

ABSTRACT
Streptococcus pneumoniae (the pneumococcus) is the leading cause of community-acquired pneumonia and is now recognized to be a direct contributor to adverse acute cardiac events. During invasive pneumococcal disease, S. pneumoniae can gain access to the myocardium, kill cardiomyocytes, and form bacterium-filled “microlesions” causing considerable acute and long-lasting cardiac damage. While the molecular mechanisms responsible for bacterial translocation into the heart have been elucidated, the initial interactions of heart-invaded S. pneumoniae with cardiomyocytes remain unclear. In this study, we used a model of low multiplicity of S. pneumoniae infection with HL-1 mouse cardiomyocytes to investigate these early events. Using adhesion/invasion assays and immunofluorescent and transmission electron microscopy, we showed that S. pneumoniae rapidly adhered to and invaded cardiomyocytes. What is more, pneumococci existed as intravacuolar bacteria or escaped into the cytoplasm. Pulse-chase assays with BrdU confirmed intracellular replication of pneumococci within HL-1 cells. Using endocytosis inhibitors, bacterial isogenic mutants, and neutralizing antibodies against host proteins recognized by S. pneumoniae adhesins, we showed that S. pneumoniae uptake by cardiomyocytes is not through the well-studied canonical interactions identified for vascular endothelial cells. Indeed, S. pneumoniae invasion of HL-1 cells occurred through clathrin-mediated endocytosis (CME) and independently of choline binding protein A (CbpA)/laminin receptor, CbpA/polymeric immunoglobulin receptor, or cell wall phosphorylcholine/platelet-activating factor receptor. Subsequently, we determined that pneumolysin and streptococcal pyruvate oxidase-derived H2O2 production were required for cardiomyocyte killing. Finally, we showed that this cytotoxicity could be abrogated using CME inhibitors or antioxidants, attesting to intracellular replication of S. pneumoniae as a key first step in pneumococcal pathogenesis within the heart.
KEYWORDS: Cardiac acute events, cardiomyocytes, invasive pneumococcal disease, Streptococcus pneumoniae, facultatively intracellular pathogens, oxidative stress
INTRODUCTION
Streptococcus pneumoniae (the pneumococcus) is a Gram-positive bacterium which typically colonizes the nasopharynx asymptomatically (1, 2). However, it is also an opportunistic pathogen, capable of causing a wide spectrum of human diseases, including otitis media, community-acquired pneumonia, bacteremia/sepsis, and meningitis (3–5). In 30% of adults with pneumococcal pneumonia, S. pneumoniae escapes the airway and causes bacteremia/invasive pneumococcal disease (IPD). This typically occurs in those who are ≥65 years of age and/or in some fashion immunocompromised (4). Despite development and use of multiple antipneumococcal vaccines for decades, S. pneumoniae remains a major cause of human morbidity and death worldwide (6).
Approximately 20% of adults hospitalized for severe pneumococcal disease experience some sort of major adverse cardiac events (MACE) (7). These include heart failure, arrhythmia, and myocardial infarction, in decreasing order of incidence. Increased risk for adverse cardiac events not only occurs in the hospital setting but also persists for up to 10 years after successful resolution of the pneumonia episode (8). An increasing body of evidence suggests that MACE during pneumonia is due in part to the ability of S. pneumoniae to directly damage the myocardium (9–11). In fact, experimental evidence from challenged mice and nonhuman primates, and from autopsied human cardiac sections, suggests that S. pneumoniae translocates into the myocardium during a bacteremic episode (10, 12–14). Once in the heart, S. pneumoniae replicates within the myocardium and causes the formation of microlesions, which have been directly implicated in heart failure (10, 13, 14). Due to the limited regenerative ability of the heart, the presence of de novo scar formation is observed at former microlesion sites after antibiotic treatment (10). The latter presumably is responsible for the heightened incidence of MACE in convalescence.
S. pneumoniae is thought to invade the heart through the coronary circulation, as most pneumococci detected within the myocardium are close to the vascular network (13, 15). Interaction of S. pneumoniae with endothelial cells has been extensively studied as a part of S. pneumoniae translocation across the blood-brain barrier (BBB), which occurs during the development of pneumococcal meningitis (16). It is now known that S. pneumoniae adheres to and crosses the BBB through transcytosis in a platelet-activating factor receptor (PAFR)- and laminin receptor (LR)-dependent manner (17, 18). The pneumococcal ligands for these host proteins are cell wall phosphorylcholine and choline binding protein A (CbpA), respectively (18, 19). More recently, polymeric immunoglobulin receptor (pIgR) has also been implicated in S. pneumoniae translocation across the BBB, with CbpA also reported as its bacterial ligand (20). During heart invasion, the same interactions are thought to allow the bacteria to translocate across the cardiac vasculature and invade the myocardium, since microlesion formation is abrogated in a PAFR−/− mouse following passive immunization with anti-LR, or when mice were challenged with a cbpA null mutant of S. pneumoniae (13). However, once bacteria are in the myocardium, very little is known about the interactions between individual pneumococci and the cardiomyocytes, the most abundant cell type within the myocardium. In this study, using an in vitro cell infection model, we examined these early interactions between S. pneumoniae and cardiomyocytes and showed that an unexpected intracellular step is most likely involved in formation of cardiac microlesions.
RESULTS
HL-1 cell killing by Streptococcus pneumoniae requires contact with live bacteria.
After crossing the vascular endothelium, S. pneumoniae damages the myocardium, creating bacterium-associated microlesions. Our published studies using immunofluorescence microscopy suggest that this begins with the interaction of a single pneumococcus with surrounding cardiomyocytes (15). A trypan blue exclusion assay showed that at a low multiplicity of infection (MOI), i.e., less than 10 bacteria per cardiomyocyte, S. pneumoniae was able to induce cell death in approximately 60% of HL-1 cardiomyocytes after 6 h of infection in vitro, without detachment (Fig. 1A). Killing required live bacteria, as neither ethanol-killed nor heat-killed pneumococci reduced the viability of HL-1 cells (Fig. 1B). Importantly, HL-1 cell cytotoxicity required direct bacterial contact with S. pneumoniae; a Transwell assay demonstrated that no cytotoxicity occurred when HL-1 cardiomyocytes and the strain TIGR4 were separated by a 0.2-μm membrane (Fig. 1C).
FIG 1.

Streptococcus pneumoniae killing of cardiomyocytes at a low MOI requires contact. (A) Cell viability (left graph) and total cell count (right graph) of noninfected (white bars) or TIGR4-infected (black bars) HL-1 cells were determined by trypan blue exclusion assay. (B) HL-1 cardiomyocyte death was measured by LDH release from cells infected by live, heat-killed, or ethanol-killed TIGR4. (C) Bacterial toxicity determined by LDH release in a Transwell permeable support system (n = 3). Errors bars represent standard errors of the means. Statistical analyses were done with t test (A) and 1-way analysis of variance (ANOVA) with Holm-Sidak correction for multiple comparison (B and C). ****, P ≤ 0.0001.
Streptococcus pneumoniae invades and multiplies inside HL-1 cardiomyocytes.
To better understand the host-pathogen interactions leading to cardiomyocyte death, the kinetics of TIGR4–HL-1 cell interactions were monitored. As shown in Fig. 2A, the number of S. pneumoniae bacteria associated with cardiomyocytes (i.e., adherent and invaded) dipped slightly but then increased 4-fold from 2 h to 6 h postinfection. Studies done in parallel, but with gentamicin treatment that killed extracellular bacteria after 1 h of adhesion, revealed that up to 20% of associated bacteria were in effect internalized by HL-1 (Fig. 2A). The presence of intracellular pneumococci was confirmed using double immunofluorescence, in which bacteria were labeled twice, before and after permeabilization of the HL-1 cell membranes, leading to double staining of extracellular bacteria and single staining of intracellular bacteria (Fig. 2B). Notably, the number of internalized bacteria peaked at 120 min (i.e., 60 min of infection plus 60 min of gentamicin treatment) and decreased thereafter (Fig. 2A). Interestingly, transmission electron microscopy (TEM) imaging raised the possibility that S. pneumoniae was able to multiply inside cardiomyocytes, as more than one pneumococcus were observed within intracellular vacuoles (Fig. 2C). Less frequently, we also observed pneumococci located in the cytoplasm of the cell without a surrounding membrane, suggesting that S. pneumoniae might also escape these vacuoles (Fig. 2C). Pulse-chase of HL-1 cells with the nucleoside analog bromodeoxyuridine (BrdU) following infection, washing, and gentamicin treatment to kill extracellular S. pneumoniae showed BrdU-positive pneumococci within HL-1 cells after 2 h of infection (Fig. 2D), thereby suggesting that pneumococcal replication within the cardiomyocytes was indeed occurring. This finding also suggests that the decline in intracellular pneumococci detected after 120 min may be due to entry of gentamicin in membrane-compromised infected HL-1 cells and not S. pneumoniae intracellular killing by the cell itself.
FIG 2.

Streptococcus pneumoniae invades and multiplies inside HL-1 cardiomyocytes. (A) Kinetics of the interaction between TIGR4 and HL-1 cardiomyocytes. Associated or internalized bacterial loads were determined by CFU counts without or with gentamicin treatment, respectively. (B) Immunofluorescence microscopy on HL-1 cardiomyocytes after 1 h of infection by S. pneumoniae. Pneumococci were stained before (rhodamine [red]) and after (FITC [green]) permeabilization of the cell membrane using anti-serotype 4 antibody. (C) Representative TEM image showing intracellular S. pneumoniae inside HL-1 cardiomyocytes after 2 h of infection. Green arrowheads show cytoplasmic S. pneumoniae. Red arrowheads show multiple intravacuolar S. pneumoniae organisms. (D) Intracellular multiplication was assayed using the BrdU incorporation assay. S. pneumoniae was stained with anti-serotype 4 antibody (red) after gentamicin treatment and a BrdU (green) pulse. DAPI staining shows nuclei. Scale bars, 10 μm.
CME is necessary for S. pneumoniae-mediated cardiomyocyte killing.
To investigate the mechanism of TIGR4 invasion, we monitored the ability of cardiomyocytes to internalize the bacteria in the presence of diverse endocytosis inhibitors. Only the two inhibitors of clathrin-mediated endocytosis (CME), i.e., methyl-beta cyclodextrin (MβCD), which blocks clathrin-coated pit budding by removal of membrane cholesterol (21), and Pitstop 2, which competitively inhibits the terminal domain of clathrin (22), showed a significant reduction of internalized bacteria despite comparable adhesion levels (Fig. 3A). In order to confirm the specificity of our inhibitor strategy (i.e., timing and concentration), we used Listeria monocytogenes, a bacterium which is known to invade cells through CME (23), and showed results similar to those observed with S. pneumoniae TIGR4 (see Fig. S1A in the supplemental material). In addition, experiments using immunofluorescence microscopy showed that clathrin localized upon S. pneumoniae after 30 min of infection (Fig. S1B). What is more, this form of cell invasion was also observed with S. pneumoniae belonging to different serotypes: serotype 2 strain D39 and serotype 6A strain 6A10. These strains showed the same significant reduction in internalization when cells were treated with CME inhibitors (Fig. S1A). Notably and consistent with our results shown in Fig. 1C, contact-mediated invasion of cardiomyocytes was observed to be requisite for TIGR4 cytotoxicity, as evidenced by the abrogation of HL-1 cell death in the presence of Pitstop 2 (Fig. 3B).
FIG 3.

Clathrin-mediated endocytosis is required for intracellular S. pneumoniae-mediated killing. Shown is the interaction between HL-1 cardiomyocytes and TIGR4 in the presence of the designated endocytosis inhibitors. (A) Adhesion was measured 30 min after infection and invasion was measured 1 h after infection followed by 1 h of gentamicin treatment. (B) TIGR4-mediated HL-1 cell death in the presence of designated endocytosis inhibitors was measured using LDH release (n = 3). Errors bars represent standard errors of the means. Statistical analyses were done with t test (methyl-beta-cyclo-dextrin [MβCD] and PitStop 2) and 1-way ANOVA with Holm-Sidak correction for multiple comparison (cytochalasin D and genistein) (A) and t test (B). ***, P ≤ 0.001; ****, P ≤ 0.0001. Endocytosis inhibitors were used at the following concentrations: MβCD, 10 mM; cytochalasin D, 10 μM; genistein, 200 μM; and PitStop 2, 30 μM.
CME being a receptor-mediated form of endocytosis, we attempted to identify the bacterial and host determinants involved in S. pneumoniae internalization by HL-1 cells. S. pneumoniae is known to interact with LR and PAFR to mediate its entry into and translocation through endothelial cells. This occurs in CbpA- and ChoP-dependent fashions, respectively (17–19). Unexpectedly, studies with isogenic mutants that lacked these virulence determinants (i.e., ΔcpbA or ΔlicA) as well as experiments with neutralizing antibodies against the host proteins (PAFR, LR, and pIgR) ruled out these canonical interactions (Fig. 4) and in some instances enhanced interaction (i.e., adhesion and invasion).
FIG 4.

Streptococcus pneumoniae interacts with cardiomyocytes independently of the canonical pathways. Shown are the interactions between HL-1 cardiomyocytes and wild-type TIGR4 (WT) and its derivative choline binding protein A (ΔcbpA) and cholinekinase (ΔlicA) mutants alone (A) or in the presence of the designated neutralizing antibodies (1 μg/ml) (B). Adhesion was measured 30 min after infection, and invasion was measured 1 h after infection followed by 1 h of gentamicin treatment (n = 3). Errors bars represent standard errors of the means. Statistical analyses were done with t tests (A) and 1-way ANOVA with Holm-Sidak correction for multiple comparison (B). **, P ≤ 0.01; ***, P ≤ 0.001.
Pneumolysin and intracellular bacterial hydrogen peroxide production induces cell death.
We next sought to identify the virulence determinants necessary for the intracellular S. pneumoniae-mediated killing of cardiomyocytes. To do this, we constructed isogenic TIGR4 mutants deficient in pneumolysin (Δply) and pyruvate oxidase (ΔspxB), and we observed that both showed nearly complete attenuation of HL-1 cell death (Fig. 5A), although again presenting higher adhesive ability and invasiveness that was similar to or higher than, respectively, those of TIGR4 (see Fig. S2). Pneumolysin is a pore-forming toxin produced by S. pneumoniae (24), and its role in cardiomyocyte killing has already been demonstrated, albeit at late stages of microlesion formation or when administered extracellularly (13, 15). In contrast, SpxB is a metabolic enzyme that generates H2O2 as a result of converting pyruvate to acetylphosphate. A role for bacterially derived H2O2 in cardiomyocyte killing has not been described. In order to test for this, we infected HL-1 cells pretreated with catalase, which neutralizes only extracellular H2O2 (25), or Tempol (26), which neutralizes total H2O2. The absence of protection against cytotoxicity seen with catalase but the significant protection observed with Tempol suggest that S. pneumoniae-mediated HL-1 killing was due to intracellular production of H2O2 after internalization of the bacteria (Fig. 5B). Consistent with our newfound requirement for intracellular H2O2-mediated killing of cardiomyocytes, we observed that D39 was less efficient in killing HL-1 cells (Fig. 5C) and produced significantly less H2O2 (Fig. 5D) than TIGR4, despite a higher invasive ability (Fig. 5E). Thus, the differences in microlesion formation previously observed between these two strains (15) could be, among other factors, due to their differential abilities to produce hydrogen peroxide and then explain the differential phenotypes of these two strains. Along such lines, we observed that TIGR4 and D39 elicited a cytokine and chemokine profile distinct from that of infected HL-1 cells (see Fig. S3 and supplemental materials and methods).
FIG 5.
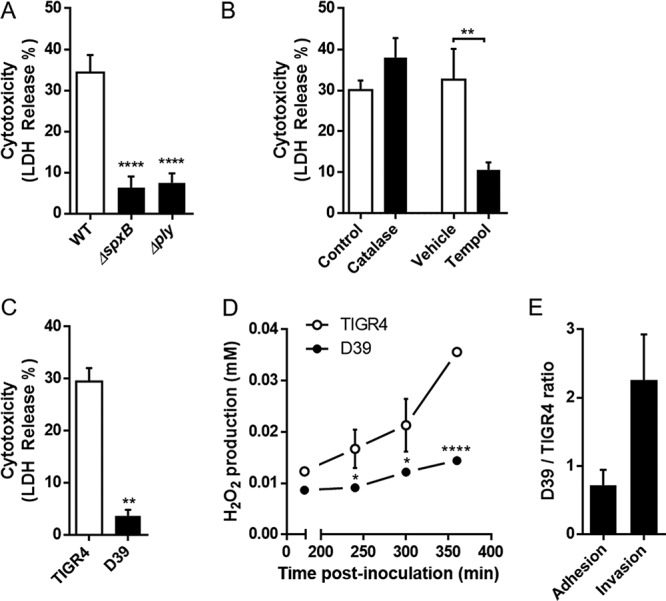
Intracellular hydrogen peroxide produced by S. pneumoniae is required to induce cytotoxicity. (A and B) HL-1 death was measured by assaying LDH release induced by pyruvate oxidase (ΔspxB) or pneumolysin (Δply) TIGR4 mutants (A) or in the presence of extracellular (catalase, 10 U/ml) or extra- and intracellular (Tempol, 3 mM) antioxidants (B). (C) LDH release induced after 6 h of infection by S. pneumoniae TIGR4 and S. pneumoniae D39. (D) Kinetics of H2O2 produced by TIGR4 and D39 during infection. (E) TIGR4 and D39 adhesive and invasive abilities (n = 3). Errors bars represent standard errors of the means. Statistical analyses were done with 1-way ANOVA with Holm-Sidak correction for multiple comparison (A) and t test (B, C, and D). *, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001. Catalase was used at 10 U/ml and Tempol at 3 mM.
Neutralization of ROS protects the heart during infection.
We subsequently explored whether inhibition of reactive oxygen species (ROS) with Tempol would confer cardiac protection in vivo following S. pneumoniae challenge.
Mice treated with Tempol (20 mg/kg) at the time of intraperitoneal (i.p.) challenge with TIGR4 and after 24 h showed a dramatic reduction in the number of cardiac microlesions that could be detected per heart section (Fig. 6A and B). This protection occurred despite the absence of significant differences in bacterial burden seen in the heart and blood of infected mice, indicating that Tempol was not protecting via an antimicrobial effect (Fig. 6C and D). These results suggest that antioxidants may have a beneficial effect as adjunct therapy to antimicrobials in preventing cardiac injury during invasive pneumococcal disease.
FIG 6.

Tempol attenuates cardiac microlesion formation in vivo without modifying bacterial burden. BALB/cJ mice were challenged using 1.0 × 103 CFU/ml of TIGR4 in the presence or absence of Tempol (20 mg/kg). Tempol was administered a second time 24 h after infection. (A) Visualization of microlesion formation in the presence or absence of Tempol. S. pneumoniae was stained with anti-serotype 4 antibody (green), and sections were counterstained with DAPI (blue). Scale bar, 250 μm. (B) Microlesions were enumerated by counting foci of S. pneumoniae in three nonconsecutive stained heart sections separated by at least 50 μm. (C) Final heart burden at the time of sacrifice (T0 + 30 h) was measured by colony counts. (D) Bacteremia was assayed every 6 h by tail bleed. Dotted lines represent detection limits (n = 5 per condition). Error bars represent standard errors of the means. Statistical analyses were done with t tests (B and C) and 2-way ANOVA with Holm-Sidak correction for multiple comparison (D). *, P < 0.05.
DISCUSSION
Recently, it has become evident that invasive pneumococcal disease is associated with MACE, and this can have long-lasting and lethal consequences (7, 13, 14). We have since shown that MACE is due in part to the translocation of S. pneumoniae into the heart, leading to the formation of bacterium-filled microlesions. Importantly, while the molecular interactions involved in translocation of S. pneumoniae across vascular endothelial cells and into the heart have been elucidated in vivo (13), up to this point nothing was known about the early interactions between S. pneumoniae and cardiomyocytes that lead to microlesion formation. Under the premise that microlesions start with individual bacteria after endothelium crossing, in this study, we used an in vitro model of low-MOI cardiomyocyte infection to investigate the early events that occur within the heart.
We observed that at a low MOI, S. pneumoniae is able to kill cardiomyocytes, yet this outcome required live bacteria and direct contact of S. pneumoniae with the cell. An explanation for the latter observation became apparent when we subsequently determined that uptake of pneumococci by cardiomyocytes occurred and was necessary for HL-1 cell killing. Importantly, bacterial uptake by cardiomyocytes was found to be CME dependent, but not through the already-characterized uptake mechanisms implicated for vascular endothelial cells: CbpA interaction with pIgR or LR (17, 20) and cell wall phosphorylcholine interaction with PAFR (18, 27). As CME typically involves a ligand-receptor interaction to be initiated, further effort is now warranted to identify the bacterial ligand(s) and host molecule(s) which are responsible for S. pneumoniae adhesion and uptake by cardiomyocytes. This represents a possible point of intervention to prevent cardiac damage following S. pneumoniae infection.
It is noteworthy that various S. pneumoniae mutants with changes in genes involved in different functions showed enhanced interaction with host cells (i.e., increase in adhesion and invasion). We speculate that these mutations somehow impacted the bacteria at a structural level, modulating the expression of or destabilizing the capsular polysaccharide, thereby allowing stronger host cell interactions due to the unmasking of what is yet the unidentified surface determinant(s) (28). Supporting this possibility, capsule levels varied in our ΔcbpA and ΔlicA mutants (unpublished data) and have been previously described to be altered in ΔspxB backgrounds (29, 30), and the localization of pneumolysin in the cell wall (31) may also play a structural role important for the interaction of the bacteria with the cell. These altered interactions may have been missed by other investigators due to the use of nonencapsulated pneumococci in most of the adhesion/invasion studies described in the literature.
Our past studies (13) and that of Alhamdi et al. (11) have shown that pneumolysin at high doses can kill cardiomyocytes in vitro and contributes to cardiac damage. However, these studies were done in a manner that most likely reflected events occurring in the heart once mature microlesions had formed and pneumococci had become again extracellular. Our results described herein confirm a critical role for pneumolysin at a low MOI and when it is intracellular but suggest that another factor is simultaneously critical for S. pneumoniae-mediated killing, i.e., SpxB-produced H2O2. Along such lines, Bewley et al. showed that pneumolysin was not the only factor responsible for S. pneumoniae killing of macrophages from within, although these other factors were not identified (32). One possibility is that H2O2 potentiates the ability of pneumolysin to kill cardiomyocytes via necroptosis, a programmed mode of necrosis. This form of cell death has recently been shown to be active in macrophages and lung epithelial cells as a result of ion dysregulation resulting from pneumolysin-mediated plasma membrane damage and damage to mitochondria (33, 34). SpxB-derived H2O2 might oxidize lipid membranes, thereby serving as a dual hit to infected cells. Ongoing studies in the laboratory are exploring this possibility. Recent studies by Bryant et al. also suggest that H2O2 can potentiate the effect of pneumolysin by increasing its activity or release from S. pneumoniae (35). These possibilities are favored by our result showing that deletion of SpxB or pneumolysin dramatically reduced HL-1 cell death in a manner that was synergetic and not additive.
SpxB is a key metabolic enzyme whose deletion most likely has major pleiotropic effects on fitness. In fact, the corresponding gene has been shown to be important for colonization (36), competence (37), and capsule production (29). Its deletion, therefore, can also attenuate the bacterium in a manner that is substantially greater than inhibition of H2O2 production. Consequently, our report that SpxB-deficient S. pneumoniae does not kill cardiomyocytes must be interpreted with caution. Supporting the notion that it is indeed the absence of bacterially derived H2O2 and not other negative effects of SpxB deletion that lead to this attenuated state, we observed that Tempol-treated HL-1 cells infected with wild-type TIGR4 remained alive. Tempol presumably has no effect on S. pneumoniae metabolism, as we did not observe any impact of Tempol on bacterial fitness (i.e., bacterial growth [data not shown]). Thus, the effect of SpxB is most likely 2-fold, necessary for metabolic fitness but also serving as the source of a potent cytotoxic product, both ultimately contributing to formation of cardiac microlesions. Importantly, these observations gave some insight into putative therapeutic use of antioxidants to reduce cardiac damage during invasive pneumococcal disease.
In summary, we have collected evidence that S. pneumoniae organisms are taken up by cardiomyocytes and subsequently killed as result of pneumolysin and SpxB-mediated H2O2 production. This most likely reflects the events that occur in the heart following bacterial translocation into the myocardium from the bloodstream and serves as a basis of a working model in which cardiomyocytes might serve as a protective niche, following cardiac invasion, that allows S. pneumoniae to replicate while avoiding immune system or host defenses. Ultimately, cardiomyocyte killing occurs from inside the cell, and as documented for mature lesions, bacteria that emerge are able to kill immune cells with extracellular release of pneumolysin (15).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Strains used in this study are described in Table S1 in the supplemental material. Isogenic mutant derivatives of S. pneumoniae serotype 4 strain TIGR4 (38) were created using splicing overlap extension PCR as described previously (39, 40) and using primers listed in Table S2 in the supplemental material. Bacteria were grown in Todd-Hewitt broth with 0.5% yeast extract (THY) or on blood agar plates (Remel) in a humidified atmosphere at 37°C with 5% CO2. Erythromycin (1 μg/ml) and spectinomycin (100 μg/ml) were added to the growth media when necessary. Listeria monocytogenes growth and infection conditions are described in the supplemental materials and methods.
Cell culture.
Mouse atrial cardiomyocytes (HL-1) were grown in Claycomb serum-free medium (Sigma) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Atlanta Biologicals), 2 mM l-glutamine (Corning), 0.1 mM norepinephrine (Sigma), and 1× penicillin-streptomycin solution (Corning; Cellgro) in a humidified atmosphere at 37°C and 5% CO2 (41).
Ethics statement.
All mouse experiments were reviewed and approved by the Institutional Animal Care and Use Committee at The University of Alabama at Birmingham (UAB; protocol IACUC-20175). Animal care and experimental protocols adhered to public law 89-544 (Animal Welfare Act) (42) and its amendments, Public Health Services guidelines, and the Guide for the Care and Use of Laboratory Animals (43).
Mouse experiments.
Female 6-week-old BALB/cJ mice (Jackson) were challenged with 1.0 × 103 pneumococci in 100 μl of phosphate-buffered saline (PBS) by intraperitoneal (i.p.) injection. Tempol (20 mg/kg [of body weight]) was injected i.p. in 100 μl of PBS with the bacteria during infection (T0) and then alone every 24 h. Blood for assessment of bacterial burden was obtained by tail bleeds every 6 h. At the final time point (T0 + 30 h) or when deemed moribund, mice were euthanized by CO2 asphyxiation and death was confirmed by pneumothorax before heart collection. Hearts collected were then washed thoroughly with PBS and longitudinally half sectioned. One half was homogenized in 1 ml of PBS for heart burden determination, and the other half was embedded in cassettes with optimal cutting temperature compound (Tissue-Tek; 4583) before sectioning and staining.
Monitoring of associated/internalized S. pneumoniae.
HL-1 cells were seeded at 5.0 × 105 per well in 12-well plates coated with fibronectin (1 mg/ml; Sigma) in 0.2% gelatin (Sigma). S. pneumoniae from an overnight culture on a blood agar plate was used to inoculate THY, and this preculture was incubated until reaching an optical density at 620 nm (OD620) of 0.3 to 0.4. Just before infection, HL-1 cells were washed twice with prewarmed PBS and infected with 5.0 × 106 CFU/ml of TIGR4 corresponding to a multiplicity of infection of 10 (MOI10) in prewarmed Dulbecco modified Eagle medium (DMEM; Corning; Cellgro), followed by 10 min of centrifugation at 500 × g. Cells were then treated as described below. For monitoring of associated S. pneumoniae and following designated times of incubation, infected cells were washed three times with warm PBS, scraped, and resuspended in 1 ml of PBS. The total bacterial count was determined by serial dilutions of bacterial suspensions, plating on blood agar plates, and extrapolation from colony counts after overnight incubation. For monitoring intracellular S. pneumoniae, after 30 min of incubation, cells were washed three times with PBS before addition of 900 μl of warm DMEM and incubation. At each required time point, extracellular bacteria were killed by addition of 100 μl of 2-mg/ml gentamicin in DMEM (final concentration = 200 μg/ml) followed by 1 h of incubation. Cells were then washed three times with 1× PBS, lysed by addition of 500 μl of chilled distilled water, and incubated for 10 min at 4°C. Bacterial counts were then determined by colony counts after overnight growth from serial dilutions on blood agar plates. In instances where drug pretreatments were required, cells were pretreated for 1 h with the appropriate drug after 1 wash with prewarmed DMEM. Drugs were then washed from the cell supernatant using PBS before TIGR4 challenge. In instances where cells where treated with antibodies, 1 μg/ml of designated antibody or isotype control (see details in supplemental materials and methods) was incubated with HL-1 cells for 1 h before challenge by TIGR4.
Transmission electron microscopy.
The day before infection, fibronectin-coated Transwell permeable inserts (12 mm and 0.2 μm; Costar) were seeded at 5.0 × 105 HL-1 cells per well. Cells were infected as described for experiments for monitoring of associated/internalized bacteria. To avoid any conditioning of the cells, the same S. pneumoniae-containing DMEM inoculum was added to the top and the bottom of the inserts. Plates were then incubated for the designated amount of time. Infected cells were fixed in 3% electron microscopy-grade glutaraldehyde in 0.1 M cacodylate buffer with 0.1 M sucrose for 1 h at room temperature (RT) and overnight at 4°C. After fixation, the specimens were rinsed several times with 0.1 M sodium cacodylate buffer (pH 7.4), followed by postfixation with 1% osmium tetroxide in cacodylate buffer for 1 h at RT in the dark. After rinsing again with cacodylate buffer for 4 changes at 15 min each, the specimens were dehydrated through a series of graded ethyl alcohols from 50 to 100%: 50% for 5 min, 2% uranyl acetate in 50% ethyl alcohol for 30 min in the dark, 50% for 5 min, 80% for 5 min, 95% for 5 min, and four changes of 100% for 15 min each. Samples were then transferred into embedding resin (Embed 812; Electron Microscopy Sciences, Hatfield, PA) for 12 to 18 h, with embedding medium changed after 1 and 2 days. Subsequently, blocks were polymerized overnight in a 70°C embedding oven and were then ready to section. The resin blocks were first thick sectioned at 1 μm with a histo diamond knife using an Ultracut UCT 7 (Leica, Bannockburn, IL). Sections were collected on slides and stained with toluidine blue. These sections were used as a reference to trim blocks for thin sectioning. The appropriate blocks were then thin sectioned using a diamond knife (Diatome; Electron Microscopy Sciences) at 70 to 90 nm (silver to pale gold using color interference), and sections were then placed on copper grids. After drying, the sections were stained with the heavy metals uranyl acetate and lead citrate for contrast. After drying, the grids were viewed on a Tecnai Spirit 120kv TEM (FEI, Hillsboro, OR). Digital images were taken with a charge-coupled-device (CCD) camera (Advanced Microscopy Techniques Corp., Woburn, MA).
Cytotoxicity assays.
Cell death was evaluated using trypan blue exclusion assay and lactate dehydrogenase (LDH) release assays. A total of 1.0 × 105 HL-1 cardiomyocytes were infected with 1.0 × 106 S. pneumoniae TIGR4 bacteria in a 24-well plate after 2 washes in prewarmed PBS. After 6 h of incubation, supernatant was collected and mixed with adherent cells from the same well, detached by 10 min of 0.05% trypsin-EDTA (Gibco) treatment at 37°C. Ten microliters of this mixture was stained using 10 μl of 0.04% trypan blue solution and loaded on counting slides (Bio-Rad), and the total number and percentage of live cells were determined using a TC20 automatic cell counter (Bio-Rad). LDH release was assayed using a Pierce cytotoxicity LDH assay kit (Thermo Scientific) according to the manufacturer's instructions. TIGR4 was heat killed by 15 min of incubation at 80°C and ethyl alcohol killed by 15 min incubation in 95% ethanol. In instances where drugs were required, after 1 wash prewarmed DMEM, the appropriate drug was added 30 min before infection, kept during all the incubation for antioxidants or 1 h, and washed before infection for endocytosis inhibitors. For Transwell experiments, 5.0 × 105 HL-1 cardiomyocytes were seeded in 12-well plates coated with fibronectin. Each well was then divided into two chambers using Transwell permeable inserts (12 mm and 0.2 μm; Costar), and 5.0 × 106 TIGR4 bacteria were then added in either the upper or the lower chamber in DMEM. Plates were incubated for 6 h at 37°C, and LDH release in the supernatant from the lower HL-1-containing chamber was measured.
Immunofluorescence on cells.
The day before infection, borosilicate coverslips (Knittel-Glass) were coated with fibronectin in a 24-well plate and seeded at 1.0 × 105 cells per well. The day of infection, cells were infected at MOI10 and incubated according to the appropriate timeline. Coverslips were then washed 3 times with PBS and fixed for 15 min at RT with 4% paraformaldehyde (PFA) in PBS. Coverslips were then washed once with PBS and aldehydes were quenched by addition of 50 mM NH4Cl in PBS for 30 min at 4°C. For double immunofluorescence, coverslips were blocked using 1% bovine serum albumin (BSA) in PBS (blocking buffer) for 1 h at RT and incubated for 1 h at RT with rabbit anti-serotype 4 (Statens Serum Institut; 1:1,000 in blocking buffer). Coverslips were then washed 3 times with 0.1% Tween 20 in PBS (PBST) and incubated with rhodamine-conjugated donkey anti-rabbit antibody (Millipore; 1:1,000 in blocking buffer) for 1 h at RT in the dark. Coverslips were then washed 3 times with PBST and permeabilized using 0.2% Triton X-100 in PBS (PBSTX) for 15 min at RT. Cells were then blocked and incubated with primary antibody as described above. After 3 washes in PBSTX, coverslips were incubated for 1 h at RT with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit antibody (Millipore; 1:1,000) supplemented with 4′,6-diamidino-2-phenylindole (DAPI; NucBlue Fixed Cell ReadyProbes reagent; Thermo Fisher). Cells were then washed 3 times with PBSTX, and coverslips were mounted using Fluorsave (Millipore) and kept at 4°C before imaging.
Immunofluorescence on cardiac sections.
Frozen 7-μm-thick cardiac sections were fixed with 10% neutral buffered formalin, permeabilized in 0.2% Triton X-100, and blocked with PBS containing 10% serum from species to which the secondary antibody belonged (blocking buffer). Sections were then incubated overnight at 4°C with blocking buffer containing a 1:1,000 dilution of primary antibody: rabbit anti-serotype 4 capsular polysaccharide antibody (Statens Serum Institut; catalog no. 16747). The next day, sections were vigorously washed with 0.2% Triton X-100 and then incubated for 1 h at RT with blocking buffer containing secondary antibody at a 1:2,000 dilution: FITC-labeled goat anti-rabbit antibody (Jackson ImmunoResearch; catalog no. 111-096-144). Slides were counterstained with DAPI (Molecular Probes by Life Technologies; R37606), and sections were mounted with FluorSave (Calbiochem; 345789) and covered with coverslips for visualization. Images of cardiac sections were captured using a Leica LMD6 microscope equipped with a DFC3000G monochrome camera. Image stitching of whole immunofluorescence microscopy stained cardiac sections was performed using Leica LASX software. When indicated, cardiac microlesions were enumerated by counting foci of pneumococci in three capsule-stained heart sections which were at least >50 μm apart. The first section was cut 300 μm into the heart from the surface.
BrdU incorporation assay.
Cells were prepared as for immunofluorescence. After 1 h of infection, HL-1 cells were washed and incubated in DMEM-gentamicin (200 μg/ml). After 30 min, the medium was replaced with DMEM-gentamicin containing BrdU (10 μM) for 30 min before PBS washes, fixation, and staining.
Supplementary Material
ACKNOWLEDGMENTS
We thank Melissa Chimento and Ed Phillips at the UAB High Resolution Imaging Facility for the transmission electron microscopy and the UAB Heflin Center for Genomic Sciences for Sanger sequencing. We also thank Norberto Gonzalez-Juarbe for technical assistance and Ashleigh N. Riegler for manuscript editing.
Financial support for this work was received from the National Institutes of Health (RO1 AI114800) and the American Heart Association (16GRNT3023007).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00569-17.
REFERENCES
- 1.Bogaert D, De Groot R, Hermans PW. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 4:144–154. doi: 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 2.Obaro S, Adegbola R. 2002. The pneumococcus: carriage, disease and conjugate vaccines. J Med Microbiol 51:98–104. doi: 10.1099/0022-1317-51-2-98. [DOI] [PubMed] [Google Scholar]
- 3.Wunderink RG, Waterer GW. 2014. Community-acquired pneumonia. N Engl J Med 370:1863. doi: 10.1056/NEJMcp1214869. [DOI] [PubMed] [Google Scholar]
- 4.Vallés J, Diaz E, Martin-Loeches I, Bacelar N, Saludes P, Lema J, Gallego M, Fontanals D, Artigas A. 2016. Evolution over a 15-year period of the clinical characteristics and outcomes of critically ill patients with severe community-acquired pneumonia. Med Intensiva 40:238–245. doi: 10.1016/j.medin.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Tonnaer EL, Graamans K, Sanders EA, Curfs JH. 2006. Advances in understanding the pathogenesis of pneumococcal otitis media. Pediatr Infect Dis J 25:546–552. doi: 10.1097/01.inf.0000222402.47887.09. [DOI] [PubMed] [Google Scholar]
- 6.Alonso De Velasco E, Verheul AF, Verhoef J, Snippe H. 1995. Streptococcus pneumoniae: virulence factors, pathogenesis, and vaccines. Microbiol Rev 59:591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musher DM, Rueda AM, Kaka AS, Mapara SM. 2007. The association between pneumococcal pneumonia and acute cardiac events. Clin Infect Dis 45:158–165. doi: 10.1086/518849. [DOI] [PubMed] [Google Scholar]
- 8.Eurich DT, Marrie TJ, Minhas-Sandhu JK, Majumdar SR. 2017. Risk of heart failure after community acquired pneumonia: prospective controlled study with 10 years of follow-up. BMJ 356:j413. doi: 10.1136/bmj.j413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown AO, Millett ER, Quint JK, Orihuela CJ. 2015. Cardiotoxicity during invasive pneumococcal disease. Am J Respir Crit Care Med 191:739–745. doi: 10.1164/rccm.201411-1951PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reyes LF, Restrepo MI, Hinojosa CA, Soni NJ, Anzueto A, Babu BL, Gonzalez-Juarbe N, Rodriguez AH, Jimenez A, Chalmers JD, Aliberti S, Sibila O, Winter VT, Coalson JJ, Giavedoni LD, Dela Cruz CS, Waterer GW, Witzenrath M, Suttorp N, Dube PH, Orihuela CJ. 2017. Severe pneumococcal pneumonia causes acute cardiac toxicity and subsequent cardiac remodeling. Am J Respir Crit Care Med doi: 10.1164/rccm.201701-0104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alhamdi Y, Neill DR, Abrams ST, Malak HA, Yahya R, Barrett-Jolley R, Wang G, Kadioglu A, Toh CH. 2015. Circulating pneumolysin is a potent inducer of cardiac injury during pneumococcal infection. PLoS Pathog 11:e1004836. doi: 10.1371/journal.ppat.1004836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reyes LF, Restrepo MI, Hinojosa CA, Soni NJ, Shenoy AT, Gilley RP, Gonzalez-Juarbe N, Noda JR, Winter VT, de la Garza MA, Shade RE, Coalson JJ, Giavedoni LD, Anzueto A, Orihuela CJ. 2016. A non-human primate model of severe pneumococcal pneumonia. PLoS One 11:e0166092. doi: 10.1371/journal.pone.0166092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown AO, Mann B, Gao G, Hankins JS, Humann J, Giardina J, Faverio P, Restrepo MI, Halade GV, Mortensen EM, Lindsey ML, Hanes M, Happel KI, Nelson S, Bagby GJ, Lorent JA, Cardinal P, Granados R, Esteban A, LeSaux CJ, Tuomanen EI, Orihuela CJ. 2014. Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog 10:e1004383. doi: 10.1371/journal.ppat.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown AO, Orihuela CJ. 2015. Visualization of Streptococcus pneumoniae within cardiac microlesions and subsequent cardiac remodeling. J Vis Exp 2015(98):e52590. doi: 10.3791/52590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilley RP, Gonzalez-Juarbe N, Shenoy AT, Reyes LF, Dube PH, Restrepo MI, Orihuela CJ. 2016. Infiltrated macrophages die of pneumolysin-mediated necroptosis following pneumococcal myocardial invasion. Infect Immun 84:1457–1469. doi: 10.1128/IAI.00007-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ring A, Weiser JN, Tuomanen EI. 1998. Pneumococcal trafficking across the blood-brain barrier. Molecular analysis of a novel bidirectional pathway. J Clin Invest 102:347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orihuela CJ, Mahdavi J, Thornton J, Mann B, Wooldridge KG, Abouseada N, Oldfield NJ, Self T, Ala'Aldeen DA, Tuomanen EI. 2009. Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J Clin Invest 119:1638–1646. doi: 10.1172/JCI36759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. 1995. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature 377:435–438. doi: 10.1038/377435a0. [DOI] [PubMed] [Google Scholar]
- 19.Rosenow C, Ryan P, Weiser JN, Johnson S, Fontan P, Ortqvist A, Masure HR. 1997. Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol Microbiol 25:819–829. doi: 10.1111/j.1365-2958.1997.mmi494.x. [DOI] [PubMed] [Google Scholar]
- 20.Iovino F, Engelen-Lee JY, Brouwer M, van de Beek D, van der Ende A, Valls Seron M, Mellroth P, Muschiol S, Bergstrand J, Widengren J, Henriques-Normark B. 2017. pIgR and PECAM-1 bind to pneumococcal adhesins RrgA and PspC mediating bacterial brain invasion. J Exp Med 214:1619–1630. doi: 10.1084/jem.20161668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilsdonk EP, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, Rothblat GH. 1995. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem 270:17250–17256. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- 22.von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, Chircop M, Sakoff J, von Kries JP, Saenger W, Krausslich HG, Shupliakov O, Robinson PJ, McCluskey A, Haucke V. 2011. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 23.Ireton K. 2007. Entry of the bacterial pathogen Listeria monocytogenes into mammalian cells. Cell Microbiol 9:1365–1375. doi: 10.1111/j.1462-5822.2007.00933.x. [DOI] [PubMed] [Google Scholar]
- 24.Braun JS, Novak R, Gao G, Murray PJ, Shenep JL. 1999. Pneumolysin, a protein toxin of Streptococcus pneumoniae, induces nitric oxide production from macrophages. Infect Immun 67:3750–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Preston TJ, Muller WJ, Singh G. 2001. Scavenging of extracellular H2O2 by catalase inhibits the proliferation of HER-2/Neu-transformed rat-1 fibroblasts through the induction of a stress response. J Biol Chem 276:9558–9564. doi: 10.1074/jbc.M004617200. [DOI] [PubMed] [Google Scholar]
- 26.Francischetti IM, Gordon E, Bizzarro B, Gera N, Andrade BB, Oliveira F, Ma D, Assumpcao TC, Ribeiro JM, Pena M, Qi CF, Diouf A, Moretz SE, Long CA, Ackerman HC, Pierce SK, Sa-Nunes A, Waisberg M. 2014. Tempol, an intracellular antioxidant, inhibits tissue factor expression, attenuates dendritic cell function, and is partially protective in a murine model of cerebral malaria. PLoS One 9:e87140. doi: 10.1371/journal.pone.0087140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuomanen EI. 1997. The biology of pneumococcal infection. Pediatr Res 42:253–258. doi: 10.1203/00006450-199709000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez CJ, Hinojosa CA, Shivshankar P, Hyams C, Camberlein E, Brown JS, Orihuela CJ. 2011. Changes in capsular serotype alter the surface exposure of pneumococcal adhesins and impact virulence. PLoS One 6:e26587. doi: 10.1371/journal.pone.0026587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Echlin H, Frank MW, Iverson A, Chang TC, Johnson MD, Rock CO, Rosch JW. 2016. Pyruvate oxidase as a critical link between metabolism and capsule biosynthesis in Streptococcus pneumoniae. PLoS Pathog 12:e1005951. doi: 10.1371/journal.ppat.1005951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carvalho SM, Farshchi Andisi V, Gradstedt H, Neef J, Kuipers OP, Neves AR, Bijlsma JJ. 2013. Pyruvate oxidase influences the sugar utilization pattern and capsule production in Streptococcus pneumoniae. PLoS One 8:e68277. doi: 10.1371/journal.pone.0068277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price KE, Camilli A. 2009. Pneumolysin localizes to the cell wall of Streptococcus pneumoniae. J Bacteriol 191:2163–2168. doi: 10.1128/JB.01489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bewley MA, Naughton M, Preston J, Mitchell A, Holmes A, Marriott HM, Read RC, Mitchell TJ, Whyte MK, Dockrell DH. 2014. Pneumolysin activates macrophage lysosomal membrane permeabilization and executes apoptosis by distinct mechanisms without membrane pore formation. mBio 5:e01710-. doi: 10.1128/mBio.01710-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.González-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, Restrepo MI, Dube PH, Bergman MA, Orihuela CJ. 2017. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ 24:917–928. doi: 10.1038/cdd.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.González-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, Orihuela CJ. 2015. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PLoS Pathog 11:e1005337. doi: 10.1371/journal.ppat.1005337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bryant JC, Dabbs RC, Oswalt KL, Brown LR, Rosch JW, Seo KS, Donaldson JR, McDaniel LS, Thornton JA. 2016. Pyruvate oxidase of Streptococcus pneumoniae contributes to pneumolysin release. BMC Microbiol 16:271. doi: 10.1186/s12866-016-0881-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Regev-Yochay G, Trzcinski K, Thompson CM, Lipsitch M, Malley R. 2007. SpxB is a suicide gene of Streptococcus pneumoniae and confers a selective advantage in an in vivo competitive colonization model. J Bacteriol 189:6532–6539. doi: 10.1128/JB.00813-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bättig P, Muhlemann K. 2008. Influence of the spxB gene on competence in Streptococcus pneumoniae. J Bacteriol 190:1184–1189. doi: 10.1128/JB.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- 39.Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 41.Claycomb WC, Lanson NA Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ Jr. 1998. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.US Congress. 1966. Animal Welfare Act. Public law 89-544.
- 43.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.