

Abstract
Lewy body disorders are characterized by the emergence of α-synucleinopathy in many parts of the central and peripheral nervous systems, including in the telencephalon. Dense α-synuclein+ pathology appears in regio inferior of the hippocampus in both Parkinson’s disease and dementia with Lewy bodies and may disturb cognitive function. The preformed α-synuclein fibril model of Parkinson’s disease is growing in use, given its potential for seeding the self-propagating spread of α-synucleinopathy throughout the mammalian brain. Although it is often assumed that the spread occurs through neuroanatomical connections, this is generally not examined vis-à-vis the uptake and transport of tract-tracers infused at precisely the same stereotaxic coordinates. As the neuronal connections of the hippocampus are historically well defined, we examined the first-order spread of α-synucleinopathy three months following fibril infusions centered in the mouse regio inferior (CA2 + CA3), and contrasted this to retrograde and anterograde transport of the established tract-tracers FluoroGold and biotinylated dextran amines (BDA). Massive hippocampal α-synucleinopathy was insufficient to elicit memory deficits or loss of cells and synaptic markers in this model of early disease processes. However, dense α-synuclein+ inclusions in the fascia dentata were negatively correlated with memory capacity. A modest compensatory increase in synaptophysin was evident in the stratum radiatum of cornu Ammonis in fibril-infused animals, and synaptophysin expression correlated inversely with memory function in fibril but not PBS-infused mice. No changes in synapsin I/II expression were observed. The spread of α-synucleinopathy was somewhat, but not entirely consistent with FluoroGold and BDA axonal transport, suggesting that variables other than innervation density also contribute to the materialization of α-synucleinopathy. For example, layer II entorhinal neurons of the perforant pathway exhibited somal α-synuclein+ inclusions as well as retrogradely labeled FluoroGold+ somata. However, some afferent brain regions displayed dense retrograde FluoroGold label and no α-synuclein+ inclusions (e.g. medial septum/diagonal band), supporting the selective vulnerability hypothesis. The pattern of inclusions on the contralateral side was consistent with specific spread through commissural connections (e.g. stratum pyramidale of CA3), but again, not all commissural projections exhibited α-synucleinopathy (e.g. hilar mossy cells). The topographical extent of inclusions is displayed here in high-resolution images that afford viewers a rich opportunity to dissect the potential spread of pathology through neural circuitry. Finally, the results of this expository study were leveraged to highlight the challenges and limitations of working with preformed α-synuclein fibrils.
Keywords: Parkinson’s disease; Lewy body; Dementia, -synuclein fibrils; Hippocampus; Tract-tracing; FluoroGold; Biotinylated dextran amines; Entorhinal cortex
1. Introduction
Parkinson’s disease is a systemic disorder affecting multiple parts of the brain and spinal cord (Del Tredici and Braak, 2016; Raudino and Leva, 2012) and many peripheral organs, such as the gastrointestinal tract (Fasano et al., 2015; Salat-Foix et al., 2011). The hallmark pathology in this condition is the eosinophilic Lewy body, a type of inclusion harboring ubiquitin and aggregated proteins such as fibrillar α-synuclein (Goedert et al., 2013; Spillantini et al., 1998). Some have speculated that the first brain regions to develop α-synuclein+ inclusions in prodromal Parkinson’s disease are the olfactory bulb/anterior olfactory nucleus (OB/AON) in the rostral telencephalon and the dorsal motor nucleus of the vagus in the medulla oblongata (Beach et al., 2009; Braak et al., 2003a; Braak et al., 2003b; Hawkes et al., 2007, 2009). According to Braak’s scheme, Lewy pathology does not appear within dopaminergic neurons of the ventral mesencephalon until stage III and may pass through the brain in a self-propagating manner by hijacking preexisting neuronal connections (Braak et al., 2003a; Braak et al., 2003b). By end stages, Lewy pathology finally encroaches upon the neocortex, and this may be associated with cognitive dysfunction (Aarsland et al., 2005; Bertrand et al., 2004; Braak et al., 2006a; Braak et al., 2005; Jellinger, 2000). Beach and colleagues have expanded upon Braak’s staging scheme by proposing a tiered system that accounts for cases in which Lewy pathology does not necessarily commence in the brainstem and is concentrated in the telencephalon (Beach et al., 2009). Telencephalic Lewy pathology is often associated with dementia (Armstrong et al., 2014; Kalaitzakis and Pearce, 2009; Mattila et al., 2000; Yang and Yu, 2016).
Estimates of the cumulative prevalence of dementia in Parkinson’s patients have ranged from 48% to 78% (Aarsland et al., 2003; Hely et al., 2005; Kalaitzakis and Pearce, 2009). Even in newly diagnosed patients, 24–36% of subjects exhibit some form of cognitive impairment (Muslimovic et al., 2005). Memory impairment in Lewy body disorders is correlated with α-synucleinopathy and/or tissue atrophy in hippocampal CA2/CA3, CA1, and the subiculum (Adamowicz et al., 2017; Beyer et al., 2013; Kalaitzakis et al., 2009), even in the absence of frank neurodegeneration in the hippocampus (Churchyard and Lees, 1997; Ince et al., 1991; Joelving et al., 2006). Of all the hippocampal subregions afflicted with α-synucleinopathy, the small CA2 field is the focus of the densest Lewy pathology in Parkinson’s disease (Bertrand et al., 2004; Churchyard and Lees, 1997; Flores-Cuadrado et al., 2016; Harding and Halliday, 2001; Mattila et al., 1999). It seems important that the CA2-predominant pattern also extends to dementia with Lewy bodies (Dickson et al., 1991; Dickson et al., 1994; Harding and Halliday, 2001; Marui et al., 2004), although the specific functional consequences of Lewy pathology commencing in CA2 have not been established.
It is not known if the materialization of α-synucleinopathy is sufficient by itself to cause memory deficits in Parkinson’s disease dementia and dementia with Lewy bodies. Although some have argued that Lewy pathology drives the cognitive dysfunction in Parkinson’s disease (Aarsland et al., 2005), others have argued that Lewy pathology does not predict memory loss (Colosimo et al., 2003; Parkkinen et al., 2005). Further complicating matters, almost 30% of Parkinson’s cases harbor concomitant Alzheimer’s pathology (Irwin et al., 2012). In addition, the presence of comorbid Alzheimer’s pathology is associated with worse prognosis (Irwin et al., 2017), although Alzheimer’s and Lewy pathology may predict dementia independently in Parkinson’s patients (Toledo et al., 2016). A causal link between Lewy pathology and dementia is difficult to assess in correlative human studies but has been scrutinized in animal models. As expected, memory loss and synaptic deficits are associated with Lewy-like pathology in the hippocampus of α-synuclein-overexpressing mice (Costa et al., 2012; Fujita et al., 2010; Goldberg et al., 2015; Hall et al., 2015; Lim et al., 2011; Paumier et al., 2013; Price et al., 2010; Rockenstein et al., 2014). Multiple investigators have also infused preformed, recombinant α-synuclein fibrils into the hippocampus in vivo (Guo et al., 2013; Hu et al., 2016; Luk et al., 2012a; Roostaee et al., 2013; Rutherford et al., 2015; Sacino et al., 2014a; Sacino et al., 2014b). For example, Luk and colleagues targeted the hippocampal formation with fibril infusions and reported inclusion pathology in this structure but did not measure behavioral deficits in those animals (Luk et al., 2012a). Sacino and colleagues also infused the hippocampal formation with fibrils, but did not measure memory deficits and reported little spread of Lewy pathology away from the infusion site in nontransgenic mice (Sacino et al., 2014b). Hu and colleagues reported that α-synuclein fibril infusions into the mouse hippocampus elicited Lewy-like inclusions and impairments in working memory during Y-maze testing (Hu et al., 2016). Although the latter findings suggest that α-synuclein fibril infusions can be employed to mimic the dementia in Lewy body disorders, it is not clear whether α-synucleinopathy was sufficient to elicit memory impairments because the Lewy-like pathology was also accompanied by hippocampal cell loss (Hu et al., 2016).
In accordance with Braak’s hypothesis on the self-propagating spread of Lewy pathology, embryonic brain tissue can acquire Lewy pathology over the course of years following transplantation into Parkinson’s patients’ brains (Braak and Del Tredici, 2008; Kordower et al., 2008; Li et al., 2008). Furthermore, many experimental studies suggest direct cell-to-cell spread of α-synucleinopathy (Braak et al., 2003a; Braak et al., 2003b; Del Tredici and Braak, 2016; Jucker and Walker, 2013; Lee et al., 2010; Lee et al., 2011; Luk et al., 2012a; Luk et al., 2012b; Mason et al., 2016; Masuda-Suzukake et al., 2013; Osterberg et al., 2015; Paumier et al., 2015; Ulusoy et al., 2013; Volpicelli-Daley et al., 2014b; Walker et al., 2006). Thus, the spread of α-synucleinopathy in the in vivo preformed fibril model is often—but not always—assumed to transpire via afferent and efferent neural circuits that either directly or indirectly connect with the infusion site (Bieri et al., 2017; Blumenstock et al., 2017; Chu and Kordower, 2015; Luk et al., 2012a; Masuda-Suzukake et al., 2013; Paumier et al., 2015; Rey et al., 2016; Sacino et al., 2014a; Sacino et al., 2014b; Shimozawa et al., 2017; Thakur et al., 2017). Some authors have specifically argued that the transmission of fibril-induced pathology does not follow established neuroanatomical links in transgenic α-synuclein over-expressors (Sorrentino et al., 2017). However, the topography of the spread is typically reported at low anatomical resolution and not vis-à-vis the uptake and transport of established retrograde and anterograde tract-tracers infused at precisely the same stereotaxic coordinates. Thus, the potential spread of α-synucleinopathy through neuronanatomical circuitry in vivo remains poorly understood.
For a rigorous examination of the potential transfer of α-synucleinopathy through preexisting neural networks, the hippocampus is an excellent target site. Hippocampal anatomy is defined better than almost all other brain regions, having been scrutinized since the work of Ramón y Cajal, Lorente de Nó, and Theodor Blackstad, among others (Andersen, 2007; Blackstad, 1956; Risold and Swanson, 1996; Swanson and Cowan, 1977). The hippocampus is heterogeneous and has a palisade-like structure, features that facilitate the dissection of topographically restricted neuronal circuitry. In our previous study, we demonstrated that 1 h-long sonication of α-synuclein fibrils was more productive in eliciting transmission of dense α-synucleinopathy than shorter-duration sonications with a probe (Mason et al., 2016). Thus, it seemed appropriate to examine in greater depth than before the transmission of α-synuclein pathology following infusions of 1 h-sonicated preformed fibrils into the hippocampal formation. On this backdrop, the primary goal of the present study was to compare the pattern of inclusion formation in animals infused unilaterally with fibrils to the uptake and transport of retrograde and anterograde tract-tracers infused at the same stereotaxic coordinates. A secondary goal was to examine the effects of bilateral hippocampal α-synucleinopathy on memory deficits. Third, the impact of fibril infusions on the numbers of α-synuclein+ inclusions, NeuN+ neurons, and Hoechst+ cell bodies was measured in seven limbic brain regions. Fourth, the potential loss of two synaptic markers was assessed. While tackling these questions, many of the strengths and limitations of the fibril model became evident, and here we take advantage of an impressive body of previous work on hippocampal anatomy to discuss some of the shortcomings of the model.
2. Methods
2.1. Animals and surgeries
All experimental procedures were approved by the Duquesne University IACUC (protocol no. 1403-03) and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Seventy male CD1 mice (Charles River, Wilmington MA) were housed in a 12:12 photoperiod with ad libitum access to food and water. At 2.5–4 months of age, mice were deeply anesthetized with 2% vaporized isoflurane and infused with the solutions listed in Table 1 at the following stereotaxic coordinates: AP −2.5 mm, ML +/− 2.8 mm, and DV −3.6 mm from Bregma and the top of the skull. These coordinates targeted the dorsal portions of regio inferior in our pilot study with infusions of blue dye. The preformed, recombinant fibrils made from wildtype mouse α-synuclein were synthesized according to previously published protocols (Volpicelli-Daley et al., 2014b; Volpicelli-Daley et al., 2011), and were sonicated for 1 h in a waterbath (Bransonic series model M1800, Branson Ultrasonics Corporation, Danbury CT) immediately before use, as described in the Supplemental Information section of our previous report (Mason et al., 2016). In the latter series of studies, the 1 h waterbath sonication protocol led to the densest Lewy-like pathology compared to all other sonication parameters. Control animals were infused in parallel with an equal volume of phosphate-buffered saline (PBS).
Table 1.
Animals and treatments.
| Target site | Number of mice | Infusate | Dose per hemisphere | Survival |
|---|---|---|---|---|
| Bilateral hippocampal CA2 + CA3 | 10 | PBS | 1 μL PBS | 3 months |
| Bilateral hippocampal CA2 + CA3 | 10 | Fibrils | 5 μg fibrils in 1 μL PBS | 3 months |
| Unilateral hippocampal CA2 + CA3 | 9 | PBS | 1 μL PBS | 3 months |
| Unilateral hippocampal CA2 + CA3 | 9 | Fibrils | 5 μg fibrils in 1 μL PBS | 3 months |
| Unilateral hippocampal CA2 + CA3 | 5 | Fibrils + 10 kDa BDA | 5 μg fibrils in 1 μL PBS
5% BDA in 0.2 μL PBS |
3 months |
| Unilateral hippocampal CA2 + CA3 | 5 | Fibrils + FluoroGold | 5 μg fibrils in 1 μL PBS
1.5% FG in 0.2 μL PBS |
3 months |
| Unilateral hippocampal CA2 + CA3 | 4 | FluoroGold | 1.5% FG in 0.2 μL PBS | 7 days |
| Unilateral hippocampal CA2 + CA3 | 5 | 3 kDa BDA | 10% BDA in 0.2 μL PBS | 10 days |
| Unilateral hippocampal CA2 + CA3 | 4 | 10 kDa BDA | 5% BDA in 0.2 μL PBS | 7 days |
| Unilateral dorsal thalamus | 5 | Fibrils | 5 μg fibrils in 1 μL PBS | 3 months |
| Unilateral cortex | 4 | Fibrils | 5 μg fibrils in 1 μL PBS | 3 months |
In addition to hippocampal infusions, separate control animals were infused with fibrils in the cortex at the following coordinates, again following verification with blue dye: AP −2.5 mm, ML +2.8 mm, and DV −2.0 mm. A second group of control animals was infused in the thalamus at the following coordinates, following verification with blue dye: AP −2.5 mm, ML +2.2 mm, and DV −4.0 mm. Every attempt was made to minimize animal suffering. All mice subjected to surgical procedures were injected with buprenorphine immediately following surgery. Furthermore, lidocaine was applied to the incision for the following three days.
2.2. Behavioral analyses
Animals were subjected to memory and olfactory tests two and three months after bilateral infusion of the α-synuclein fibrils. For the former test, animals were subjected to a habituation phase consisting of 10 min spent in an open field (45 × 60 × 60 cm) (Cai et al., 2012). On the next day, the animals were subjected to a familiarization phase, during which two identical objects were fixed in opposite corners of the same arena, each 10 cm away from the wall. The familiarization phase lasted five minutes and was followed the next day by recognition testing, during which animals were exposed to only one familiar object from the day before and a second novel object. The novel object exploration ratio was defined as the time spent exploring the novel object as a fraction of total exploration time (i.e. novel + familiar). The head of the animal had to be within 4 cm of the object for exploration scoring, and the animal must have faced the object within a 45° angle. On the day after the novel object test, the animal was subjected to the novel place recognition test, in which one familiar object from the familiarization phase was moved to a new location in the arena. The novel place recognition ratio was defined as the time spent exploring the familiar object in the novel place as a fraction of total exploration time. In addition, the buried food test was performed to measure olfactory function, according to previously described methods (Lehmkuhl et al., 2014). On the first day, animals were exposed to a peanut on top of corncob bedding and the latency to contact the exposed peanut immediately following entry into the cage was measured. On the second day, the latency to find a peanut buried ~2 cm deep in clean corncob bedding was measured. Animals that showed no interest in the exposed peanut on day 1 were excluded from the analysis (n = 2 PBS-infused cases).
2.3. Thioflavin S staining
Brains were removed after the survival periods listed in Table 1 and cryoprotected in 30% sucrose in 10 mM phosphate-buffered saline (PBS). Free-floating brain sections were cut in the sagittal or coronal plane and stored in cryoprotectant at −20 °C (Watson et al., 1986). The Thioflavin S staining protocol was adapted from work by Paumier and colleagues (Paumier et al., 2015). Sections were mounted onto glass slides and dried. Slides were then immersed in 10 mM PBS (5 min), followed by 0.05% KMnO4/PBS (20 min). Following two rinses in PBS (2 min each), sections were immersed in 0.2% K2S2O5/0.2% oxalic acid/PBS (~1 min). After five PBS rinses (2 min each), slides were immersed in freshly filtered 0.0125% Thioflavin S/40% EtOH/60% PBS for 3 min in the dark. This was followed by immersion in 50% EtOH/50% PBS (10 min) and again by four rinses in PBS (5 min each). Next, nuclei were stained with the Hoechst reagent (0.003 μM; Hoechst 33258, bisBenzimide) in 0.3% Triton X-100, followed by a final 5-min rinse in water. Stained sections were then dried, coverslipped, and viewed with epifluorescent microscopy (Olympus IX73, Pittsburgh PA).
2.4. Immunostaining and microscopy
Standard immunofluorescence procedures were performed with antibodies listed in Table 2. Briefly, a 1-in-5 series of adjacent free-floating sagittal or coronal brain sections was collected and blocked in 50% Odyssey Block (LI-COR, Lincoln, NE) in 10 mM PBS with 0.3% Triton X-100 for 1 h. This was followed by overnight exposure at 4 °C to primary antibodies diluted in the same blocking solution. On the next day, sections were washed in three rinses of PBS and exposed to secondary antibodies for 1 h at room temperature. Some sections were exposed to the fluorescent nuclear markers Hoechst (0.005 μM) or DRAQ5 (0.5 μM) during the secondary antibody incubation period. This incubation period was followed by three washes in PBS. Washed sections were then mounted onto glass slides and coverslipped. Immunostaining was visualized on the epifluorescent microscope mentioned above and large, stitched images were generated using 4× or 10× objectives. Higher-resolution images were captured with 40× or 100× objectives. A blinded observer manually counted the number of inclusions, Hoechst+ nuclei (all cells), and NeuN+ nuclei (neurons only) with a 20× objective (field of view = 447.1 μm × 337.0 μm in area) through CA1, CA2, CA3, the granular and molecular layers of the dentate gyrus, the presubiculum, and the posteromedial cortical amygdala. Sections from control and experimental groups were always processed, photographed, and analyzed in parallel. Photomicrographs for each hippocampal subregion were captured at the same anatomical level for every brain. If this specific subregion was not present in the 1-in-5 sagittal series due to loss of tissue during batch processing, those animals were excluded from analyses.
Table 2.
Antibodies.
| Primary antibody | Host | Company | Cat. no. | Lot no. | Dilution |
|---|---|---|---|---|---|
| Anti-pSer129 (aa 124–134; AYEMPSpEEGYQ conjugated to KLH via a C-terminal Cysteine) | Mouse | BioLegend | 825,701 | B213120 | 1:1000 |
| Anti-pSer129 (aa 127–131; MPSpEE) | Rabbit | Abcam | Ab59264 | GR52476-25 | 1:300 |
| Anti-Synaptophysin | Rabbit | Millipore | YE269 | JBC1794853 | 1:1000 |
| Anti-Synapsin I/II | Rabbit | Synaptic Systems | 106,002 | 22 | 1:500 |
| Anti-K48-linked ubiquitin | Rabbit | Millipore | 07307 | 2,299,608 | 1:500 |
| Anti-NeuN | Guinea Pig | Millipore | ABN90 | 2,031,353 | 1:6000 |
| Secondary antibody | Host | Company | Cat. no. | Lot no. | Dilution |
|---|---|---|---|---|---|
| Anti-mouse @ 488 | Donkey | Life Technologies | A21202 | 1423052 | 1:700 |
| Anti-mouse @ 546 | Donkey | Life Technologies | A10036 | 1736962 | 1:700 |
| Anti-mouse @ 647 | Donkey | Jackson ImmunoResearch | 715-605-150 | 127706 | 1:700 |
| Anti-rabbit @ 790 | Donkey | Jackson ImmunoResearch | 711-655-152 | 115149 | 1:700 |
| Anti-rabbit @ 790 | Donkey | Jackson ImmunoResearch | 711-655-152 | 129291 | 1:1000 |
| Anti-rabbit @ 546 | Goat | Life Technologies | A11035 | 1579044 | 1:700 |
| Anti-rabbit @ 488 | Donkey | Jackson ImmunoResearch | 711-545-152 | 120705 | 1:700 |
| Anti-guinea pig @ 488 | Donkey | Jackson ImmunoResearch | 706-545-148 | 108077 | 1:1000 |
| Anti-guinea pig @ 647 | Donkey | Jackson ImmunoResearch | 706-605-148 | 123960 | 1:1000 |
A high-sensitivity, low-resolution 16-bit infrared imager (Odyssey Classic, LI-COR Biosciences) was used to measure synaptophysin and synapsin I/II immunostaining in the hippocampal formation using ImageStudio Lite software (Version 5.2, LI-COR). For the latter two measurements, the boundaries of CA2/CA3 and the dentate gyrus were traced by a blinded observer in all sagittal sections containing the temporal pole of the hippocampus (i.e. the infusion epicenter) in the 1-in-5 section series. Next, background fluorescence was subtracted from the total fluorescent signal within the enclosed target region of interest and calculated with the formula (IB/AB) × AX, where IB is the total fluorescence intensity of the background region, AB is the area of the background region, and AX is the area of the target region of interest. This background fluorescence intensity was calculated on control brain sections treated with all solutions except for the primary antibodies, which always led to loss of staining in all of our assays. In addition, our previous work supports the specificity of the primary antibodies (Mason et al., 2016).
FluoroGold-injected cases were labeled with DRAQ5 to delineate cytoarchitectonic boundaries, whereas BDA cases were labeled with Hoechst or DRAQ5. FluoroGold was visualized with UV illumination and BDA was visualized after incubation in streptavidin-conjugated fluorophores (1:500; AlexaFluor 488 or 546, ThermoFisher Scientific, Waltham, MA) in 0.3% Triton X-100 in 10 mM PBS for 1 h at room temperature, followed by three washes, tissue mounting, and slide coverslipping.
For the delineation of anatomical boundaries, the following rodent brain atlases were consulted: Paxinos and Franklin’s “The Mouse Brain in Stereotaxic Coordinates” (Franklin and Paxinos, 2013), Swanson’s “Brain Maps III: Structure of the Rat Brain (Swanson, 2004), and the Richard Allen Mouse Brain Atlas online portal (2017).
2.5. Statistics and blinding
Data are presented in figures as unnormalized, raw numbers and were analyzed by the two-tailed Student’s t-test, two-tailed Mann-Whitney U test, or a two-tailed Pearson correlation analysis (GraphPad Prism, Version 6.0). Alpha was always set at 0.05. All histological and behavioral measurements were performed by blinded investigators. Animals that died before behavioral testing or sacrifice were excluded from analyses.
3. Results
3.1. α-synuclein fibril infusions into the hippocampus do not elicit behavioral deficits within three months
In order to model the archicortical Lewy pathology in Parkinson’s disease dementia and dementia with Lewy bodies, we infused PBS or α-synuclein fibrils in Cajal’s regio inferior (CA2 and CA3 of Lorente de Nó) (Blackstad, 1956). Fibrils were infused in both hemispheres because unilateral hippocampal injury models are not as likely to result in behavioral impairments as bilateral models (Li et al., 1999). All animals were tested and sacrificed within three months postinfusion, because 1) Hu et al. reported working memory deficits two months after hippocampal fibril infusions (Hu et al., 2016), and 2) we attempted to limit the neuroanatomical spread of pathology to first-order circuitry only, as Lewy-like pathology becomes too widespread at longer survival times to make precise conclusions about the route of transmission (Mason et al., 2016; Paumier et al., 2015; Rey et al., 2016). The novel object and novel place recognition tests were performed at two and three months postinfusion. There was no evidence of memory impairments in animals infused with fibrils at either time point (Fig. 1A–B, D–E).
Fig. 1.
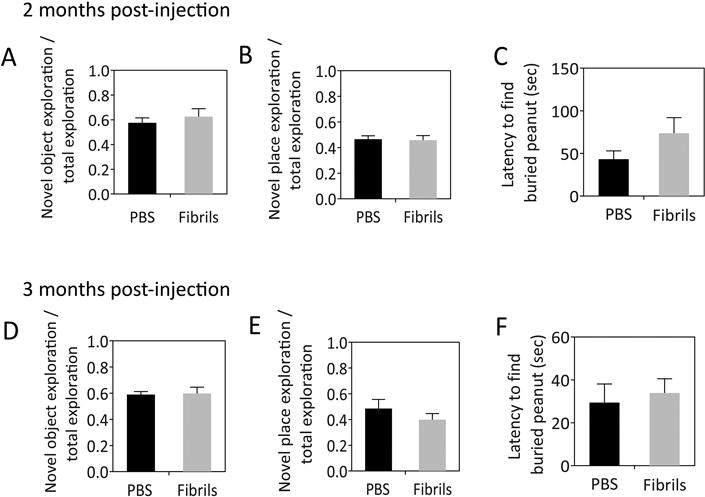
α-synuclein fibril infusions do not lead to memory or olfactory deficits within three months. CD1 mice were bilaterally infused with phosphate-buffered saline (1 μL PBS) or an equal volume of α-synuclein fibrils (5 μg) into regio inferior (CA2 + CA3) of the hippocampal formation. Two months postinfusion, animals were subjected to the novel object and novel place recognition tests for memory (A–B) and buried pellet test for olfaction (C). All three tests were repeated at three months postinfusion (D–F), immediately before sacrifice. In the memory tests, the time spent contacting a novel object in a familiar place or a familiar object in a novel place was divided by the total exploration time to yield a ratio. For the olfaction test, the latency to contact a peanut buried in clean corncob bedding was measured. All measurements were completed by a blinded investigator. n = 8–10 mice per group. The two-tailed t-test revealed no significant changes.
Previously, we employed the same 1 h waterbath sonication protocol and infused fibrils into the hippocampus of four mice in a small control study designed to optimize sonication parameters (Mason et al., 2016). Brains from those animals were restained and examined in the present study, and dense pathology was observed in their amygdalae. The amygdala is considered part of the extended olfactory network (Soudry et al., 2011) and the amygdalar α-synucleinopathy in Parkinson’s patients has been hypothesized to contribute to olfactory deficits (Harding et al., 2002b). Therefore, we also measured the latency to contact a peanut buried within cage bedding (Fig. 1C, F). According to this behavior test, no significant olfactory deficits were elicited by hippocampal fibril infusions. In addition, there were no significant differences between PBS and fibril-infused animals in the latency to contact an exposed peanut placed on top of the bedding (not shown), suggesting equivalent motivation to find food. Taken together, these results demonstrate that bilateral fibril infusions centered in CA2/CA3 do not elicit overt memory or smell impairments within three months postinfusion.
3.2. α-synuclein fibril infusions into the hippocampus elicit dense inclusions in the limbic telencephalon
Despite not exhibiting a behavioral syndrome, animals sacrificed at three months following fibril infusions exhibited massive somal and neuritic inclusions in the hippocampal formation, as measured by immunostaining for pathologically phosphorylated α-synuclein (pSer129; Fig. 2A–C), an established marker of Lewy pathology in human postmortem tissue (Anderson et al., 2006; Fujiwara et al., 2002). Three distinct sagittal levels from two brains are displayed in Fig. 2—one infused with PBS and one infused with fibrils. The anatomical sublocalization of pathology, its morphological features, and the distinction of pSer129 label from background staining can be judged by the reader by downloading higher resolution versions of this and other histology figures at the following link, either in the original Adobe Illustrator form (preferred) or in TIFF format (viewable without special software): Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0. In all figures, background staining was brightened to aid the visualization of cytoarchitectonic boundaries (e.g. see background staining in PBS-infused case in Fig. 2A–C). The disturbance of tissue in the overlying cortex in Fig. 2 identifies the path of the needle in both animals (white arrows). A quantitative analysis of the number of pSer129+ structures revealed a dramatic increase in inclusion numbers in fibril-infused animals, including in extrahippocampal structures, such as the posteromedial cortical nucleus of the amygdala and the presubiculum (Fig. 2D). By far the densest pathology was evident in the dentate gyrus (compare Y-axes in Fig. 2D). Although there were outliers among the fibril-infused groups, we have not deleted those animals and shown all the individual values on the boxplot graphs in Fig. 2D to illustrate the full variance of the data. Similarly high variability in inclusion counts was also evident in our previous work (Mason et al., 2016). We leveraged this variability and performed linear regression analyses to test for correlations between inclusion counts and each of the behavioral measures (Table 4). These analyses revealed a significant positive correlation between inclusion numbers in the molecular layer of the dentate gyrus and the latency to find a buried peanut at two months postinfusion. This effect disappeared at three months postinfusion, perhaps because the animals became accustomed to the presence of a buried peanut somewhere in the cage by the second testing round, as is evident from the reduction in raw latency values in Fig. 1F compared to Fig. 1C. Inclusion counts in the granule cell layer of the dentate gyrus were negatively correlated with the novel object exploration ratio at two months but not three months postinfusion. Inclusion counts in the molecular layer of the dentate gyrus were negatively correlated with the novel place exploration ratio at three months postinfusion. These results suggest that higher α-synucleinopathy in the dentate gyrus is positively associated with olfactory dysfunction and negatively associated with memory function. The switch in direction of the statistically significant correlations of inclusion counts with olfactory versus memory function is unlikely to be coincidental, as they are all consistent with an association between α-synucleinopathy and poor neurological outcomes.
Fig. 2.
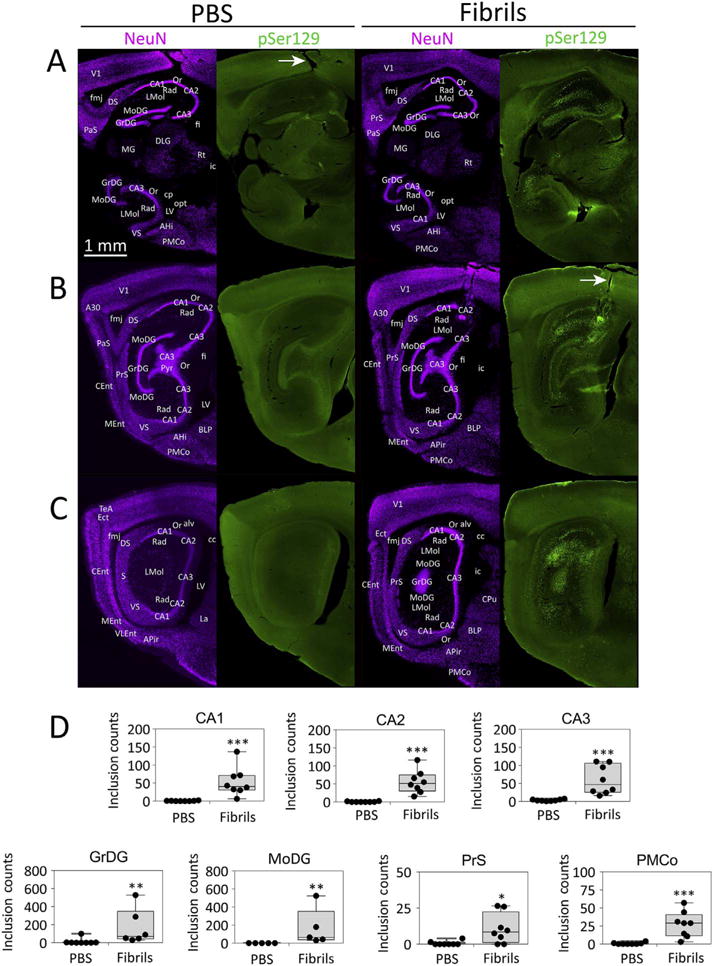
α-synuclein fibril infusions lead to the emergence of dense inclusions in the hippocampal formation. CD1 mice were bilaterally infused with phosphate-buffered saline (1 μL PBS) or an equal volume of α-synuclein fibrils (5 μg) into regio inferior (CA2 + CA3) of the hippocampal formation. Three months later, sagittal brain sections were collected and stained with antibodies against the neuronal nuclear marker NeuN and pathologically phosphorylated α-synuclein (mouse anti-pSer129). Stitched images of immunostained sections at three sagittal levels of the hippocampal formation in one PBS-infused mouse and one fibril-infused mouse are displayed in A–C. Note that there may be slight imperfections at the boundaries of the computerized stitched images. The PBS-infused case is included to show the extent of background staining with the phospho-Ser129 antibody. The disruption of tissue in the overlying cortex reveals the path of the needle in panel A or B (white arrows). The adjacent NeuN and pSer129 images are from the same section viewed in different fluorescent channels; therefore, the anatomical labels are only placed on the NeuN image. The boxplots in panel D show a dramatic increase in the number of pSer129+ inclusions in Ammon’s horn (CA1, CA2, and CA3), the stratum granulosum of the dentate gyrus (GrDG), the molecular layer of the dentate gyrus (MoDG), the presubiculum (PrS), and the posteromedial cortical amygdala (PMCo). The units shown on the Y axes are raw, unnormalized counts. **p ≤ 0.01, ***p ≤ 0.001 for PBS versus fibrils; two-tailed Mann-Whitney U test. n = 5–8 mice per group. For abbreviations, please consult Table 3. A higher-resolution version of this figure is necessary to view the label and can be downloaded from the following links: Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0.
Table 4.
Two-tailed correlation matrix for the number of inclusions in limbic regions versus olfactory and memory function.
| CA1 | CA2 | CA3 | GrDG | MoDG | PMCo | PrS | |
|---|---|---|---|---|---|---|---|
| Latency to find buried peanut at 2 months | r = 0.2713 | r = 0.1644 | r = 0.2356 | r = 0.03041 | r = 0.9710 | r = −0.0042 | r = 0.3093 |
| p = 0.5157 | p = 0.6973 | p = 0.5744 | p = 0.9375 | p = 0.0059** | p = 0.9921 | p = 0.4996 | |
| Latency to find buried peanut at 3 months | r = −0.5920 | r = −0.4976 | r = −0.5396 | r = −0.3916 | r = −0.4420 | r = −0.5203 | r = −0.3456 |
| p = 0.1614 | p = 0.2558 | p = 0.2113 | p = 0.4426 | p = 0.4561 | p = 0.2313 | p = 0.5022 | |
| Novel object exploration ratio at 2 months | r = −0.4187 | r = −0.5720 | r = −0.3107 | r = −0.8162 | r = −0.5528 | r = 0.3658 | r = 0.3743 |
| p = 0.3019 | p = 0.1385 | p = 0.4538 | p = 0.0251* | p = 0.3339 | p = 0.3728 | p = 0.4082 | |
| Novel place exploration ratio at 2 months | r = 0.4428 | r = 0.3640 | r = 0.3536 | r = −0.0155 | r = 0.2014 | r = 0.1986 | r = 0.3851 |
| p = 0.2718 | p = 0.3754 | p = 0.3902 | p = 0.9736 | p = 0.745 | p = 0.6373 | p = 0.3937 | |
| Novel object exploration ratio at 3 months | r = 0.4628 | r = 0.3688 | r = 0.2697 | r = −0.1681 | r = 0.6115 | r = 0.4454 | r = 0.5994 |
| p = 0.2957 | p = 0.4156 | p = 0.5587 | p = 0.7503 | p = 0.2731 | p = 0.3165 | p = 0.2086 | |
| Novel place exploration ratio at 3 months | r = 0.3923 | r = 0.3756 | r = 0.4631 | r = −0.1895 | r = −0.8981 | r = 0.2348 | r = 0.1948 |
| p = 0.3364 | p = 0.3592 | p = 0.2479 | p = 0.6840 | p = 0.0384* | p = 0.5756 | p = 0.6755 |
3.3. α-synuclein fibril infusions into the hippocampus do not elicit cell loss
Several studies suggest that there is no neuron loss in CA1, CA2/CA3, the dentate gyrus, or subiculum in Parkinson’s disease (Churchyard and Lees, 1997; Ince et al., 1991; Joelving et al., 2006). However, there is selective neuron loss in lower presubiculum pyramidal neurons of the hippocampal formation in dementia with Lewy bodies (Harding et al., 2002a). Hu and colleagues reported apoptosis in the hippocampus two months following fibril infusions into this structure (Hu et al., 2016). In order to compare neuronal viability in our model to that of previous studies and to the human α-synucleinopathies, we counted the numbers of NeuN+ and Hoechst+ cells in many subregions of the hippocampal formation and in the amygdala (Fig. 3). The results clearly show that the α-synucleinopathy was not accompanied by any neuron loss (i.e. NeuN+ cell counts) or overall cell loss (i.e. Hoechst+ cell counts), including in the presubiculum. The low variance of these data confirm that our definitions of anatomical substructures are consistent across animals and that the variance in inclusion counts in Fig. 2 did not arise during tissue processing and imaging. Considered together with the behavioral findings and inclusion count data, these results demonstrate that the bilateral infusion protocol elicits intense α-synucleinopathy in the hippocampal formation and surrounding limbic allocortex but that this is insufficient to elicit either cell loss or memory impairments.
Fig. 3.
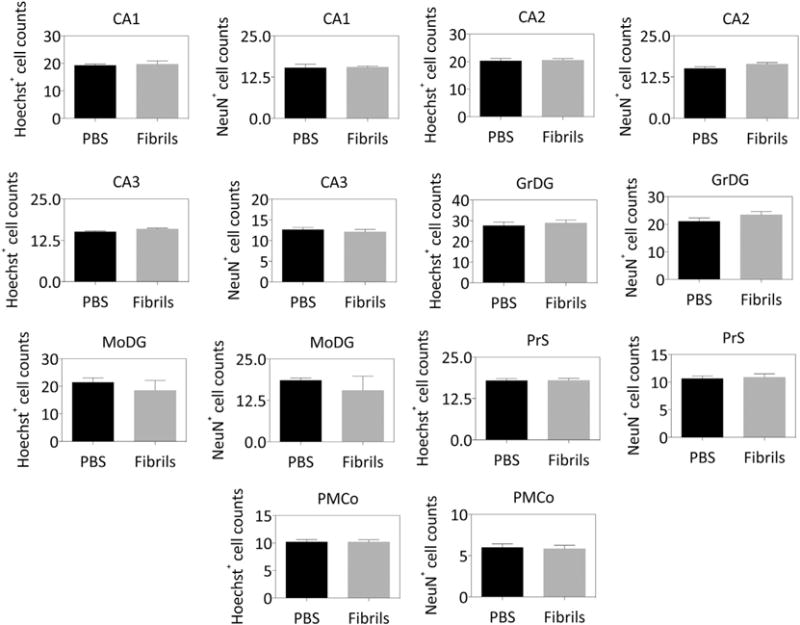
α-synuclein fibril infusions do not elicit any cell or neuron loss in the hippocampal formation. CD1 mice were bilaterally infused with phosphate-buffered saline (1 μL PBS) or an equal volume of α-synuclein fibrils (5 μg) into regio inferior (CA2 + CA3) of the hippocampal formation. Three months later, sagittal brain sections were collected and stained with the nuclear marker Hoechst and antibodies against the neuronal marker NeuN. A blinded observer counted the overall number of cells (Hoechst+ counts) and specifically the neuronal cells (NeuN+ counts) in Ammon’s horn (CA1, CA2, and CA3), the stratum granulosum of the dentate gyrus (GrDG), the molecular layer of the dentate gyrus (MoDG), the presubiculum (PrS), and the posteromedial cortical amygdala (PMCo). The units shown on the Y axes are raw, unnormalized counts. Graphed are the mean + SEM. Note the low variance of the cell counts, in contrast to the pSer129+ inclusions shown in Fig. 2. n = 5–8 mice per group. The two-tailed t-test revealed no significant changes.
3.4. α-synuclein fibril infusions into the hippocampus elicit a modest increase in synaptophysin immunoreactivity in CA2/CA3 without changes in hippocampal size
Beginning with Cajal’s predictions, a large body of literature suggests that loss of synapses may precede and even drive the loss of somata in neurodegenerative disorders (Ramón et al., 1991; Shankar and Walsh, 2009). Previous work has suggested that Lewy pathology may lead to defects in trafficking along axons (Chu et al., 2012; Volpicelli-Daley et al., 2014a) and is associated with synaptic dysfunction (Bellucci et al., 2016; Nikolaus et al., 2009; Phan et al., 2017; Schulz-Schaeffer, 2010). Therefore, we examined synaptophysin and synapsin I/II immunoreactivity in the hippocampus using an ultrasensitive, low-resolution infrared imager. We discovered that fibril infusions elicited a modest increase in synaptophysin levels in the temporal pole of CA2/CA3—but not the dentate gyrus—at three months postinfusion (Fig. 4A). These unexpected observations were confirmed by an independent, second blinded observer (not shown). Levels of the second synaptic marker, synapsin I/II, were not similarly affected (Fig. 4A). The images displayed in Fig. 4C reveal that this increase was concentrated in the stratum radiatum adjacent to the CA3 stratum pyramidale (white arrows), close to the infusion epicenter. A two-tailed correlation matrix in Table 5 reveals that synaptophysin levels in Ammon’s horn were weakly negatively correlated with memory function in the novel object recognition test at three months after fibril infusions. Notably, PBS-infused animals failed to exhibit a similar correlation.
Fig. 4.
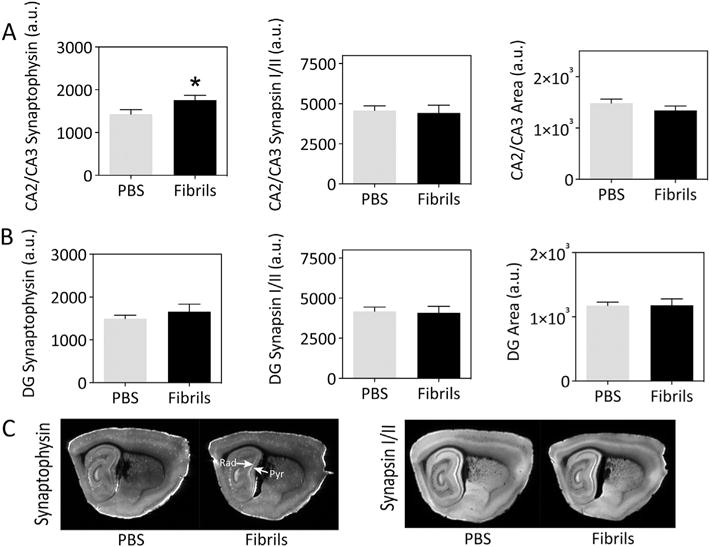
α-synuclein fibril infusions lead to a modest increase in synaptophysin levels in Ammon’s horn. CD1 mice were bilaterally infused with phosphate-buffered saline (1 μL PBS) or an equal volume of α-synuclein fibrils (5 μg) into regio inferior (CA2 + CA3) of the hippocampal formation. Three months later, sagittal brain sections were collected and stained with antibodies against synaptophysin (A) or synapsin I/II (B). Staining was visualized on a low-resolution, ultrasensitive Odyssey Imager and a blinded observer traced the boundaries of CA2/CA3 or the dentate gyrus. The arbitrary units (a.u.) shown on the Y axes are raw, unnormalized fluorescence levels for protein content or pixel values for area. Representative low-resolution images are illustrated in C. Graphed are the mean + SEM. n = 8–10 mice per group. **p ≤ 0.05, **p ≤ 0.01 for PBS versus fibrils; two-tailed t-test. Rad = stratum radiatum; Pyr = stratum pyramidale. A higher-resolution version of this figure can be downloaded from the following links: Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0.
Table 5.
Two-tailed correlation matrix for synaptophysin levels and memory function.
| PBS-treated mice | Ammon’s horn | Dentate gyrus |
|---|---|---|
| Novel object exploration ratio at 2 months | r = 0.0500 | r = 0.2336 |
| p = 0.9064 | p = 0.5777 | |
| Novel place exploration ratio at 2 months | r = 0.3710 | r = −0.1050 |
| p = 0.3256 | p = 0.7881 | |
| Novel object exploration ratio at 3 months | r = 0.3190 | r = 0.1340 |
| p = 0.4027 | p = 0.7311 | |
| Novel place exploration ratio at 3 months | r = 0.5796 | r = −0.0612 |
| p = 0.1321 | p = 0.8858 | |
| Fibril-treated mice | Ammon’s horn | Dentate gyrus |
| Novel object exploration ratio at 2 months | r = −0.1712 | r = −0.2862 |
| p = 0.6597 | p = 0.4919 | |
| Novel place exploration ratio at 2 months | r = −0.2368 | r = −0.3474 |
| p = 0.5396 | p = 0.3991 | |
| Novel object exploration ratio at 3 months | r = −0.7651 | r = −0.6784 |
| p = 0.0269* | p = 0.0938 | |
| Novel place exploration ratio at 3 months | r = −0.1703 | r = −0.2755 |
| p = 0.6613 | p = 0.5091 |
Joelving and coworkers failed to observe volume changes in any hippocampal subregion in Parkinson’s disease (Joelving et al., 2006), but the results of other studies suggest volume loss in the hippocampus in Parkinson’s disease or in dementia with Lewy bodies, especially in CA1, CA2, CA3, and the subiculum (Beyer et al., 2013; Camicioli, 2002; Churchyard and Lees, 1997; Cordato et al., 2000; Laakso et al., 1996). Although we had not collected any evidence of neurodegeneration, tissue atrophy can occur in the absence of any cell death, as it is only defined as a loss in parenchymal volume. Therefore, we also report the area of CA2/CA3 and the dentate gyrus in Fig. 4. However, the sizes of these structures were not affected by fibril infusions. The area of the entire hippocampal formation as a whole was also unaffected, as confirmed by a second blinded observer (not shown).
3.5. α-synuclein fibril infusions in the hippocampus elicit somal and neuritic inclusions that are ubiquitin- and Thioflavin-positive
In order to determine if the pathology observed in our model was Lewy-like in nature, we performed several control experiments. First, we used two antibodies raised against pSer129 (polyclonal and monoclonal; Fig. 5A). Slight differences in their epitopes are listed in Table 2. In the present study, the mouse pSer129 monoclonal antibody labeled more Lewy-like inclusions than the rabbit polyclonal. Nevertheless, all of the structures labeled with both antibodies exhibited the same morphology: 1) punctae or short neuritic threads and 2) somal staining confined to part of the cytoplasm or ensconcing the outer perimeter of the nucleus. Thus, the morphology of pSer129+ structures was distinct from the diffuse staining throughout the cytoplasm that one would expect of a soluble, nonaggregated protein, in agreement with previous work employing the fibril model (Blumenstock et al., 2017; Hu et al., 2016; Luk et al., 2012a; Mason et al., 2016; Paumier et al., 2015; Rey et al., 2016; Sacino et al., 2014b).
Fig. 5.
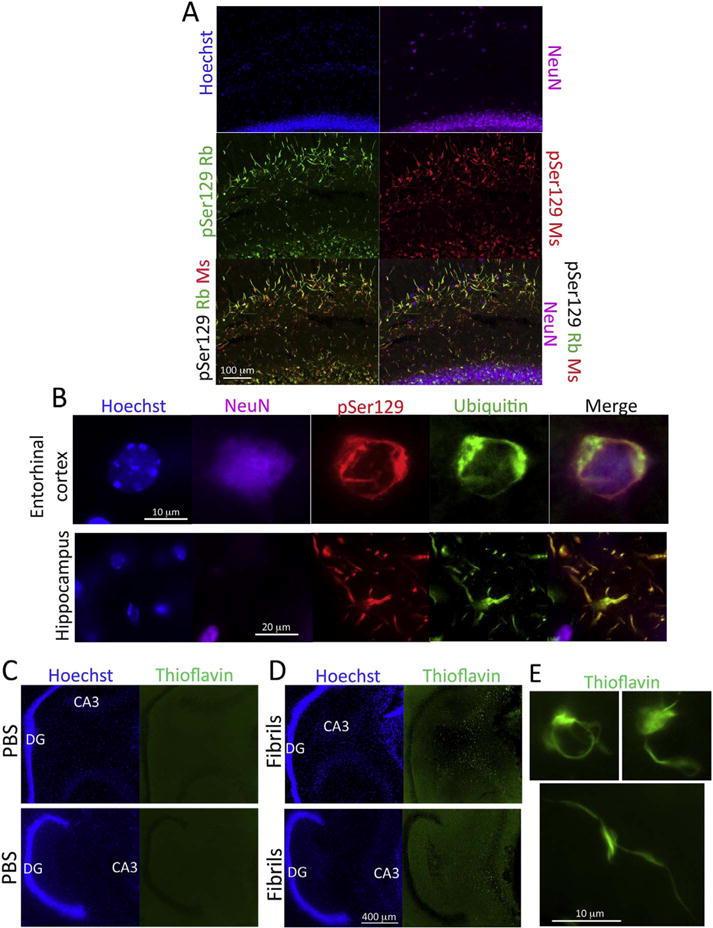
α-synuclein fibril infusions lead to the formation of ubiquitin and Thioflavin S-positive inclusions. CD1 mice were infused with α-synuclein fibrils (5 μg) into regio inferior (CA2 + CA3) of the hippocampal formation. Three months later, sagittal brain sections were collected and stained with mouse monoclonal and rabbit polyclonal antibodies against pathologically phosphorylated α-synuclein (pSer129) and guinea pig antibodies against NeuN (A). Nuclei were labeled with the Hoechst reagent. Note that the structures labeled in green in the merged image actually emit both red and green fluorescence, but that the greater green fluorescence intensity overwhelms the red label and the colocalization therefore does not appear yellow. (B) Quadruple staining with the Hoechst reagent and antibodies against pSer129, NeuN, and ubiquitin. (C–D) Sagittal sections from two representative vehicle-infused and two fibril-infused animals are shown after staining with Thioflavin S and the nuclear marker Hoechst. Note the dense Thioflavin staining in CA3 but not the fascia dentata. High magnification views of somal and neuritic Thioflavin S-stained inclusions in fibril-infused mice are shown in panel E. For abbreviations, please consult Table 3. A higher-resolution version of this figure is necessary to view the label and can be downloaded from the following links: Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Next, we dual-labeled brain tissue for pSer129 and the well-established Lewy pathology marker ubiquitin (Spillantini et al., 1998; Tofaris et al., 2003). As expected, both neurites and cell bodies were dual-labeled for these markers (Fig. 5B). However, more inclusions were labeled with α-synuclein antibodies than ubiquitin antibodies, as is also the case in postmortem tissue from Parkinson’s brains (Spillantini et al., 1998) and brain tissue from transgenic mice overexpressing α-synuclein (Lim et al., 2011). Many of the somal pSer129+ inclusions were housed in NeuN+ structures (Fig. 5B), demonstrating a neuronal origin of the pathology, consistent with previous reports (Mason et al., 2016; Rey et al., 2016), but not ruling out other cell types that may also house inclusions (Sorrentino et al., 2017).
At the site of infusion, dense Thioflavin S staining was observed in somata and neurites, as illustrated at both low magnification (Fig. 5C–D) and high magnification (Fig. 5E), suggesting that some inclusions were indeed amyloid in nature. As in our previous work, however, not all sites bearing pSer129+ pathology exhibited Thioflavin S label (Mason et al., 2016), as is evident from a close examination of the unlabeled granule cell layer of the dentate gyrus in two fibril-infused mice depicted in Fig. 5D (see higher resolution version of this figure at the abovementioned Dropbox link). This was unexpected because the stratum granulosum of the dentate gyrus sends massive efferents to CA3 and contained the densest pSer129+ inclusions according to the quantification and images in Fig. 2. Thus, Thioflavin S only stained inclusions near the infusion epicenter. These observations suggest that pSer129-labeled structures are at varying stages of maturity and confirm previous work that pSer129 antibodies label more inclusions than other Lewy markers (Osterberg et al., 2015; Spillantini et al., 1998).
3.6. Topographical pattern of α-synucleinopathy transmission after hippocampal infusions of fibrils
In order to study the topography of α-synucleinopathy transmission out of the hippocampal formation, we examined the pattern of inclusion formation in animals injected unilaterally with the fibrils. Some animals were also injected unilaterally with fibrils plus the retrograde tracer FluoroGold (Schmued and Fallon, 1986; Wessendorf, 1991) or the anterograde tracer 10 kDa biotinylated dextran amines (BDA) (Reiner et al., 2000; Veenman et al., 1992; Wouterlood and Jorritsma-Byham, 1993). We commenced this analysis by tracing the pSer129 immunostaining on top of stitched images of entire brain sections. Multiple cases were drawn in this fashion, because the inclusion count values in Fig. 2 demonstrated considerable variance. Two representative cases are displayed in Fig. 6A–B, and one unique case with dense pathology in additional structures beyond other cases is included in Fig. 6C. As expected, these schematics reveal dense α-synucleinopathy in Ammon’s horn at the site of infusion. Somal inclusions were observed in the pyramidal layers of Ammon’s horn and the stratum granulosum of the dentate gyrus. Neuritic threads were observed in the stratum radiatum and oriens as well the lacusonum moleculare and molecular layer of the area dentata. In general, the densest inclusions were formed in the stratum granulosum of the dentate gyrus, consistent with the raw inclusion count values in Fig. 2 (see Y-axes).
Fig. 6.
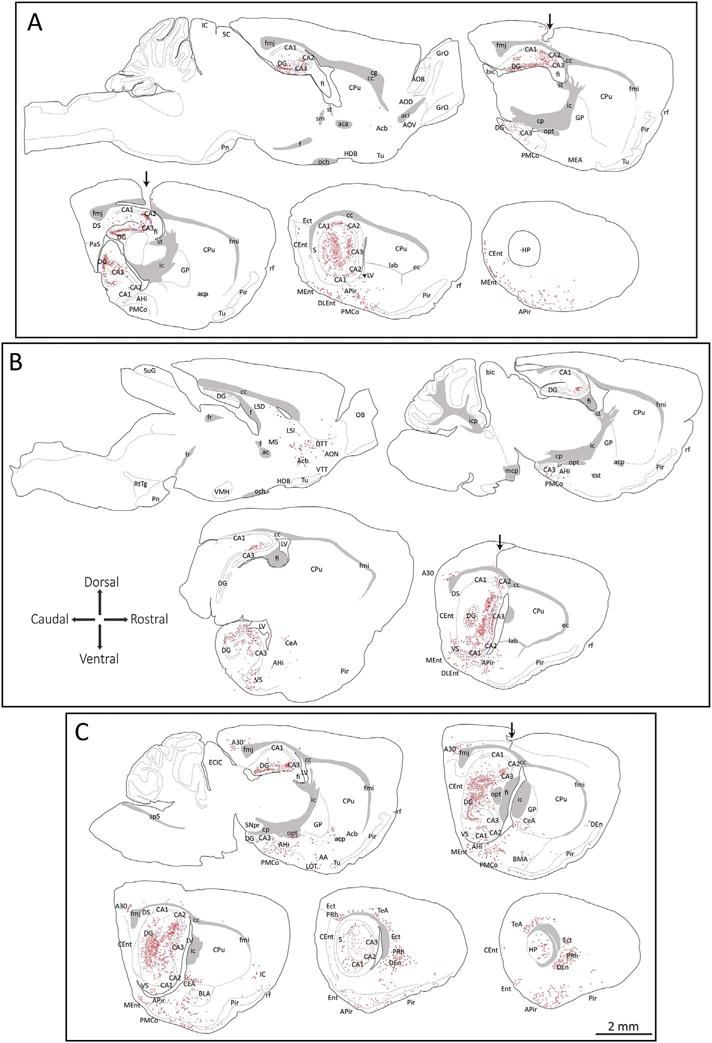
Schematic map of α-synucleinopathy transmission through the brain. CD1 mice were infused with α-synuclein fibrils (5 μg) into regio inferior (CA2 + CA3) of the hippocampal formation. Three months later, sagittal brain sections were collected and stained with antibodies against pathologically phosphorylated α-synuclein (mouse anti-pSer129) and NeuN. Nuclei were stained with the Hoechst reagent. Schematized somal (red dots) and neuritic (red flourishes) pSer129+ inclusions were drawn on top of high-quality stitched images of whole brain sections. Only the most obvious anatomical boundaries were traced, after consulting the NeuN and Hoechst labeling. Two representative cases are shown in panels A and B and the case in panel C is shown because it harbored unusually dense pathology, perhaps from diffusion into the overlying cortex. The path of the needle in all three cases is illustrated by a black arrow. For abbreviations, please consult Table 3. A higher-resolution version of this figure is necessary to view the label and can be downloaded from the following links: Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In addition to dense labeling of the hippocampus proper, moderate to dense inclusions were consistently located in the subiculum, the entorhinal cortex, the amygdalopiriform transition area, and the posteromedial cortical amygdala (Fig. 6). Sparse somal and/or neuritic inclusions were found in the nucleus accumbens, the intermediate lateral septum, and the olfactory peduncle. Most of the inclusions within the entorhinal cortex were housed within layer II and layer III somata, which project into the dentate gyrus and other hippocampal subfields such as CA3 and CA1 via the perforant or temporoammonic pathway (Cappaert et al., 2015; Witter, 2007; Witter et al., 1988).
In the animal with the densest inclusions, some cortical diffusion of fibrils may have transpired, as there were dense somal inclusions in deeper layers of the temporal association cortex and the dorsal endopiriform nucleus, unlike in the other cases (Fig. 6C). In this animal there were also dense inclusions in many nuclei of the amygdaloid complex, such as the central and basolateral nuclei, in the ectorhinal and perirhinal areas, and in the cingulate cortex. Although inclusions were formed in great quantities in this animal, the densest pathology was still concentrated in the limbic allocortex, especially the archicortex, as with all the other cases.
Images of the pSer129 label on which the schematics in Fig. 6 are based can be viewed in Supplementary Fig. S1. The center of the infusion in regio inferior is evident in Fig. S1A–B. These images reveal that somal inclusions in the entorhinal cortex are concentrated in superficial layers II and III, although sparse labeling of neurites was on occasion observed in deeper layers (Fig. S1E–F). There was dense labeling of fibers in the hilus (defined as CA4 by Lorente de Nó or the polymorphic zone (Scharfman and Myers, 2012)), (Fig. S1J, 1SR), in the area of the mossy fiber projection. Moderate to sparse pSer129+ inclusions were housed in the posteromedial cortical nucleus of the amygdala and amygdalopirifom transition area (Fig. S1G–H, S1S–T), the lateral septum and accumbens (Fig. S1K–L), and the cingulate cortex (Fig. S1O–P). In the animal with the densest pathology (schematically illustrated in Fig. 6C), there was also dense pSer129 label in the dorsal endopiriform nucleus (Fig. S1U–V).
In addition to the label presented schematically in Fig. 6, we observed pSer129+ inclusions in the laterodorsal tegmentum, locus coeruleus, dorsal and ventral subdivisions of the lateral septum, and posterior hypothalamic area (see coronal sections in Fig. 7A–J). We failed to observe much, if any labeling in the medial septum (see Fig. 7C–D) and diagonal band with pSer129 antibodies. Additional sparse inclusions were apparent in the lateral hypothalamic area and mammillary region. High magnification examples of inclusions in multiple areas are presented in Fig. 7K. A higher resolution version of this figure can be downloaded at the abovementioned Dropbox link.
Fig. 7.
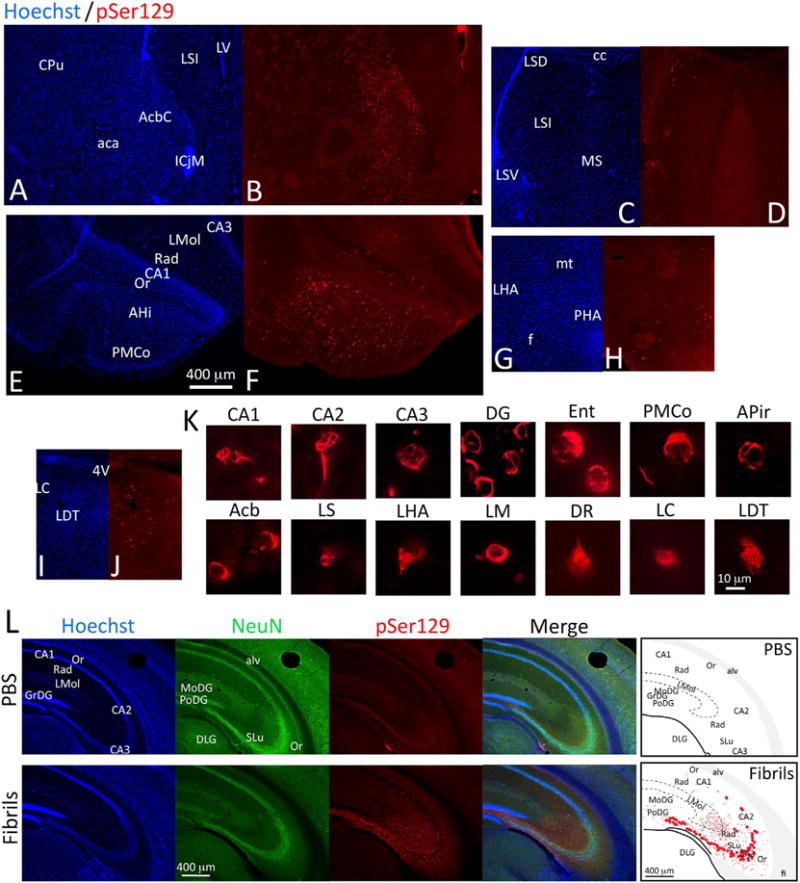
α-synucleinopathy transmission to regions connected with the hippocampus. CD1 mice were infused with α-synuclein fibrils (5 μg) into regio inferior (CA2 + CA3) of the hippocampal formation. Three months later, coronal brain sections were collected and stained with antibodies against pathologically phosphorylated α-synuclein (mouse anti-pSer129). Nuclei were stained with the Hoechst reagent. Moderate to dense inclusions were found in the nucleus accumbens core (A–B) and the cortical amygdala (E–F). Small numbers of inclusions were found in the dorsal and ventral divisions of the lateral septum (C–D), the posterior hypothalamus (G–H), and the locus coeruleus and surrounding tegmentum (I–J). (K) High-magnification images of individual inclusions in various brain regions were captured with a 100× oil objective. (L) The pattern of contralateral pSer129, NeuN, and Hoechst staining in coronal sections of the hippocampus in unilateral vehicle and fibril-infused animals is illustrated with images and schematics. Note that the majority of somal inclusions are in the stratum pyramidale of contralateral CA3. The PBS-infused case is included to show the extent of contralateral background staining with the phospho-Ser129 antibody. For abbreviations, please consult Table 3. A higher-resolution version of this figure is necessary to view the label and can be downloaded from the following links: Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0.
3.7. Cross-hemispheric transmission of α-synucleinopathy through commissural circuitry
Commissural projections connecting the two rodent hippocampi were reported many decades ago by Blackstad (Blackstad, 1956). Subsequent studies with modern tract-tracing techniques confirm and extend his observations that commissural hippocampal efferents originate largely in CA3 and hilar mossy cells, with additional projections from CA2 and the granule cells (Cui et al., 2013; Laurberg and Sorensen, 1981; Ribak et al., 1986; Zappone and Sloviter, 2001). Some studies report that homotopic commissural connections also exist between the CA1 fields (Buchhalter et al., 1990; Shiosaka and Tohyama, 1984; van Groen and Wyss, 1990), although the commissural connections of CA1 are considered weaker than those of CA3 (Cappaert et al., 2015). It has also been suggested that the presubiculum and parasubiculum—but not the subiculum itself—project bilaterally, as does the entorhinal cortex (Cappaert et al., 2015). Thus, if α-synucleinopathy travels across neuroanatomic circuits, it should be densest in the contralateral hemisphere in the CA3 fields and hilar mossy cells. As expected, the densest somal inclusions in the contralateral hemisphere were present in CA3, as visualized in sections cut in the coronal plane from animals infused unilaterally with fibrils (Fig. 7L). In the contralateral hippocampus, neuritic threads were observed in the stratum lucidum and stratum radiatum, as well as the stratum lacusonum-moleculare. Raisman and Gottlieb both suggested that the rostral portions of CA1 do not project densely to the contralateral side (Gottlieb and Cowan, 1973; Raisman et al., 1965), consistent with our illustrations in Fig. 7L.
As commissural connections are important in understanding the anatomical transmission of α-synucleinopathy, Fig. 8 depicts three coronal sections of the hippocampus labeled with both monoclonal and polyclonal pSer129 antibodies, to show the full topographical extent of α-synucleinopathy (see higher resolution version of this figure at the abovementioned Dropbox link). These dual-labeling studies revealed denser labeling in CA1 with the mouse pSer129 monoclonal than the rabbit pSer129 polyclonal antibody. The polyclonal antibody densely labeled contralateral CA3 pyramidal cells and neurites but did not densely label CA1, consistent with the abovementioned reports that CA1 sends sparser contralateral projections than CA3 (Cappaert et al., 2015). Given the prominence of the commissural projections of hilar mossy cells and the less dense commissural projections from granule cells of the area dentata (Zappone and Sloviter, 2001), it is notable that hilar mossy cells do not exhibit as much pSer129 label as the neighboring stratum granulosum in the contralateral hemisphere (Fig. 8). The stratum oriens adjacent to the pyramidal layer of CA3 was densely labeled with both pSer129 antibodies, as one would predict based on Gottlieb and Cowan’s work showing dense commissural projections to this area (Gottlieb and Cowan, 1973). In the ipsilateral hemisphere of the case shown in Fig. 8, the pSer129 label in the entorhinal cortex was concentrated in layers II and III, probably reflecting fibril uptake by the perforant or temporoammonic projections to the dentate gyrus and Ammon’s horn (Cappaert et al., 2015). The entorhinal cortex was also labeled on the side contralateral to the fibril infusions, as would be expected from the bilateral nature of entorhinal projections to CA3, CA1, and the subiculum and of layer II projections to the dentate gyrus (Steward and Scoville, 1976; van Groen et al., 2003). Some entorhinal cortical projections to the contralateral side also originate in deeper layers (Steward and Scoville, 1976; van Groen et al., 2003), but those did not appear to be as densely labeled with pSer129 antibodies. Bilateral pSer129 label was also evident in the perirhinal neocortex, which is known to harbor reciprocal connections with CA1 (Kealy and Commins, 2011). Furthermore, CA1 sends projections to the contralateral subiculum as well as perirhinal and entorhinal cortices (van Groen and Wyss, 1990), all of which exhibited inclusions in the hemisphere contralateral to fibril infusions.
Fig. 8.
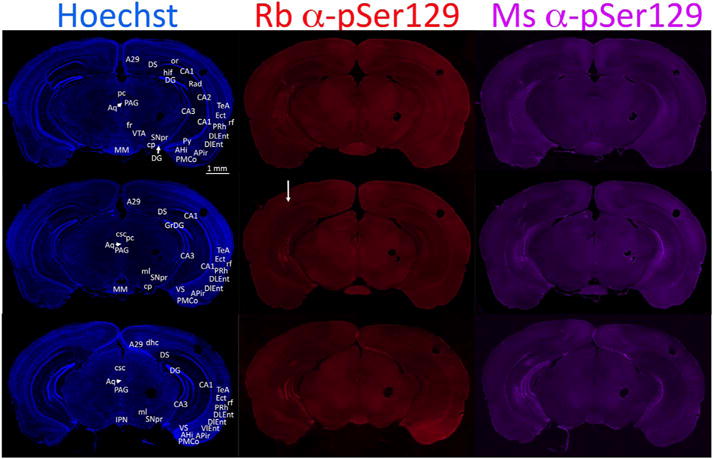
α-synucleinopathy transmission through the ipsilateral and contralateral hemispheres, as shown in coronal sections. CD1 mice were infused with α-synuclein fibrils (5 μg) into regio inferior (CA2 + CA3) of the hippocampal formation. Three months later, coronal brain sections were collected and co-stained with rabbit and mouse antibodies against pathologically phosphorylated α-synuclein (pSer129) and the nuclear marker Hoechst. Three coronal levels of the hippocampus are shown in this figure. The infused (right) hemisphere is on the left side of the images and a white arrow in the middle panel shows the path of the needle, with some disturbance in the tissue in the dorsal hippocampus and overlying cortex. The circular fiducial marks were made in the cortex and mesencephalon on the side opposite to the infusion. This infusion was centered more caudally than intended and exhibited some evidence of fibril diffusion up the needle track (see the inclusions in the cortex below the arrow indicating the needle path). The mouse monoclonal antibody elicited denser labeling of inclusions than the rabbit polyclonal. Note that there may be slight imperfections at the boundaries of the computerized stitched images. For abbreviations, please consult Table 3. A higher-resolution version of this figure is necessary to view the label and can be downloaded from the following links: Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0.
Finally, the dearth of labeling in the breadth of the mesencephalon sandwiched between the two crescent-shaped hippocampi in Fig. 8 favors specific transport across commissural circuits, instead of nonspecific uptake via the cerebrospinal fluid and interstitial space, although the latter cannot be ruled out. A set of higher resolution images of the ipsilateral hippocampus in Supplemental Fig. S2 illustrates the massive quantities of hippocampal inclusions and the specific, layered configuration of the inclusions in somata of the stratum granulosum, fibers of the lacusonum moleculare, and somata of the ventral subiculum.
3.8. Comparison of α-synucleinopathy to retrograde and orthograde labeling with tract-tracers
In studies of the neuroanatomical transmission of Lewy-like pathology, it is important to infuse established tract-tracers at the same stereotaxic coordinates as the fibrils, particularly in brain regions where connections are highly topographically organized, such as the tri-layered hippocampus. First, we compared the topography of the inclusions with that of afferent FluoroGold+ neurons (Fig. 9; see higher resolution, version of this figure at the abovementioned Dropbox link). Animals simultaneously injected with FluoroGold plus fibrils exhibited little retrograde label, as they were sacrificed three months later and the FluoroGold labeling intensity had waned over this time. Therefore, we report here the pattern of FluoroGold label in animals sacrificed within a week of tracer infusion. Consistent with the literature on hippocampal neuroanatomy, dense FluoroGold+ cells were observed in superficial layers of the entorhinal cortex (Fig. 9A–D), the medial septal nuclei, and ventral diagonal band of Broca (Fig. 9E–F) (Cappaert et al., 2015). Given the dense FluoroGold retrograde label in the latter two structures, it is worth noting that dense α-synucleinopathy was not observed at these sites in fibril-infused mice. In general, the extent of FluoroGold labeling was much more extensive than that of pSer129 labeling, with numerous labeled neurons in the expected hypothalamic, thalamic, cortical, and brainstem afferents (Cappaert et al., 2015), not all of which exhibited pSer129+ inclusion pathology in the material examined here. This is especially noteworthy given the smaller volume of FluoroGold than fibril infusions. Nevertheless, some structures exhibiting pSer129 label did not contain any FluoroGold label. For example, FluoroGold label was present in the amygdala (Fig. 9G–H) but not in the nucleus accumbens, both of which harbored pSer129+ label in fibril-infused cases. Notably, diffusion of FluoroGold into the lateral ventricular space was evident from dense ependymal label in a few animals and therefore may have also transpired in some of the fibril-infused cases. Despite the proximity of the infusion site to the lateral ventricles, it is worth noting that there was little to no α-synucleinopathy in underlying thalamic structures in animals infused with fibrils in the hippocampus, although label was observed in these regions in some FluoroGold and BDA cases, as discussed further below.
Fig. 9.
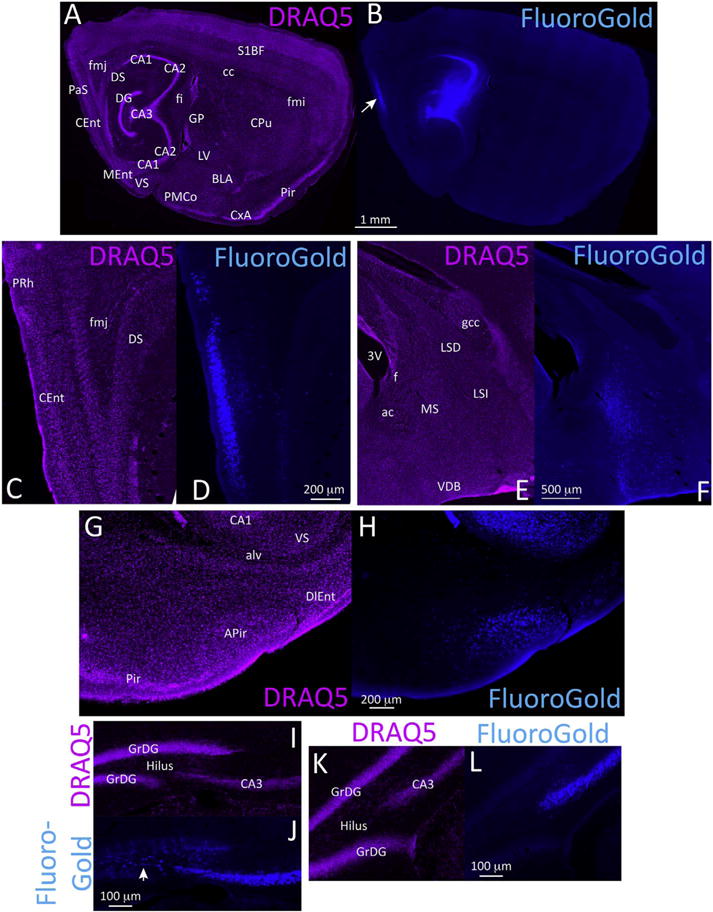
Retrograde transport of the tract-tracer FluoroGold from the septal pole of the hippocampus. CD1 mice were infused with the retrograde tract-tracer FluoroGold in regio inferior (CA2 + CA3) of the dorsal hippocampus. One week later, sagittal brain sections were collected and nuclei were stained with the infrared marker DRAQ5 (purple). FluoroGold labeling was visualized in the UV channel. The center of the infusion site is shown in A–B. Dense retrograde labeling in the superficial layers of the caudomedial entorhinal cortex is shown in C–D. Retrograde label in the medial septum and ventral diagonal band of Broca is shown in E–F. Retrograde label in the lateral portions of the amygdalopiriform transition area is shown in G–H. (I–L) Commissural projection neurons in the contralateral hemisphere in two FluoroGold-infused mice. Panels I–J are from a case with the infusion centered in the dentate gyrus and CA3, leading to contralateral retrograde label of hilar mossy cells in the polymorphic layer and CA3 pyramidal neurons. Panels K–L are from a case with the infusion centered in CA3 without dentate involvement, resulting in homotopic label in contralateral CA3 but not in hilar mossy cells. A higher-resolution version of this figure is necessary to view the label and can be downloaded from the following links: Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Commissural projection neurons terminating in CA2/CA3 were confirmed by FluoroGold labeling in the contralateral hilus, CA3, and perirhinal and entorhinal cortices. The topography varied depending on the center of the infusion site. If the FluoroGold injection encompassed CA3 as well as the hilus, homotopic retrograde label was seen in contralateral hilar mossy cells and the pyramidal cell layer of CA3 (Fig. 9I–J). If the injection was centered in CA3 and did not extend into the hilus, the contralateral side exhibited retrograde label in CA3 but not within the hilus (Fig. 9K–L).
Previous work has shown that 10 kDa BDA is transported preferentially (but not exclusively) in the anterograde direction, whereas 3 kDa BDA is transported mostly in the retrograde direction, although this has been contested (Heimer et al., 2006; Reiner et al., 2000; Veenman et al., 1992). Therefore, in the present study we infused animals with both BDA tracers. In our hands, BDA was preferentially transported largely in the orthograde direction no matter the molecular weight. Fig. 10 illustrates dense anterograde label with the 10 kDa form of BDA (a higher resolution version of this figure can be downloaded at the abovementioned Dropbox link). A range of BDA infusion centers are shown in this figure, once again to display the variability in the placement of injection, as expected from the limits of resolution of the stereotaxic machine, inter-animal variations in the appearance of Bregma, and the thinly layered organization of the hippocampal palisade. Nevertheless, in all cases there was dense labeling of Ammon’s horn. The Schaffer collateral pathway that originates in CA3 and terminates in CA1 was densely anterogradely labeled by BDA. Retrograde BDA label was evident in the parasubiculum (Fig. 10F) and entorhinal cortex (Fig. 10J). Some animals exhibited dense anterograde label of the deeper layers of the entorhinal cortex in addition to moderate retrograde label in layer II of this structure (Supplemental Fig. S3A–B) and anterograde label in the molecular layer of the dentate gyrus (Supplemental Fig. S3C–E). The topography of BDA labeling in the molecular layer of the area dentata in panels S3C–E can be contrasted with the dense pSer129 label in the stratum granulosum in previous figures. Thick fiber bundles were also observed penetrating the lateral septum (Supplemental Fig. S3C–E), which are pictorially represented in Supplemental Fig. S3F, also consistent with the literature on the efferent projections of CA3 (Cappaert et al., 2015; Cui et al., 2013). This massive BDA-labeled efferent fiber tract was not densely labeled in any of the pSer129 material. Anterograde BDA label was also observed in the horizontal limb of the diagonal band of Broca (Fig. S3F), where no pSer129 label was found. Higher magnification views of the dense anterograde and occasionally retrograde label in the lateral septum are depicted in Fig. S3G–J. Anterograde BDA label was not observed in the amygdala, which harbored moderate to dense FluoroGold and pSer129 staining.
Fig. 10.
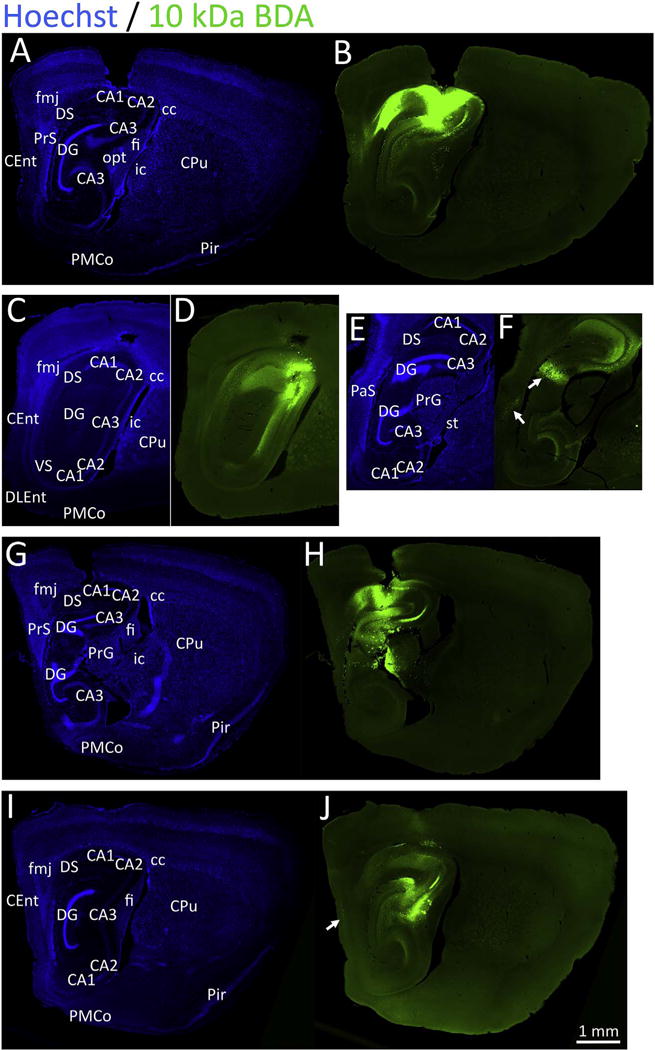
Injections of 10 kDa biotinylated dextran amines (BDA) in the septal pole of the hippocampus. CD1 mice were infused with the tract-tracer BDA (10 kDa) in regio inferior (CA2 + CA3) of the hippocampus. One week later, sagittal brain sections were collected and nuclei were stained with the Hoechst reagent (A, C, E, G, I). BDA was visualized with a streptavidin-conjugated fluorophore (B, D, F, H, J). Four separate animals are shown (A–B, C–F, G–H, and I–J) in order to illustrate the range of tracer diffusion. The case in panels A-B is centered in CA1 and CA2, the case in panels C–F is centered in CA2/CA3, the case in panels G–H is centered in CA1 and the dentate gyrus with diffusion into the thalamus, and the case in panels I–J is centered in CA3. The case in panels C–F is shown at two distinct sagittal levels to illustrate the topography of the retrograde label in the granule cell layer of the dentate gyrus (northeast arrow in panel F). The northwest arrow in panel F points to retrograde label in the parasubiculum and the arrow in panel J points to retrograde label in deeper layers of the entorhinal cortex. A higher-resolution version of this figure is necessary to view the label and can be downloaded from the following links: Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0.
Given the ventral location of some of our injection sites, we show a series of images from one 3 kDa BDA case in Fig. 11, in order to demonstrate the full potential extent of the diffusion and uptake of the tract-tracers. This case was particularly instructive, as it revealed dense involvement of the optic tract in the thalamus, leading to extremely dense retrograde labeling of the optic chiasm on the ventral surface of the brain as well as anterograde labeling of retinal projections radiating into the superior colliculus (white arrows in Fig. 11B). Although we did not observe dense pSer129+ inclusions in the superior colliculus in fibril-infused cases, the animal schematically illustrated in Fig. 6C (with the most extensive inclusions of all) also showed some neuritic pSer129 labeling in the ventral portion of the optic tract. However, we emphasize that most cases failed to exhibit this label and that white matter often displays high background with pSer129 antibodies (Sacino et al., 2014b). The 3 kDa BDA case in Fig. 11 exhibited dense anterograde and retrograde transport to the deep and superficial layers of the entorhinal cortex (see panel H), respectively, as well as label in the lateral septum, supramammillary nucleus, nucleus accumbens, ventromedial hypothalamic nucleus, and the thalamus. This case was further noteworthy in its absence of amygdalar label. A higher resolution version of this figure can be downloaded at the abovementioned Dropbox link.
Fig. 11.
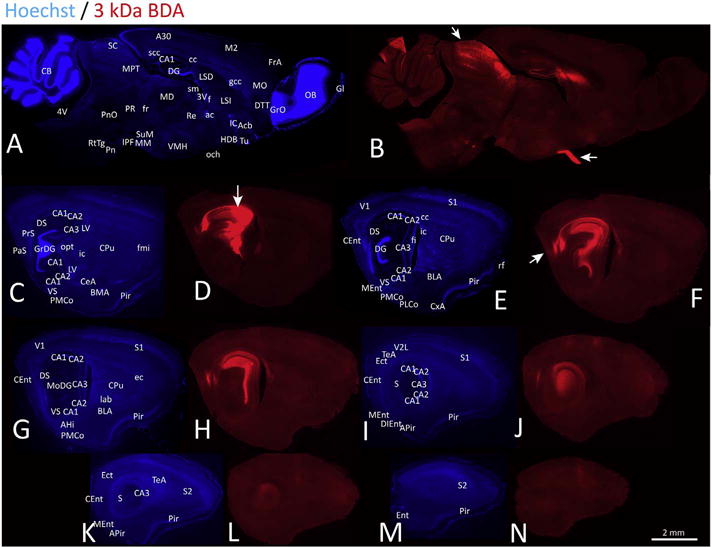
Anterograde and retrograde transport of 3 kDa biotinylated dextran amines (BDA) from the septal pole of the hippocampus. CD1 mice were infused with the tract-tracer BDA (3 kDa) in regio inferior (CA2 + CA3) of the hippocampus. Ten days later, sagittal brain sections were collected and nuclei were stained with the Hoechst reagent (A, C, E, G, I, K, M). BDA was visualized with a streptavidin-conjugated fluorophore (B, D, F, H, J, L, N). All brain sections shown in this figure are from one instructive case with diffusion into the optic tract of the underlying thalamus. The path of the needle is displayed with a white arrow in panel D. Although the injection was centered in CA2 + CA3, diffusion into the optic tract led to dense anterograde label of retinal projections to the superior colliculus and dense retrograde label of the optic chiasm in panel B (see two white arrows). This panel also shows anterograde label in the thalamus and retrograde label in the ventromedial hypothalamus. Dense anterograde label was observed in deep layers of the caudomedial entorhinal cortex in panel F, in addition to moderate retrograde label in superficial layers (white arrow). A higher-resolution version of this figure is necessary to view the label and can be downloaded from the following links: Either http://ddc.duq.edu/pharmacology/2/ or https://www.dropbox.com/sh/gfk20tkzpfaf9q2/AAC8Ac4LJQFqpzFMEOYmyecba?dl=0.
3.9. Control experiments to assess extraparenchymal diffusion of α-synuclein fibrils
In the Franklin and Paxinos brain atlas for C57BL/J6 mice (weight range 26–30 g), our stereotaxic coordinates place the needle tip in the pyramidal layer of CA3 on the left side of the coronal section and in the stratum oriens and fimbria on the right side, as the hippocampus is slightly asymmetrically sectioned in this atlas (Fig. 52 of this atlas) (Franklin and Paxinos, 2013). In our pilot work using blue dye in CD1 mice, the dye diffused throughout CA2 and CA3 of the regio inferior at these coordinates and did not enter the thalamus but did diffuse dorsally in some cases into CA1 and the overlying cortex. Compared to the C57 animals used to craft the Franklin and Paxinos atlas, the range of weights for the CD1 mice used in the present study was much higher (38 to 56 g), corresponding to the greater skull thickness in the latter strain. This makes our infusions in CD1 mice much more dorsal than the coordinates in the Franklin and Paxinos atlas would indicate. Nevertheless, the BDA tracer injections showed involvement of the optic tract and thalamus in some cases that were accidentally too rostral and/or centered too close to the midline, due to the caudolateral (i.e. septotemporal) curvature of the hippocampal crescent. Thus, we cannot exclude the potential for diffusion of fibrils into the overlying cortex and underlying thalamus. Therefore, to control for the diffusion of fibrils into these regions, separate animals were injected in the overlying cortex and the underlying thalamus, in order to identify pathology that might be attributable to extrahippocampal diffusion. Three months postinfusion, thalamic fibril infusions elicited sparse inclusions in the thalamus (Supplemental Fig. S4A–C), which were not observed following hippocampal injections. pSer129 label was observed in the claustrum (Supplemental Fig. S4D–F), which projects to dorsal thalamic nuclei in many species (Mathur, 2014). Very sparse pSer129 labeling was observed in the parabrachial nucleus and laterodorsal tegmentum, which also innervate the thalamus (Holmstrand and Sesack, 2011; Krout and Loewy, 2000). The cortex-infused cases only revealed sparse inclusion pathology in the overlying cortex, concentrated within the deeper layers (Supplemental Fig. S4G–I), not unlike the case with dorsal diffusion of fibrils up the needle track in Fig. 6C. Layer VI of the cortical edifice shares dense connections with the thalamus, but also projects intracortically and receives cortical afferents (Briggs, 2010). Neither cortical nor thalamic infusions led to the robust pattern of pSer129+ inclusions seen following hippocampal fibril infusions. For example, cortical and thalamic injections did not lead to pSer129+ structures in the accumbens, lateral septum, and cortical amygdala nuclei. The limited inclusions forming after thalamic and cortical injections suggests that the patterns reported in previous figures arose largely from the hippocampal formation itself and not from surrounding brain regions.
4. Discussion
The preformed α-synuclein fibril model works in rats, mice, and monkeys and is growing in use (Abdelmotilib et al., 2017; Blumenstock et al., 2017; Chu and Kordower, 2015; Luk et al., 2012a; Masuda-Suzukake et al., 2013; Osterberg et al., 2015; Paumier et al., 2015; Rey et al., 2016; Sacino et al., 2014a; Sacino et al., 2014b; Shimozawa et al., 2017; Thakur et al., 2017; Zhao et al., 2017). In the present study, we closely examined the topography of α-synucleinopathy in mice infused three months earlier with preformed α-synuclein fibrils in CA2/CA3 of the hippocampal formation, with the objectives of 1) tracing the spread of pathology through the brain at early time points and 2) mimicking the early Lewy pathology that surfaces in this structure in Lewy body dementias. The early survival period facilitated the analysis of first-order transmission of pathology from only the infusion site; if and when pathology is transmitted transneuronally, identifying the nodes through which it passes becomes increasingly difficult (Card and Enquist, 2014). Massive pSer129+ inclusions materialized in the hippocampal formation by three months postinfusion, but this was not sufficient to elicit memory loss, olfactory impairments, loss of synaptic markers, or any type of cell loss. As expected, fibril-infused animals exhibited moderate to dense α-synucleinopathy in structures outside of the hippocampus proper (e.g. subiculum) and other components of the limbic system (e.g. entorhinal cortex, amygdala, lateral septum, nucleus accumbens), in addition to sparse label in the hypothalamus and brainstem. Some, but not necessarily all of the pathology might be attributed to transmission through neuroanatomical connections. The predominantly limbic pathology is consistent with a model of incidental Lewy body disease or prodromal dementia with Lewy bodies, although it is possible that behavioral deficits would not emerge even with longer incubation periods. At three months postinfusion, early compensatory changes in synaptic function may have resulted in the modest increase in synaptophysin expression in the stratum radiatum, perhaps to overcome the detrimental effects of proteinopathy within neuritic processes. If this stress response compensates against the toxicity of Lewy pathology as speculated, it may have contributed to a delay in the onset of dementia-like symptoms. This change probably did not reflect a global change in synapse numbers, as synapsin I/II levels were not similarly increased. The majority, if not all, brain regions harboring pSer129+ structures in fibril-infused animals share first-order connections with the site of infusion, as confirmed by studying the transport of anterograde and retrograde tract-tracers from the same stereotaxic coordinates. However, the pSer129 labeling did not overlap perfectly in either density or topographical extent with FluoroGold or BDA label.
4.1. Potential passage through some, but not all, established neuroanatomical connections
Several studies employing the α-synuclein fibril model are consistent with the view that α-synucleinopathy spreads into neuronal populations that are directly or indirectly connected with the infusion site (Abdelmotilib et al., 2017; Blumenstock et al., 2017; Luk et al., 2012a; Paumier et al., 2015; Rey et al., 2013; Rey et al., 2016; Sacino et al., 2014a; Sacino et al., 2014b; Shimozawa et al., 2017). In our previous study, we employed the tracer FluoroGold to determine that α-synuclein+ inclusions emerge at early time points in first-order afferent structures that project directly to the fibril infusion site in the olfactory bulb and anterior olfactory nucleus (Mason et al., 2016). A study examining the spread of Lewy pathology from the gut to the brain also employed FluoroGold infusions, but only as a positive control (Holmqvist et al., 2014). However, the present study was conducted at higher anatomical resolution than previous reports, involved bilateral infusions and behavioral tests, and employed both anterograde and retrograde tracers. After regio inferior infusions, the densest pSer129 pathology was observed within the area dentata. The efferent projections of the dentate gyrus are concentrated in the mossy fiber pathway that terminates in nearby CA3 (Amaral et al., 2007; Cappaert et al., 2015). Thus, uptake of fibrillar material by granule cells of the fascia dentata may have transpired at the site of infusion in CA3. The mossy fiber projection is unmyelinated and originates in glutamatergic neurons (Amaral et al., 2007; Cappaert et al., 2015), features that are strongly associated with vulnerability to Lewy pathology in human tissue and in experimental models (Braak and Del Tredici, 2004; Braak et al., 2003a; Braak et al., 2003b; Braak et al., 2006b; Taguchi et al., 2014).
The major arguments favoring specific neuroanatomical transmission of α-synucleinopathy in our material include 1) dense somal pSer129+ inclusions in superficial layers of the entorhinal cortex and a few neuritic inclusions in the deeper layers of the entorhinal cortex, consistent with retrograde BDA and FluoroGold labeling of somata in superficial layers and anterograde BDA labeling of fibers in deeper layers, respectively, and 2) somal and neuritic pSer129+ inclusions in the contralateral hippocampus that are consistent with known commissural projections. The emergence of α-synucleinopathy in cell bodies in the superficial layers of the entorhinal cortex is consistent with potential retrograde transport because layer II of this structure sends projections into the dentate gyrus via the perforant pathway (Witter, 2007; Witter et al., 1988). In addition, entorhinal layer III and a few cells in the deeper layers project to CA1, CA3, and the subiculum (Cappaert et al., 2015; van Groen et al., 2003). The dense efferent projection from CA1 to the deeper layers of the entorhinal cortex (layers IV and V) (Cappaert et al., 2015) might underlie the labeling of pSer129+ fibers at the latter site. However, the neuritic pSer129 label in deep entorhinal layers was much sparser than the labeling of somata in superficial layers, especially when contrasted to the pattern of anterograde and retrograde entorhinal BDA label depicted in Fig. S3B. These observations are consistent with greater retrograde than anterograde spread of pathology.
The densest commissural projections of the hippocampi arise in CA3 (Cui et al., 2013; Zappone and Sloviter, 2001), a pattern similarly evident in the α-synucleinopathy of the contralateral hemisphere after unilateral fibril infusions. In addition, the surrounding regions of the temporal cortex also dispatch commissural efferents (van Groen et al., 2003), and harbored pSer129 labeling on the side contralateral to the infusion. Far less pSer129+ pathology was observed in the mesencephalic structures lying between the two hippocampal crescents in the coronal sections of Fig. 8, even though this area contains vulnerable dopamine neurons expressing high levels of α-synuclein protein (Taguchi et al., 2015). These observations are not consistent with the argument that fibrils induce nonspecific pathology by traveling through the cerebrospinal fluid, periventricular zones, and then the interstitial space of deeper parenchyma. Given the striking vulnerability of granule cells of the area dentata in the ipsilateral hippocampus, mass diffusion of fibrils through the parenchyma and uptake in the contralateral hippocampus would be expected to fill contralateral granule cells with inclusions more densely than the neighboring CA3 field, but this was also not the case. It therefore seems unlikely that the fibrils reached the distant contralateral hippocampus solely via diffusion and not commissural connections.
Unlike previous reports by Sacino et al. and Hu et al., we observed pSer129+ inclusions in many extrahippocampal regions (Hu et al., 2016; Sacino et al., 2014b), perhaps because of differences in fibril synthesis and sonication parameters. Nevertheless, we were surprised by the dearth of inclusions in the septal/diagonal band complex. Regio inferior is known to receive a substantial projection from the septal nuclei and diagonal band (Cappaert et al., 2015). Other than inputs from the basal forebrain, subcortical inputs to the fascia dentata and CA2 originate in the supramammillary nucleus, perifornical and posterior hypothalamic areas, locus coeruleus, and raphe (Aznar et al., 2004; Cappaert et al., 2015; Cui et al., 2013; Haglund et al., 1984; Jones and Moore, 1977; Moore et al., 1978; Samuels and Szabadi, 2008). Furthermore, CA3 receives extrahippocampal projections from the endopiriform nucleus, amygdala, lateral septum, locus coeruleus, and raphe (Cappaert et al., 2015). Accordingly, many of these known afferents harbored pSer129+ inclusions. Consistent with our findings of locus coeruleus inclusions, Guo and colleagues reported tau pathology in the locus coeruleus following hippocampal infusions of α-synuclein fibrils (Guo et al., 2013). In some animals we observed pSer129+ inclusions in the mammillary body, which used to be considered an important target of the hippocampus, until Swanson and Cowan demonstrated that the hippocampal projection to the hypothalamus originates in the subiculum and not in Ammon’s horn (Swanson and Cowan, 1975). These findings suggest that the effective area of transmission of pathology in our model is not limited to the hippocampus proper (Ammon’s horn) but that it extends into the subiculum.
The lateral septum is the major extrahippocampal target of Ammon’s horn, especially of CA3 (Cappaert et al., 2015; Swanson, 1981). In the present study, sparse pSer129 staining but dense anterograde BDA label was observed in the lateral septum. The subcortical projections of the septal pole of CA1 include the lateral septum, diagonal band, and nucleus accumbens (Cappaert et al., 2015). The projections of the temporal pole of CA1 include, among others, the anterior olfactory structures, the nucleus accumbens, and the amygdala, especially the basal nuclei (Cappaert et al., 2015; Swanson, 1981). Passage of α-synucleinopathy into the lateral septum, accumbens, and olfactory peduncle in the present study is therefore consistent with some, albeit limited anterograde transport. It is worth noting that we failed to observe retrograde labeling of accumbens neurons with the FluoroGold tracer, making retrograde passage of pathology to the accumbens unlikely. In our previous study on the transmission of olfactory α-synucleinopathy, we did not collect convincing evidence of anterograde transport (Mason et al., 2016), suggesting that the direction of transport may vary depending upon the induction site and the circuits available for fibril uptake.
We observed dense labeling in the amygdala following fibril and FluoroGold (but not BDA) infusions in the hippocampus. Some of the amygdalar labeling observed in our material may have originated from CA1, as the afferents of CA1 include the posteromedial cortical and accessory basal amygdala (Cappaert et al., 2015). However, amygdalar efferents also terminate in the stratum oriens and stratum radiatum of CA3. In addition, the ventral subiculum harbored pSer129+ inclusions and is known to receive a dense projection from the amygdala, including the posteromedial cortical amygdala (Krettek and Price, 1974, 1977). In some cases, we observed diffusion of FluoroGold into the cerebrospinal fluid, as evidenced by intense labeling of the ependymal lining of the lateral ventricle. These results suggest that some fibrils may also have reached the amygdala via the cerebrospinal fluid. FluoroGold is small and hydrophilic (Wessendorf, 1991), and we have observed in this and previous studies that it diffuses quite readily into the ventricles from parenchymal injection sites relative to other tracers (Leak and Moore, unpublished). α-synuclein is present in the ventricular compartment in humans, albeit at low concentrations in Parkinson’s patients (El-Agnaf et al., 2003; Stewart et al., 2014), and may seed pathology in distant sites via diffusion if it reaches those destinations at sufficient concentrations. However, this would likely not be the major mode of transmission of human Lewy pathology and is discussed further below. The targeted hippocampal palisade is narrow relative to more spherical brain regions infused in previous preformed fibril studies, such as the striatum and the olfactory bulb (Luk et al., 2012a; Mason et al., 2016; Paumier et al., 2015; Rey et al., 2016). Variability in the precise placement of the infusate is common in stereotaxic surgery on mice, as illustrated by the range of BDA infusion epicenters displayed in Fig. 10. Therefore, we do not rule out ventricular diffusion followed by entry of fibrils into the interstitial space of neighboring parenchyma as one of the possible routes of ipsilateral fibril transport. Rey and colleagues performed control infusions into the cerebrospinal fluid to bolster their argument that the transmission of α-synuclein from the olfactory bulb is not attributable to ventricular diffusion (Rey et al., 2013). In previous studies, we infused tract-tracers into the lateral ventricles adjacent to the hippocampus and noted densely labeled neurons in medial hypothalamic nuclei that contact the periventricular zones of the third ventricle, as well as particularly dense labeling of the raphe complex (Leak and Moore, 2012). Serotonergic neurons of the raphe have long been known to elaborate an extensive cerebrospinal fluid-contacting terminal plexus (Aghajanian and Gallager, 1975; Chan-Palay, 1976; Richards, 1977). These specific patterns were not evident in our pSer129-immunopositive material. It is worth noting that phosphorylated α-synuclein has been shown to accumulate in subpial and periventricular astrocytes in multiple system atrophy (Nakamura et al., 2016), suggesting that periventricular cells are capable of displaying Lewy pathology but failed to do so in our model, suggesting limited periventricular uptake of fibrils.
In order to control against the extrahippocampal diffusion and uptake of fibrils, we infused the thalamus and cortex—instead of the lateral ventricles—as we reasoned that any escape of fibrils into the nearby ventricular compartment would result in the entry of fibrils into the periventricular regions of precisely these two areas. Therefore, we injected these two regions with even higher concentrations of fibrils than would be afforded had we targeted the ventricular space, as the cerebrospinal fluid would quickly dilute the fibrils. Furthermore, our previous experience with infusing the ventricular space taught us that the lateral ventricles are narrower in vivo than in formalin-fixed (shrunken) postmortem tissue, and therefore unlikely to be injected without also involving nearby parenchyma. None of the thalamic or cortical injections of fibrils led to robust pSer129+ pathology, and the patterns were notably different from the fibril-infused cases centered in regio inferior. Nevertheless, future studies with highly concentrated fibril solutions delivered in tiny volumes, perhaps together with a long-lasting dye, might limit the ventricular diffusion caveat, although it is possible that smaller infusion zones would be insufficient to seed massive Lewy pathology, as observed here.
We did not collect strong evidence of transneuronal passage of α-synucleinopathy. For example, the anterograde projections of CA1 to the lateral septum did not lead to transneuronal involvement of the dense lateral septal projections to the anterior hypothalamus and preoptic area (Risold and Swanson, 1996). Rey and colleagues reported widespread dissemination of α-synucleinopathy at longer survival times (one year) in an effort to model a fuller extent of the spread (Rey et al., 2016).
To view the full topographical extent of pathology and avoid a Type II error, it was important to employ the mouse antibody in the present study, as it is the most sensitive Lewy marker. Our examination of the contralateral hemisphere in coronal sections suggests the differences in monoclonal versus polyclonal pSer129 staining may adhere to differences in innervation density, with dense projections (commissural CA3 efferents) being labeled with both mouse and rabbit antibodies but sparse projections (commissural CA1 efferents) being labeled more readily with mouse antibodies. It is also important to note that the injected fibrils are not phosphorylated and do not stain with pSer129 antibodies (Luk et al., 2009; Mason et al., 2016). Thus, the pSer129 inclusions reported here likely form from endogenous α-synuclein aggregation. In this context, it is also important to note that α-synuclein knockout mice fail to develop pSer129+ inclusions in the preformed fibril model (Volpicelli-Daley et al., 2016).
4.2. Selective resistance against the formation of α-synuclein-containing protein aggregations
Given that the area of fibril uptake may have been larger than that of the tracers, and that FluoroGold or BDA were taken up and transported by some neural circuits that exhibited little to no pSer129 label (e.g. medial septum/diagonal band, contralateral hilar mossy cells), neuronal subpopulations may exhibit differences in inherent vulnerabilities to proteinopathic stress, consistent with our previous work (Posimo et al., 2013; Posimo et al., 2015). In Parkinson’s disease, the septal complex and diagonal band of the basal forebrain do develop Lewy pathology in Braak stage III (Braak et al., 2003a), but the present study suggests that the neurons affected in this manner might be a different set than those that project into the hippocampi, provided our data can be extrapolated to humans. Had Lewy pathology settled in the basal forebrain in our model, dementia-like symptoms might have surfaced, because cholinergic dysfunction is linked with Parkinson’s disease dementia (Hall et al., 2014).
Several of our BDA infusions led to dense anterograde label of visual projections radiating into the superior colliculus and intense retrograde label of the optic tract, chiasm, and nerve. These features were not similarly evident in our fibril-infused cases. Most axons in the optic tract are myelinated, and myelinated neurons seem to refrain from developing Lewy pathology (Braak and Del Tredici, 2004; Braak et al., 2003a; Braak et al., 2003b; Braak et al., 2006b). Second, the concentration of fibrils in the infusion center may be too low for uptake by fibers passing through the nearby optic tract. Nevertheless, uptake of tract-tracers by fibers en passant has been reported in multiple systems (Dado et al., 1990; Heimer et al., 2006), and needs to be considered as a potential caveat in all fibril infusion studies.
Aside from the basal forebrain and visual system, the FluoroGold and/or BDA cases displayed dense labeling in multiple other areas that exhibited little to moderate inclusion pathology, including many nuclei in the thalamus, hypothalamus, and brainstem. For example, sparse or no pSer129 label was seen in the suprammillary and ventromedial hypothalamic regions and the thalamic nucleus reuniens, which are connected with the hippocampi or send fibers through the fimbria (Canteras et al., 1994; Haglund et al., 1984; Varela et al., 2014) and therefore transported our tract-tracers.
4.3. Hippocampal α-synucleinopathy is correlated with, but does not cause major cognitive deficits within three months postinfusion
Memory deficits in Parkinson’s disease dementia and in dementia with Lewy bodies are generally attributed to limbic and neocortical Lewy pathology (Armstrong et al., 2014; Kalaitzakis and Pearce, 2009; Mattila et al., 2000; Yang and Yu, 2016). Specifically, dementia in Parkinson’s disease is traditionally associated with α-synucleinopathy in the anterior cingulate gyrus, superior frontal gyrus, temporal and entorhinal cortices, the amygdaloid complex, and hippocampal CA2 and CA1 (Adamowicz et al., 2017; Churchyard and Lees, 1997; Cordato et al., 2000; Kalaitzakis et al., 2009). In the present study, we collected no evidence of memory loss despite inclusions so dense that they could be visualized with low-power microscope objectives. Many authors have argued that extensive cortical and limbic Lewy pathology does not necessarily cause dementia (Colosimo et al., 2003; Harding and Halliday, 2001; Kalaitzakis and Pearce, 2009; Parkkinen et al., 2005). Alternatively, Parkinson’s disease dementia and dementia with Lewy bodies may be more accurately modeled with a different injection target, such as the basal forebrain, entorhinal cortex, or amygdala, which might better mimic the specific routes of transmission of human Lewy pathology. A recent study by Adamowics and colleagues reported that the Lewy pathology in CA1 correlates well with memory deficits in dementia with Lewy bodies compared to CA2 inclusion numbers (Adamowicz et al., 2017). Hu and colleagues reported memory loss two months after α-synuclein fibril infusions into the mouse hippocampus, but they targeted the dentate gyrus, infused a five-fold greater volume of fibrils than in the present study, and reported hippocampal apoptosis (Hu et al., 2016). In the present study, α-synucleinopathy in the dentate gyrus was significantly negatively correlated with memory function according to the novel object and novel place recognition tests, but the association was correlative without being causal, because animals infused with fibrils did not spend less time exploring the novel object or novel place than PBS-infused controls. We interpret our results to suggest that the underlying cellular vulnerability to α-synucleinopathy may influence memory and olfactory function, but that the α-synucleinopathy itself does not, at least not in early disease timeframes, or else we would have observed an increase in memory and olfactory impairments in the fibril-infused group. In other words, animals with hippocampi that are more prone to α-synuclein aggregation may harbor other underlying vulnerabilities that render them less able to perform well on the memory and olfactory tests.
4.4. Impact of dense α-synucleinopathy on hippocampal cell numbers
Korbo and colleagues suggested that dementia is not necessarily associated with hippocampal neurodegeneration (Korbo et al., 2004). Accordingly, several studies show that there is no neuron loss in CA1, CA2/CA3, the dentate gyrus, or subiculum in Parkinson’s disease (Ince et al., 1991; Joelving et al., 2006), although there is loss of presubicular pyramidal neurons in dementia with Lewy bodies (Harding et al., 2002a). The present study clearly demonstrated a lack of neurodegeneration at sites with dense Lewy-like pathology, including the presubiculum and amygdala. The α-synuclein pathology we observed in the limbic telencephalon may have been below a critical threshold for emergence of cognitive dysfunction and may need to be accompanied by tissue atrophy or synaptic loss at later survival time points. There are conflicting reports of atrophy of hippocampal subregions in Parkinson’s disease (Beyer et al., 2013; Camicioli, 2002; Churchyard and Lees, 1997; Cordato et al., 2000; Joelving et al., 2006; Laakso et al., 1996). Joelving and coworkers reported higher variability in hippocampal volume and cell numbers in Parkinson’s patients than control subjects, confirming the heterogeneous nature of this disorder and perhaps explaining the inconsistencies across studies (Joelving et al., 2006). In this context, it seems significant that hippocampal atrophy is mild in cases with Parkinson’s disease and greater in cases with dementia with Lewy bodies (Cordato et al., 2000). Alternatively, our findings of dense α-synuclein pathology in the absence of memory deficits are consistent with reports of a dissociation between behavior and histopathology in experimental models of dementia as well as in dementia patients (Sekiyama et al., 2016). In sum, the emergence of Lewy-like pathology is not necessarily accompanied by a behavioral syndrome, although it certainly may precede poor neurological outcomes.
4.5. Impact of α-synucleinopathy on presynaptic markers
Pre and postmortem clinical work suggests that behavioral deficits—such as dementia—can surface before cell loss, perhaps due to early changes in synaptic transmission (Selkoe, 2002; Shankar and Walsh, 2009). For example, loss of synapses in CA2/CA3 is significantly correlated with dementia in Alzheimer’s disease (Samuel et al., 1994). α-synucleinopathy is known to cause deficits in axon trafficking and synaptic impairments (Bellucci et al., 2016; Nikolaus et al., 2009; Schulz-Schaeffer, 2010; Volpicelli-Daley, 2017; Volpicelli-Daley et al., 2014a; Volpicelli-Daley et al., 2016). Indeed, most α-synuclein aggregates are located in presynaptic terminals in Lewy body disorders and their distribution overlaps with the presynaptic marker synaptophysin (Schulz-Schaeffer, 2010). There may be a reduction in synaptic markers in dementia with Lewy bodies—including in synaptophysin levels—particularly when patients present with concomitant Alzheimer’s pathology (Hansen et al., 1998; Nikolaus et al., 2009; Revuelta et al., 2008). Unexpectedly, we observed an increase in synaptophysin levels in Ammon’s horn in fibril-treated animals, and speculate that this may partly underlie their lack of functional impairments. Synaptophysin levels are augmented by inflammatory processes (Zhang et al., 2012) and increase in the hippocampus with aging, perhaps to compensate against age-related memory decline (Benice et al., 2006). In the present study, synaptophysin levels in Ammon’s horn were negatively correlated with memory function in the novel object recognition test in fibril but not PBS-infused animals. Therefore, we speculate that animals with higher brain fitness (i.e. superior cognitive function) and less vulnerability to proteinopathic stress exhibited less need for a compensatory response in synaptic function relative to control animals. Without the emergence of this compensatory change, fibril-infused animals might have performed worse on the behavioral tests than vehicle controls. We observed no changes in a second synaptic marker, synapsin I/II, which was decreased in abundance by oligomeric α-synuclein in previous studies (Larson et al., 2017). Nevertheless, future studies are warranted to identify 1) whether the modest increase in synaptophysin reflects an increase in synaptophysin levels per axon terminal, and 2) the functional consequences of this putatively compensatory change, as they might shed light on the delayed onset and slow progression of most Lewy body disorders. Future studies on protein levels and posttranslational changes in other presynaptic and postsynaptic markers are also warranted, as some have argued that presynapses are preserved in dementia with Lewy bodies whereas dendritic spines are retracted (Schulz-Schaeffer, 2010). Furthermore, α-synuclein fibril infusions in the striatum lead to derangement of spines in the cerebral cortex and α-synuclein overexpression leads to decreased cortical spine density (Blumenstock et al., 2017).
4.6. Limitations of exogenous delivery of fibrillar α-synuclein material in vivo
An important technical caveat of the present study is the difference in injection volume between the tracers and the fibrils. However, even if we had injected a larger volume of tract-tracers, the sphere of diffusion would largely be determined by the concentration of the stock solution anyway. The diffusion of a tracer or any other compound into the brain parenchyma is also dependent on its chemical structure and ability to bind cell membranes and the extracellular matrix, and not simply on the volume delivered. Our fibril infusions were of a relatively large volume because the stock solution was dilute and previous studies employing the same source of fibrils delivered similar or greater doses and volumes (Luk et al., 2012a; Osterberg et al., 2015; Paumier et al., 2015; Rey et al., 2016). Indeed, some studies in the preformed α-synuclein fibril model injected fibril volumes in mice that were two to five times larger than in the present study (Blumenstock et al., 2017; Hu et al., 2016; Osterberg et al., 2015). Given the mammillary body labeling discussed above, it is likely that the fibril sphere of diffusion in some animals was larger than that of the tracers, although this is difficult to predict accurately, as visualization is entirely dependent upon histochemical staining (e.g. antibody affinity, streptavidin/biotin affinity) and camera exposure and contrast settings, even if the fibrils had been directly tagged with a fluorophore. It is also likely that the rates of transport of the fibrils and the tracers differ significantly; for precisely this reason, we report the tracer labeling at early time points and the pSer129 labeling at longer time points. These are important but unavoidable limitations of using tract-tracers and histology to comprehend the spread of pSer129+ labeling and estimate the area of uptake.
We note that the lack of behavioral deficits at three months is not a shortcoming of this model, as Lewy body disorders are characterized by long prodromal phases and memory loss might unfold with longer incubation periods. Aside from the technical caveats identified above, other shortcomings of the present study can be summarized as follows: 1) we observed dense pathology in the dentate gyrus, whereas in human α-synucleinopathies, the dentate gyrus has notably few inclusions and most hippocampal Lewy pathology is concentrated in CA2 (Armstrong et al., 2014; Braak et al., 2003a; Gomez-Tortosa et al., 2000). 2) We failed to observe any atrophy in hippocampal subregions, although there may be tissue atrophy in CA1, CA3, and the subiculum in Parkinson’s disease and this is correlated with memory disruption (Beyer et al., 2013). Perhaps we failed to observe behavioral deficits for this very reason. 3) We did not observe loss of presubicular neurons, as reported in human cases of dementia with Lewy bodies (Harding et al., 2002a). 4) Previous studies demonstrate Lewy neurites in CA2 and CA3 in dementia with Lewy bodies, but did not report Lewy bodies (i.e. somal inclusions) in these structures (Churchyard and Lees, 1997; Dickson et al., 1994). This was interpreted to suggest that the origin of the neurites was extrahippocampal, perhaps cortical. Adamowicz and colleagues state that Lewy inclusions in the entorhinal cortex are housed in somata whereas the inclusions in Ammon’s horn are within neurites, suggesting involvement of entorhinal cortex-to-cornu Ammonis efferent circuitry (Adamowicz et al., 2017). Similarly, other authors conclude that terminals of the perforant pathway develop α-synucleinopathy in humans (Iseki et al., 1998). Our observation of somal inclusions in CA2 and CA3 likely reflects the direct infusion of fibrils in these structures, but, nevertheless, leads to a pattern not evident in humans. 5) Our studies were performed in young animals to save on cost. It is possible that we would not have observed any compensatory increase in synaptophysin had we challenged older mice with fibril exposure. Furthermore, behavioral deficits would probably develop at an accelerated rate in older animals. 6) Finally, our experiments were not designed to distinguish definitively between first and second-order neuronal label, and we cannot rule out that some of the pSer129 label was transneuronal, even at three months. 7) The background staining with pSer129 antibodies is high (Sacino et al., 2014b), as with all phospho-antibodies, although the inclusions reported here are morphologically distinct from background label and labeled much more intensely, consistent with previous studies in the fibril model (Blumenstock et al., 2017; Luk et al., 2012a; Mason et al., 2016; Paumier et al., 2015; Rey et al., 2016). Nevertheless, it is important to include high-resolution images of large swaths of the brain as we did, including all the areas of high background, such as white matter. 8) The Thioflavin and ubiquitin staining is more limited than that of pSer129, suggesting that only a fraction of α-synuclein+ inclusions house amyloid material. This is consistent with previous reports suggesting that α-synuclein+ inclusions are at various stages of maturity (Mason et al., 2016; Osterberg et al., 2015). It is difficult to place the Thioflavin results in the context of the literature as the topography is not generally discussed in detail, and the Thioflavin images are usually only shown at high magnification and not at both high and low magnification as we have. Mouse α-synuclein aggregates may exhibit lower Thioflavin fluorescence than human amyloid material (Kang et al., 2011; Luk et al., 2016), and, therefore, a Type II error may further confound our interpretations of Thioflavin label. We stained our inclusions with ubiquitin antibodies because this was the preferred method of visualizing inclusions before pSer129 antibodies became widely available (Spillantini et al., 1998; Tofaris et al., 2003). In our hands, ubiquitin staining was present both at the site of infusion and extrahippocampal sites such as the entorhinal cortex, but even this labeling did not have as wide a sweep as the α-synuclein-stained structures. Other authors have discussed additional important caveats of the preformed fibril model (Rey et al., 2015; Volpicelli-Daley et al., 2016).
We speculate that α-synucleinopathy is generally determined by at least some of the following criteria: 1) density of axon terminals from afferent neurons and densities of somata at the site of infusion, affecting the likelihood of retrograde and anterograde transport, respectively. 2) Expression of receptors that transport α-synuclein, such as LAG3 (Mao et al., 2016) and probably others. 3) The capacity of the fibrils to bind to lipid membranes (Iyer et al., 2016) versus the hydrophobic portions of extracellular matrix at the site of infusion. 4) Inherent vulnerability differences in loss of protein quality control, such as the integrity of proteasomal and autophagic systems (Chu et al., 2009; Ciechanover and Kwon, 2015; Domert et al., 2016; Karpowicz et al., 2017; Sacino et al., 2017). 5) Proximity to the ventricular space and diffusion into neighboring parenchymal structures via the cerebrospinal fluid and interstitial space or glymphatic system. 6) Firing rate of the fibril-exposed neurons, which might influence rates of endocytosis of extracellular material during vesicle membrane recycling. Bowen et al. reported that neurons are capable of strong phagocytic activity and that this may underlie cell-to-cell spread of disease (Bowen et al., 2007) and recent studies by Flavin and colleagues suggest that amyloid fibrils tend to invade neurons by endocytic vesicle rupture (Flavin et al., 2017). 7) The density of phagocytic astrocytes and microglia at the site of injection, as they might clear fibrillar debris, as well as the degree of myelination by local oligodendrocytes, which may provide a physical barrier against entry of α-synuclein through the axon or may decrease energetic demands and oxidative stress (Braak and Del Tredici, 2004; Braak et al., 2003a; Braak et al., 2003b; Braak et al., 2006b). 8) The strain of the fibrils and the sonication parameters are likely to influence their seeding/templating properties (Guo et al., 2013; Mason et al., 2016; Peng et al., 2017; Volpicelli-Daley et al., 2014b). 9) Endogenous α-synuclein expression, which determines how much protein is available for templating and propagation (Taguchi et al., 2014). 10) The ability of the affected cell to form exosomes and tunneling nanotubes and thereby transfer α-synuclein to neighboring cells (Abounit et al., 2016; Bieri et al., 2017; Okuda et al., 2017). 11) In addition to these determinants, Surmeier and colleagues argue that pacemaking properties, low intrinsic calcium buffering, and cytosolic calcium oscillations contribute to selectively vulnerable phenotypes (Surmeier et al., 2017). The obvious strength of the fibril model is that it can be leveraged to assess these potential mechanisms in the future.
5. Conclusion
There are multiple explanations for the spread of α-synucleinopathy in the preformed fibril model. First, the proteinopathy may be transmitted via anatomical connections, perhaps after synaptic or somal uptake and classic retrograde or anterograde transport. Alternatively, the fibrils might diffuse along the interstitial space surrounding the axolemma and subsequently be endocytosed at distant afferent and efferent sites in the absence of specific cytoplasmic transport (Valdinocci et al., 2017). Fibrils might also diffuse through the glymphatic space or the cerebrospinal fluid and enter the parenchymal space at distant locations but only lead to inclusion formation in the most vulnerable neuron subpopulations. Future studies are warranted to determine if there is loss of transmission of Lewy-like pathology in transgenic animals with selective deficits in synaptic uptake or axoplasmic transport mechanisms.
In summary, we report here an extensive characterization of inclusion numbers and cell counts in multiple subregions of the hippocampal formation in the early period following α-synuclein fibril infusions centered in regio inferior. The pattern of pathology transmission from the regio inferior is largely but not entirely predictable from the uptake and transport of well-established tract-tracers. The infusions of tract-tracers at the same stereotaxic coordinates permits the identification of neuron types that have access to fibrils but refrain from developing pathology. Studies of regional differences in vulnerability to proteotoxic stress are important because they facilitate the identification of differentially expressed molecules that promote or obstruct endogenous defenses. Collectively, the present findings demonstrate that dense bilateral α-synucleinopathy in the hippocampal archicortex is not necessarily sufficient to elicit memory deficits, although inclusions within the dentate gyrus were positively correlated with olfactory deficits and negatively correlated with memory function, suggesting some association of these behaviors with underlying vulnerability to proteotoxicity. The fibril-infused stratum radiatum displayed a slight increase in synaptophysin, suggestive of compensatory changes within axon terminals in response to proteotoxic stress. Thus, our model is one of incidental or prodromal Lewy body disease, where frank cell loss and overt behavioral deficits have not emerged, but where early compensatory changes perhaps responsible for the slow onset and protracted progression of proteinopathic diseases can be scrutinized.
Supplementary Material
Table 3.
Abbreviations.
| 3V third ventricle | ic internal capsule |
| 4V fourth ventricle | IC inferior colliculus |
| 5N motor trigeminal nucleus | ICjM large island of Calleja |
| 7n facial nerve | icp inferior cerebellar peduncle |
| 7N facial nucleus | IPF interpeduncular fossa |
| 8n vestibulocochlear nerve | IPN interpeduncular nucleus |
| A29 cingulate cortex, area 29 | LH lateral habenula |
| A30 cingulate cortex, area 30 | LHA lateral hypothalamic area |
| AA anterior amygdaloid area | lab longitudinal association bundle |
| ac anterior commissure | LC locus coeruleus |
| aca anterior commissure, anterior part | LDT laterodorsal tegmental nucleus |
| Acb nucleus accumbens | LM lateral mammillary nucleus |
| AcbC accumbens nucleus, core | LMol stratum lacunosum-moleculare |
| aci anterior commissure, intrabulbar | lo lateral olfactory tract |
| acp anterior commissure, posterior part | LOT nucleus of the lateral olfactory tract |
| AHi amygdalohippocampal area | Lrt lateral reticular nucleus |
| alv alveus | LS lateral septal nucleus |
| AMG amygdala | LSD lateral septal nucleus, dorsal part |
| AOB accessory olfactory bulb | LSI lateral septal nucleus, intermediate |
| AOD anterior olfactory area, dorsal | LSS lateral stripe of the striatum |
| AOE anterior olfactory nucleus, external | LSV lateral septal nucleus, ventral part |
| AOM anterior olfactory nucleus, medial | LV lateral ventricle |
| AON anterior olfactory nucleus | M2 secondary motor cortex |
| AOP anterior olfactory area, posterior | mcp middle cerebellar peduncle |
| AOV anterior olfactory area, ventral | MD mediodorsal thalamic nucleus |
| APir amygdalopiriform transition area | MEA medial amygdala |
| Aq aqueduct | MEnt medial entorhinal cortex |
| AV anteroventral thalamic nucleus | MG medial geniculate body |
| bic brachium of inferior colliculus | ml medial lemniscus |
| BLA basolateral nucleus of amygdala | mlf medial longitudinal fasciculus |
| BLP basolateral amygdaloid nucleus, posterior part | MoDG molecular layer of dentate gyrus |
| BMA basomedial nucleus of amygdala | MM medial mammillary nucleus |
| CA1 cornu Ammonis, field 1 | MO medial orbital cortex |
| CA2 cornu Ammonis, field 2 | MPT medial pretectal nucleus |
| CA3 cornu Ammonis, field 3 | MS medial septal nucleus |
| CB cerebellum | mt medial terminal nucleus |
| cc corpus callosum | OB olfactory bulb |
| CeA central nucleus of amygdala | och optic chiasm |
| CeL central amygdaloid nucleus, lateral part | Or oriens layer of the hippocampus |
| CEnt caudomedial entorhinal cortex | ot optic tract |
| cg cingulum | PAG periaquadyctal gray |
| CL claustrum | PaS parasubiculum |
| cp cerebral peduncle | pc posterior commissure |
| CPu caudoputamen | PH posterior hypothalamic nucleus |
| csc commissure of superior colliculi | Pir piriform cortex |
| cst commissural stria terminalis | PLCo posterolateral cortical amygdala |
| CxA cortex-amygdala transition zone | PMCo posteromedial cortical amygdala |
| DCIC pericentral nucleus of inferior colliculus | PoDG polymorph layer of the dentate gyrus |
| DEn dorsal endopiriform nucleus | Pn pontine nuclei |
| DG dentate gyrus | PnO pontine reticular nucleus, oral part |
| dhc dorsal hippocampal commissure | PR prerubral field |
| DIEnt dorsolateral entorhinal cortex | Pr5 principal trigeminal nucleus |
| DLG dorsal nucleus of lateral geniculate body | PrG pregeniculate nucleus of prethalamus |
| DR dorsal raphe | PRh perirhinal cortex |
| DS dorsal subiculum | Pyr stratum pyramidale of the hippocampus |
| DTT dorsal tenia tecta | Rad stratum radiatum of the hippocampus |
| ec external capsule | Re reuniens thalamic nucleus |
| ECIC external nucleus of inferior colliculus | rf rhinal fissure |
| Ect ectorhinal cortex | rms rostral migratory stream |
| Eml external medullary lamina | Rt reticular thalamic nucleus |
| Ent entorhinal cortex | RtTg reticulotegmental nucleus of pons |
| EPl external plexiform layer, olfactory bulb | S subiculum |
| f fornix | S1 primary somatosensory cortex |
| FC frontal cortex | S1BF somatosensory 1, barrel field |
| fi fimbria | S2 second somatosensory cortex |
| fmi forceps minor of the corpus callosum | SC superior colliculus |
| fmj forceps major of the corpus callosum | scc splenium of corpus callosum |
| fr fasciculus retroflexus | SLu stratum lucidum of the hippocampus |
| FrA frontal association cortex | sm stria medullaris |
| gcc genu of corpus callosum | SNpr substantia nigra, pars reticulata |
| Gl glomerular layer, olfactory bulb | sp spinal trigeminal tract |
| GP globus pallidus | Sp spinal trigeminal nucleus |
| GrDG granule layer of dentate gyrus | sp5 spinal trigeminal tract |
| GrO granule cell layer of olfactory bulb | st stria terminalis |
| HDB horizontal limb of the diagonal band | STN subthalamic nucleus |
| hif hippocampal fissure | SuG superficial gray layer, superior colliculus |
| HP hippocampus | SVZ subventricular zone |
| TeA temporal association cortex | |
| TT tenia tecta | |
| Tu olfactory tubercle | |
| V2L secondary visual cortex, lateral part | |
| VDB ventral diagonal band | |
| VM ventromedial thalamic nucleus | |
| VMH ventromedial hypothalamic nucleus | |
| VP ventral pallidum | |
| VS ventral subiculum | |
| VTA ventral tegmental area | |
| VTT ventral tenia tecta | |
| ZI zona incerta |
Acknowledgments
Designed the experiments, drew the brain maps, and wrote the paper: RKL. Performed the experiments and generated the figures: NN, DMM, RKL, TNB, and BKD. Commented on the paper: DAJ and KCL. Synthesized the fibrils: KCL. Analyzed the data: NN, DMM, RKL, MAC, KMM, TNB, BKD, and DS. Funded by NINDS grant R15NS093539 (RKL) and Hillman Foundation GRANT109033 (RKL).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.expneurol.2017.10.017.
- None of the authors have any conflicts to disclose.
- This research involved the use of animals with formal approval from the Duquesne University Institutional Animal Care and Use Committee.
- This research does not involve human subjects.
References
- Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sorensen P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol. 2003;60:387–392. doi: 10.1001/archneur.60.3.387. [DOI] [PubMed] [Google Scholar]
- Aarsland D, Perry R, Brown A, Larsen JP, Ballard C. Neuropathology of dementia in Parkinson’s disease: a prospective, community-based study. Ann Neurol. 2005;58:773–776. doi: 10.1002/ana.20635. [DOI] [PubMed] [Google Scholar]
- Abdelmotilib H, Maltbie T, Delic V, Liu Z, Hu X, Fraser KB, Moehle MS, Stoyka L, Anabtawi N, Krendelchtchikova V, Volpicelli-Daley LA, West A. Alpha-synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic neurodegeneration. Neurobiol Dis. 2017;105:84–98. doi: 10.1016/j.nbd.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, Olivo-Marin JC, Melki R, Zurzolo C. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016;35:2120–2138. doi: 10.15252/embj.201593411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamowicz DH, Roy S, Salmon DP, Galasko DR, Hansen LA, Masliah E, Gage FH. Hippocampal alpha-synuclein in dementia with Lewy bodies contributes to memory impairment and is consistent with spread of pathology. J Neurosci. 2017;37:1675–1684. doi: 10.1523/JNEUROSCI.3047-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanian GK, Gallager DW. Raphe origin of serotonergic nerves terminating in the cerebral ventricles. Brain Res. 1975;88:221–231. doi: 10.1016/0006-8993(75)90386-8. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Scharfman HE, Lavenex P. The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies) Prog Brain Res. 2007;163:3–22. doi: 10.1016/S0079-6123(07)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P. The Hippocampus Book. Oxford University Press; Oxford; New York: 2007. [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Armstrong RA, Kotzbauer PT, Perlmutter JS, Campbell MC, Hurth KM, Schmidt RE, Cairns NJ. A quantitative study of alpha-synuclein pathology in fifteen cases of dementia associated with Parkinson disease. J Neural Transm. 2014;121:171–181. doi: 10.1007/s00702-013-1084-z. (Vienna). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas, T.R.A.M.B. Richard Allen, Richard Allen mouse brain atlas online portal. 2017 http://mouse.brain-map.org/static/atlas.
- Aznar S, Qian ZX, Knudsen GM. Non-serotonergic dorsal and median raphe projection onto parvalbumin- and calbindin-containing neurons in hippocampus and septum. Neuroscience. 2004;124:573–581. doi: 10.1016/j.neuroscience.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL, 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG, Arizona Parkinson’s Disease, C Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009;117:613–634. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci A, Mercuri NB, Venneri A, Faustini G, Longhena F, Pizzi M, Missale C, Spano P. Review: Parkinson’s disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol. 2016;42:77–94. doi: 10.1111/nan.12297. [DOI] [PubMed] [Google Scholar]
- Benice TS, Rizk A, Kohama S, Pfankuch T, Raber J. Sex-differences in age-related cognitive decline in C57BL/6J mice associated with increased brain micro-tubule-associated protein 2 and synaptophysin immunoreactivity. Neuroscience. 2006;137:413–423. doi: 10.1016/j.neuroscience.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Bertrand E, Lechowicz W, Szpak GM, Lewandowska E, Dymecki J, Wierzba-Bobrowicz T. Limbic neuropathology in idiopathic Parkinson’s disease with concomitant dementia. Folia Neuropathol. 2004;42:141–150. [PubMed] [Google Scholar]
- Beyer MK, Bronnick KS, Hwang KS, Bergsland N, Tysnes OB, Larsen JP, Thompson PM, Somme JH, Apostolova LG. Verbal memory is associated with structural hippocampal changes in newly diagnosed Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2013;84:23–28. doi: 10.1136/jnnp-2012-303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieri G, Gitler AD, Brahic M. Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol Dis. 2017 doi: 10.1016/j.nbd.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstad TW. Commissural connections of the hippocampal region in the rat, with special reference to their mode of termination. J Comp Neurol. 1956;105:417–537. doi: 10.1002/cne.901050305. [DOI] [PubMed] [Google Scholar]
- Blumenstock S, Rodrigues EF, Peters F, Blazquez-Llorca L, Schmidt F, Giese A, Herms J. Seeding and transgenic overexpression of alpha-synuclein triggers dendritic spine pathology in the neocortex. EMBO Mol Med. 2017 doi: 10.15252/emmm.201607305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen S, Ateh DD, Deinhardt K, Bird MM, Price KM, Baker CS, Robson JC, Swash M, Shamsuddin W, Kawar S, El-Tawil T, Roos J, Hoyle A, Nickols CD, Knowles CH, Pullen AH, Luthert PJ, Weller RO, Hafezparast M, Franklin RJ, Revesz T, King RH, Berninghausen O, Fisher EM, Schiavo G, Martin JE. The phagocytic capacity of neurones. Eur J Neurosci. 2007;25:2947–2955. doi: 10.1111/j.1460-9568.2007.05554.x. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. Poor and protracted myelination as a contributory factor to neurodegenerative disorders. Neurobiol Aging. 2004;25:19–23. doi: 10.1016/j.neurobiolaging.2003.04.001. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. Assessing fetal nerve cell grafts in Parkinson’s disease. Nat Med. 2008;14:483–485. doi: 10.1038/nm0508-483. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003a;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Braak H, Rub U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003b;110:517–536. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- Braak H, Rub U, Jansen Steur EN, Del Tredici K, de Vos RA. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology. 2005;64:1404–1410. doi: 10.1212/01.WNL.0000158422.41380.82. [DOI] [PubMed] [Google Scholar]
- Braak H, Rub U, Del Tredici K. Cognitive decline correlates with neuro-pathological stage in Parkinson’s disease. J Neurol Sci. 2006a;248:255–258. doi: 10.1016/j.jns.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Braak H, Rub U, Schultz C, Del Tredici K. Vulnerability of cortical neurons to Alzheimer’s and Parkinson’s diseases. J Alzheimers Dis. 2006b;9:35–44. doi: 10.3233/jad-2006-9s305. [DOI] [PubMed] [Google Scholar]
- Briggs F. Organizing principles of cortical layer 6. Front Neural Circuits. 2010;4:3. doi: 10.3389/neuro.04.003.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchhalter JR, Fieles A, Dichter MA. Hippocampal commissural connections in the neonatal rat. Brain Res Dev Brain Res. 1990;56:211–216. doi: 10.1016/0165-3806(90)90084-c. [DOI] [PubMed] [Google Scholar]
- Cai L, Gibbs RB, Johnson DA. Recognition of novel objects and their location in rats with selective cholinergic lesion of the medial septum. Neurosci Lett. 2012;506:261–265. doi: 10.1016/j.neulet.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camicioli R. Identification of Parkinsonism and Parkinson’s disease. Drugs Today (Barc) 2002;38:677–686. doi: 10.1358/dot.2002.38.10.740195. [DOI] [PubMed] [Google Scholar]
- Canteras NS, Simerly RB, Swanson LW. Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris-leucoagglutinin study in the rat. J Comp Neurol. 1994;348:41–79. doi: 10.1002/cne.903480103. [DOI] [PubMed] [Google Scholar]
- Cappaert NLM, Van Strien NM, Witter MP. Hippocampal Formation. 4th. Elsevier Academic Press; Amsterdam, Boston: 2015. [Google Scholar]
- Card JP, Enquist LW. Transneuronal circuit analysis with pseudorabies viruses. Curr Protoc Neurosci. 2014;68(1 5):1–39. doi: 10.1002/0471142301.ns0105s68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Palay V. Serotonin axons in the supra- and subependymal plexuses and in the leptomeninges; their roles in local alterations of cerebrospinal fluid and vasomotor activity. Brain Res. 1976;102:103–130. doi: 10.1016/0006-8993(76)90578-3. [DOI] [PubMed] [Google Scholar]
- Chu Y, Kordower JH. The prion hypothesis of Parkinson’s disease. Curr Neurol Neurosci Rep. 2015;15:28. doi: 10.1007/s11910-015-0549-x. [DOI] [PubMed] [Google Scholar]
- Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-sy-nuclein inclusions. Neurobiol Dis. 2009;35:385–398. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Chu Y, Morfini GA, Langhamer LB, He Y, Brady ST, Kordower JH. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain. 2012;135:2058–2073. doi: 10.1093/brain/aws133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchyard A, Lees AJ. The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson’s disease. Neurology. 1997;49:1570–1576. doi: 10.1212/wnl.49.6.1570. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegen-erative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo C, Hughes AJ, Kilford L, Lees AJ. Lewy body cortical involvement may not always predict dementia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2003;74:852–856. doi: 10.1136/jnnp.74.7.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordato NJ, Halliday GM, Harding AJ, Hely MA, Morris JG. Regional brain atrophy in progressive supranuclear palsy and Lewy body disease. Ann Neurol. 2000;47:718–728. [PubMed] [Google Scholar]
- Costa C, Sgobio C, Siliquini S, Tozzi A, Tantucci M, Ghiglieri V, Di Filippo M, Pendolino V, de Iure A, Marti M, Morari M, Spillantini MG, Latagliata EC, Pascucci T, Puglisi-Allegra S, Gardoni F, Di Luca M, Picconi B, Calabresi P. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain. 2012;135:1884–1899. doi: 10.1093/brain/aws101. [DOI] [PubMed] [Google Scholar]
- Cui Z, Gerfen CR, Young WS., 3rd Hypothalamic and other connections with dorsal CA2 area of the mouse hippocampus. J Comp Neurol. 2013;521:1844–1866. doi: 10.1002/cne.23263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dado RJ, Burstein R, Cliffer KD, Giesler GJ., Jr Evidence that fluoro-gold can be transported avidly through fibers of passage. Brain Res. 1990;533:329–333. doi: 10.1016/0006-8993(90)91358-n. [DOI] [PubMed] [Google Scholar]
- Del Tredici K, Braak H. Review: sporadic Parkinson’s disease: development and distribution of alpha-synuclein pathology. Neuropathol Appl Neurobiol. 2016;42:33–50. doi: 10.1111/nan.12298. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Ruan D, Crystal H, Mark MH, Davies P, Kress Y, Yen SH. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer’s disease: light and electron microscopic immunocytochemistry of CA2-3 neurites specific to DLBD. Neurology. 1991;41:1402–1409. doi: 10.1212/wnl.41.9.1402. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Schmidt ML, Lee VM, Zhao ML, Yen SH, Trojanowski JQ. Immunoreactivity profile of hippocampal CA2/3 neurites in diffuse Lewy body disease. Acta Neuropathol. 1994;87:269–276. doi: 10.1007/BF00296742. [DOI] [PubMed] [Google Scholar]
- Domert J, Sackmann C, Severinsson E, Agholme L, Bergstrom J, Ingelsson M, Hallbeck M. Aggregated alpha-synuclein transfer efficiently between cultured human neuron-like cells and localize to lysosomes. PLoS One. 2016;11:e0168700. doi: 10.1371/journal.pone.0168700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, Cookson MR, Hardy J, Allsop D. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003;17:1945–1947. doi: 10.1096/fj.03-0098fje. [DOI] [PubMed] [Google Scholar]
- Fasano A, Visanji NP, Liu LW, Lang AE, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015;14:625–639. doi: 10.1016/S1474-4422(15)00007-1. [DOI] [PubMed] [Google Scholar]
- Flavin WP, Bousset L, Green ZC, Chu Y, Skarpathiotis S, Chaney MJ, Kordower JH, Melki R, Campbell EM. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017 doi: 10.1007/s00401-017-1722-x. [DOI] [PubMed] [Google Scholar]
- Flores-Cuadrado A, Ubeda-Banon I, Saiz-Sanchez D, de la Rosa-Prieto C, Martinez-Marcos A. Hippocampal alpha-synuclein and interneurons in Parkinson’s disease: data from human and mouse models. Mov Disord. 2016 doi: 10.1002/mds.26586. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates. Fourth. Academic Press, an imprint of Elsevier; Amsterdam: 2013. [Google Scholar]
- Fujita M, Sugama S, Sekiyama K, Sekigawa A, Tsukui T, Nakai M, Waragai M, Takenouchi T, Takamatsu Y, Wei J, Rockenstein E, Laspada AR, Masliah E, Inoue S, Hashimoto M. A beta-synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat Commun. 2010;1:110. doi: 10.1038/ncomms1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. Alpha-synuclein is phosphorylated in synu-cleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- Goldberg NR, Caesar J, Park A, Sedgh S, Finogenov G, Masliah E, Davis J, Blurton-Jones M. Neural stem cells rescue cognitive and motor dysfunction in a transgenic model of dementia with Lewy bodies through a BDNF-dependent mechanism. Stem Cell Rep. 2015;5:791–804. doi: 10.1016/j.stemcr.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT. Alpha-synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol. 2000;99:352–357. doi: 10.1007/s004010051135. [DOI] [PubMed] [Google Scholar]
- Gottlieb DI, Cowan WM. Autoradiographic studies of the commissural and ip-silateral association connection of the hippocampus and detentate gyrus of the rat. I The commissural connections. J Comp Neurol. 1973;149:393–422. doi: 10.1002/cne.901490402. [DOI] [PubMed] [Google Scholar]
- van Groen T, Miettinen P, Kadish I. The entorhinal cortex of the mouse: or-ganization of the projection to the hippocampal formation. Hippocampus. 2003;13:133–149. doi: 10.1002/hipo.10037. [DOI] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund L, Swanson LW, Kohler C. The projection of the supramammillary nucleus to the hippocampal formation: an immunohistochemical and anterograde transport study with the lectin PHA-L in the rat. J Comp Neurol. 1984;229:171–185. doi: 10.1002/cne.902290204. [DOI] [PubMed] [Google Scholar]
- Hall H, Reyes S, Landeck N, Bye C, Leanza G, Double K, Thompson L, Halliday G, Kirik D. Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain. 2014;137:2493–2508. doi: 10.1093/brain/awu193. [DOI] [PubMed] [Google Scholar]
- Hall K, Yang S, Sauchanka O, Spillantini MG, Anichtchik O. Behavioural deficits in transgenic mice expressing human truncated (1–120 amino acid) alpha-synuclein. Exp Neurol. 2015;264:8–13. doi: 10.1016/j.expneurol.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Hansen LA, Daniel SE, Wilcock GK, Love S. Frontal cortical synaptophysin in Lewy body diseases: relation to Alzheimer’s disease and dementia. J Neurol Neurosurg Psychiatry. 1998;64:653–656. doi: 10.1136/jnnp.64.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AJ, Halliday GM. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol. 2001;102:355–363. doi: 10.1007/s004010100390. [DOI] [PubMed] [Google Scholar]
- Harding AJ, Lakay B, Halliday GM. Selective hippocampal neuron loss in dementia with Lewy bodies. Ann Neurol. 2002a;51:125–128. doi: 10.1002/ana.10071. [DOI] [PubMed] [Google Scholar]
- Harding AJ, Stimson E, Henderson JM, Halliday GM. Clinical correlates of selective pathology in the amygdala of patients with Parkinson’s disease. Brain. 2002b;125:2431–2445. doi: 10.1093/brain/awf251. [DOI] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: the dual hit theory revisited. Ann N Y Acad Sci. 2009;1170:615–622. doi: 10.1111/j.1749-6632.2009.04365.x. [DOI] [PubMed] [Google Scholar]
- Heimer L, Záborszky LS, Wouterlood FG, Lanciego JL. Neuroanatomical Tract-Tracing 3: Molecules, Neurons, and Systems. 3rd. Springer; New York: 2006. [Google Scholar]
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney multicenter study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord. 2005;20:190–199. doi: 10.1002/mds.20324. [DOI] [PubMed] [Google Scholar]
- Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Bjorklund T, Wang ZY, Roybon L, Melki R, Li JY. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805–820. doi: 10.1007/s00401-014-1343-6. [DOI] [PubMed] [Google Scholar]
- Holmstrand EC, Sesack SR. Projections from the rat pedunculopontine and la-terodorsal tegmental nuclei to the anterior thalamus and ventral tegmental area arise from largely separate populations of neurons. Brain Struct Funct. 2011;216:331–345. doi: 10.1007/s00429-011-0320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Ren X, Liu Y, Li Z, Zhang L, Chen X, He C, Chen JF. Aberrant adenosine A2A receptor signaling contributes to neurodegeneration and cognitive impairments in a mouse model of synucleinopathy. Exp Neurol. 2016;283:213–223. doi: 10.1016/j.expneurol.2016.05.040. [DOI] [PubMed] [Google Scholar]
- Ince P, Irving D, MacArthur F, Perry RH. Quantitative neuropathological study of Alzheimer-type pathology in the hippocampus: comparison of senile dementia of Alzheimer type, senile dementia of Lewy body type, Parkinson’s disease and non-demented elderly control patients. J Neurol Sci. 1991;106:142–152. doi: 10.1016/0022-510x(91)90251-2. [DOI] [PubMed] [Google Scholar]
- Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V, Lee VM, Leverenz JB, Montine TJ, Duda JE, Hurtig HI, Trojanowski JQ. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72:587–598. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, Lee EB, Van Deerlin VM, Lopez OL, Kofler JK, Nelson PT, Jicha GA, Woltjer R, Quinn JF, Kaye J, Leverenz JB, Tsuang D, Longfellow K, Yearout D, Kukull W, Keene CD, Montine TJ, Zabetian CP, Trojanowski JQ. Neuropathological and genetic correlates of survival and dementia onset in synu-cleinopathies: a retrospective analysis. Lancet Neurol. 2017;16:55–65. doi: 10.1016/S1474-4422(16)30291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseki E, Marui W, Kosaka K, Akiyama H, Ueda K, Iwatsubo T. Degenerative terminals of the perforant pathway are human alpha-synuclein-immunoreactive in the hippocampus of patients with diffuse Lewy body disease. Neurosci Lett. 1998;258:81–84. doi: 10.1016/s0304-3940(98)00856-8. [DOI] [PubMed] [Google Scholar]
- Iyer A, Schilderink N, Claessens MM, Subramaniam V. Membrane-bound alpha synuclein clusters induce impaired lipid diffusion and increased lipid packing. Biophys J. 2016;111:2440–2449. doi: 10.1016/j.bpj.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Morphological substrates of mental dysfunction in Lewy body disease: an update. J Neural Transm Suppl. 2000;59:185–212. doi: 10.1007/978-3-7091-6781-6_21. [DOI] [PubMed] [Google Scholar]
- Joelving FC, Billeskov R, Christensen JR, West M, Pakkenberg B. Hippocampal neuron and glial cell numbers in Parkinson’s disease—a stereological study. Hippocampus. 2006;16:826–833. doi: 10.1002/hipo.20212. [DOI] [PubMed] [Google Scholar]
- Jones BE, Moore RY. Ascending projections of the locus coeruleus in the rat. II Autoradiographic study. Brain Res. 1977;127:25–53. [PubMed] [Google Scholar]
- Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaitzakis ME, Pearce RK. The morbid anatomy of dementia in Parkinson’s disease. Acta Neuropathol. 2009;118:587–598. doi: 10.1007/s00401-009-0597-x. [DOI] [PubMed] [Google Scholar]
- Kalaitzakis ME, Christian LM, Moran LB, Graeber MB, Pearce RK, Gentleman SM. Dementia and visual hallucinations associated with limbic pathology in Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:196–204. doi: 10.1016/j.parkreldis.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Kang L, Wu KP, Vendruscolo M, Baum J. The A53T mutation is key in de-fining the differences in the aggregation kinetics of human and mouse alpha-synu-clein. J Am Chem Soc. 2011;133:13465–13470. doi: 10.1021/ja203979j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpowicz RJ, Jr, Haney CM, Mihaila TS, Sandler RM, Petersson EJ, Lee VM. Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem. 2017 doi: 10.1074/jbc.M117.780296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kealy J, Commins S. The rat perirhinal cortex: a review of anatomy, physiology, plasticity, and function. Prog Neurobiol. 2011;93:522–548. doi: 10.1016/j.pneurobio.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Korbo L, Amrein I, Lipp HP, Wolfer D, Regeur L, Oster S, Pakkenberg B. No evidence for loss of hippocampal neurons in non-Alzheimer dementia patients. Acta Neurol Scand. 2004;109:132–139. doi: 10.1034/j.1600-0404.2003.00182.x. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Krettek JE, Price JL. Projections from the amygdala to the perirhinal and en-torhinal cortices and the subiculum. Brain Res. 1974;71:150–154. doi: 10.1016/0006-8993(74)90199-1. [DOI] [PubMed] [Google Scholar]
- Krettek JE, Price JL. Projections from the amygdaloid complex and adjacent olfactory structures to the entorhinal cortex and to the subiculum in the rat and cat. J Comp Neurol. 1977;172:723–752. doi: 10.1002/cne.901720409. [DOI] [PubMed] [Google Scholar]
- Krout KE, Loewy AD. Parabrachial nucleus projections to midline and in-tralaminar thalamic nuclei of the rat. J Comp Neurol. 2000;428:475–494. doi: 10.1002/1096-9861(20001218)428:3<475::aid-cne6>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Laakso MP, Partanen K, Riekkinen P, Lehtovirta M, Helkala EL, Hallikainen M, Hanninen T, Vainio P, Soininen H. Hippocampal volumes in Alzheimer’s disease, Parkinson’s disease with and without dementia, and in vascular dementia: an MRI study. Neurology. 1996;46:678–681. doi: 10.1212/wnl.46.3.678. [DOI] [PubMed] [Google Scholar]
- Larson ME, Greimel SJ, Amar F, LaCroix M, Boyle G, Sherman MA, Schley H, Miel C, Schneider JA, Kayed R, Benfenati F, Lee MK, Bennett DA, Lesne SE. Selective lowering of synapsins induced by oligomeric alpha-synuclein exacerbates memory deficits. Proc Natl Acad Sci U S A. 2017;114:E4648–E4657. doi: 10.1073/pnas.1704698114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurberg S, Sorensen KE. Associational and commissural collaterals of neurons in the hippocampal formation (hilus fasciae dentatae and subfield CA3) Brain Res. 1981;212:287–300. doi: 10.1016/0006-8993(81)90463-7. [DOI] [PubMed] [Google Scholar]
- Leak RK, Moore RY. Innervation of ventricular and periventricular brain compartments. Brain Res. 2012;1463:51–62. doi: 10.1016/j.brainres.2012.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol. 2010;6:702–706. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Lim HS, Masliah E, Lee HJ. Protein aggregate spreading in neuro-degenerative diseases: problems and perspectives. Neurosci Res. 2011;70:339–348. doi: 10.1016/j.neures.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmkuhl AM, Dirr ER, Fleming SM. Olfactory assays for mouse models of neurodegenerative disease. J Vis Exp. 2014:e51804. doi: 10.3791/51804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Matsumoto K, Watanabe H. Different effects of unilateral and bilateral hippocampal lesions in rats on the performance of radial maze and odor-paired associate tasks. Brain Res Bull. 1999;48:113–119. doi: 10.1016/s0361-9230(98)00157-9. [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Lim Y, Kehm VM, Lee EB, Soper JH, Li C, Trojanowski JQ, Lee VM. Alpha-Syn suppression reverses synaptic and memory defects in a mouse model of dementia with Lewy bodies. J Neurosci. 2011;31:10076–10087. doi: 10.1523/JNEUROSCI.0618-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurode-generation in nontransgenic mice. Science. 2012a;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012b;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Covell DJ, Kehm VM, Zhang B, Song IY, Byrne MD, Pitkin RM, Decker SC, Trojanowski JQ, Lee VM. Molecular and biological compatibility with host alpha-synuclein influences fibril pathogenicity. Cell Rep. 2016;16:3373–3387. doi: 10.1016/j.celrep.2016.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Goncalves RA, Liang Y, Zhang S, Qi C, Lam S, Keiler JA, Tyson J, Kim D, Panicker N, Yun SP, Workman CJ, Vignali DA, Dawson VL, Ko HS, Dawson TM. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353 doi: 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marui W, Iseki E, Kato M, Akatsu H, Kosaka K. Pathological entity of dementia with Lewy bodies and its differentiation from Alzheimer’s disease. Acta Neuropathol. 2004;108:121–128. doi: 10.1007/s00401-004-0869-4. [DOI] [PubMed] [Google Scholar]
- Mason DM, Nouraei N, Pant DB, Miner KM, Hutchison DF, Luk KC, Stolz JF, Leak RK. Transmission of alpha-synucleinopathy from olfactory structures deep into the temporal lobe. Mol Neurodegener. 2016;11:49. doi: 10.1186/s13024-016-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M. Prion-like spreading of pathological alpha-synu-clein in brain. Brain. 2013;136:1128–1138. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur BN. The claustrum in review. Front Syst Neurosci. 2014;8:48. doi: 10.3389/fnsys.2014.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PM, Rinne JO, Helenius H, Roytta M. Neuritic degeneration in the hippocampus and amygdala in Parkinson’s disease in relation to Alzheimer pathology. Acta Neuropathol. 1999;98:157–164. doi: 10.1007/s004010051064. [DOI] [PubMed] [Google Scholar]
- Mattila PM, Rinne JO, Helenius H, Dickson DW, Roytta M. Alpha-synu-clein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 2000;100:285–290. doi: 10.1007/s004019900168. [DOI] [PubMed] [Google Scholar]
- Moore RY, Halaris AE, Jones BE. Serotonin neurons of the midbrain raphe: ascending projections. J Comp Neurol. 1978;180:417–438. doi: 10.1002/cne.901800302. [DOI] [PubMed] [Google Scholar]
- Muslimovic D, Post B, Speelman JD, Schmand B. Cognitive profile of patients with newly diagnosed Parkinson disease. Neurology. 2005;65:1239–1245. doi: 10.1212/01.wnl.0000180516.69442.95. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Mori F, Kon T, Tanji K, Miki Y, Tomiyama M, Kurotaki H, Toyoshima Y, Kakita A, Takahashi H, Yamada M, Wakabayashi K. Accumulation of phosphorylated alpha-synuclein in subpial and periventricular as-trocytes in multiple system atrophy of long duration. Neuropathology. 2016;36:157–167. doi: 10.1111/neup.12243. [DOI] [PubMed] [Google Scholar]
- Nikolaus S, Antke C, Muller HW. In vivo imaging of synaptic function in the central nervous system: I. Movement disorders and dementia. Behav Brain Res. 2009;204:1–31. doi: 10.1016/j.bbr.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Okuda S, Uemura N, Takahashi R. Alpha-synuclein fibrils propagate through tunneling nanotubes. Mov Disord. 2017;32:394. doi: 10.1002/mds.26909. [DOI] [PubMed] [Google Scholar]
- Osterberg VR, Spinelli KJ, Weston LJ, Luk KC, Woltjer RL, Unni VK. Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015;10:1252–1260. doi: 10.1016/j.celrep.2015.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkkinen L, Kauppinen T, Pirttila T, Autere JM, Alafuzoff I. Alpha-synu-clein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol. 2005;57:82–91. doi: 10.1002/ana.20321. [DOI] [PubMed] [Google Scholar]
- Paumier KL, Sukoff Rizzo SJ, Berger Z, Chen Y, Gonzales C, Kaftan E, Li L, Lotarski S, Monaghan M, Shen W, Stolyar P, Vasilyev D, Zaleska MWDH, Dunlop J. Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson’s disease. PLoS One. 2013;8:e70274. doi: 10.1371/journal.pone.0070274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, Sandoval IM, Fleming S, Dirr E, Polinski NK, Trojanowski JQ, Lee VM, Sortwell CE. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synu-clein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis. 2015;82:185–199. doi: 10.1016/j.nbd.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Gathagan RJ, Lee VM. Distinct alpha-synuclein strains and implications for heterogeneity among alpha-synucleinopathies. Neurobiol Dis. 2017 doi: 10.1016/j.nbd.2017.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan JA, Stokholm K, Zareba-Paslawska J, Jakobsen S, Vang K, Gjedde A, Landau AM, Romero-Ramos M. Early synaptic dysfunction induced by alpha-synuclein in a rat model of Parkinson’s disease. Sci Rep. 2017;7:6363. doi: 10.1038/s41598-017-06724-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posimo JM, Titler AM, Choi HJ, Unnithan AS, Leak RK. Neocortex and allocortex respond differentially to cellular stress in vitro and aging in vivo. PLoS One. 2013;8:e58596. doi: 10.1371/journal.pone.0058596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posimo JM, Weilnau JN, Gleixner AM, Broeren MT, Weiland NL, Brodsky JL, Wipf P, Leak RK. Heat shock protein defenses in the neocortex and allo-cortex of the telencephalon. Neurobiol Aging. 2015;36:1924–1937. doi: 10.1016/j.neurobiolaging.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Rockenstein E, Ubhi K, Phung V, MacLean-Lewis N, Askay D, Cartier A, Spencer B, Patrick C, Desplats P, Ellisman MH, Masliah E. Alterations in mGluR5 expression and signaling in Lewy body disease and in transgenic models of alpha-synucleinopathy—implications for excitotoxicity. PLoS One. 2010;5:e14020. doi: 10.1371/journal.pone.0014020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Raisman G, Cowan WM, Powell TPS. The extrinsic afferent, commissural and association fibres of the hippocampus. Brain. 1965;88:963–996. [Google Scholar]
- Ramón Y, Cajal S, DeFelipe J, Jones EG. Cajal’s Degeneration and Regeneration of the Nervous System. Oxford University Press; New York: 1991. [Google Scholar]
- Raudino F, Leva S. Involvement of the spinal cord in Parkinson’s disease. Int J Neurosci. 2012;122:1–8. doi: 10.3109/00207454.2011.613551. [DOI] [PubMed] [Google Scholar]
- Reiner A, Veenman CL, Medina L, Jiao Y, Del Mar N, Honig MG. Pathway tracing using biotinylated dextran amines. J Neurosci Methods. 2000;103:23–37. doi: 10.1016/s0165-0270(00)00293-4. [DOI] [PubMed] [Google Scholar]
- Revuelta GJ, Rosso A, Lippa CF. Neuritic pathology as a correlate of synaptic loss in dementia with lewy bodies. Am J Alzheimers Dis Other Demen. 2008;23:97–102. doi: 10.1177/1533317507310565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Petit GH, Bousset L, Melki R, Brundin P. Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol. 2013;126:555–573. doi: 10.1007/s00401-013-1160-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, George S, Brundin P. Spreading the word: precise animal models and validated methods are vital when evaluating prion-like behaviour of alpha-synuclein. Neuropathol Appl Neurobiol. 2015 doi: 10.1111/nan.12299. [DOI] [PubMed] [Google Scholar]
- Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med. 2016 doi: 10.1084/jem.20160368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribak CE, Seress L, Peterson GM, Seroogy KB, Fallon JH, Schmued LC. A GABAergic inhibitory component within the hippocampal commissural pathway. J Neurosci. 1986;6:3492–3498. doi: 10.1523/JNEUROSCI.06-12-03492.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JG. Autoradiographic evidence for the selective accumulation of [3H]5-HT by supra-ependymal nerve terminals. Brain Res. 1977;134:151–157. doi: 10.1016/0006-8993(77)90933-7. [DOI] [PubMed] [Google Scholar]
- Risold PY, Swanson LW. Structural evidence for functional domains in the rat hippocampus. Science. 1996;272:1484–1486. doi: 10.1126/science.272.5267.1484. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Nuber S, Overk CR, Ubhi K, Mante M, Patrick C, Adame A, Trejo-Morales M, Gerez J, Picotti P, Jensen PH, Campioni S, Riek R, Winkler J, Gage FH, Winner B, Masliah E. Accumulation of oligomer-prone alpha-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain. 2014;137:1496–1513. doi: 10.1093/brain/awu057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roostaee A, Beaudoin S, Staskevicius A, Roucou X. Aggregation and neuro-toxicity of recombinant alpha-synuclein aggregates initiated by dimerization. Mol Neurodegener. 2013;8:5. doi: 10.1186/1750-1326-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford NJ, Sacino AN, Brooks M, Ceballos-Diaz C, Ladd TB, Howard JK, Golde TE, Giasson BI. Studies of lipopolysaccharide effects on the induction of alpha-synuclein pathology by exogenous fibrils in transgenic mice. Mol Neurodegener. 2015;10:32. doi: 10.1186/s13024-015-0029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, McKinney AB, Thomas MA, Shaw G, Golde TE, Giasson BI. Brain injection of alpha-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. J Neurosci. 2014a;34:12368–12378. doi: 10.1523/JNEUROSCI.2102-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, Thomas MA, McKinney AB, McGarvey NH, Rutherford NJ, Ceballos-Diaz C, Robertson J, Golde TE, Giasson BI. Amyloidogenic alpha-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 2014b;127:645–665. doi: 10.1007/s00401-014-1268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks MM, Chakrabarty P, Saha K, Khoshbouei H, Golde TE, Giasson BI. Proteolysis of alpha-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J Neurochem. 2017;140:662–678. doi: 10.1111/jnc.13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salat-Foix D, Andrews CN, Meddings J, Suchowersky O. Gastrointestinal symptoms in Parkinson disease: clinical aspects and management. Can J Neurol Sci. 2011;38:557–564. doi: 10.1017/s0317167100012099. [DOI] [PubMed] [Google Scholar]
- Samuel W, Masliah E, Hill LR, Butters N, Terry R. Hippocampal connectivity and Alzheimer’s dementia: effects of synapse loss and tangle frequency in a two-component model. Neurology. 1994;44:2081–2088. doi: 10.1212/wnl.44.11.2081. [DOI] [PubMed] [Google Scholar]
- Samuels ER, Szabadi E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr Neuropharmacol. 2008;6:235–253. doi: 10.2174/157015908785777229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Myers CE. Hilar mossy cells of the dentate gyrus: a historical perspective. Front Neural Circuits. 2012;6:106. doi: 10.3389/fncir.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Fallon JH. Fluoro-gold: a new fluorescent retrograde axonal tracer with numerous unique properties. Brain Res. 1986;377:147–154. doi: 10.1016/0006-8993(86)91199-6. [DOI] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010;120:131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiyama K, Takamatsu Y, Koike W, Waragai M, Takenouchi T, Sugama S, Hashimoto M. Insight into the dissociation of behavior from histology in synucleinopathies and in related neurodegenerative diseases. J Alzheimers Dis. 2016;52:831–841. doi: 10.3233/JAD-151015. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M, Higuchi M, Yanai K, Hisanaga SI, Hasegawa M. Propagation of pathological alpha-synuclein in marmoset brain. Acta Neuropathol Commun. 2017;5:12. doi: 10.1186/s40478-017-0413-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiosaka S, Tohyama M. Evidence for an alpha-melanocyte stimulating hormonergic hippocampal commissural connection in the rat, revealed by a double-labeling technique. Neurosci Lett. 1984;49:213–216. doi: 10.1016/0304-3940(84)90162-9. [DOI] [PubMed] [Google Scholar]
- Sorrentino ZA, Brooks MMT, Hudson V, 3rd, Rutherford NJ, Golde TE, Giasson BI, Chakrabarty P. Intrastriatal injection of alpha-synuclein can lead to widespread synucleinopathy independent of neuroanatomic connectivity. Mol Neurodegener. 2017;12:40. doi: 10.1186/s13024-017-0182-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudry Y, Lemogne C, Malinvaud D, Consoli SM, Bonfils P. Olfactory system and emotion: common substrates. Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128:18–23. doi: 10.1016/j.anorl.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Scoville SA. Cells of origin of entorhinal cortical afferents to the hippocampus and fascia dentata of the rat. J Comp Neurol. 1976;169:347–370. doi: 10.1002/cne.901690306. [DOI] [PubMed] [Google Scholar]
- Stewart T, Liu C, Ginghina C, Cain KC, Auinger P, Cholerton B, Shi M, Zhang J, Parkinson Study Group, D.I. Cerebrospinal fluid alpha-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. Am J Pathol. 2014;184:966–975. doi: 10.1016/j.ajpath.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. 2017;18:101–113. doi: 10.1038/nrn.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW. A direct projection from Ammon’s horn to prefrontal cortex in the rat. Brain Res. 1981;217:150–154. doi: 10.1016/0006-8993(81)90192-x. [DOI] [PubMed] [Google Scholar]
- Swanson LW. Brain Maps III: Structure of the Rat Brain: An Atlas With Printed and Electronic Templates for Data, Models, and Schematics. 3rd. Elsevier Academic Press; Amsterdam, Boston: 2004. [Google Scholar]
- Swanson LW, Cowan WM. Hippocampo-hypothalamic connections: origin in subicular cortex, not Ammon’s horn. Science. 1975;189:303–304. doi: 10.1126/science.49928. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Cowan WM. An autoradiographic study of the organization of the efferent connections of the hippocampal formation in the rat. J Comp Neurol. 1977;172:49–84. doi: 10.1002/cne.901720104. [DOI] [PubMed] [Google Scholar]
- Taguchi K, Watanabe Y, Tsujimura A, Tatebe H, Miyata S, Tokuda T, Mizuno T, Tanaka M. Differential expression of alpha-synuclein in hippocampal neurons. PLoS One. 2014;9:e89327. doi: 10.1371/journal.pone.0089327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Watanabe Y, Tsujimura A, Tanaka M. Brain region-dependent differential expression of alpha-synuclein. J Comp Neurol. 2015 doi: 10.1002/cne.23901. [DOI] [PubMed] [Google Scholar]
- Thakur P, Breger LS, Lundblad M, Wan OW, Mattsson B, Luk KC, Lee VMY, Trojanowski JQ, Bjorklund A. Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1710442114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem. 2003;278:44405–44411. doi: 10.1074/jbc.M308041200. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, Waits K, Boluda S, Grossman M, Van Deerlin VM, Lee EB, Arnold SE, Duda JE, Hurtig H, Lee VM, Adler CH, Beach TG, Trojanowski JQ. Pathological alpha-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol. 2016;131:393–409. doi: 10.1007/s00401-015-1526-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy A, Rusconi R, Perez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, Monte DA. Caudo-rostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol Med. 2013;5:1119–1127. doi: 10.1002/emmm.201302475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdinocci D, Radford RA, Siow SM, Chung RS, Pountney DL. Potential modes of intercellular alpha-synuclein transmission. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18020469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Groen T, Wyss JM. Extrinsic projections from area CA1 of the rat hippocampus: olfactory, cortical, subcortical, and bilateral hippocampal formation projections. J Comp Neurol. 1990;302:515–528. doi: 10.1002/cne.903020308. [DOI] [PubMed] [Google Scholar]
- Varela C, Kumar S, Yang JY, Wilson MA. Anatomical substrates for direct interactions between hippocampus, medial prefrontal cortex, and the thalamic nucleus reuniens. Brain Struct Funct. 2014;219:911–929. doi: 10.1007/s00429-013-0543-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenman CL, Reiner A, Honig MG. Biotinylated dextran amine as an ante-rograde tracer for single- and double-labeling studies. J Neurosci Methods. 1992;41:239–254. doi: 10.1016/0165-0270(92)90089-v. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA. Effects of alpha-synuclein on axonal transport. Neurobiol Dis. 2017;105:321–327. doi: 10.1016/j.nbd.2016.12.008. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VM. Formation of alpha-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell. 2014a;25:4010–4023. doi: 10.1091/mbc.E14-02-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Lee VM. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc. 2014b;9:2135–2146. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Kirik D, Stoyka LE, Standaert DG, Harms AS. How can rAAV-alpha-synuclein and the fibril alpha-synuclein models advance our understanding of Parkinson’s disease? J Neurochem. 2016;139(Suppl. 1):131–155. doi: 10.1111/jnc.13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Levine H, 3rd, Mattson MP, Jucker M. Inducible proteopathies. Trends Neurosci. 2006;29:438–443. doi: 10.1016/j.tins.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RE, Jr, Wiegand SJ, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–159. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- Wessendorf MW. Fluoro-gold: composition, and mechanism of uptake. Brain Res. 1991;553:135–148. doi: 10.1016/0006-8993(91)90241-m. [DOI] [PubMed] [Google Scholar]
- Witter MP. The perforant path: projections from the entorhinal cortex to the dentate gyrus. Prog Brain Res. 2007;163:43–61. doi: 10.1016/S0079-6123(07)63003-9. [DOI] [PubMed] [Google Scholar]
- Witter MP, Griffioen AW, Jorritsma-Byham B, Krijnen JL. Entorhinal projections to the hippocampal CA1 region in the rat: an underestimated pathway. Neurosci Lett. 1988;85:193–198. doi: 10.1016/0304-3940(88)90350-3. [DOI] [PubMed] [Google Scholar]
- Wouterlood FG, Jorritsma-Byham B. The anterograde neuroanatomical tracer biotinylated dextran-amine: comparison with the tracer Phaseolus vulgaris-leu-coagglutinin in preparations for electron microscopy. J Neurosci Methods. 1993;48:75–87. doi: 10.1016/s0165-0270(05)80009-3. [DOI] [PubMed] [Google Scholar]
- Yang W, Yu S. Synucleinopathies: common features and hippocampal manifestations. Cell Mol Life Sci. 2016 doi: 10.1007/s00018-016-2411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappone CA, Sloviter RS. Commissurally projecting inhibitory interneurons of the rat hippocampal dentate gyrus: a colocalization study of neuronal markers and the retrograde tracer fluoro-gold. J Comp Neurol. 2001;441:324–344. doi: 10.1002/cne.1415. [DOI] [PubMed] [Google Scholar]
- Zhang HH, Zhang XQ, Wang WY, Xue QS, Lu H, Huang JL, Gui T, Yu BW. Increased synaptophysin is involved in inflammation-induced heat hyper-algesia mediated by cyclin-dependent kinase 5 in rats. PLoS One. 2012;7:e46666. doi: 10.1371/journal.pone.0046666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HT, John N, Delic V, Ikeda-Lee K, Kim A, Weihofen A, Swayze EE, Kordasiewicz HB, West AB, Volpicelli-Daley LA. LRRK2 antisense oligo-nucleotides ameliorate alpha-synuclein inclusion formation in a Parkinson’s disease mouse model. Mol Ther Nucleic Acids. 2017;8:508–519. doi: 10.1016/j.omtn.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.