

Abstract
Endothelial cell (EC) dysfunction has been associated with inflammatory and autoimmune diseases; however, the factors contributing to this dysfunction have not been fully explored. Because activation of TLRs has been implicated in autoimmune diseases, the goal of this study was to determine the effects of TLR ligands on EC function. Human dermal microvascular ECs (HDMECs) treated with TLR3 (Poly(I:C)), -4 (LPS) and -7 (Imiquimod) agonists showed decreased proliferation and a reduced total number of branching tubules in 3D human dermal organoid ex vivo culture. In contrast, the TLR9 ligand class C, ODN2395, increased angiogenesis. The anti-proliferative effects of TLR3, -4 and -7 ligands correlated with significant downregulation of a key regulator of vascular homeostasis, Fli1, while TLR9 increased Fli1 levels. Furthermore, Poly(I:C) and LPS induced endothelial to mesenchymal transition (EndoMT) that was reversed by the pretreatment with TGFβ neutralizing antibody or re-expression of Fli1. We showed, that Fli1 was required for the HDMEC proliferation by transcriptionally repressing FOXO3A. In contrast to TLR9, which suppressed activation of the FOXO3A pathway, TLR3, -4, and -7 ligands activated FOXO3A as indicated by decreased phosphorylation and increased nuclear accumulation. The inverse correlation between Fli1 and FOXO3A was also observed in the vasculature of scleroderma patients. This work revealed opposing effects of TLR9 and TLR3, -4, and -7 on the key angiogenic pathways, Fli1 and FOXO3A. Our results provide a novel mechanistic insight into the regulation of angiogenesis by TLRs and confirm a central role of Fli1 in regulating vascular homeostasis.
Keywords: ECs, TLRs, Fli1, FOXO3A
Introduction
Endothelial cells (ECs) control important vascular functions, including blood pressure, permeability, coagulation, and angiogenesis (1). Various pathological conditions including autoimmune diseases, such as Systemic lupus erythematosus (SLE) and Systemic sclerosis (SSc) are associated with dysfunctional endothelium (2, 3). In addition to its important role in regulating vascular homeostasis, endothelial cells participate in inflammatory response by increasing production of proinflammatory factors including MCP-1, IL-6, TNFα and interferons. While inappropriate activation of ECs could contribute to tissue damage and defective angiogenesis (2), the mechanism underlying the interactions of ECs with the immune system, especially the role of Toll-like receptors (TLRs) in this process, is still poorly understood.
Innate immunity constitutes the first line of defense against invading pathogens and endogenous danger signals. TLRs belong to a family of pattern recognition receptors (PRRs) that play a critical role in the activation of the innate immune system (4). TLRs can be recognized by a wide variety of exogenous ligands, including microbial cell wall components, proteins, and nucleic acids, as well as endogenous ligands released by injured or stressed cells. TLRs are expressed ubiquitously in immune cells and are less widespread in non-immune cells such as fibroblasts, epithelial or endothelial cells. The presence of TLRs on ECs depends on EC origin/location and may reflect specialized functions of different ECs (5). In response to their ligands, TLRs can activate the NF-κB signaling pathway and trigger secretion of the pro-inflammatory cytokines, including IL-6, or production of Type 1 interferons (IFN) (4). Recent studies demonstrated the importance of TLR signaling in the development of several diseases characterized by endothelial dysfunction, including atherosclerosis, hypertension and autoimmune diseases. In particular, endosomal TLR-3, 7, 9, that recognize dsRNA, ssRNA and unmethylated CpG oligonucleotide sequences, as well as plasma membrane TLR4, essential in recognition of bacterial pathogens, were suggested to be important in the initiation and progression of vascular dysfunction (6–8). Importantly, although TLR receptors share similar signaling pathways, they may have divergent effects on cellular function. For example, TLR7 and TLR9 displayed opposing effects on the development of systemic lupus erythematous, through different effects on the autoantibody production (9, 10). Despite significant progress in delineating the role of selected TLR ligands in EC activation and injury, the signaling pathways involved in this processes are still poorly understood.
Relevant to these studies, we have recently observed that expression levels of Fli1, a key regulator of vascular homeostasis, were significantly reduced by treatments with IFN-α in HDMECs (11). Fli1 is a member of the Ets family of transcription factors and is characterized by the presence of the evolutionary conserved DNA-binding (ETS) domain. Fli1 has been shown to play a major role in hematopoiesis, embryonic development, and vasculogenesis. Additionally, in SSc, reduced levels of Fli1 in endothelial cells and fibroblasts (12) were correlated with vascular dysfunction, inflammation, and upregulated matrix production (13–16). Mice with a conditional knockout of Fli1 in endothelial cells showed abnormal skin vasculature, with greatly compromised vessel integrity and markedly increased vessel permeability (17). Notably, vascular changes resulting from Fli1 deficiency overlapped to a great extent with the abnormalities observed in the vasculature of SSc patients, suggesting that persistently reduced levels of Fli1 in endothelial cells could be important in disease pathogenesis. While activation of the immune system in SSc is likely to contribute to vascular pathology, the role of immune mediators in the regulation of Fli1 remains largely unexplored. Given the important role of TLRs in the pathogenesis of autoimmune diseases, including SSc, the goal of this study was to determine the role of selected TLR ligands in modulating endothelial cell function focusing on a crosstalk between the TLR and the Fli1 signaling pathways.
Material and methods
Cells
Human dermal microvascular endothelial cells (HDMECs) were isolated from human foreskin as previously described (11, 18). Cells were cultured on bovine collagen-coated 6-well plates in EBM medium supplemented with 10% FBS, and EC growth supplement mix at 37°C with 5% CO2 in air. The culture medium was changed every other day. All the experiments were performed on cell from early passages.
siRNA Transient Transfections
HDMECs were transfected with siRNA specific to human Fli1 (ON-TARGETplus SMART pool; GE Dharmacon, Lafayette, CO) or negative control siRNA at the concentration of 10 nM using Lypofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol.
Luciferase Activity Assay
HDMECs were transfected with expression vector encoding Fli1FLAG and empty vector (pSG5) or treated with Fli1 Adenovirus and G0 control for 24h (12). Next, cells were co-transfected with the FOXO3A reporter plasmid FHRE-luc using Fugene6 (Roche Applied Science), according to the manufacturer’s instructions. FOXO3A-dependent transcriptional activity was measured by the Thermo Scientific Pierce Firefly Luciferase Glow Assay Kit (Thermo Fisher). Promoter/reporter plasmids were co-transfected with pCMV-βGal (Clontech) to adjust for differences in transfection efficiencies between samples.
Western Blot
For Western blot, whole-cell extracts were prepared from HDMECs using lysis buffer with the following composition: 1% Triton X-100, 50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 3 mmol/L MgCl2, 1 mmol/L CaCl2, proteinase inhibitor mixture (Roche), and 1 mmol/L phenylmethyl sulfonyl fluoride. Protein extracts were subjected to SDS-PAGE and transferred to nitrocellulose membranes. Membranes were incubated overnight with primary antibodies, washed, and incubated for 1 hour with appropriate horseradish peroxidase-conjugated secondary antibody. After washing, visualization was performed by enhanced chemiluminescence (Pierce, Rockford, IL). The following primary antibodies were used: mouse anti-human FLI1 (BD Biosciences, Billerica, MA), rabbit anti-human FOXO3A (Abcam, Cambridge, MA), rabbit polyclonal SNAI1 (Santa Cruz, CA), goat anti-Collagen type I (Southern Biotech, Birmingham, AL) rabbit anti-human FOXO3A (p Ser253) (Novus Biologicals, Littleton, CO) at a dilution of 1:1,000 dilution, and a control mouse monoclonal anti β-actin (Sigma, St Louis, MO) at a dilution of 1:5,000. Proteins levels were quantified using Image J software.
Chromatin Immunoprecipitation Assay
The chromatin immunoprecipitation (ChiP) assay was carried out essentially as described previously (19). Briefly, cells were treated with 1% formaldehyde for 10 minutes. The cross-linked chromatin was then prepared and sonicated to an average size of 300–500 bp. The DNA fragments were immunoprecipitated overnight with or without polyclonal anti-Fli1 antibody at 4°C. After reversal of cross-linking, the immunoprecipitated chromatin was amplified by PCR amplification of specific regions of the FOXO3A genomic locus. The primers were as follows: FOXO3A/F-605∶5′-TAGCGGTTTTGTCGTCATTTAC-3′, FOXO3A/R-772; 5′-CACAGTCGTGTTTCACTTTTCA-3′. The amplified DNA products were resolved by agarose gel electrophoresis.
Quantitative RT-PCR analysis
Total RNA was isolated using TRIzol reagent (MRC, Inc., Cincinnati, OH). Real-time PCR assays were performed using the StepOnePlus Real-Time PCR system (Applied Biosystems, Foster City, CA). Briefly, 1 μg of total RNA was reverse transcribed with random hexamers using the Transcriptor First Strand complementary DNA Synthesis kit (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s protocol. The amplification mixture (10 μl) contained 1 μl of complementary DNA, 0.5 μM of each primer, and 5 μl of SYBR Green PCR Master Mix. The primers are listed in Supplementary Table I. Relative changes in the levels of genes of interest were determined by the 2−ΔΔCT method.
Immunofluorescence staining on adherent cell cultures
For immunofluorescence, cultured HDMECs grown on collagen-coated cover slips were treated with Fli1siRNA for 48h or Poly(I:C), LPS and Imiquimod for 24h. Cells were fixed with 4% paraformaldehyde for 15 minutes followed by incubation with 0.15 M Glycine for 30 min. Non-specific protein binding was blocked with 3% BSA for 1 h. Next, cells were incubated at 4°C overnight with primary antibodies: goat anti-mouse VE-cadherin (Santa Cruz, CA) and rabbit anti-human FOXO3A (Abcam, Cambridge, MA). After washing, cell cultures were incubated with an Alexa fluor 488 donkey anti-goat (Invitrogen, Grand Island, NY) and an Alexa fluor 594 donkey anti-rabbit (Invitrogen, Grand Island, NY) for 1.5 h. Cells were mounted on slides using Vectashield with DAPI (Vector Laboratories, Burlingame, CA) and examined using a FluoView FV10i confocal microscope system (Olympus, Center Valley, PA) at 488 nm (green), 594 nm (red) and 405 nm (blue).
Proliferation assay
Proliferative responses were examined using the Essen BioScience IncuCyteTM Live-Cell Imaging system. Briefly, scrambled or Fli1siRNA treated cells were plated on an ImageLock 96-well plate and grown overnight. 5–10% confluent cells were treated additionally with 1μg/ml of Poly(I:C), LPS, Imiquimod and ODN2395. Images were captured every 3h for a total of 72h. Area under curves was measured using the GraphPad Prism software.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded skin tissue sections using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions. Briefly, sections (8-μm thick) were mounted on APES (aminopropyltriethoxy silane solution)-coated slides, deparaffinized with Histo-Clear (National Diagnostics, Atlanta, GA), and rehydrated through a graded series of ethanol. Endogenous peroxidase was blocked by incubation in 3% hydrogen peroxide for 30 minutes, followed by incubation with 0.15 M Glycine for 45 min, and normal blocking serum for 1 hour. The sections were then incubated overnight at 4 °C with antibodies against FOXO3A, or Fli1, diluted 1:100 in blocking buffer, followed by incubation for 30 minutes with a biotinylated secondary antibody solution. A solution containing avidin:biotin:peroxidase complexes was applied to the sections subsequently. Immunoreactivity was visualized with diaminobenzidine (Vector Laboratories, Burlingame, CA), and the sections were counterstained with hematoxylin. Images were collected using a microscope (BH-2; Olympus, Center Valley, PA).
3D human dermal organoid culture ex vivo angiogenesis assay
Dermal tissues obtained from foreskins were treated with scrambled or Fli1siRNA for 24h and then embedded in matrigel in 96 well plates or directly embedded in matrigel and treated with 1μg/ml of Poly(I:C), LPS, Imiquimod and ODN2395 in EGM-2 medium. Outgrowth of sprouts was quantified at day 3,4,5 and 6. Data represent an n=8 well each point with 3 different cell cultures. *p<0.05.
Apoptosis assay
Apoptosis was evaluated using the Essen BioScience IncuCyteTM Live-Cell Imaging system. Briefly, 70–80% confluent cells on ImageLock 96-well plate were treated with SCR or Fli1siRNA for 48h or Poly(I:C), LPS, Imiquimod and ODN2395 for 24h. Next, 50μl of Anexin V reagent (eBioscience, San Diego, CA) was added on each well. Images were captured every 3h for total of 48h.
Statistical analyses
All data were analyzed by Student’s t-Test. The level for statistical significance was set at p≤0.05.
Results
Distinct effects of selected TLR ligands on Fli1 protein level in HDMECs
To determine whether TLR ligands can regulate expression of Fli1, HDMECs were treated with 1ug/ml of each: LTA (Lipoteichoic acid) (TLR2), Poly(I:C) (TLR3), LPS (TLR4), Flagellin (TLR5), Imiquimod (TLR7), and ODN2395 (TLR9 - class C) for 24 hours. VEGF (25ng/ml) was used as a positive control. TGFβ (1ng/ml) and IFN-α (100U/ml) were used as a negative control for Western blot. We did not observe any changes in mRNA levels of Fli1 in response to any of the tested TLR ligands (data not shown). Dose and time dependent responses are shown in the Supplementary Figure 1. Treatment with Poly(I:C), LPS, Imiquimod, LTA, and Flagellin significantly decreased protein levels of Fli1, while TLR9 ligand, ODN2395, led to a significant increase of Fli1 (Figure 1 and Supplementary Fig. 1). CpG ODNs (small synthetic oligodeoxynucleotides with unmethylated CpG dinucleotides) are able to mimic the immunostimulatory activity of bacterial DNA and activate TLR9. The three different classes of CpG ODNs, (A, B and C), which differ in their immunostimulatory activities were recently characterized. To further characterize the effect of specific ODNs on Fli1 level, HDMECs were treated with ODN2336 (class A), ODN684 (class B) and ODN2395 (class C). We confirmed increased protein levels of Fli1 in response to class C TLR9 ligand. On the other hand, treatment with TLR9 ligands from class A and class B had no effect on Fli1 protein levels (Figure 1B), suggesting distinct regulation of Fli1 in response to ODNs from class C. Overall, these data indicate that Fli1 protein levels are regulated differently by specific TLR ligands in HDMVECs. Because of the potential relevance to scleroderma, we focused on Poly(I:C), LPS, Imiquimod and OD2395 in the subsequent experiments (20).
Figure 1. The effect of TGFβ and selected TLR ligands on the Fli1 protein level in HDMECs.
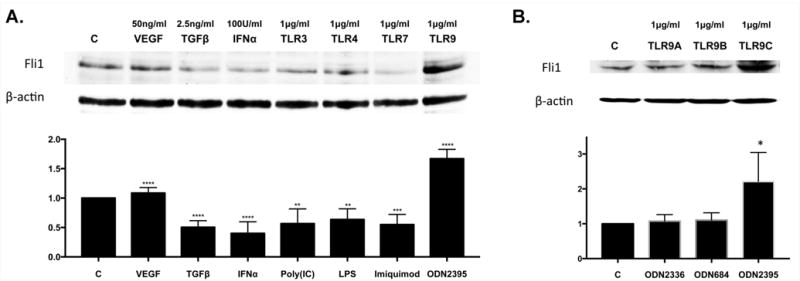
A. Confluent HDMECs were treated with VEGF, TGFβ, IFNα, Poly(I:C), LPS, Imiquimod and ODN2395 (A) or with ODN2336, ODN684 and ODN2395 (B) for 24h and collected for analysis by western blot. Densitometry of the western blot was analyzed with the Image J software (NIH). Results are shown as mean ± SD, n=3. Significant difference was accepted at *p<0.05 against the control group and calculated according to the Student t-test.
Downregulation of Fli1 in response to selected TLR ligands is mediated by TGFβ, ET-1, and type I interferon
It was previously reported that selected TLR ligands could modulate TGFβ and ET-1 signaling (21–23). To test the effect of selected TLR ligands on TGFβ and ET-1 mRNA levels, HDMECs were treated with Poly(I:C), LPS, Imiquimod and ODN2395 for 3 hours. Real-time PCR analysis showed a statistically significant increase in mRNA levels of TGFβ2 and -3 isoforms in cells treated with Poly(I:C) and LPS (Figure 2A), while treatment with Imiquimod only moderately increased TGFβ3 isoform. Likewise, mRNA levels of ET-1 were significantly upregulated in cells treated with Poly(I:C), LPS and Imiquimod (Figure 2A). In contrast, TLR9 ligand had no effect on TGFβ or ET-1 mRNA expression. Both TGFβ and ET-1 have been previously shown to downregulate Fli1 protein levels (24, 25). Therefore, we wished to determine if downregulation of Fli1 protein level in HDMECs is TGFβ or ET-1 dependent. Confluent cells were pretreated with specific inhibitor of TGFβRI kinase (SB431542) for 1h or Bosentan (BOS- dual endothelin receptor types A and B antagonist) for 3h and then treated with Poly(I:C), LPS and Imiquimod for another 24h. SB431542 pretreatment did not affect Fli1 protein levels, however, Bosentan consistently increased basal expression of Fli1, suggesting that the endogenously produced ET-1 negatively regulates the basal Fli1 expression levels. The blockade of TGFβ signaling reversed the effects of Poly(I:C), LPS and Imiquimod on Fli1 protein levels in HDMECs (Figure 2B). Similar effects were observed in cells pretreated with Bosentan. Consistent with our previous findings demonstrating downregulation of Fli1 by IFNα (11), pretreatment with B18R, a decoy receptor for type I interferon, reversed the effects of Poly(I:C), LPS, and Imiquimod on Fli1 protein levels (Supplementary Figure 2). Together, these data suggest that TLR ligands downregulate Fli1 protein levels by several interdependent pathways in endothelial cells (26–28).
Figure 2. TGFβ and ET-1 mediates Fli1 downregulation in HDMECs.
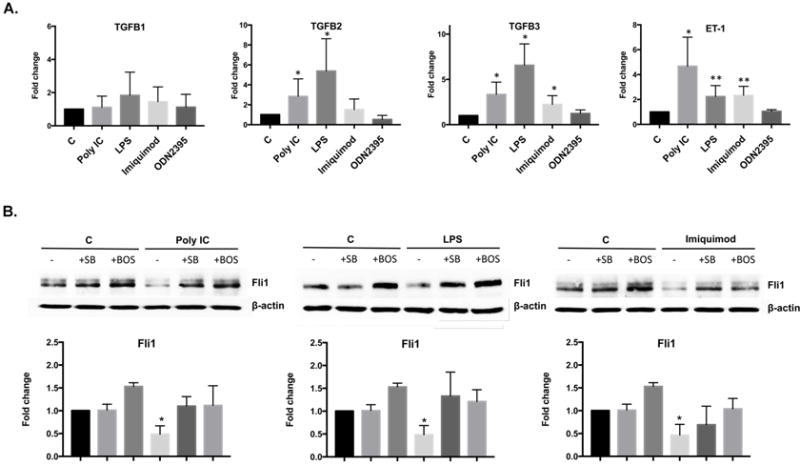
A. HDMVECs were treated for 3 h with Poly(I:C), LPS, Imiquimod and ODN2395 and analyzed by real-time qPCR for TGFβ1, TGFβ2, TGFβ3 and ET-1 gene expression. B. HDMECs were pretreated with TGFβRI kinase inhibitor (SB431542) for 1h or Bosentan (BOS- dual endothelin receptor types A and B antagonist) for 3h and then treated with Poly(I:C), LPS and Imiquimod for another 24h. Fli1, protein level was analyzed by western blot. Densitometry of the western blot was analyzed with the Image J software (NIH). Results are shown as mean ± SD, n=3. Significant difference was accepted at *p<0.05 against the control group and calculated according to the Student t-test.
Fli1 is required for HDMEC proliferation
Our previous studies have demonstrated the key role of Fli1 in regulating vessel permeability, however other functions of Fli1 in ECs have not yet been thoroughly investigated (17). To assess the effects of Fli1 and/or selected TLR ligands on EC proliferation, control siRNA (SCR) and Fli1siRNA-treated HDMECs were stimulated with Poly(I:C) (1ug/ml), LPS (1ug/ml), Imiquimod (1ug/ml) and ODN2395 (1ug/ml) for 72 hours and examined with the Essen BioScience IncuCyteTM Live-Cell Imaging system. As shown in Figure 3A, cells stimulated with Poly(I:C), LPS, or Imiquimod showed significantly decreased proliferation, while ODN2395 significantly increased cell proliferation. Depletion of Fli1 also significantly reduced proliferation. Moreover, we observed further reduction of proliferation in Fli1 deficient cells treated with Poly(I:C), LPS, Imiquimod. Treatment with the ODN2395 ligand did not reverse the inhibitory effect of Fli1siRNA. Pretreatment with B18R, reversed the inhibitory effect of Poly(I:C), LPS and Imiquimod, and further enhanced the pro-proliferative properties of ODN2395, however it had no effect on the basal proliferation level (Figure 3B). These data demonstrate that Fli1 deficiency is sufficient to impair proliferation of HDMECs. Furthermore, anti-proliferative effects of Poly(I:C), LPS, or Imiquimod in HDMECs are mediated by the induction of type I IFN.
Figure 3. The effect of Fli1 downregulation and/or selected TLR ligands on the proliferation of HDMECs.
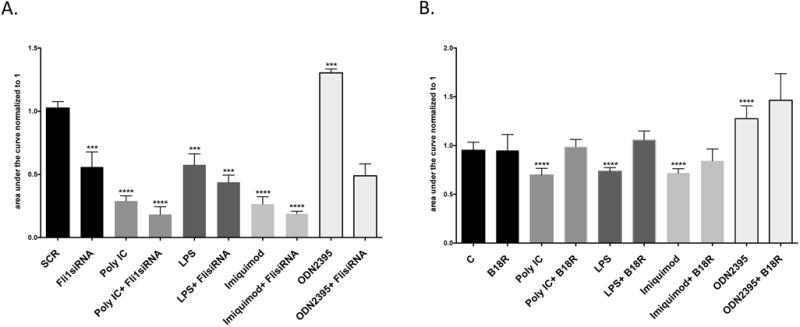
Proliferation was examined with the Essen BioScience IncuCyteTM Live-Cell Imaging system. 5-10% confluent cells on ImageLock 96-well plate were treated with Poly(I:C), LPS, Imiquimod and ODN2395 together with SCR or Fli1siRNA (A) or B18R (B). Images with an estimated confluence were captured every 3h for total of 72h. Area under curves was measured in the GraphPad Prism software. Data represent an n=3 well each point with 3 different cells culture. Student t-test *p<0.05
To further verify the role of Fli1 deficiency and selected TLR ligands in the process of angiogenesis, we employed an ex vivo 3D dermal organoid culture system. As shown on Figure 4A, Fli1siRNA treated tissue explants displayed significantly reduced total number of branching tubules compared to explants treated with control siRNA. Likewise, Poly(I:C), LPS and Imiquimod blocked outgrowth of sprouts in dermal organoid cultures (Figure 4B). In contrast, ODN2395 treatment increased number of growing tubules. These results are consistent with the proliferation assay using a monolayer cell culture and provide additional confirmation for the important role of Fli1 and selected TLR ligands in angiogenesis.
Figure 4. Human dermal ex vivo angiogenesis assay.
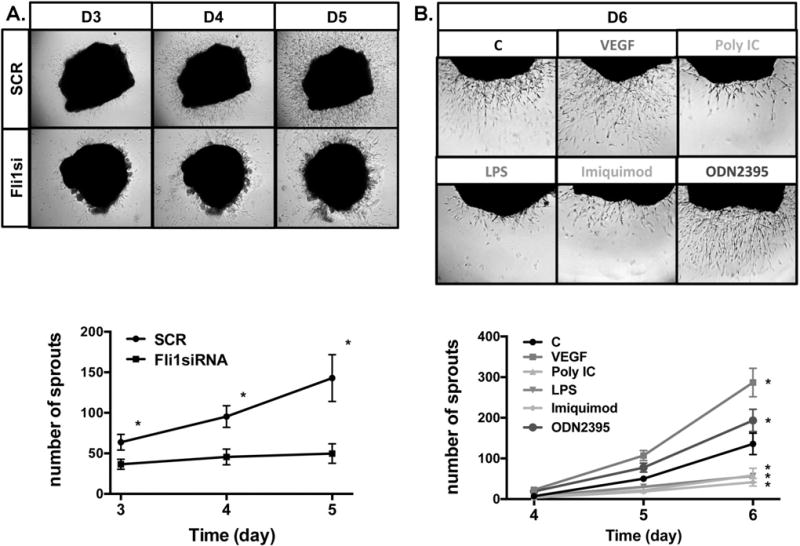
A. Dermal tissues obtained from foreskins were treated with SCR or Fli1siRNA for 24h, embedded in matrigel and cultured in EGM-2 medium. B. Dermal tissues were directly embedded in matrigel and cultured with EGM-2 medium. Sprout outgrowth was quantified at day 3,4,5 and 6. Data represent an n=8 well each point with 3 different cultures. Student t-test, *p<0.05
Because apoptosis of endothelial cells has been implicated as a prerequisite to the development of many inflammatory and immune diseases (29), we next sought to determine whether apoptosis contributed to the anti-proliferative effect of selected TLRs or Fli1 deficiency. To detect apoptosis in real time we used IncuCyte system in combination with Annexin V reagents. Treatment with Poly(I:C) showed modest increase in the number of apoptotic cells (Supplemental Figure 3), while the number of apoptotic cells in HDMECs treated with LPS, Imiquimod and ODN2395 was comparable to the control levels. Likewise, HDMECs treated with either control siRNA (SCR) or Fli1siRNA for up to 48h did not show any significant changes in the number of apoptotic cells (Supplemental Figure 3).
Fli1 deficiency or stimulation with selected TLR ligands regulate Endothelial to Mesenchymal Transition (EndoMT)
Recent studies have established the importance of EndoMT during development and in various pathological processes (30). To determine the effect of selected TLR ligands on EndoMT, we performed double-fluorescence staining for VE-cadherin and phalloidin on HDMECs treated with Poly(I:C), LPS, Imiquimod and ODN2395 for 24 hours (Figure 5A). In contrast to untreated cells that showed prominent VE-cadherin staining and exhibited a cortical actin staining below the cell membranes, Poly(I:C) and LPS treated cells showed markedly decreased expression of VE-cadherin, as well as elongated F-actin stress fibers. In contrast, Imiquimod and ODN2395 treated cells did not show any alterations in VE-cadherin cellular localization or expression, and exhibited a cortical actin staining below the cell membranes similar to observed in untreated cells. Consistent with the previously published study (22), Fli1 depletion reduced VE-cadherin expression and induced morphological rearrangements of the actin filaments (Figure 5A). Additionally, mRNA levels of EndoMT regulatory transcription factor, SNAIL1, was moderately, but significantly increased in HDMECs treated with Poly(I:C), but was markedly increased in HDMECs treated with LPS (Figure 5B). LPS also increased SNAIL1 protein levels at the 24 hour timepoint, while only a small increase of SNAIL1 protein levels was observed in some, but not all, cell lines treated with Poly(I:C) (Fig. 5C). Imiquinod did not affect SNAIL1 mRNA or protein levels, while OD2395 decreased SNAIL1 protein, only. We did not observe any changes in mRNA levels of other EndoMT genes including: SNAIL2, SNAIL3, ZEB1 and TWIST in response to all tested TLR ligands (data not shown). Furthermore, we observed increased collagen type I secretion in the medium from cells treated with Poly(I:C), LPS and Imiquimod in a time dependent manner (Figure 5D). No change in collagen secretion was observed in ODN2395 treated cells (Figure 5D). In a rescue experiment, EndoMT-related changes induced by Poly(I:C) and LPS were largely reversed by treating cells with the TGFβ neutralizing antibody and partially reversed by re-expression of Fli1 via Andenovirus (Figure 6). Together, these data suggest that Poly(I:C) and LPS, as well as deficiency of Fli1 could contribute to the process of EndoMT. Furthermore, these effects were dependent on the autocrine TGFβ.
Figure 5. Effect of Fli1 downregulation and selected TLR ligands on EndoMT.
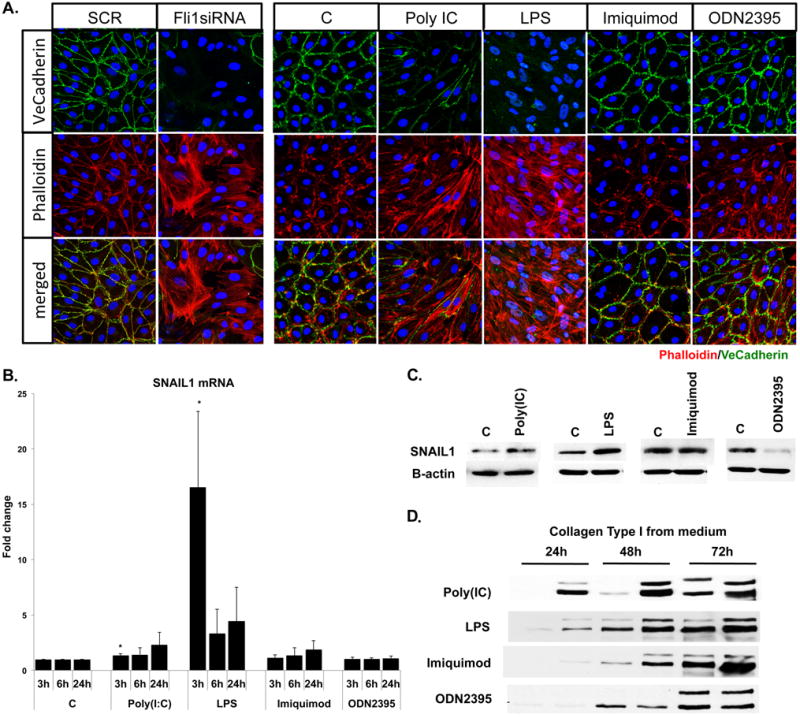
A. Immunofluorescence staining of VE-cadherin (green) and Phalloidin (red) in HDMECs treated with SCR, Fli1siRNA, Poly(I:C), LPS, Imiquimod and ODN2395. Representative images are shown from three different experiments. B. mRNA levels of SNAIL1 at 3h, 6h, and 24h were analyzed by qPCR, n=3. C. SNAIL protein levels were analyzed at 24h timepoint by Western Blot, n=2. D. Collagen Type I protein levels from medium was analyzed by Western Blot, n=2.
Figure 6. Effect of TGFβ neutralizing antibody and Fli1 adenovirus on Poly(I:C) and LPS induced EndoMT.
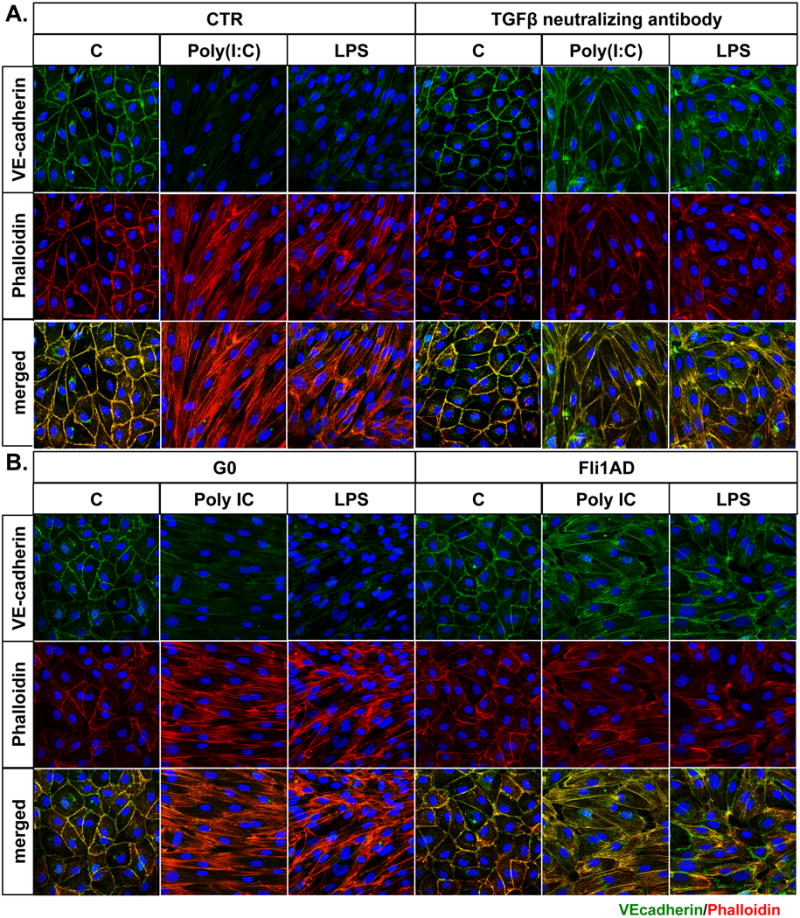
HDMECs were pretreated with TGFβ neutralizing antibody for 1h (A) or FliAd for 24h (B) and then treated with Poly(I:C) and LPS. Expression levels of VE-cadherin (green) and Phalloidin (red) were analyzed by immunofluorescent staining. Representative images are shown from three different experiments.
Selected TLR ligands and/or absence of Fli1 activate FOXO3A in HDMECs
Data presented in the previous sections implicated downregulation of Fli1 in the anti-proliferative effects of selected TLR ligands, suggesting that Fli1 regulates expression of genes that are critically involved in proliferative responses. One such gene is FOXO3A, a member of FOXO family of transcription factors, that was shown to be involved in regulation of metabolism, autophagy, cell cycle arrest and apoptosis (31). Phosphorylation of FOXO3A, in the presence of survival factors, results in nuclear exclusion, retention and degradation of FOXO3A in the cytoplasm. Conversely, the presence of anti-proliferative and proapoptotic factors leads to FOXO3A dephosphorylation, nuclear translocation, and activation of FOXO target genes. To test the hypothesis that deficiency of Fli1 regulates expression of FOXO3A, HDMECs were treated with SCR and Fli1siRNA for 48h. We observed upregulation of FOXO3A in HDMECs treated with Fli1siRNA at the mRNA and protein level, however Fli1 deficiency did not affect phosphorylation levels of FOXO3A (Figure 7A, B). Furthermore, we demonstrated that Fli1 transcriptionally regulated the FOXO3A gene. CHiP assay showed Fli1 binding to the FOXO3A promoter (Figure 7C). Moreover, we confirmed increased FOXO3A transcription activity in cells treated with Fli1siRNA and decreased FOXO3A activity in cells transfected with Fli1FLAG plasmid using luciferase assay (Figure 7D). On the other hand, at the 24h time point, treatment with Poly(I:C), LPS and Imiquimod did not affect total protein levels of FOXO3A (Figure 7F). This may be due to the effect of TLR ligands on the protein stability of Fli1, however further studies are needed to explain this finding. Instead, Poly(I:C), LPS and Imiquimod decreased phosphorylation of FOXO3A (S253) (Figure 7F). In contrast, treatment with ODN2395 resulted in increased phosphorylation of FOXO3A (S253) (Figure 7F). Double immunofluorescence staining of FOXO3A/VE-cadherin confirmed nuclear accumulation of FOXO3A in cells treated with Fli1siRNA, as well as Poly(I:C), LPS and Imiquimod (Figure 7E, G). Cells stimulated with ODN2395 showed diminished nuclear FOXO3A staining (Figure 7F). These data suggest that Fli1 deficiency as well as treatment with Poly(I:C), LPS and Imiquimod, impairs proliferation of HDMECs by activating the FOXO3A pathway, while ODN2395 elicits a protective effect by having an opposite effect on the activity of FOXO3A.
Figure 7. The effect of Fli1 downregulation and selected TLR ligands on the FOXO3A levels in HDMECs.
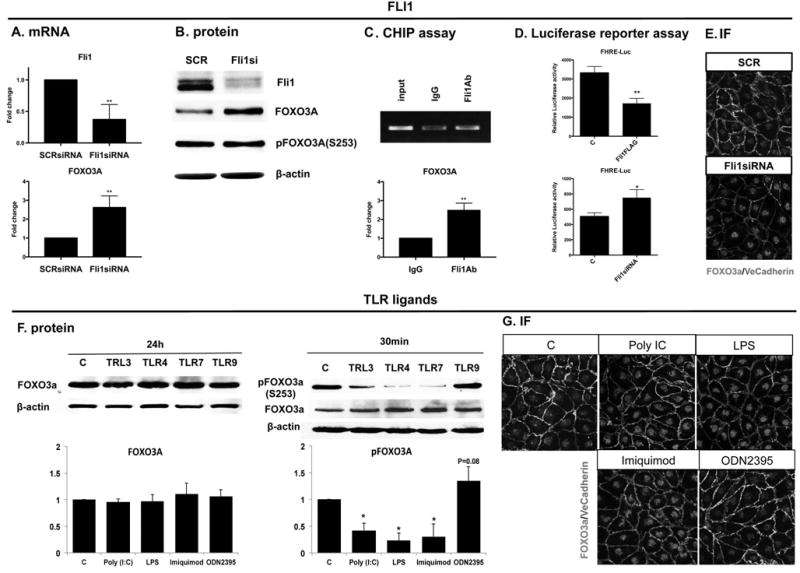
Confluent HDMECs were treated with SCR or Fli1siRNA for 48h (upper panel) or with Poly(I:C), LPS, Imiquimod and ODN2395 for 24h (lower panel). Samples were collected for analysis by qPCR (A), Western blot (B, F), CHiP assay (C) and Luciferase Assay (D). Double IF staining of FOXO3A/VeCadherin was performed on paraformaldehyde fixed HDMECs (E, G). Representative images are shown from three different experiments. Densitometry of the western blot was analyzed with the Image J software (NIH). Results are shown as mean ± SD, n=3. Student t-test, *p<0.05
Endothelial FOXO3A is elevated in the inflammatory regions of SSc skin
To illustrate the distribution of FOXO3A and Fli1 expression in pathological conditions we performed IHC staining on SSc skin biopsies. We have previously reported that Fli1 protein is expressed at reduced levels in SSc skin vasculature (12). As shown in Figure 8A, we observed elevated endothelial FOXO3A levels in the skin of SSc patients. Moreover the number of FOXO3A positive vessels was especially increased in SSc biopsies characterized by immune cell infiltrates (Figure 8C). Furthermore, IHC staining in the corresponding skin regions showed decreased Fli1 levels. In the healthy skin biopsies, we observed the opposite result with increased Fli1 and decreased FOXO3A levels in endothelial cells (Figure 8B). SSc patient data are included in Table I.
Figure 8. Distribution of FOXO3A and Fli1 in the SSc skin.
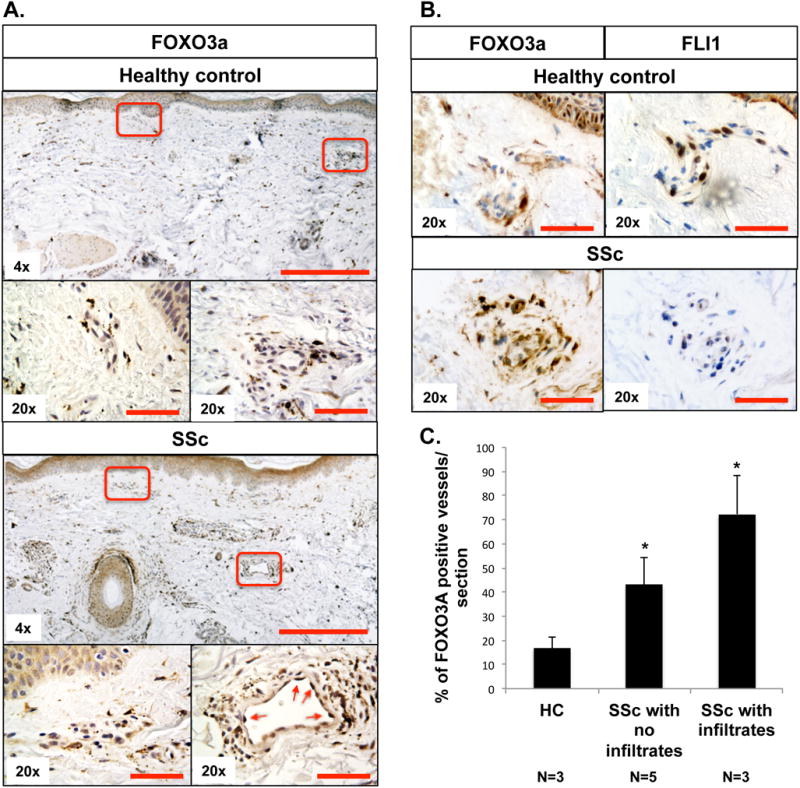
(A, B) IHC staining of FOXO3A and FLI1 was performed on paraffin sections from the skin of healthy controls and SSc patients. Red arrows indicate positive FOXO3A staining in endothelial cells. C. The bar graph represents percentage of FOXO3A positive vessels per section. Results are shown as mean ± SD (Student t-test*p<0.05). HC group, n=3. SSc with no infiltrates group, n=5. SSc with infiltrates group, n=3. Scale bar: 200μm for 4× and 25μm for 20× images.
Table I.
SSc patient data.
| # | Sex | Age | Limited/diffuse | lesional/nonlesional | Disease duration | Inflamatory Foci |
|---|---|---|---|---|---|---|
| SD14-12 | M | 60 | Diffuse SSc | lesional | 4 years | + |
| SD14-31 | F | 34 | Diffuse SSc | lesional | 13 months | + |
| SD15-01 | F | 57 | Limmited SSc | lesional | 1 year | + |
| SD14-53 | M | 55 | Diffuse SSc | lesional | 5 years | − |
| SD14-17 | M | 48 | Limited SSc | lesional | 17 months | − |
| SD15-02 | F | 51 | Limited SSc | lesional | 2 months | − |
| SD15-04 | F | 48 | Limited SSc | nonlesional | 3.5 months | − |
| SD15-05 | F | 62 | Limited SSc | nonlesional | 19 months | − |
Discussion
Endothelial cells play a crucial role in inflammatory processes by maintaining the vessel integrity and immune cell trafficking. Excessive endothelial cell activation in chronic inflammatory settings can lead to EC dysfunctions and development of a broad spectrum of human diseases (2, 3). This study focused on delineating the TLR-mediated molecular mechanisms involved in regulating EC function. We showed that activation of TLR3, -4, and -7 elicited potent anti-angiogenic effects, while TLR9 promoted angiogenesis in HDMECs. We further demonstrated that activation of the TGFβ, ET-1, and type I IFN signaling pathways and subsequent downregulation of Fli1 protein levels played a key role in the anti-angiogenic effects of selected TLR ligands. Fli1 transcriptionally repressed FOXO3A, a well-known inhibitor of cell proliferation. In addition, TLR3, -4, and -7 agonists rapidly decreased phosphorylation and increased nuclear accumulation of FOXO3A, while TLR9 enhanced Fli1 protein levels and induced FOXO3A phosphorylation, thus preventing its nuclear accumulation. These results implicate Fli1 and FOXO3A in regulation of angiogenesis by the TLRs through the rapid modulation of the FOXO3A activity status, and through the Fli1-mediated increased FOXO3A expression levels (Figure 9).
Figure 9. TLRs regulate EC functions by FLI1/FOXO3A pathway.
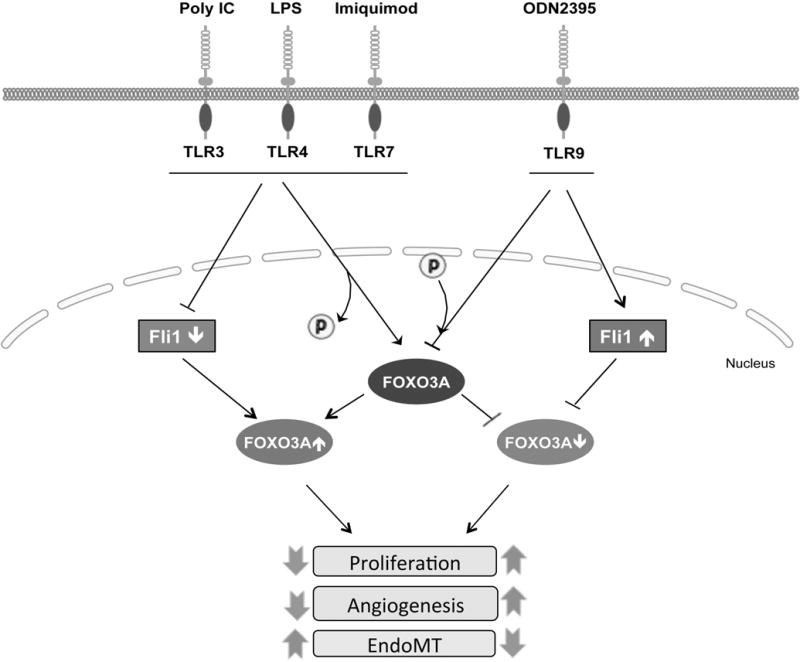
TLRs regulate EC functions through the rapid modulation of FOXO3A activity status, and through the FLI1-mediated increase of FOXO3A expression levels.
In addition to a widely investigated role of TLRs in inducing inflammatory responses, TLR-mediated signaling pathways, including NFκβ, MAPK and PI3K, have also been studied in the context of cell proliferation (32). However these extended functions of the TLR family are not well understood and may depend on a specific cell type and physiological context. Hence, previous studies describing the effects of selected TLRs in angiogenesis produced conflicting results with limited mechanistic insights. On one hand, it has been demonstrated that the TLR regulated inflammation is often associated with increased angiogenesis. For example LPS was shown to promote angiogenesis in the inflammatory setting (33). On the other hand, selected TLR ligands including Poly(I:C), LPS and Imiquimod as well as ODNs were also shown to have a negative effect on endothelial cell proliferation (34–38). Here we show that depletion of Fli1 or stimulation with Poly(I:C), LPS and Imiquimod greatly reduced the basal level of proliferation of HDMECs. Additionally, co-stimulation with Fli1siRNA and these TLR ligands completely blocked the ability of cells to proliferate. These anti-proliferative effects can be explained, at least in part, by activation of the TGFβ and IFNα signaling pathways that have previously been shown to inhibit proliferation of ECs (26, 39). On the other hand, ET-1 has been shown to stimulate EC proliferation, therefore it is unlikely to mediate the anti-proliferative effects of these TLR ligands (40). In contrast, TLR9 ligand, ODN2395, increased basal proliferation levels of HDMECs. Our observation differs from a recently published study describing anti-angiogenic effects of TLR9 agonist from class B, ODN1826, in corneal neovascularization (41). However, in a different study, TLR9 expression in mononuclear cells infiltrating lung cancer was associated with increased angiogenesis and progression of tumor growth (42). These conflicting observations have recently been clarified by Wu et al (38). The authors showed that the angiogenic effects of CpG motifs were dependent on the backbone structure (38). The ODN2395 with a phosphodiester (PD) backbone exhibited proangiogenic effects, while ODN2395 with a phosphorothioate (PS) backbone were antiangiogenic (38). Our study, which used ODN2395 with a PD backbone is consistent with these observations. Interestingly, Wu et al have also suggested that the effects of synthetic ODNs in endothelial cells did not require TLR9, but the binding site for CpG-ODN in endothelial cells remains unclear.
We showed that anti-angiogenic effects of selected TLR ligands correlated with downregulation of Fli1 protein levels. Fli1 is highly expressed in ECs and plays an important role in controlling vascular homeostasis, however its regulation is incompletely understood. Previous studies on fibroblasts revealed that Fli1 levels are regulated via multiple mechanisms including, transcriptional regulation, post-translational modifications, as well as epigenetic mechanisms (24, 43–45). Furthermore, IFN-α and -γ were shown to reduce Fli1 mRNA and protein levels in HDMECs (11). A similar observation was reported in macrophages, where the mRNA level of Fli1 was significantly downregulated in response to IFN-α and other inflammatory mediators such as LPS, and the differentiation-inducing agents (46–48). The findings of this study indicate that selected TLR ligands, including Poly(I:C), LPS and Imiquimod, significantly reduced Fli1 protein levels by activating the ET-1 and TGFβ, as well as interferon signaling pathways. TGFβ and ET-1 were previously shown to promote Fli1 protein degradation by inducing posttranslational modifications in the Fli1 protein (24, 25). In this study, we show that dual ET-1 receptor inhibitor, bosentan, as well as specific inhibitor of TGFβRI kinase (SB431542) prevented downregulation of Fli1 in response to Poly(I:C), LPS and Imiquimod. Importantly, only bosentan consistently upregulated Fli1 at the basal level, suggesting that endogenously produced ET-1 could contribute to the regulation of Fli1 levels in ECs. Consistent with its proangiogenic effect, TLR9 ligand from class C, ODN2395, increased Fli1 protein level, whereas agonists from class A and B had no effect. The mechanism involved in the upregulation of Fli1 protein levels by ODN2395 is presently unclear. In contrast to other TLR ligands examined in this study, ODN2395 did not stimulate expression levels of TGFβ or ET-1. One possibility is that ODN2395, through an unknown mechanism, counteracts the inhibitory effects of the endogenous ET-1. Alternatively, ODN2395 may upregulate Fli1 protein levels either directly or through another mediator. For example, it was previously reported that stimulation with TLR9 agonist from class B resulted in increased release of VEGF in a mouse model of lung carcinoma (49). Fli1 was among the ETS transcription factor family members that were increased in response to VEGF (50), suggesting that VEGF may be responsible for the upregulation of Fli1 in response to ODN2395.
Furthermore we showed that some TLR ligands could modulate EC plasticity and trigger the differentiation to mesenchymal phenotype. This process is called endothelial to mesenchymal transition (EndoMT) and has been implicated in many pathological processes, including cancer, PAH, artherosclerosis and fibrosis (30, 51). Interestingly, cells that undergo EndoMT support increased transendothelial migration of the immune cells by regulating adhesion and permeability (52). Moreover, transformed ECs may participate in fibrotic responses and therefore contribute to the defective angiogenic process (53, 54). TGFβ is one of the most widely studied inducers of EndoMT (55), however other factors, especially ET-1 (56), and several inflammatory mediators, including LPS, have also been shown to induce EndoMT in vitro and in animal models (57). We have previously reported that several EndoMT-related genes, including VE-cad, SNAIL1, and FSP1 were transcriptionally regulated by Fli1 (58, 59). This study has confirmed the critical role of Fli1 in the process of EndoMT. Additionally, we showed that LPS and Poly(I:C) also induced EndoMT in HDMECs. Even though the TLR7 ligand, Imiquimod, downregulated Fli1 protein levels and upregulated ET-1, it did not induce the morphological changes in HDMECs. Unlike Poly(I:C) or LPS Imiquimod only weakly induced TGFβ2 and -3, suggesting that the balance between the TGFβ and interferon signaling pathways determines the EndMT outcome. On the other hand, only LPS, but not Poly(I:C), consistently upregulated SNAIL1 at the mRNA and protein levels. This suggests that Poly(I:C) can induce EndoMT through a yet unknown mechanism. Likewise, it was recently demonstrated that nuclear/cytosolic protein ratios of SNAIL1 were unchanged in Poly(I:C) induced EMT in keratinocytes (60). However, Poly(I:C) upregulated SNAIL1 at the mRNA and protein levels in human small-airway epithelial cells, indicating cell type specific responses (61). Together, these data suggest that deficiency of Fli1, as well as exposure to LPS or Poly(I:C) can induce morphological changes in ECs and may be important in initiating pro-fibrogenic changes in the vascular compartment.
To confirm the inverse correlation between Fli1 and FOXO3A expression in the disease setting, we analyzed skin biopsies obtained from patients with SSc. Consistent with our in vitro findings, FOXO3A was highly expressed in ECs, primarily in the regions characterized by immune cell infiltrates, and its expression correlated with decreased endothelial expression of Fli1. Interestingly, it was previously reported that scleroderma sera induces apoptosis of endothelial progenitor cells (EPCs) via FOXO3A/Bim pathway (62). These data suggest that the activated Fli1/FOXO3A pathway is present in SSc ECs and may be important in the disease pathogenesis.
In conclusion, our work demonstrates that selected TLR ligands can regulate protein levels of Fli1 in HDMECs. Furthermore, this study shows that particular TLR ligands can have either detrimental or protective effects on HDMECs function. Poly(I:C), LPS and Imiquimod impaired proliferation through downregulation of Fli1 and activation of the FOXO3A pathway, while TLR9 ligand, ODN2395, elicited a protective effect by having an opposite effect on those pathways. In addition, our data suggest that different effects of specific TLR ligands were associated with TGFβ expression levels. Together these findings confirm a central role of TLRs in endothelial dysfunction and suggest that selected TLR ligands may contribute to endothelial cell injury in autoimmune diseases such as Systemic Sclerosis.
Supplementary Material
Acknowledgments
We wish to thank Dr. Hans Dooms and Dr. Jeffrey Browning for help with editing the manuscript.
Bibliography
- 1.Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, Nishigaki I. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9:1057–1069. doi: 10.7150/ijbs.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salvador B, Arranz A, Francisco S, Cordoba L, Punzon C, Llamas MA, Fresno M. Modulation of endothelial function by Toll like receptors. Pharmacol Res. 2016;108:46–56. doi: 10.1016/j.phrs.2016.03.038. [DOI] [PubMed] [Google Scholar]
- 3.Murdaca G, Colombo BM, Cagnati P, Gulli R, Spano F, Puppo F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis. 2012;224:309–317. doi: 10.1016/j.atherosclerosis.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 5.Fitzner N, Clauberg S, Essmann F, Liebmann J, Kolb-Bachofen V. Human skin endothelial cells can express all 10 TLR genes and respond to respective ligands. Clin Vaccine Immunol. 2008;15:138–146. doi: 10.1128/CVI.00257-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fullard N, O’Reilly S. Role of innate immune system in systemic sclerosis. Semin Immunopathol. 2015;37:511–517. doi: 10.1007/s00281-015-0503-7. [DOI] [PubMed] [Google Scholar]
- 7.Trivedi S, Greidinger EL. Endosomal Toll-like receptors in autoimmunity: mechanisms for clinical diversity. Therapy. 2009;6:433–442. doi: 10.2217/thy.09.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol. 2014;306:H184–196. doi: 10.1152/ajpheart.00328.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, Shlomchik MJ. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184:1840–1848. doi: 10.4049/jimmunol.0902592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chrobak I, Lenna S, Stawski L, Trojanowska M. Interferon-gamma promotes vascular remodeling in human microvascular endothelial cells by upregulating endothelin (ET)-1 and transforming growth factor (TGF) beta2. J Cell Physiol. 2013;228:1774–1783. doi: 10.1002/jcp.24337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kubo M, Czuwara-Ladykowska J, Moussa O, Markiewicz M, Smith E, Silver RM, Jablonska S, Blaszczyk M, Watson DK, Trojanowska M. Persistent down-regulation of Fli1, a suppressor of collagen transcription, in fibrotic scleroderma skin. Am J Pathol. 2003;163:571–581. doi: 10.1016/S0002-9440(10)63685-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czuwara-Ladykowska J, Shirasaki F, Jackers P, Watson DK, Trojanowska M. Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. J Biol Chem. 2001;276:20839–20848. doi: 10.1074/jbc.M010133200. [DOI] [PubMed] [Google Scholar]
- 14.Asano Y, Czuwara J, Trojanowska M. Transforming growth factor-beta regulates DNA binding activity of transcription factor Fli1 by p300/CREB-binding protein-associated factor-dependent acetylation. J Biol Chem. 2007;282:34672–34683. doi: 10.1074/jbc.M703907200. [DOI] [PubMed] [Google Scholar]
- 15.Romano E, Chora I, Manetti M, Mazzotta C, Rosa I, Bellando-Randone S, Blagojevic J, Soares R, Avouac J, Allanore Y, Ibba-Manneschi L, Matucci-Cerinic M, Guiducci S. Decreased expression of neuropilin-1 as a novel key factor contributing to peripheral microvasculopathy and defective angiogenesis in systemic sclerosis. Ann Rheum Dis. 2016;75:1541–1549. doi: 10.1136/annrheumdis-2015-207483. [DOI] [PubMed] [Google Scholar]
- 16.Looney AP, Han R, Stawski L, Marden G, Iwamoto M, Trojanowska M. Synergistic Role of Endothelial ERG and FLI1 in Mediating Pulmonary Vascular Homeostasis. Am J Respir Cell Mol Biol. 2017;57:121–131. doi: 10.1165/rcmb.2016-0200OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asano Y, Stawski L, Hant F, Highland K, Silver R, Szalai G, Watson DK, Trojanowska M. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. Am J Pathol. 2010;176:1983–1998. doi: 10.2353/ajpath.2010.090593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richard L, Velasco P, Detmar M. Isolation and culture of microvascular endothelial cells. Methods Mol Med. 1999;18:261–269. doi: 10.1385/0-89603-516-6:261. [DOI] [PubMed] [Google Scholar]
- 19.Nakerakanti SS, Bujor AM, Trojanowska M. CCN2 is required for the TGF-beta induced activation of Smad1-Erk1/2 signaling network. PLoS One. 2011;6:e21911. doi: 10.1371/journal.pone.0021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dowson C, Simpson N, Duffy L, O’Reilly S. Innate Immunity in Systemic Sclerosis. Curr Rheumatol Rep. 2017;19:2. doi: 10.1007/s11926-017-0630-3. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Kam CS, Liu FQ, Liu Y, Lui VC, Lamb JR, Tam PK. LPS-induced up-regulation of TGF-beta receptor 1 is associated with TNF-alpha expression in human monocyte-derived macrophages. J Leukoc Biol. 2008;83:1165–1173. doi: 10.1189/jlb.0807521. [DOI] [PubMed] [Google Scholar]
- 22.Farina GA, York MR, Di Marzio M, Collins CA, Meller S, Homey B, Rifkin IR, Marshak-Rothstein A, Radstake TR, Lafyatis R. Poly(I:C) drives type I IFN- and TGFbeta-mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Invest Dermatol. 2010;130:2583–2593. doi: 10.1038/jid.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farina G, York M, Collins C, Lafyatis R. dsRNA activation of endothelin-1 and markers of vascular activation in endothelial cells and fibroblasts. Ann Rheum Dis. 2011;70:544–550. doi: 10.1136/ard.2010.132464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asano Y, Trojanowska M. Phosphorylation of Fli1 at threonine 312 by protein kinase C delta promotes its interaction with p300/CREB-binding protein-associated factor and subsequent acetylation in response to transforming growth factor beta. Mol Cell Biol. 2009;29:1882–1894. doi: 10.1128/MCB.01320-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akamata K, Asano Y, Aozasa N, Noda S, Taniguchi T, Takahashi T, Ichimura Y, Toyama T, Sato S. Bosentan reverses the pro-fibrotic phenotype of systemic sclerosis dermal fibroblasts via increasing DNA binding ability of transcription factor Fli1. Arthritis Res Ther. 2014;16:R86. doi: 10.1186/ar4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castanares C, Redondo-Horcajo M, Magan-Marchal N, ten Dijke P, Lamas S, Rodriguez-Pascual F. Signaling by ALK5 mediates TGF-beta-induced ET-1 expression in endothelial cells: a role for migration and proliferation. J Cell Sci. 2007;120:1256–1266. doi: 10.1242/jcs.03419. [DOI] [PubMed] [Google Scholar]
- 27.Fang F, Ooka K, Sun X, Shah R, Bhattacharyya S, Wei J, Varga J. A synthetic TLR3 ligand mitigates profibrotic fibroblast responses by inducing autocrine IFN signaling. J Immunol. 2013;191:2956–2966. doi: 10.4049/jimmunol.1300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koda M, Bauer M, Krebs A, Hahn EG, Schuppan D, Murawaki Y. Endothelin-1 enhances fibrogenic gene expression, but does not promote DNA synthesis or apoptosis in hepatic stellate cells. Comp Hepatol. 2006;5:5. doi: 10.1186/1476-5926-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winn RK, Harlan JM. The role of endothelial cell apoptosis in inflammatory and immune diseases. J Thromb Haemost. 2005;3:1815–1824. doi: 10.1111/j.1538-7836.2005.01378.x. [DOI] [PubMed] [Google Scholar]
- 30.Lin F, Wang N, Zhang TC. The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life. 2012;64:717–723. doi: 10.1002/iub.1059. [DOI] [PubMed] [Google Scholar]
- 31.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49:1–9. doi: 10.1016/j.cyto.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grote K, Schutt H, Schieffer B. Toll-like receptors in angiogenesis. ScientificWorldJournal. 2011;11:981–991. doi: 10.1100/tsw.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimmer S, Steinmetz M, Asdonk T, Motz I, Coch C, Hartmann E, Barchet W, Wassmann S, Hartmann G, Nickenig G. Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ Res. 2011;108:1358–1366. doi: 10.1161/CIRCRESAHA.111.243246. [DOI] [PubMed] [Google Scholar]
- 35.Yang M, Xiao Z, Lv Q, Liu X, Zhou L, Chen X, Chen M, Fang L, Xie X, Hu J. The functional expression of TLR3 in EPCs impairs cell proliferation by induction of cell apoptosis and cell cycle progress inhibition. Int Immunopharmacol. 2011;11:2118–2124. doi: 10.1016/j.intimp.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 36.Sun RJ, Wang QY, Zhang JB, Guo YF, Zhao XD. Regulation of proliferation and apoptosis of human vascular endothelial cell by Acheron. Zhonghua Shao Shang Za Zhi. 2011;27:156–160. [PubMed] [Google Scholar]
- 37.Kuznetsova SA, Starikova EA, Freidlin IS, Smirnov VS. In vitro studies of changes in human endothelial cell functions under the effect of imiquimod. Bull Exp Biol Med. 2012;154:237–240. doi: 10.1007/s10517-012-1921-3. [DOI] [PubMed] [Google Scholar]
- 38.Wu J, Su W, Powner MB, Liu J, Copland DA, Fruttiger M, Madeddu P, Dick AD, Liu L. Pleiotropic action of CpG-ODN on endothelium and macrophages attenuates angiogenesis through distinct pathways. Sci Rep. 2016;6:31873. doi: 10.1038/srep31873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Indraccolo S. Interferon-alpha as angiogenesis inhibitor: learning from tumor models. Autoimmunity. 2010;43:244–247. doi: 10.3109/08916930903510963. [DOI] [PubMed] [Google Scholar]
- 40.Kuhlmann CR, Most AK, Li F, Munz BM, Schaefer CA, Walther S, Raedle-Hurst T, Waldecker B, Piper HM, Tillmanns H, Wiecha J. Endothelin-1-induced proliferation of human endothelial cells depends on activation of K+ channels and Ca+ influx. Acta Physiol Scand. 2005;183:161–169. doi: 10.1111/j.1365-201X.2004.01378.x. [DOI] [PubMed] [Google Scholar]
- 41.Wu J, Cui H, Dick AD, Liu L. TLR9 agonist regulates angiogenesis and inhibits corneal neovascularization. Am J Pathol. 2014;184:1900–1910. doi: 10.1016/j.ajpath.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Belmont L, Rabbe N, Antoine M, Cathelin D, Guignabert C, Kurie J, Cadranel J, Wislez M. Expression of TLR9 in tumor-infiltrating mononuclear cells enhances angiogenesis and is associated with a worse survival in lung cancer. Int J Cancer. 2014;134:765–777. doi: 10.1002/ijc.28413. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Fan PS, Kahaleh B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006;54:2271–2279. doi: 10.1002/art.21948. [DOI] [PubMed] [Google Scholar]
- 44.Fang F, Marangoni RG, Zhou X, Yang Y, Ye B, Shangguang A, Qin C, Wang W, Bhattacharyya S, Wei J, Tourtellotte WG, Varga J. TLR9 signaling is augmented in systemic sclerosis and elicits TGF-beta-dependent fibroblast activation. Arthritis Rheumatol. 2016 doi: 10.1002/art.39655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan ES, Liu H, Fernandez P, Luna A, Perez-Aso M, Bujor AM, Trojanowska M, Cronstein BN. Adenosine A(2A) receptors promote collagen production by a Fli1- and CTGF-mediated mechanism. Arthritis Res Ther. 2013;15:R58. doi: 10.1186/ar4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klemsz MJ, Maki RA, Papayannopoulou T, Moore J, Hromas R. Characterization of the ets oncogene family member, fli-1. J Biol Chem. 1993;268:5769–5773. [PubMed] [Google Scholar]
- 47.Ho HH, Ivashkiv LB. Downregulation of Friend leukemia virus integration 1 as a feedback mechanism that restrains lipopolysaccharide induction of matrix metalloproteases and interleukin-10 in human macrophages. J Interferon Cytokine Res. 2010;30:893–900. doi: 10.1089/jir.2010.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Darby TG, Meissner JD, Ruhlmann A, Mueller WH, Scheibe RJ. Functional interference between retinoic acid or steroid hormone receptors and the oncoprotein Fli-1. Oncogene. 1997;15:3067–3082. doi: 10.1038/sj.onc.1201503. [DOI] [PubMed] [Google Scholar]
- 49.Sorrentino R, Morello S, Giordano MG, Arra C, Maiolino P, Adcock IM, Pinto A. CpG-ODN increases the release of VEGF in a mouse model of lung carcinoma. Int J Cancer. 2011;128:2815–2822. doi: 10.1002/ijc.25626. [DOI] [PubMed] [Google Scholar]
- 50.Heo SH, Choi YJ, Ryoo HM, Cho JY. Expression profiling of ETS and MMP factors in VEGF-activated endothelial cells: role of MMP-10 in VEGF-induced angiogenesis. J Cell Physiol. 2010;224:734–742. doi: 10.1002/jcp.22175. [DOI] [PubMed] [Google Scholar]
- 51.Medici D. Endothelial-Mesenchymal Transition in Regenerative Medicine. Stem Cells Int. 2016;2016:6962801. doi: 10.1155/2016/6962801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Varga J, Trojanowska M, Kuwana M. Pathogenesis of systemic sclerosis: recent insights of molecular and cellular mechanisms and therapeutic opportunities. Journal of Scleroderma and Related Disorders. 2017:0–0. [Google Scholar]
- 54.Manetti M, Romano E, Rosa I, Guiducci S, Bellando-Randone S, De Paulis A, Ibba-Manneschi L, Matucci-Cerinic M. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann Rheum Dis. 2017;76:924–934. doi: 10.1136/annrheumdis-2016-210229. [DOI] [PubMed] [Google Scholar]
- 55.Li Z, Jimenez SA. Protein kinase Cdelta and c-Abl kinase are required for transforming growth factor beta induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. 2011;63:2473–2483. doi: 10.1002/art.30317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, Yagi K, Miyagawa K, Rikitake Y, Suzuki T, Kisanuki YY, Yanagisawa M, Hirata K. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 121:2407–2418. doi: 10.1161/CIRCULATIONAHA.110.938217. [DOI] [PubMed] [Google Scholar]
- 57.Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM, Gopalan B, Stylianou E, Fiocchi C. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. 179:2660–2673. doi: 10.1016/j.ajpath.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asano Y, Stawski L, Hant F, Highland K, Silver R, Szalai G, Watson DK, Trojanowska M. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. Am J Pathol. 176:1983–1998. doi: 10.2353/ajpath.2010.090593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taniguchi T, Asano Y, Akamata K, Noda S, Takahashi T, Ichimura Y, Toyama T, Trojanowska M, Sato S. Fibrosis, vascular activation, and immune abnormalities resembling systemic sclerosis in bleomycin-treated Fli-1-haploinsufficient mice. Arthritis Rheumatol. 2015;67:517–526. doi: 10.1002/art.38948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takada K, Komine-Aizawa S, Hirohata N, Trinh QD, Nishina A, Kimura H, Hayakawa S. Poly I:C induces collective migration of HaCaT keratinocytes via IL-8. BMC Immunol. 2017;18:19. doi: 10.1186/s12865-017-0202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tian B, Patrikeev I, Ochoa L, Vargas G, Belanger KK, Litvinov J, Boldogh I, Ameredes BT, Motamedi M, Brasier AR. NF-kappaB Mediates Mesenchymal Transition, Remodeling, and Pulmonary Fibrosis in Response to Chronic Inflammation by Viral RNA Patterns. Am J Respir Cell Mol Biol. 2017;56:506–520. doi: 10.1165/rcmb.2016-0259OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu S, Evans S, Yan B, Povsic TJ, Tapson V, Goldschmidt-Clermont PJ, Dong C. Transcriptional regulation of Bim by FOXO3a and Akt mediates scleroderma serum-induced apoptosis in endothelial progenitor cells. Circulation. 2008;118:2156–2165. doi: 10.1161/CIRCULATIONAHA.108.787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.