Human obesity is a major worldwide health crisis (Flegal et al., 2013) that promotes metabolic syndrome (characterized by insulin resistance, hyperglycemia, and hypertension) (Lusis et al., 2008), β-cell dysfunction, and, ultimately, type 2 diabetes (Kahn et al., 2006). The liver is an insulin-sensitive organ that is critical for the maintenance of normal glucose homeostasis (Michael et al., 2000). Insulin promotes increased uptake of glucose in peripheral tissues (primarily skeletal muscle) and reduces hepatic gluconeogenesis (DeFronzo et al., 1985). Insulin resistance suppresses these normal regulatory mechanisms and, thus, promotes hyperglycemia. Consumption of a high-fat diet (HFD) causes insulin resistance, which prevents insulin-mediated inhibition of hepatic gluconeogenesis (Pilkis and Granner, 1992). Moreover, peripheral insulin resistance (e.g., in adipose tissue) causes increased lipolysis that promotes hepatic gluconeogenesis (Perry et al., 2015; Titchenell et al., 2015, 2016). The critical role of the liver in glycemic regulation is particularly highlighted by the widespread use of the drug metformin to treat type 2 diabetes, which principally acts in the liver to inhibit gluconeogenesis and reduce plasma triglyceride levels (Viollet and Foretz, 2013). Thus, understanding the molecular mechanisms of hepatic insulin resistance may provide a basis for the design of therapeutic interventions.
The intracellular pathways that promote and maintain insulin resistance and type 2 diabetes are highly complex and still not fully understood. Perplexingly, diabetics experience “selective insulin resistance” whereby insulin fails to suppress hepatic glucose production but still promotes hepatic lipogenesis (Brown and Goldstein, 2008; Shimomura et al., 2000; Titchenell et al., 2016). Surprisingly, triple-knockout Akt1/Akt2/Foxo1 and double-knockout Insr/Foxo1 mice still suppress hepatic glucose production in response to insulin (Lu et al., 2012; Perry et al., 2015; Titchenell et al., 2015). As a result, systems biology approaches are increasingly being recognized as vital to the study of metabolic diseases (Zhao et al., 2015). Systems biology embraces the inherent complexities of disease and draws upon the wealth of available knowledge from molecular biology and biochemistry to facilitate comprehensive, multi-dimensional analysis and modeling of disease-relevant systems and processes (Kitano, 2002).
Available omic technologies enable rapid and comprehensive analysis of many biological regulatory levels. Epigenomic and transcriptomic methodologies (e.g., chromatin immunoprecipitation sequencing [ChIP-seq] and mRNA sequencing [mRNA-seq]) rapidly profile full genomic regulatory and gene expression landscapes (Metzker, 2010). Proteomic analysis via mass spectrometry is increasingly becoming more sensitive and comprehensive, allowing for detailed analysis of global and modified proteomes (Azimifar et al., 2014). Metabolomics, the collective study of small-molecule species, is now being used extensively to identify new mechanisms and biomarkers of metabolic disease in both targeted and untargeted fashions (Dunn et al., 2011).
Few studies, to date, have attempted to analyze and integrate multiple types of omic data in the context of diet-induced metabolic disease. Those that have used simple correlative statistics (Miraldi et al., 2013; Oberbach et al., 2011), overlaid proteomic and metabolomic data onto known pathways with genome-scale metabolic reconstructions (Yizhak et al., 2010), or combined transcriptomic and metabolomic data with known pathway and regulatory data for analysis within local interaction neighborhoods (Eckel-Mahan et al., 2013). By contrast, we integrate matched multi-omic data into a tractable network model that is not biased toward interactions occurring in well-established signaling or metabolic pathways. Instead, we collate diverse types of interactions from databases of literature-curated and high-throughput data to build a large network of physical associations. We then use advanced network optimization methods to prune the possible interaction space to only the most relevant connections that model the input data. Our results are, thus, more interpretable and provide clearer directions for follow-up studies.
We present a comprehensive integrative analysis of high-fat-diet (HFD)-induced hepatic insulin resistance in the mouse liver. We fed male C57BL/6J mice a 16-week HFD to induce obesity and insulin resistance and compared these animals to normal chow-diet (CD)-fed controls. We collected genome-wide epigenomic data using histone modification ChIP-seq to interrogate active genomic regulatory regions, performed mRNA-seq to quantify hepatic transcriptomes, utilized an untargeted shotgun proteomic profiling methodology to quantify >6,000 hepatic proteins, and quantified nearly 400 small molecules to interrogate molecular responses to high-fat feeding. We identified genes, proteins, and metabolites altered between CD and HFD and jointly analyzed our epigenomic and transcriptomic data to predict transcriptional regulators that likely influence gene expression changes between the diets. We then developed a network modeling approach based on the prize-collecting Steiner forest (PCSF) algorithm (Tuncbag et al., 2013, 2016) to analyze all the omic data in the context of known protein-protein and protein-metabolite interactions. For this purpose, we constructed a vast interactome of such associations and developed computational methods to avoid biases from well-studied, highly connected proteins and metabolites. The PCSF model revealed a richly interconnected network of biological species and processes perturbed by HFD that could be divided into functional sub-networks. This analysis uncovered well-established features of hepatic insulin resistance, including glucose, lipid, and amino acid metabolism. Importantly, it also revealed poorly characterized aspects of the condition, including hepatocellular injury, cell-cell interactions, extracellular matrix (ECM) organization, and apoptosis. Finally, we validated several network modeling predictions with additional experiments on frozen liver sections from CD and HFD livers. We showed that HFD feeding leads to disrupted hepatic architecture and tight junctions, altered bile acid handling, and enhanced cellular apoptosis.
RESULTS
High-Fat-Diet Feeding Induces Obesity and Insulin Resistance in Mouse
We examined diet-induced obesity and insulin resistance by feeding 8-week-old male C57BL/6J mice an HFD for 16 weeks (Figure 1). Control mice were fed a standard chow diet (CD) for the same 16-week period, and all animals were euthanized at the 24-week time point. This model is particularly suited for the study of human metabolic diseases, as HFD consumption by mice induces complications consistent with the progression of human metabolic syndrome (Collins et al., 2004). Indeed, we found that HFD-fed mice exhibited obesity, hepatic steatosis, hyperglycemia, insulin resistance, and glucose intolerance compared with CD-fed mice (Figure S1).
Figure 1. Overview of Systems Biology Study of HFD-Induced Insulin Resistance.
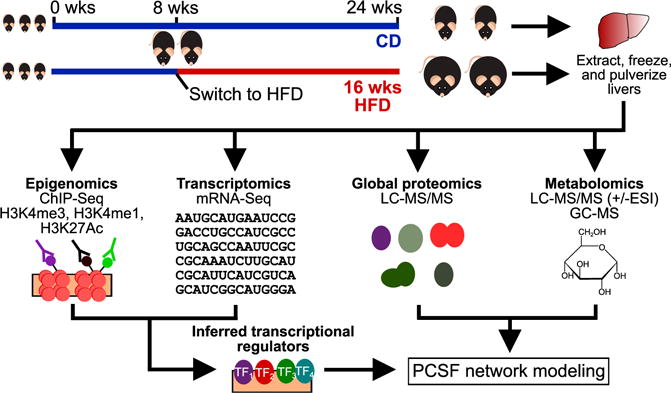
We fed 8-week-old male C57BL/6J mice a 16-week standard laboratory chow diet (CD) or a high-fat diet (HFD) to induce obesity and insulin resistance. At 24 weeks, we sacrificed the mice and extracted, flash froze, and pulverized their livers. We used these tissue samples to assay epigenomes, transcriptomes, proteomes, and metabolomes. We then used mRNA-seq (differential genes) and histone modification ChIP-seq (valleys within enriched peaks) data with known DNA-binding motifs to infer active transcriptional regulators. These regulators, along with differential proteins and metabolites, were used as input to the prize-collecting Steiner forest (PCSF) algorithm to uncover a network of interconnections among the data. ESI, electrospray ionization; GC-MS, gas chromatography-mass spectrometry; LC-MS/MS, liquid chromatography-tandem mass spectrometry; TF, transcription factor.
Omic Datasets Demonstrate Wide-Ranging Effects of HFD on Mouse Liver Biology
We collected an array of datasets using high-throughput omic experimental methods to broadly capture the effects of HFD in the liver (Figures 1 and 2). We used the information obtained from analysis of these datasets to inform our subsequent integrative network modeling efforts.
Figure 2. HFD Induces Perturbations to Hepatic Omic Levels.
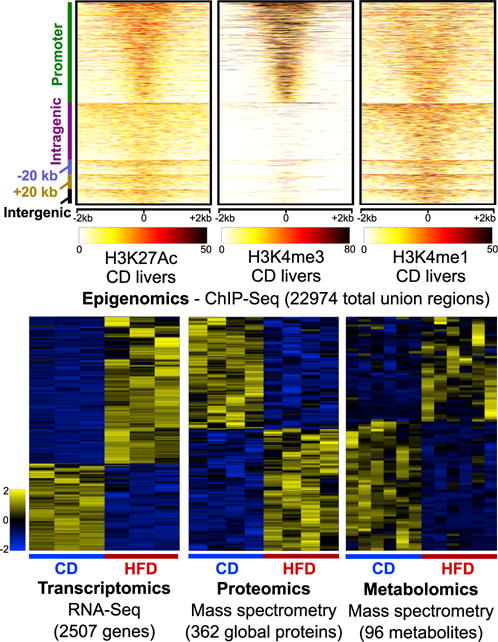
(Top panels) Smoothed read density profiles in ±2-kb windows around the union of all identified enrichment regions (22,974 total) for histone marks H3K27Ac, H3K4me3, and H3K4me1 from CD liver samples. The mappings on the left are with respect to the closest RefSeq gene start site: promoter (±2 kb to start site); intragenic, −20 kb (within 20 kb upstream), +20 kb (within 20 kb downstream), and intergenic (>20 kb away from nearest gene). (Lower panels) We found 2,507 genes (n = 3 for CD and HFD), 362 global proteins (n = 4 for CD and HFD), and 96 metabolites (n = 6 for CD and HFD) perturbed by HFD consumption. Clustergrams show individual Z-scored values for species from CD and HFD replicates. Only the most significantly changing peptide is shown as a representative for each of the differential global proteins, though full statistics were performed on all peptides.
See also Figures S1, S2, S3, and S4.
Epigenomics
We profiled the epigenomes of CD and HFD livers with histone modification ChIP-seq experiments for H3K27Ac, which marks active enhancers (Creyghton et al., 2010); H3K4me3, which marks active and poised promoters (Santos-Rosa et al., 2002); and H3K4me1, which marks active and poised enhancers (Creyghton et al., 2010) (Figure 2, top panels). We tested for differences in histone modification levels between the diets but found few significant differential regions (<1%). Overall, these data provide a comprehensive map of >22,000 active regulatory regions, of which 89% map within ±20 kb of >14,000 expressed liver genes.
Transcriptomics
We used mRNA-seq to identify 2,507 genes differentially expressed between CD and HFD livers. Of these, 1,572 genes are upregulated, and 935 genes are downregulated in HFD livers (Figure 2, bottom left; Figure S2A). Genes upregulated by HFD are enriched in lipid metabolism (Aacs, Ldlr, and Srebf1) and carbohydrate metabolism (Gck, Hk2, and Pfkl), while genes downregulated by HFD are enriched in amino-acid catabolism (Arg1, Gldc, Got1, and Hdc) and small-molecule catabolism (Aadat, Aass, Cps1, Csad). Shared biological enrichments between the two classes of genes include carboxylic acid and oxoacid metabolism. We also performed TaqMan assays on additional CD and HFD samples (8 or more livers per condition) to further test for evidence of immune cell infiltration in HFD livers (as observed in our mRNA-seq results) (Figure S3). We found up-regulation of Cd3e (T cells), Cd11c (dendritic cells/monocytes/macrophages), Emr1 (monocytes/macrophages), and Nos2 (M2-like macrophages), together with downregulation of Arg1 (M2-like macrophages). These results suggest that immune cell infiltration, indeed, plays a role in promoting and maintaining the insulin-resistant state of HFD mice.
Proteomics
We used mass spectrometry (Zhou et al., 2013) to quantify CD and HFD liver global proteomes, identifying 51,689 unique peptides that mapped to 6,384 unique proteins. We used a weighted least-squares regression procedure to find 362 differentially expressed proteins, with 189 upregulated and 173 downregulated in HFD livers (Figure 2, bottom middle; Figure S2B). Proteins up-regulated by HFD are uniquely enriched in fatty acid β-oxidation (CROT, ECI1, and HADH), fatty acid transport (CD36, FABP1, and FABP2) and carbohydrate biosynthesis (FBP1, GBE1, GCK, and GYS2), while the proteins downregulated by HFD are uniquely enriched in cholesterol biosynthesis (CYP51, DHCR7, FDPS, and IDI1) and the urea cycle (CPS1, NAGS, and OTC). Both sets of proteins are enriched in amino acid metabolism, carboxylic acid metabolism, and oxidation-reduction processes (Data S1).
Metabolomics
We obtained metabolomic measurements by mass spectrometry of 381 metabolites in CD and HFD livers (Figure 2, bottom right; Figure S2C); 96 metabolites are significantly different between the two diets, with 43 upregulated and 53 downregulated by HFD. These metabolites include amino acids (11 upregulated and 22 downregulated by HFD), lipids (11 upregulated and 21 downregulated by HFD), carbohydrates (10 upregulated and 1 downregulated by HFD), and peptides (2 upregulated and 2 downregulated by HFD) (Data S2). We also observed expected increases in the levels of glucose and other carbohydrate molecules, as hyperglycemia is a well-established feature of hepatic insulin resistance.
Overall changes in gene and protein expression induced by HFD consumption are only weakly to moderately correlated (r = 0.2–0.4), even when we restrict our analyses to genes and proteins that were called significantly different between both conditions (Figures S4A and S4B). This modest correlation is generally consistent with other systems (Schwanhäusser et al., 2011) and is also consistent with prior observations from CD and HFD mouse livers on a smaller, targeted set of ~200 matched species (Wu et al., 2014). We also observed specific biological processes that are enriched in the set of differential mRNAs but not in the set of differential proteins (and vice versa). For example, proteins upregulated by HFD are uniquely enriched in fatty acid β-oxidation and carboxylic acid catabolism (Figure S4C). These comparisons demonstrate how individual omic datasets can highlight different aspects of disease processes.
Epigenomic and Transcriptomic Dataset Integration Uncovers Transcriptional Regulators Influencing Differential Gene Expression
We collected epigenomic and transcriptomic data with the goal of uncovering changes in transcriptional regulation between CD and HFD livers. To reconstruct this transcriptional regulatory network, we inferred the genomic binding locations of potential transcriptional regulators using our ChIP-seq datasets and DNA-binding motif data from TRANSFAC (Wingender et al., 1996). As we found little evidence for changes in these histone modifications between diets, we used the set of significant ChIP-seq regions in CD livers for our analyses. We searched each dataset for histone “valleys,” or regions between peaks of local modification enrichment where histones are depleted and where regulators likely bind (Figure 3A) (Ramsey et al., 2010; Wamstad et al., 2012). We merged these discovered valleys into one set of 123,974 total loci and scanned the underlying genomic sequences for matches to a set of 1,588 DNA-binding motifs that map to at least one human or mouse transcriptional regulator (Figure 3B). For each regulator (motif) and each differentially expressed gene, a transcription factor affinity (TFA) score was derived as a distance-weighted sum of individual motif enrichment scores in regions near the gene’s annotated transcription start site. We then used the linear regression of each motif’s TFA scores against the expression levels of all the differentially expressed genes and took significant regression coefficients (false discovery rate [FDR] < 0.01) as evidence for active regulators (Figures 3C and 3D).
Figure 3. Motif Regression Procedure Identifies Transcriptional Regulators.
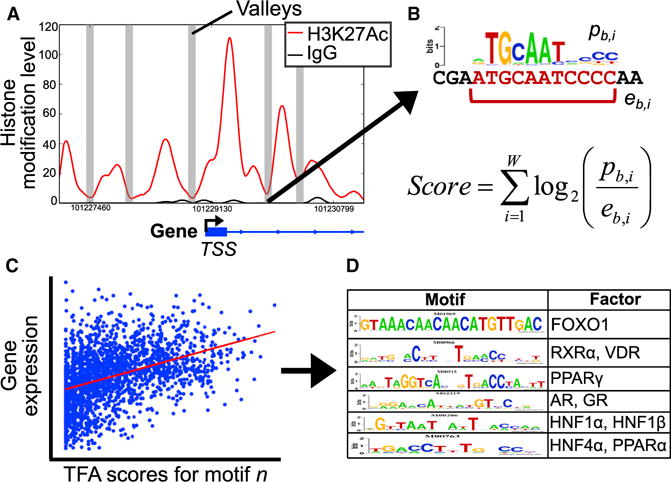
(A) We extracted read density profiles for significantly enriched histone modification levels, smoothed the profiles, and scanned for “histone valleys,” or regions of local signal depletion (an H3K27Ac enrichment region is shown here as an example). TSS, transcription start site.
(B) For each valley, we scanned the underlying genomic sequence for matches to a library of DNA-binding factor motifs. Against each differential gene, we computed a transcription factor affinity (TFA) score for all motifs as a distance-weighted sum of individual match scores.
(C) For each motif, we used linear regression to predict gene expression levels from the motif TFA scores.
(D) This procedure found 358 significant motifs that map to 272 regulatory proteins; select results are shown in the table.
In total, we identified 358 significant DNA-binding motifs that mapped to 272 unique transcriptional regulatory proteins (Data S3), including known liver-enriched transcription factors such as hepatic nuclear factors 1α, 1β, and 4α; retinoid X receptors α and β; peroxisome proliferator-activated receptor α; and C/EBP α (Schrem et al., 2002, 2004). We also found strong enrichment for nuclear factor I proteins (A, B, C, and X), SOX4, FOXO1, and the vitamin D receptor (VDR). These significant factors served as the core transcriptional regulatory data that we incorporated into our network models.
Prize-Collecting Steiner Forest Model Integrates Multiple Omic Datasets
Each type of omic data provides a glimpse into the effect of HFD on a particular regulatory level. To obtain a more comprehensive view of the data, we expanded upon an established network modeling algorithm called the prize-collecting Steiner forest (PCSF) (Tuncbag et al., 2013, 2016). We built a combined protein-protein and protein-metabolite interactome from the iRefIndex (v.13) database (Razick et al., 2008) for protein-protein interactions and obtained protein-metabolite interactions from the Human Metabolome Database (HMDB; v.3.6) (Wishart et al., 2013) and the human metabolic reconstruction Recon 2 (v.3) (Thiele et al., 2013). To account for differences in reliability of the various types of interactions, we assigned to each an “edge cost” that scaled inversely with our confidence in the interaction (see Experimental Procedures for details). We used this interaction network and the omic data as input to the PCSF algorithm to identify interactions that connect the omic data (Figure S5).
As part of the PCSF approach, omic results (e.g., differential proteins) are assigned prizes (e.g., as log2 fold changes), and the algorithm attempts to maximize the inclusion of these prize nodes while avoiding low-confidence edges, which have high edge costs. Thus, the algorithm is not constrained to include all data in the final network but, at the same time, is capable of introducing species not present in the original set of data. These interactome-derived species are called “Steiner” nodes and are included when necessary to create connections between the data. We also implemented a method that assigns “negative prizes” to interactome nodes with many interactions. These highly connected species, or “hubs,” have a high likelihood of appearing in network models run with almost any input data (e.g., ubiquitin or water). Negative prizes discourage the algorithm from using such nodes in the PCSF solution and allow for more specific interactions to explain the data (Figures S6A and S6B).
We used as input data—or “terminals” in PCSF parlance—83 differential metabolites, 329 differential proteins, and the 272 transcriptional regulators identified by our motif regression analysis (Data S4). We generated and merged multiple solutions by running the algorithm on the same data multiple times with random noise added to the edge costs. This procedure produced a richer set of possible connections explaining the data and enabled the assessment of network components’ robustness. We also assessed node specificity to hepatic insulin resistance by comparing how many times each node appears in networks generated with random input data (i.e., random interactome nodes that match the degree distribution of the real input data).
The full PCSF solution (Figure 4; Data S5) includes 907 species connected by 2,365 interactions (see also Table 1). We found that the vast majority of nodes included in the final network are very specific to our system (Figure S6C). To increase interpretability of the network model, we identified smaller sub-networks and performed gene, small-molecule, and pathway enrichment analyses on these. We computed rank scores for these sub-networks according to their prize densities (the sum of prizes multiplied by the fractional size of the sub-network; Figure S7). Among the top ranked sub-networks are those enriched for amino acid and pyruvate metabolism, fatty acid oxidation, apoptosis, transcription, ECM, and bile acid metabolism. Additionally, we devised a scheme to rank interactome-derived Steiner nodes by their likely importance in the model according to several features, including the robustness and specificity of nodes. We used a weighted summation of scores based on these features to perform this ranking (see Experimental Procedures for details).
Figure 4. Multi-omic PCSF Model Uncovers Features of Hepatic Insulin Resistance.
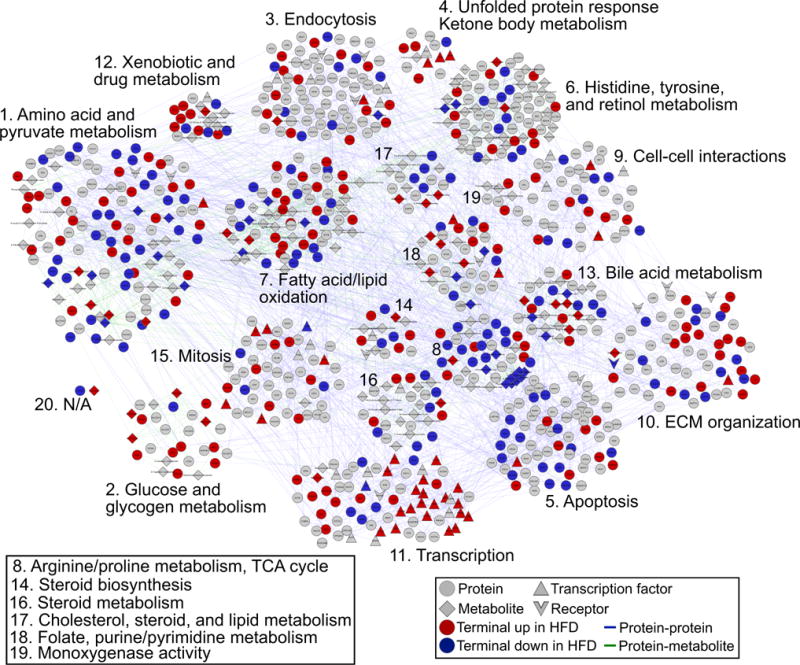
The full PCSF model includes 398 terminal nodes and 509 predicted Steiner nodes connected by 2,365 interactions. We divided the solution into 20 sub-networks and highlight the specific biological processes contained within these. Colored nodes (red or blue) represent terminal nodes, gray nodes represent Steiner nodes, and shapes indicate node types (proteins, metabolites, transcription factors, or receptors).
See also Figures S5 and S6.
Table 1.
PCSF Model Terminal Node Inclusion Statistics
| Terminal Type | Number of Terminals | Number Included in Final Model | % Included |
|---|---|---|---|
| Metabolites | 83 | 63 | 75.9 |
| Global proteins | 329 | 301 | 91.5 |
| Transcription factors | 272 | 34 | 12.5 |
This table shows numbers of terminals (input data) supplied to the PCSF algorithm for metabolite, global protein, and transcription factor data types. The numbers of terminals for each data type present in the final solution are shown, along with percentages.
See also Figure S5.
The PCSF Model Introduces Species with Known Relevance to Metabolic Disease
We developed an automated strategy to identify network nodes that have not been previously reported as associated with insulin resistance and related complications. For this purpose, we used the DisGeNET database (Piñero et al., 2015), which collates gene-disease information from public data as well as from literature via natural language processing, to determine which of the predicted molecules introduced by the PCSF into the network (Steiner nodes) are known to be associated with obesity, insulin resistance, and/or type 2 diabetes. Of 394 Steiner proteins, 121 (~30%) possess some known disease link according to DisGeNET (Data S4). Some examples include: clusterin (CLU), in which polymorphisms are associated with type 2 diabetes (Daimon et al., 2011) and where knockout in C57BL/6J mice exacerbates HFD-induced insulin resistance (Kwon et al., 2014); L-arginine:glycine amidinotransferase (GATM, a.k.a. AGAT), where knockout in C57BL/6J mice depletes creatine, enhances glucose tolerance, and protects from diet-induced obesity (Choe et al., 2013); and nuclear receptor co-activator 1 (NCOA1, a.k.a. SRC-1), depletion of which can result in increased glucose uptake, enhanced insulin sensitivity, and resistance to age-associated obesity and glucose intolerance (Wang et al., 2012). A literature review revealed additional Steiner nodes with known relevance to disease, including the metabolite glyoxylic acid, which has been characterized as a marker metabolite for type 2 diabetes (Nikiforova et al., 2014). Thus, our model incorporates many predicted nodes with known relevance to these conditions, though many are not well-established actors in these contexts.
The PCSF Model Identifies Biological Features of Obesity-Induced Hepatic Insulin Resistance
Among the 20 sub-networks we identified from the full PCSF model are networks enriched in glucose and glycogen metabolism (sub-network 2), amino acid metabolism (sub-network 1), fatty acid and lipid oxidation (sub-network 7), transcriptional regulation (sub-network 11), and bile acid metabolism (sub-network 13). These sub-networks all describe well-established aspects of hepatic insulin resistance (Data S5 and S6). Specific details for some of the biological mechanisms contained in these sub-networks are included in the Supplemental Information.
Importantly, we also identified sub-networks enriched in biological processes not typically associated with hepatic insulin resistance. One such sub-network is enriched in ECM organizational and structural proteins (sub-network 10; Figure 5). Proteins associated with the ECM in this sub-network include collagens 1A1, 1A2, and 6A1 (COL1A1/1A2/6A1), as well as endoglin (ENG), fibronectin 1 (FN1), intergrin α5 (ITGA5), and the transforming growth factor β(TGF-β) receptor 1 (TGFB1). At the center of this sub-network is FN1, which connects, among other nodes, most of the collagen proteins and ITGA5. Both ENG and TGFBR1 are predicted Steiner nodes connected through ITGA5. Several Steiner nodes in this sub-network rank very highly by our criteria, including CD79A, 5′-3′ exoribonuclease 1 (XRN1), and CLU.
Figure 5. PCSF Sub-networks for Select Biological Processes.
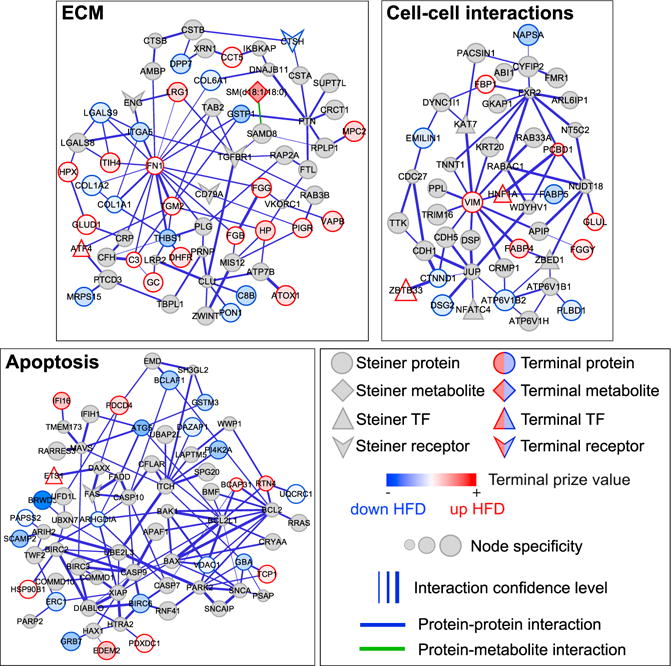
We highlight PCSF model sub-networks that are enriched in extracellular matrix (ECM, top left), cell-cell interactions (top right), and apoptosis (bottom left). Note that node specificities should only be compared within sub-networks, as overall panel sizes differ for clarity. TF, transcription factor.
See also Figure S7.
Changes to the hepatic ECM may also implicate altered cell-cell communication between hepatocytes in response to ECM and liver architectural disruption. Indeed, we found a sub-network enriched in proteins related to cell-cell interactions (sub-network 9; Figure 5). Included in this sub-network are the proteins E-cadherin (CDH1), cadherin 5 (CDH5), junction plakoglobin (JUP), and vimentin (VIM). These enrichments strongly suggest that changes to liver structure and the composition of the ECM are relevant to hepatic insulin resistance.
We also identified a sub-network enriched in apoptotic processes (sub-network 5; Figure 5). Terminal proteins involved in apoptosis here include autophagy-related 5 (ATG5, a late-apoptosis protein that interacts with FADD; Pyo et al., 2005), BCL-2-associated transcription factor 1 (BCLAF1), and interferon (IFN)-γ -inducible protein 16 (IFI16). The majority of the apoptosis-related proteins are predicted nodes, including BCL2; BCL2L1; caspases 7, 9, and 10; FAS; the FAS-associated death domain (FADD); and BAD. The model captures aspects of the extrinsic apoptotic pathway, whereby the death-inducing signaling complex composed of FAS, FADD, and pro-caspase 8 or −10 signals to downstream effectors (Wang et al., 2001), as well as the intrinsic pathway, which involves the pro-apoptotic Bcl-2 family member BAX and anti-apoptotic members BCL2 and BCL2L1 (Lee and Pervaiz, 2007). The model includes both initiator (CASP8 and CASP10) and effector caspases (CASP7) linked to these initiator proteins (Wang, 2014). Thus, our PCSF model, overall, suggests a role for apoptosis in maintaining hepatic insulin resistance.
Liver Tissue Analysis Confirms Global Alterations in Hepatic Processes Identified by the PCSF Model
The network results imply roles for unexpected processes related to diet-induced insulin resistance. To test these predictions, we performed imaging studies on frozen liver sections from CD and HFD mice. First, we tested the prediction that HFD livers would display altered cell-cell interactions and overall structural deficiencies. We stained liver sections for Zo1, a cytoplasmic membrane protein of intercellular tight junctions, and cytokeratins 8 and 18, which are dimerized intermediate filaments present in epithelial cells that help maintain cellular structural integrity. Using DAPI staining to identify nuclei, we found cellular boundaries and tight junctions around bile ducts in the liver of CD-fed mice. By contrast, tight junctions and structure near bile ducts of HFD livers were highly disorganized (Figure 6A). In larger fields of view, we saw highly structured hepatocyte borders and normal architecture in CD livers (Figure 6B). In contrast, HFD livers displayed irregular cytokeratin 8/18 staining, with few discernable cell borders, indicating overall disruption of the hepatic tissue architecture in response to the long-term dietary challenge.
Figure 6. Hepatic Imaging Validates Global PCSF Model Predictions.
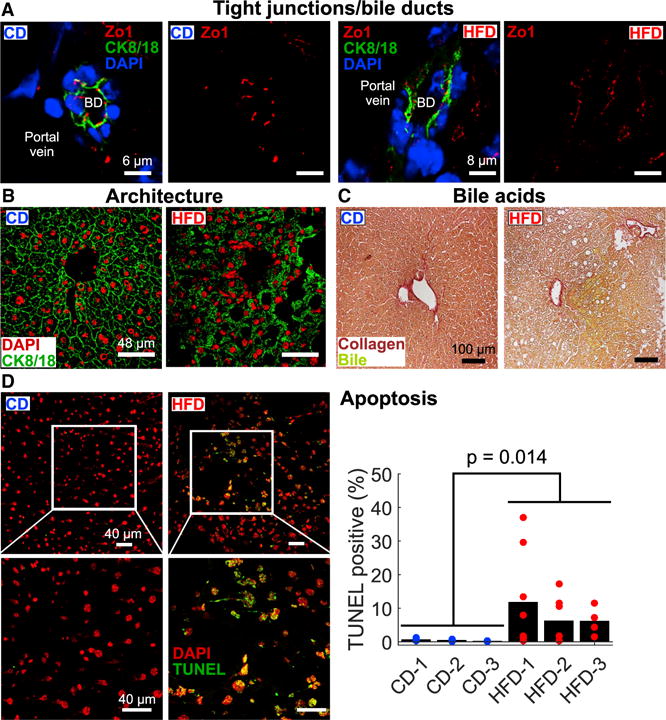
(A) HFD-induced changes in tight junction structure near bile ducts (BD) as assessed by cytokeratin 8/18 (CK8/18) and Zo1 staining. Scale bars: for CD, 6 μm; for HFD, 8 μm.
(B) CK8/18 staining revealed overall hepatic architectural defects in HFD samples. Scale bars, 48 μm.
(C) We observed enhanced bile acid leakage in HFD livers stained for collagen and bile/bilirubin compared to CD livers. Scale bars, 100 μm.
(D) TUNEL imaging revealed enhanced regions of hepatocyte apoptosis in HFD samples. Points on graph represent values from individual fields of view (ns = 9, 7, and 4 for HFD livers; ns = 4, 5, and 5 for CD livers), and bars indicate overall TUNEL-positive fraction (total TUNEL-positive cells over total cells) based on all fields of view. We found that the overall difference in TUNEL staining between the diets is statistically significant by two-tailed t test (p = 0.014). Scale bars, 40 μm.
We also tested the prediction that HFD livers would display abnormal bile acid handling by staining liver sections for collagen and bile/bilirubin (Figure 6C). As expected, we found no bile acid leakage or accumulation in CD livers. However, we observed significant bile accumulation in HFD livers. These results corroborate our prediction that HFD livers possess defects in bile acid maintenance and are consistent with the altered cellular structures we found surrounding the bile ducts of HFD-fed mice.
Finally, we tested whether consumption of an HFD enhances the number of hepatocytes undergoing apoptosis in the liver. We used DAPI and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) to assess the number of apoptotic cells. The fraction of TUNEL-positive cells in CD livers was very low (~1%), whereas HFD livers displayed regions of high TUNEL positivity (as high as 37%; Figure 6D). While not prevalent in all regions, overall apoptosis was higher in HFD samples (Figure 6D, p = 0.014). Thus, we show here evidence for enhanced hepatocyte apoptosis as a feature of HFD-induced hepatic insulin resistance.
DISCUSSION
We undertook a large-scale integrative systems analysis of HFD-induced hepatic insulin resistance. We used ChIP-seq and mRNA-seq to interrogate epigenomic regulation and transcription, untargeted shotgun proteomics to quantify >6,000 hepatic proteins, and metabolomics to profile nearly 400 small-molecule species. Using a network approach, we integrated these data-sets and highlighted major biological processes perturbed by HFD. The algorithm also incorporated disease-relevant proteins and metabolites from the interactome that were either not measured or found to be differentially expressed in our omic data. We validated several high-level model predictions by examining mouse livers for markers of specific physical features and biological processes. We found that HFD consumption perturbs hepatic architecture, disrupts bile acid handling, and enhances hepatocyte apoptosis.
The liver is a major contributor to overall glycemic regulation. Indeed, insulin-stimulated clearance of blood glucose is mediated, in part, by the inhibition of hepatic gluconeogenesis (Pilkis and Granner, 1992) and consumption of an HFD causes hepatic insulin resistance, which disrupts this process (Pilkis and Granner, 1992). As expected, we found that the HFD feeding in mice caused obesity, insulin resistance, and impaired glucose homeostasis. These features were accompanied by HFD-induced changes in >2,000 genes, 362 global proteins, and 96 metabolites. We note that these findings derive from molecules measured from whole-liver extracts. While hepatocytes represent the dominant cell type in liver tissue, hepatic stellate cells, vascular endothelial cells, and various immune cells do, indeed, influence liver function and molecular concentrations. Thus, our results must be interpreted within this framework.
We used a motif regression procedure that incorporated epigenomic, transcriptomic, and motif data to identify transcriptional regulators relevant to insulin resistance, identifying 272 significant factors. Several of these top predictions are consistent with factors identified in a prior study that used different epigenomic techniques to find regulatory regions (Leung et al., 2014). Interestingly, both our study and theirs did not observe many changes in histone modification levels between the diets, despite significant gene expression changes. An advantage of our integrative modeling approach is that, even if a pathway is not detected as changing by one experimental method, it may emerge in the network based on evidence from other types of data.
To integrate all the omic datasets we collected, we built on the established PCSF network modeling approach (Tuncbag et al., 2013, 2016). The PCSF method is not required to include all omic data yet is capable of introducing predicted nodes that are critical for establishing connections between the detected molecules. PCSF networks are generally much smaller and more tractable than solutions derived from more naive methods and reveal interpretable sub-networks enriched in specific biological processes and pathways. Here, we have significantly expanded the scope of the PCSF methods by adding physical associations of proteins and metabolites to the protein-protein interactome. This unified approach allowed us to capture a wider range of biological pathways and processes relevant to insulin resistance. We also used several strategies to improve the accuracy of our networks. Prior studies have noted that network methods can be biased toward highly studied proteins that appear as “hubs” in the interactome (Paull et al., 2013). To reduce this bias, we applied a soft penalty to highly connected nodes to discourage their inclusion while still allowing for their use when particularly necessary. We additionally tested our networks for robustness to noise and assessed the specificity of network nodes to our system.
Our integrated approach can identify many different types of links among the omic data. We found pathways that were largely dominated by proteomic data (e.g., cell-cell interactions, ECM, apoptosis) but also found several sub-networks almost entirely composed of protein-metabolite connections (e.g., bile acid metabolism and glucose metabolism). The inclusion of direct metabolomic data along with protein-metabolite interactions was critical to capturing, for instance, relevant connections among differential proteins whose roles are best explained in the context of metabolic processes (e.g., GCK and CYP7B1).
Increasingly, systems biology and omic approaches are being recognized for their utility to the study of insulin resistance and type 2 diabetes (Zhao et al., 2015). To date, however, few studies have formally integrated multiple types of omic data in these contexts, with even fewer including metabolomics. Prior studies attempting such joint analyses used correlative statistical routines (Miraldi et al., 2013; Oberbach et al., 2011), methods that overlay proteomic and metabolomic data onto genome-scale metabolic reconstructions (Yizhak et al., 2010), or methods that map metabolomic and transcriptomic data onto known pathway and transcriptional regulatory data without identifying high-confidence sub-networks (e.g., the CircadiOmics resource) (Eckel-Mahan et al., 2013). Our approach goes well beyond these previous methods by incorporating multiple data types from the same samples, allowing for interactions that occur outside well-established signaling or metabolic pathways, and using advanced approaches to reduce the possible interaction space to only the most relevant connections, thus increasing the interpretability of results and providing clear guidance for designing experiments.
Our model uncovered a highly interconnected network associated with the insulin-resistant state in the liver. We predicted that changes to the ECM, cell-cell interactions, and overall hepatic architecture are features of insulin resistance. Subsequent experiments confirmed that the overall structure of HFD mouse livers is highly disrupted, especially near bile ducts. Consistent with this observation, we also found enhanced bile acid leakage (cholestasis) into the tissue of HFD-fed mouse livers. These structural abnormalities likely also contribute to the increased apoptosis that we observed in insulin-resistant livers. The link between hepatic ECM and architectural structural remodeling with insulin resistance has been studied sparingly (Williams et al., 2015a). In one study, tail-vein injection of HFD-fed mice with a hydrolase for hyaluronan, an ECM component, reduces features of muscle and liver insulin resistance (Kang et al., 2013). Moreover, integrin-α1-subunit-deficient mice (Itga1−/−) fed an HFD display reduced fatty liver content but also severe hepatic insulin resistance, compared to wild-type HFD-fed controls (Williams et al., 2015b).
The hepatic structural changes detected in HFD-fed mice may be related to changes in apoptosis. Crosstalk between proteins relevant to insulin resistance and hepatocellular injury, including tumor necrosis factor (TNF), nuclear factor κB (NF-κB), and JNK, has been proposed as a potential driver of apoptosis in the liver (Schattenberg and Schuchmann, 2009). Indeed, apoptosis is associated with severe hepatocellular injury and steatohepatitis (Guicciardi and Gores, 2005). Here, we report increased hepatic apoptosis in HFD-fed mice. This increased hepatic apoptosis may be related to dysregulation of the hepatobiliary system (Wang, 2014) and promotes low-grade inflammation and hepatic insulin resistance.
Previous studies have associated changes in liver architecture with late stages of hepatic steatosis that lead to non-alcoholic steatohepatitis and the development of hepatic fibrosis (Ramachandran and Henderson, 2016). The results of the present study demonstrate that defects in hepatic architecture precede fibrosis and can be detected in early stages of the response to the consumption of a HFD. It is likely that these early changes in hepatic architecture contribute to the long-term development of hepatic dysfunction.
To summarize, we undertook a large-scale systems biology approach to study HFD-induced hepatic insulin resistance. We integrated multiple types of omic datasets into a network model that uncovered altered biological processes associated with the condition. By incorporating metabolites into the protein-protein interaction network, we were able to identify a wide range of molecular changes. We validated several global predictions from our network model with additional experiments and highlighted components relevant to the hepatic response to HFD consumption. The pathways and processes we found to be altered by HFD present a wide range of directions for future research. Our methods are easily applicable to other large-scale omic analyses of diverse biological systems and diseases.
EXPERIMENTAL PROCEDURES
Animals
We obtained male C57BL/6J mice (stock number 000664) from the Jackson Laboratory. All mice were housed in a specific pathogen-free facility accredited by the American Association for Laboratory Animal Care. We fed the mice either (1) a standard CD (Prolab Isopro RMH 3000, Purina) for 24 weeks or (2) 8 weeks of standard CD followed by 16 weeks of HFD (S3282, Bio-serve). We measured fat and lean mass noninvasively using 1H-MRS (Echo Medical Systems). We euthanized all mice at 24 weeks after an overnight fast and froze the livers prior to removal using clamps cooled in liquid nitrogen. The frozen livers were then pulverized into a powder using a CryoPREP impactor (Covaris). We prepared aliquots of pulverized liver for all samples for subsequent analyses. All experiments were carried out in accordance with guidelines for the use of laboratory animals and were approved by the Institutional Animal Care and Use Committees (IACUCs) of the University of Massachusetts Medical School.
Glucose and Insulin Tolerance Tests
We performed glucose and insulin tolerance tests by intraperitoneal injection of mice with glucose (1 g/kg) or insulin (1.5 U/kg) using methods described previously (Sabio et al., 2008).
Immunoblot Analysis
Protein extracts from pulverized liver were prepared in Triton lysis buffer (20 mM Tris [pH 7.4], 1% Triton X-100, 10% glycerol, 137 mM NaCl, 2 mM EDTA, 25 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM PMSF, and 10 μg/mL each of aprotinin and leupeptin). We quantified protein content by the Bradford method (Bio-Rad). Standard techniques were used to separate cell extracts (15–80 μg protein) by SDS-PAGE for immunoblot analysis using antibodies from Cell Signaling Technology (AKT and pSer473-AKT). The primary antibodies were detected by incubation with anti-mouse or anti-rabbit immunoglobulin G (IgG) conjugated to infrared dyes (IRDye, LI-COR Biosciences). We detected immune complexes using the Odyssey Infrared Imaging system (LI-COR Biosciences).
mRNA-Seq and Analysis
We prepared mRNA-seq libraries from three CD and three 16-week HFD mouse livers using the TruSeq RNA Sample Prep Kit v.1 (Illumina). This was followed by 180 ± 25 bp insert size selection using 2% agarose gel electrophoresis. We multiplexed mRNA-seq libraries and paired-end sequenced samples for 40–50 bp on an Illumina Hi-Seq 2000 machine. On average, we obtained ~20–30 million raw paired-end sequencing reads. Read alignment, gene quantification, and differential analysis details are provided in the Supplemental Information. Briefly, we aligned reads to the mm9 genome using TopHat (v.1.4.0) (Trapnell et al., 2009) and used DESeq2 (v.1.0.18) (Love et al., 2014) to perform differential expression analyses. We considered a gene to be differentially expressed if it possessed an absolute log2 fold change between conditions ≥ 0.5 and an FDR-adjusted p value (q value) ≤ 0.05 and was expressed in at least one tested condition (i.e., ≥0.1 fragments per kilo-base of transcript per million mapped reads [FPKM]).
ChIP-Seq and Analysis
Histone modification ChIP experiments were performed using the MAGnify Chromatin Immunoprecipitation System Kit (Life Technologies, Carlsbad, CA), with antibodies against H3K4me1 (17-676, Millipore), H3K4me3 (17-614, Millipore), and H3K27ac (ab4729, Abcam, Cambridge, MA). ChIP-seq libraries were constructed using the NEBNext DNA Library Prep Master Mix Set for Illumina (New England Biolabs, Ipswich, MA, USA) and sequenced on an Illumina Hi-Seq 2000 machine. We aligned raw reads using Bowtie (v.0.12.7) (Langmead, 2010) and performed peak calling using model-based analysis of ChIP-seq (MACS) (v.1.4.0rc2) (Zhang et al., 2008) against an IgG control. We considered significant MACS peaks to be those possessing a p value < 1e-6 and an FDR < 10%. We also performed differential peak analyses between conditions; details of these methods are provided in the Supplemental Information. We considered regions possessing an FDR-corrected p value < 0.05 as significant.
Global Proteomics
We collected global proteomic data from four CD and four 16-week HFD mouse livers using mass-spectrometry-based methods. Full details of the experimental methods and statistical analyses are provided in the Supplemental Information. We deemed proteins possessing an FDR < 0.1 between CD and HFD livers as differentially expressed.
Metabolomics and Analysis
We extracted and split samples (6 independent livers per condition, per Metabolon recommendations for appropriate statistical power) into equal parts for analysis on gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-tandem mass spectrometry with electrospray ionization in positive and negative ion modes (LC-MS/MS, ESI ±). A total of 381 metabolites were identified and quantitated. We imputed missing values with a k-nearest-neighbors procedure (k = 10), normalized samples according to the procedure in Anders and Huber (2010), and tested for differences using two-tailed t tests, correcting p values for multiple hypotheses. We observed strong intra-sample correlations between CD (Pearson’s r > 0.923) and HFD (r > 0.85) replicate abundances (Figure S2C). Metabolites possessing an FDR < 0.1 were deemed significant. The raw data and differential expression results for these data are provided as Data S2.
Motif Regression Analysis
General methods are described in the main text. Full details of these procedures are provided in the Supplemental Information.
Prize-Collecting Steiner Forest Modeling
Full details of all methods related to the prize-collecting Steiner forest (PCSF) modeling approach are included in the Supplemental Information. Briefly, the prize-collecting Steiner forest (Tuncbag et al., 2013, 2016) aims to find a forest F(VF, EF) from the graph G(V, E, c(e), p(v)), with nodes V, edges E, edge costs c(e) ≥ 0, and node prizes p(v) for v ∈ V, that minimizes the objective function:
where κ represents the number of trees in the forest, ω represents a tuning parameter that influences the number of trees included in the final forest, and:
The β parameter scales the importance of node prizes versus edge costs. We used a “negative prize” scaling scheme to each node in G proportional to its degree, or number of connections in the interactome, to reduce the influence of highly connected, well-studied nodes. The parameter μ scales the influence of the negative prizes, and the exponent n allows for non-linearity in the scaling. We built a combined protein-protein and protein-metabolite interactome using v.13 of the iRefIndex database (Razick et al., 2008) for protein-protein interactions (scored with the MIscore system; Villaveces et al., 2015) and v.3.6 of the HMDB (Wishart et al., 2013), supplemented with manually curated interactions from the human metabolic reconstruction Recon 2 (v.3) (Thiele et al., 2013), for protein-metabolite interactions. We used a community clustering algorithm (Blondel et al., 2008) to break the full PCSF model into smaller sub-networks and visualized all networks with Cytoscape (Shannon et al., 2003).
Liver Tissue Section Analysis and Imaging
Histology was performed using liver fixed in 10% formalin for 24 hr, dehydrated, and embedded in paraffin. Dewaxed and rehydrated sections (7 μm) were cut and stained for bile acids (product #KTHBI, American Master Tech Scientific) or with H&E (American Master Tech Scientific). Sections (7 μm) prepared from liver frozen in O.C.T. Compound (Tissue-Tek) were stained with oil red O (Sigma) to visualize lipid droplets. We acquired images using a Zeiss Axiovert 200M microscope. Liver architecture was assessed using frozen sections fixed with 4% paraformaldehyde and stained with an antibody to cytokeratin 8 (TROMA-1-c, Developmental Studies Hybridoma Bank [DSHB], University of Iowa). Immune complexes were detected using anti-rat Ig conjugated to Alexa Fluor 488. Liver damage was assessed in frozen (7-μm) sections fixed with cold ethanol/acetic acid (2:1) using an in situ cell death kit (Roche). Bile duct architecture was assessed in frozen (7-μm) sections fixed with cold methanol by staining with antibodies to Zo-1 (sc-10804, Santa Cruz Biotechnology) and cytokeratin 8/18 (sc-52325, Santa Cruz). Immune complexes were detected using anti-mouse Ig conjugated to Alexa Fluor 488 and anti-rabbit Ig conjugated to Alexa Fluor 633 (Life Technologies). DNA was detected by staining with DAPI (Life Technologies). Fluorescence was visualized using a Leica TCS SP2 confocal microscope equipped with a 405-nm diode laser.
TUNEL Imaging Analysis
We used CellProfiler (v.2.1.1) (Carpenter et al., 2006) with a custom-built analysis pipeline from modules included in the program to analyze TUNEL images. All images across CD and HFD samples were analyzed in a single run of the program at the same settings. Pipeline details are described in the Supplemental Information. The TUNEL-positive percentage per field of view was calculated as the number of positive nuclei over the total. For each liver, we calculated a single TUNEL-positive fraction by dividing the total number of TUNEL-positive nuclei by the total number of nuclei across all fields of view (ns = 9, 7, and 4 for HFD livers; ns = 4, 5, and 5 for CD livers). We used a two-tailed t test to test for statistical significance between CD and HFD livers.
Clustering and Enrichment Analyses
All hierarchical clustering analysis was done with the clustergram function in MATLAB with Euclidean distance and average linkage. For enrichment analyses, we used custom MATLAB code, implementing the hypergeometric distribution for enrichment p value calculations and used the Benjamini-Hochberg FDR procedure to correct for multiple hypotheses. In general, an FDR < 0.1 was deemed significant.
DATA AND SOFTWARE AVAILABILITY
The accession number for the mRNA-seq and ChIP-seq raw and processed data reported in this paper is GEO: GSE77625.
Supplementary Material
Highlights.
Multi-omic profiling of high-fat-diet-induced hepatic insulin resistance
Integrative modeling of omic data identifies features of hepatic insulin resistance
Obesity induces hepatocellular injury and disrupts hepatic cell-cell interactions
Enhanced hepatocyte apoptosis is a feature of obesity-induced insulin resistance
Acknowledgments
We acknowledge members of the MIT BioMicro Center for assistance with sequencing data collection. This work was supported by NIH grants R24 DK-090963 (to E.F., R.J.D., F.M.W., and D.A.L.), R01 NS-089076 (to E.F.), R01 DK107220 (to R.J.D.), P01 NS047572, and P50 HG004233, the Dana-Farber Strategic Research Initiative, and the Juvenile Diabetes Research Foundation (to J.A.M.), and used computing resources funded by the National Science Foundation under Award No. DB1-0821391 and sequencing support from the NIH (P30-ES002109).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization and methodology, A.R.S., N.J.K., X.X., D.A.L., F.M.W., R.J.D., and E.F.; Software and formal analysis, A.R.S.; Investigation, N.J.K., X.X., F.Z., S.B.F., Y.S.Y., and B.J.M.; Resources, R.J.D., E.F., and J.A.M.; Writing, A.R.S., E.F., and R.J.D.; Visualization, A.R.S. and N.J.K.; Supervision, E.F., R.J.D., F.M.W., and D.A.L.; Project administration, R.J.D.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and six data files and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.11.059.
References
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimifar SB, Nagaraj N, Cox J, Mann M. Cell-type-resolved quantitative proteomics of murine liver. Cell Metab. 2014;20:1076–1087. doi: 10.1016/j.cmet.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Blondel VD, Guillaume JL, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech. 2008;2008:P10008. [Google Scholar]
- Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7:95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe CU, Nabuurs C, Stockebrand MC, Neu A, Nunes P, Morellini F, Sauter K, Schillemeit S, Hermans-Borgmeyer I, Marescau B, et al. L-arginine:glycine amidinotransferase deficiency protects from metabolic syndrome. Hum Mol Genet. 2013;22:110–123. doi: 10.1093/hmg/dds407. [DOI] [PubMed] [Google Scholar]
- Collins S, Martin TL, Surwit RS, Robidoux J. Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: physiological and molecular characteristics. Physiol Behav. 2004;81:243–248. doi: 10.1016/j.physbeh.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daimon M, Oizumi T, Karasawa S, Kaino W, Takase K, Tada K, Jimbu Y, Wada K, Kameda W, Susa S, et al. Association of the clusterin gene polymorphisms with type 2 diabetes mellitus. Metabolism. 2011;60:815–822. doi: 10.1016/j.metabol.2010.07.033. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Gunnarsson R, Björkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest. 1985;76:149–155. doi: 10.1172/JCI111938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WB, Broadhurst DI, Atherton HJ, Goodacre R, Griffin JL. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem Soc Rev. 2011;40:387–426. doi: 10.1039/b906712b. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, Dilag-Penilla SA, Dyar KA, Baldi P, Sassone-Corsi P. Reprogramming of the circadian clock by nutritional challenge. Cell. 2013;155:1464–1478. doi: 10.1016/j.cell.2013.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta-analysis. JAMA. 2013;309:71–82. doi: 10.1001/jama.2012.113905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024–1033. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Kang L, Lantier L, Kennedy A, Bonner JS, Mayes WH, Bracy DP, Bookbinder LH, Hasty AH, Thompson CB, Wasserman DH. Hyaluronan accumulates with high-fat feeding and contributes to insulin resistance. Diabetes. 2013;62:1888–1896. doi: 10.2337/db12-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- Kwon MJ, Ju TJ, Heo JY, Kim YW, Kim JY, Won KC, Kim JR, Bae YK, Park IS, Min BH, et al. Deficiency of clusterin exacerbates high-fat diet-induced insulin resistance in male mice. Endocrinology. 2014;155:2089–2101. doi: 10.1210/en.2013-1870. [DOI] [PubMed] [Google Scholar]
- Langmead B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. 2010:7. doi: 10.1002/0471250953.bi1107s32. Chapter 11, Unit 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Pervaiz S. Apoptosis in the pathophysiology of diabetes mellitus. Int J Biochem Cell Biol. 2007;39:497–504. doi: 10.1016/j.biocel.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Leung A, Parks BW, Du J, Trac C, Setten R, Chen Y, Brown K, Lusis AJ, Natarajan R, Schones DE. Open chromatin profiling in mice livers reveals unique chromatin variations induced by high fat diet. J Biol Chem. 2014;289:23557–23567. doi: 10.1074/jbc.M114.581439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, Ahima RS, Ueki K, Kahn CR, Birnbaum MJ. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis AJ, Attie AD, Reue K. Metabolic syndrome: from epidemiology to systems biology. Nat Rev Genet. 2008;9:819–830. doi: 10.1038/nrg2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- Miraldi ER, Sharfi H, Friedline RH, Johnson H, Zhang T, Lau KS, Ko HJ, Curran TG, Haigis KM, Yaffe MB, et al. Molecular network analysis of phosphotyrosine and lipid metabolism in hepatic PTP1b deletion mice. Integr Biol. 2013;5:940–963. doi: 10.1039/c3ib40013a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforova VJ, Giesbertz P, Wiemer J, Bethan B, Looser R, Liebenberg V, Ruiz Noppinger P, Daniel H, Rein D. Glyoxylate, a new marker metabolite of type 2 diabetes. J Diabetes Res. 2014;2014:685204. doi: 10.1155/2014/685204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberbach A, Blüher M, Wirth H, Till H, Kovacs P, Kullnick Y, Schlichting N, Tomm JM, Rolle-Kampczyk U, Murugaiyan J, et al. Combined proteomic and metabolomic profiling of serum reveals association of the complement system with obesity and identifies novel markers of body fat mass changes. J Proteome Res. 2011;10:4769–4788. doi: 10.1021/pr2005555. [DOI] [PubMed] [Google Scholar]
- Paull EO, Carlin DE, Niepel M, Sorger PK, Haussler D, Stuart JM. Discovering causal pathways linking genomic events to transcriptional states using Tied Diffusion Through Interacting Events (TieDIE) Bioinformatics. 2013;29:2757–2764. doi: 10.1093/bioinformatics/btt471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol. 1992;54:885–909. doi: 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- Piñero J, Queralt-Rosinach N, Bravo À, Deu-Pons J, Bauer-Mehren A, Baron M, Sanz F, Furlong LI. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford) 2015;2015:bav028. doi: 10.1093/database/bav028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- Ramachandran P, Henderson NC. Antifibrotics in chronic liver disease: tractable targets and translational challenges. Lancet Gastroenterol Hepatol. 2016;1:328–340. doi: 10.1016/S2468-1253(16)30110-8. [DOI] [PubMed] [Google Scholar]
- Ramsey SA, Knijnenburg TA, Kennedy KA, Zak DE, Gilchrist M, Gold ES, Johnson CD, Lampano AE, Litvak V, Navarro G, et al. Genome-wide histone acetylation data improve prediction of mammalian transcription factor binding sites. Bioinformatics. 2010;26:2071–2075. doi: 10.1093/bioinformatics/btq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razick S, Magklaras G, Donaldson IM. iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformatics. 2008;9:405. doi: 10.1186/1471-2105-9-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322:1539–1543. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Schattenberg JM, Schuchmann M. Diabetes and apoptosis: liver. Apoptosis. 2009;14:1459–1471. doi: 10.1007/s10495-009-0366-2. [DOI] [PubMed] [Google Scholar]
- Schrem H, Klempnauer J, Borlak J. Liver-enriched transcription factors in liver function and development. Part I: the hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol Rev. 2002;54:129–158. doi: 10.1124/pr.54.1.129. [DOI] [PubMed] [Google Scholar]
- Schrem H, Klempnauer J, Borlak J. Liver-enriched transcription factors in liver function and development. Part II: the C/EBPs and D site-binding protein in cell cycle control, carcinogenesis, circadian gene regulation, liver regeneration, apoptosis, and liver-specific gene regulation. Pharmacol Rev. 2004;56:291–330. doi: 10.1124/pr.56.2.5. [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- Thiele I, Swainston N, Fleming RM, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, et al. A community-driven global reconstruction of human metabolism. Nat Biotechnol. 2013;31:419–425. doi: 10.1038/nbt.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titchenell PM, Chu Q, Monks BR, Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun. 2015;6:7078. doi: 10.1038/ncomms8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C, Chen H, Monks BR, Chen J, Rabinowitz JD, Birnbaum MJ. Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab. 2016;23:1154–1166. doi: 10.1016/j.cmet.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncbag N, Braunstein A, Pagnani A, Huang SS, Chayes J, Borgs C, Zecchina R, Fraenkel E. Simultaneous reconstruction of multiple signaling pathways via the prize-collecting steiner forest problem. J Comput Biol. 2013;20:124–136. doi: 10.1089/cmb.2012.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncbag N, Gosline SJ, Kedaigle A, Soltis AR, Gitter A, Fraenkel E. Network-based interpretation of diverse high-throughput datasets through the Omics Integrator software package. PLoS Comput Biol. 2016;12:e1004879. doi: 10.1371/journal.pcbi.1004879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villaveces JM, Jiménez RC, Porras P, Del-Toro N, Duesbury M, Dumousseau M, Orchard S, Choi H, Ping P, Zong NC, et al. Merging and scoring molecular interactions utilising existing community standards: tools, use-cases and a case study. Database (Oxford) 2015;2015:bau131. doi: 10.1093/database/bau131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Foretz M. Revisiting the mechanisms of metformin action in the liver. Ann Endocrinol (Paris) 2013;74:123–129. doi: 10.1016/j.ando.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis. 2014;5:e996. doi: 10.1038/cddis.2013.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ. Caspase-10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA. 2001;98:13884–13888. doi: 10.1073/pnas.241358198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Shah OJ, Hunter T. The transcriptional coactivators p/CIP and SRC-1 control insulin resistance through IRS1 in obesity models. PLoS ONE. 2012;7:e36961. doi: 10.1371/journal.pone.0036961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AS, Kang L, Wasserman DH. The extracellular matrix and insulin resistance. Trends Endocrinol Metab. 2015a;26:357–366. doi: 10.1016/j.tem.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AS, Kang L, Zheng J, Grueter C, Bracy DP, James FD, Pozzi A, Wasserman DH. Integrin α1-null mice exhibit improved fatty liver when fed a high fat diet despite severe hepatic insulin resistance. J Biol Chem. 2015b;290:6546–6557. doi: 10.1074/jbc.M114.615716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E, Dietze P, Karas H, Knüppel R. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996;24:238–241. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, et al. HMDB 3.0–The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Williams EG, Dubuis S, Mottis A, Jovaisaite V, Houten SM, Argmann CA, Faridi P, Wolski W, Kutalik Z, et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell. 2014;158:1415–1430. doi: 10.1016/j.cell.2014.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K, Benyamini T, Liebermeister W, Ruppin E, Shlomi T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics. 2010;26:i255–i260. doi: 10.1093/bioinformatics/btq183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Barrere-Cain RE, Yang X. Nutritional systems biology of type 2 diabetes. Genes Nutr. 2015;10:481. doi: 10.1007/s12263-015-0481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Lu Y, Ficarro SB, Adelmant G, Jiang W, Luckey CJ, Marto JA. Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat Commun. 2013;4:2171. doi: 10.1038/ncomms3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.