

ABSTRACT
The Gram-positive bacterium Enterococcus faecalis is both a colonizer of the gastrointestinal tract (GIT) and an agent of serious nosocomial infections. Although it is typically required for pathogenesis, GIT colonization by E. faecalis is poorly understood. E. faecalis tolerates high concentrations of GIT antimicrobials, like cholate and lysozyme, leading us to hypothesize that resistance to intestinal antimicrobials is essential for long-term GIT colonization. Analyses of E. faecalis mutants exhibiting defects in antimicrobial resistance revealed that IreK, a determinant of envelope integrity and antimicrobial resistance, is required for long-term GIT colonization. IreK is a member of the PASTA kinase protein family, bacterial transmembrane signaling proteins implicated in the regulation of cell wall homeostasis. Among several determinants of cholate and lysozyme resistance in E. faecalis, IreK was the only one found to be required for intestinal colonization, emphasizing the importance of this protein to enterococcal adaptation to the GIT. By studying ΔireK suppressor mutants that recovered the ability to colonize the GIT, we identified two conserved enterococcal proteins (OG1RF_11271 and OG1RF_11272) that function antagonistically to IreK and interfere with cell envelope integrity, antimicrobial resistance, and GIT colonization. Our data suggest that IreK, through its kinase activity, inhibits the actions of these proteins. IreK, OG1RF_11271, and OG1RF_11272 are found in all enterococci, suggesting that their effect on GIT colonization is universal across enterococci. Thus, we have defined conserved genes in the enterococcal core genome that influence GIT colonization through their effect on enterococcal envelope integrity and antimicrobial resistance.
KEYWORDS: Enterococcus, IreK, antimicrobial resistance, cell envelope integrity, colonization
INTRODUCTION
Enterococcus faecalis is a gastrointestinal tract (GIT) commensal found at relatively low abundance in the healthy human gut. Although it is harmless under normal conditions, E. faecalis can cause life-threatening infections during antibiotic-induced dysbiosis (1). Enterococci are among the most common agents of hospital-acquired infections. Due to their intrinsic resistance to commonly used antibiotics, like cephalosporins, enterococci proliferate and dominate the GIT during antibiotic therapy; they subsequently disseminate to internal organs, where they can cause damage (2, 3). GIT colonization is therefore critical for the pathogenesis of these organisms. Interfering with GIT colonization could represent an innovative strategy to prevent enterococcal infections; however, development of such therapies requires a better understanding of GIT colonization by commensals.
Previous studies identified enterococcal genes involved in various processes, such as biofilm formation, sugar transport, and the synthesis of cell wall polysaccharides, to promote GIT colonization (4–6). The genes identified in these studies are located on mobile genetic elements or are enriched in clinical isolates, suggesting that they enhance GIT colonization under some circumstances but do not represent the essential determinants of GIT colonization in the core enterococcal genome that evolved over millions of years to enable enterococci to colonize the GIT. Although these studies made important contributions to our understanding of GIT colonization, they were conducted in animal models that harbored an antibiotic-disrupted gut microbiota. To understand colonization in the unperturbed GIT, our group previously established a model that achieves long-term colonization (>11 weeks) in antibiotic-naive mice (7). As previously described, with this model, we can establish stable colonization using various E. faecalis strains, such as the laboratory strain OG1RF, and multidrug-resistant (MDR) strains, like V583. OG1RF is devoid of the plasmids and pathogenicity islands (8) typically found in MDR clinical isolates and therefore allows us to interrogate the function of core enterococcal genes in GIT colonization. Using this model, we sought to identify genetic determinants of long-term colonization of the unperturbed GIT.
To prevent overgrowth of the intestinal microbiota and regulate its composition, mammalian hosts secrete antimicrobials, like bile acids and antimicrobial peptides (9, 10). Commensals must be able to tolerate these antimicrobials in order to survive the intestinal environment (11). Typically, bacteria employ signal transduction systems to monitor their environment for antimicrobials and initiate adaptive biological responses. Because the bacterial cell envelope is a target for many GIT antimicrobials, sensory systems that monitor the integrity of the cell envelope and promote envelope repair and homeostasis are likely critical for GIT colonization. In our past work, we identified a signaling protein, IreK (previously known as PrkC), that is critical for resistance toward cell envelope-active antimicrobials in E. faecalis (12, 13).
IreK is a transmembrane protein exhibiting a conserved domain architecture comprised of an intracellular eukaryote-like Ser/Thr kinase domain and extracellular PASTA domains, defining it as a member of the PASTA kinase family. PASTA kinases are conserved across the Firmicutes phylum and are involved in various fundamental processes, such as sporulation (14, 15), energy metabolism (16, 17), and cell wall homeostasis (16, 18). Although the function of the PASTA domains is not well understood, they are thought to sense cell wall stress (15, 19). IreK promotes resistance toward cell envelope-damaging antimicrobials, such as cephalosporins, nisin, and cholate (an intestinal bile acid) (12, 20–22).
IreK conferred a modest advantage to E. faecalis in an experiment assessing short-term (16-h) persistence in the mouse GIT (12). We therefore hypothesized that IreK would promote long-term GIT colonization by E. faecalis. Analysis of an E. faecalis ΔireK mutant in our mouse colonization model (7) revealed that IreK is required for long-term colonization of the antibiotic-naive GIT. Additionally, we found that the intestinal environment can select for suppressor mutants that recover the ability to colonize, despite the absence of IreK. Examination of the suppressor mutants led us to identify novel enterococcal proteins that, along with IreK, modulate envelope integrity, antimicrobial resistance, and intestinal colonization.
RESULTS
IreK is required for long-term GIT colonization.
Numerous studies suggest that bile acids, such as cholate, due to their antimicrobial properties, are important regulators of the GIT microbiota's composition (9, 23, 24). Consistent with this idea, we previously observed that IreK promotes both the cholate resistance and short-term GIT persistence of E. faecalis (12). However, ΔireK mutants exhibit several defects, in addition to cholate susceptibility, including a marked loss of resistance to antibiotics that affect the integrity of the cell envelope (12, 20), so it remains unclear if cholate susceptibility per se accounts for the observed defect in GIT persistence. To assess the relationship between cholate resistance and long-term intestinal colonization, we asked whether cholate-sensitive E. faecalis mutants could colonize the GIT.
Two transposon insertion mutants that exhibit severe cholate resistance defects were identified (Fig. 1A) by screening a transposon library (25). One mutant has an insertion in a gene predicted to encode the Brp/Blh family beta-carotene 15,15-monooxygenase, and the other has an insertion in the putative signal peptidase II gene ispA. To our knowledge, there is no direct relationship between the proteins disrupted in these transposon mutants or between any of those proteins and IreK. Importantly, compared to the parental wild-type (WT) strain OG1RF, both of those mutants and the ΔireK mutant exhibited normal growth in culture media. Moreover, despite their defect in cholate resistance, the transposon mutants did not exhibit a defect in cell envelope integrity, as determined by permeability to chlorophenol red β-d-galactoside (CPRG). CPRG is normally excluded from the cytoplasm of wild-type E. faecalis cells. However, mutants with defects in cell envelope integrity (such as the ΔireK mutant) allow CPRG into the cytoplasm, where it is hydrolyzed by LacZ to release the red chlorophenol chromophore (Fig. 1B) (26).
FIG 1.

Loss of cholate resistance alone does not cause the ΔireK colonization defect. (A) Cholate resistance was determined for wild-type (WT) E. faecalis (OG1RF) and cholate-sensitive mutant ΔireK (CK119), as well as the brp/blh (23J13) and ispA (28M17) transposon mutants. The MICs reported represent the median values from three independent biological replicates. (B) CPRG hydrolysis was measured for the strains listed in panel A. CPRG hydrolysis in cultures (representative cultures shown directly above bar for each indicated strain) was quantified by measuring the absorbance at 570 nm after the removal of bacteria by centrifugation and normalization to the absorbance at 630 nm. The reported measurements represent averages from three independent cultures, and error bars represent standard deviations. Statistical significance was evaluated by t test. ****, P < 0.0001 versus WT. (C to F) Groups of mice (5 per group) were colonized with the strains indicated in panels A and B. Bacterial loads were determined by enumerating the enterococci in feces by culture on rifampin-supplemented BHI agar. Dashed lines, the limit of detection. Symbols for mice with undetectable colonization levels were omitted, and instead, the number of mice for which colonization was not detected (ND)/total number of mice tested is shown underneath the dashed lines.
We tested these cholate-sensitive mutants alongside the ΔireK mutant in our long-term GIT colonization model. Although these three strains had comparable levels of cholate sensitivity, they exhibited variable colonization phenotypes (Fig. 1C to F). All mice that were fed the brp/blh and the ispA mutants retained detectable levels of colonization at 5 weeks postfeeding, indicating that neither of these genes is required for long-term colonization and that cholate resistance alone does not dictate intestinal colonization fitness. In contrast, ΔireK mutants exhibited a profound colonization defect. None of the mice fed the ΔireK mutant harbored detectable levels of colonization after 4 weeks. Similarly, a ΔireK mutant of E. faecalis V583, an MDR clinical isolate, exhibited substantially impaired GIT colonization relative to its wild-type parent (see Fig. S1 in the supplemental material). These observations implicate IreK as a determinant of long-term GIT colonization by E. faecalis.
In addition to the cholate resistance defect, ΔireK mutants were found to be defective at lysozyme resistance (Fig. 1A). Lysozyme is secreted by Paneth cells in the small intestine and is implicated in regulating the composition of the microbiota through its antimicrobial activity (27, 28). To determine if the lysozyme resistance defect could account for the inability of the ΔireK mutant to colonize the GIT, we used two cholate-resistant but lysozyme-sensitive E. faecalis mutants for GIT colonization experiments. One mutant had a transposon insertion in sigV, which encodes an extracellular function sigma factor known to mediate lysozyme resistance (29, 30). The other mutant had a deletion of the croR and croS genes, which encode a two-component system that responds to cell wall stress (26, 31, 32). Although a defect specifically in lysozyme resistance of mutants lacking CroR and CroS has not been previously described, we found the ΔcroR ΔcroS mutant to be substantially less resistant to lysozyme than its parent (Fig. 2A). Despite their lysozyme sensitivity, these mutants do not exhibit a defect in cell envelope integrity (Fig. 2A and B). Although these two mutants have lost much of their natural resistance to lysozyme, they do not exhibit a colonization defect (Fig. 2C and D), suggesting that lysozyme resistance alone does not determine GIT colonization by E. faecalis.
FIG 2.

Loss of lysozyme resistance alone does not cause the ΔireK colonization defect. (A) Cholate resistance was determined for the WT E. faecalis strain (OG1RF), the cholate-sensitive ΔireK (CK119) and ΔcroR ΔcroS (SB6) mutants, and the sigV transposon mutant (35H2). The reported MICs represent the median values from three independent biological replicates. (B) CPRG hydrolysis was measured for the strains listed in panel A. The reported measurements represent averages from three independent cultures, and error bars represent standard deviations. Statistical significance was evaluated by t test. ****, P < 0.0001 versus WT. (C and D) Intestinal colonization of the lysozyme-sensitive mutants listed in panel A. Groups of 5 mice were colonized with either the ΔireK mutant or one of the tested lysozyme-sensitive mutants. Colonization levels were determined by enumerating the enterococci in feces by culture on rifampin-supplemented BHI agar. Dashed lines, the limit of detection.
To verify that the colonization defect exhibited by the ΔireK mutants is indeed due to a lack of IreK, we performed complementation analysis by expressing IreK from a plasmid. This experiment is technically challenging, because the activities (and, presumably, expression) of IreK and its cognate phosphatase IreP (which opposes IreK activity through dephosphorylation) must be properly balanced to achieve phenotypic complementation but avoid the fitness costs associated with unregulated IreK activity (13). To achieve this, we coexpressed the adjacent ireP and ireK genes from a constitutively active promoter in plasmid pJRG8 in an E. faecalis host lacking both ireP and ireK. As a control, we used an equivalent plasmid coexpressing ireP with a catalytically impaired ireK (ireK K41R), known from previous studies to be equally as defective for antimicrobial resistance as ΔireK mutants (13, 20).
We attempted to colonize the GIT of mice using these two strains. To avoid altering the resident GIT microbiota, the mice were not treated with any antibiotic for plasmid selection. We observed that mice retained GIT colonization with the strain expressing wild-type IreK for 2 weeks, during which the complementation plasmid was progressively lost. In contrast, when we fed mice ΔireK ΔireP mutants complemented with IreP and the kinase-impaired (K41R) IreK, we observed that E. faecalis colonization was undetectable for four out of five mice within a week and the complementation plasmid was entirely lost (Fig. S2) (13, 20). Thus, expression of IreK from a plasmid enhances GIT colonization by an E. faecalis mutant lacking ireK. These results indicate that the ΔireK colonization defect is due to a loss of the kinase activity of IreK. In addition, the plasmid retention kinetics suggest that the presence of WT IreK can act as a selection factor in the GIT, emphasizing the importance of this protein during GIT colonization.
The intestinal environment selects for ΔireK suppressor mutants.
During one GIT colonization experiment, one mouse fed the ΔireK mutant retained colonization at levels comparable to those observed for wild-type E. faecalis (Fig. 3A). Rifampin (Rif)-resistant E. faecalis isolates were recovered from that mouse and designated ΔireK*. Unlike the parental ΔireK mutant, ΔireK* isolates are not permeable to CPRG, indicating recovery of their cell envelope integrity (Fig. 3B). ΔireK* isolates were also resistant to cholate and lysozyme (Fig. 3C) and able to colonize the GIT (Fig. 3D).
FIG 3.

The intestinal environment can select for suppressor mutants that recover envelope integrity, antimicrobial resistance, and the ability to colonize. (A) Groups of mice (5 per group) were colonized with either WT E. faecalis (OG1RF) or the ΔireK mutant (CK119). Colonization levels were determined by enumerating the enterococci in feces by culture on rifampin-supplemented BHI agar. Arrows, mice harboring ΔireK* clones; dashed line, the limit of detection. Symbols for mice with undetectable colonization levels were omitted, and instead, the number of mice for which colonization was not detected (ND)/total number of mice tested is shown underneath the dashed line. (B) CPRG hydrolysis was measured for the ΔireK* suppressor mutant, the WT, and the ΔireK mutant. Reported measurements represent averages from three independent cultures, and error bars represent standard deviations. Statistical significance was evaluated by t test. ****, P < 0.0001 versus WT. (C) The cholate and lysozyme resistance of the strains listed in panel B was determined. The reported MICs represent the median values from three independent biological replicates. (D) Groups of mice (5 per group) were colonized with either WT E. faecalis (OG1RF), the ΔireK mutant (CK119), or the ΔireK* suppressor mutant. Intestinal colonization was assessed as described in the legend to panel A.
PCR analysis for ireK determined that ΔireK* isolates did not recover the ireK gene (data not shown), and Western blot analysis confirmed that these isolates do not express IreK (Fig. S3). This suggested that ΔireK* isolates acquired a mutation that suppressed the envelope integrity and antimicrobial resistance defects exhibited by ΔireK mutants. Importantly, these phenotypic changes were associated with the recovery of GIT colonization (Fig. 3).
To determine the genetic basis for the suppressor mutant phenotype, we sequenced the genomes from 3 ΔireK* isolates obtained from distinct segments of the GIT. Two nucleotide variants common to all three ΔireK* isolates (summarized in Fig. 4A) were identified and confirmed by Sanger sequencing to be present in ΔireK* isolates but not the parental ΔireK mutant. We also resequenced the genomes of 12 wild-type E. faecalis isolates recovered from mice after 4 weeks of stable colonization and did not find genetic alterations, indicating that colonization of the GIT does not select for mutants of colonization-competent E. faecalis.
FIG 4.
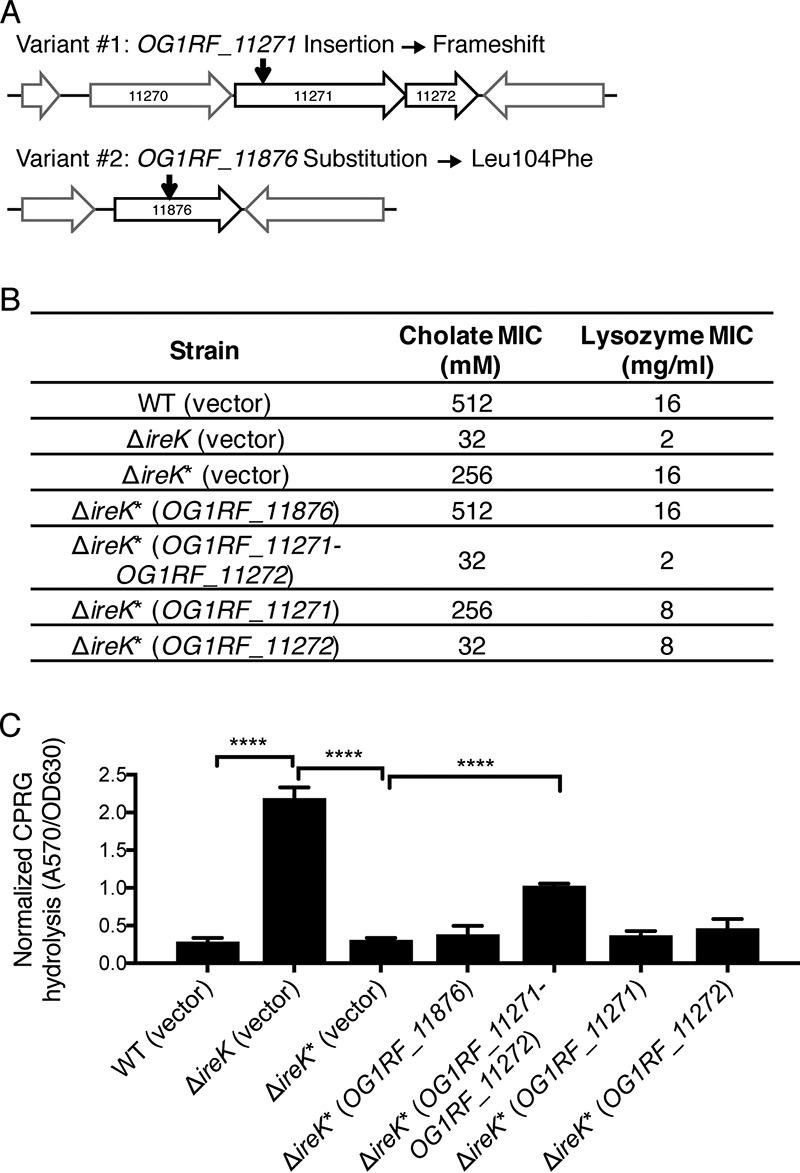
Disruption of OG1RF_11271 and OG1RF_11272 drives the phenotype of ΔireK* suppressor mutants. (A) Genetic architecture near the nucleotide variants uncovered by whole-genome sequencing (the drawing is not to scale). The locations of the nucleotide variants are indicated by black arrows. (B) The cholate and lysozyme resistance of the WT E. faecalis strain (OG1RF), the ΔireK mutant (CK119), and the ΔireK* suppressor mutant carrying the empty vector (pJRG9) or expressing wild-type copies of the indicated genes was determined. The reported MICs represent the median values from three independent biological replicates. (C) CPRG hydrolysis was measured for the strains listed in panel B. The reported measurements represent averages from three independent cultures, and error bars represent standard deviations. Statistical significance was evaluated by t test. ****, P < 0.0001.
One nucleotide variant in ΔireK* isolates is a single-nucleotide insertion in OG1RF_11271, which results in a frameshift that is predicted to disrupt the translation of OG1RF_11271 and potentially interfere with expression of the gene immediately downstream, OG1RF_11272, due to polarity. There were no mutations in the open reading frame of OG1RF_11272. The other variant encodes an L104F substitution in OG1RF_11876. To determine which variant was responsible for the suppressor mutant phenotype, a wild-type copy of each gene was independently expressed from a constitutive promoter in the vector pJRG9 (33). Carriage of this vector did not alter the antimicrobial resistance (Fig. 4B) or cell envelope integrity (Fig. 4C) of the WT, ΔireK, or ΔireK* strain. Expression of wild-type OG1RF_11876 in the ΔireK* strain did not affect cholate and lysozyme resistance levels, suggesting that the variant in OG1RF_11876 is not involved in these phenotypes. However, coexpression of OG1RF_11271-OG1RF_11272 in the ΔireK* suppressor mutant resulted in a loss of resistance to both antimicrobials, restoring the phenotype of the ΔireK mutant (Fig. 4B). Thus, the loss of OG1RF_11271 and/or OG1RF_11272 function suppresses the phenotypic defects of the ΔireK mutant.
To specifically determine if OG1RF_11271 or OG1RF_11272 was responsible, each gene was individually expressed in the ΔireK* suppressor mutant. Expression of OG1RF_11272 alone resulted in a complete loss of resistance to cholate but not to lysozyme, while expression of OG1RF_11271 alone had a minimal impact on antimicrobial resistance. Moreover, only coexpression of OG1RF_11271 and OG1RF_11272 resulted in significant permeabilization of the ΔireK* suppressor mutant to CPRG (Fig. 4C). Taken together, these complementation studies indicate that the overall phenotype of the ΔireK* suppressor mutant is caused by a disruption of both OG1RF_11271 and OG1RF_11272.
Deletion of OG1RF_11272 is sufficient to elicit the suppressor mutant phenotype in E. faecalis ΔireK.
To directly test the roles of OG1RF_11271 and OG1RF_11272, a series of in-frame deletion mutants lacking either one or both genes in the ΔireK parent was constructed. The antimicrobial resistance phenotypes of these mutants (Fig. 5A) revealed that the ΔireK ΔOG1RF_11271 ΔOG1RF_11272 mutant was fully resistant to cholate and lysozyme, corroborating our findings from the complementation studies. While deletion of OG1RF_11271 alone did not alter antimicrobial resistance, deletion of OG1RF_11272 alone in the ΔireK parent was sufficient to fully restore resistance to both cholate and lysozyme. Consistent with this, the ΔireK ΔOG1RF_11271 mutant exhibited a colonization defect similar to that of the ΔireK mutant, while deletion of OG1RF_11272 alone was sufficient to suppress the colonization defect of the ΔireK mutant (Fig. 5C). One of the mice fed ΔireK ΔOG1RF_11271 mutants retained colonization (Fig. 5C). Isolates obtained from the GIT of that mouse did not exhibit increased cholate resistance relative to the ΔireK ΔOG1RF_11271 parent (data not shown), indicating that they were not suppressor mutants. These isolates were not further examined. As expected, the ΔireK ΔOG1RF_11271 ΔOG1RF_11272 triple mutant also recovered the ability to colonize (Fig. S4). Collectively, these studies indicate that functional OG1RF_11272 interferes with antimicrobial resistance and GIT colonization in the ΔireK mutant.
FIG 5.
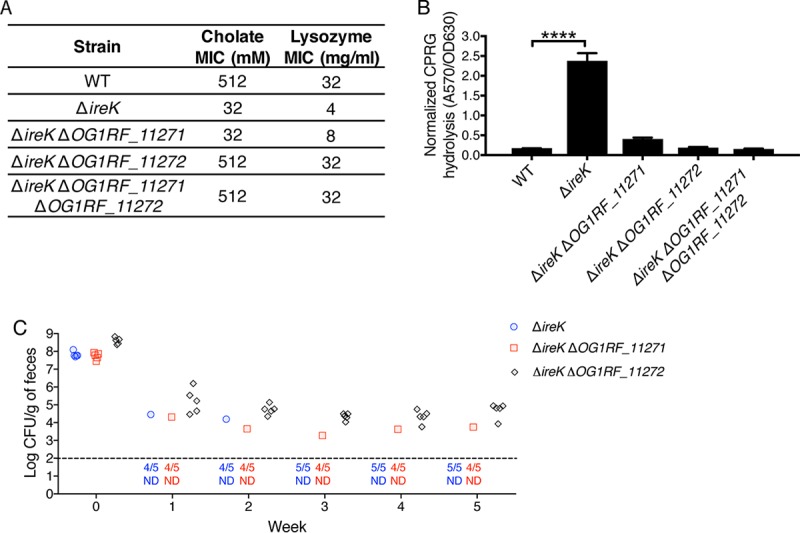
Deletion of OG1RF_11272 in the ΔireK mutant background is sufficient to elicit the suppressor mutant phenotype. (A) The cholate and lysozyme resistance of the WT E. faecalis strain (OG1RF) and the ΔireK (CK119), ΔireK ΔOG1RF_11271 (IB21), ΔireK ΔOG1RF_11272 (IB22), and ΔireK ΔOG1RF_11271 ΔOG1RF_11272 (IB23) mutants was determined. The reported MICs represent the median values from three independent biological replicates. (B) CPRG hydrolysis was measured for the strains listed in panel A. The reported measurements represent averages from three independent cultures, and error bars represent standard deviations. Statistical significance was evaluated by t test. ****, P < 0.0001. (C) Groups of mice (5 per group) were colonized with the ΔireK (CK119), ΔireK ΔOG1RF_11271 (IB21), or ΔireK ΔOG1RF_11272 (IB22) mutant. Colonization loads were determined by enumerating the enterococci in feces by culture on rifampin-supplemented BHI agar. Dashed line, the limit of detection. Symbols for mice with undetectable colonization levels were omitted, and instead, the number of mice for which colonization was not detected (ND)/total number of mice tested is shown underneath the dotted line.
The potential roles for OG1RF_11271 and OG1RF_11272 in cell envelope integrity were assessed using CPRG hydrolysis assays. Deletion of either OG1RF_11271 or OG1RF_11272 restored cell envelope integrity in the ΔireK mutant (Fig. 5B). Thus, the activities of both OG1RF_11271 and OG1RF_11272 are required to elicit the cell envelope integrity defect of the ΔireK mutant.
OG1RF_11271 and OG1RF_11272 appear to exist in an operon that also contains OG1RF_11270 (Fig. 4A). To address the possibility that OG1RF_11270 is involved in antimicrobial resistance, we deleted OG1RF_11270 in the ΔireK mutant. Deletion of OG1RF_11270 suppressed the antimicrobial resistance defect of the ΔireK mutant, as seen for deletion of OG1RF_11272. However, expression of OG1RF_11270 in the ΔireK ΔOG1RF_11270 double mutant did not complement the deletion (Fig. S5), suggesting that the suppression was due to polarity on expression of OG1RF_11271 or OG1RF_11272. Indeed, expression of OG1RF_11271 and OG1RF_11272 led to the loss of antimicrobial resistance in the ΔireK ΔOG1RF_11270 mutant, consistent with the hypothesis that deletion of OG1RF_11270 exhibits polarity on the expression of OG1RF_11271 and OG1RF_11272.
OG1RF_11271 and OG1RF_11272 do not alter expression or phosphorylation of the IreK substrate IreB.
IreB is a negative regulator of antimicrobial resistance in E. faecalis whose activity is inhibited by IreK-mediated phosphorylation. Deletion of ireB in the ΔireK mutant suppresses antimicrobial resistance defects (20). Given the similarities in phenotype with the mutants described here, it was important to determine whether OG1RF_11271 and OG1RF_11272 modulate the expression or phosphorylation levels of IreB. Immunoblot analysis revealed that deletion of OG1RF_11271 and OG1RF_11272 in the ΔireK mutant neither decreased the expression of IreB nor resulted in the aberrant phosphorylation of IreB relative to its expression and phosphorylation in the ΔireK mutant (Fig. S6). Thus, OG1RF_11271 and OG1RF_11272 modulate antimicrobial resistance through a mechanism independent of IreB expression or phosphorylation.
IreK does not control expression of OG1RF_11271 and OG1RF_11272.
To investigate the roles of OG1RF_11271 and OG1RF_11272 in wild-type E. faecalis, we examined the phenotypes of mutants lacking either one or both genes. Deletion of OG1RF_11271 and/or OG1RF_11272 did not alter cholate or lysozyme resistance in WT E. faecalis (Fig. 6A), suggesting that the endogenous levels of these proteins do not interfere with antimicrobial resistance in WT cells. Expression of OG1RF_11271 and OG1RF_11272 from a plasmid in wild-type cells was largely without effect as well. Although there was a modest decrease in lysozyme resistance upon coexpression of OG1RF_11271 and OG1RF_11272, cholate resistance and cell envelope integrity were essentially unaltered (Fig. 6B and C), Hence, the activities of OG1RF_11271 and OG1RF_11272 do not perturb antimicrobial resistance or cell envelope integrity in the presence of functional IreK. The results in Fig. 6 are consistent with a model in which IreK restricts the ability of OG1RF_11271 and OG1RF_11272 to interfere with antimicrobial resistance and envelope integrity (Fig. 7C).
FIG 6.
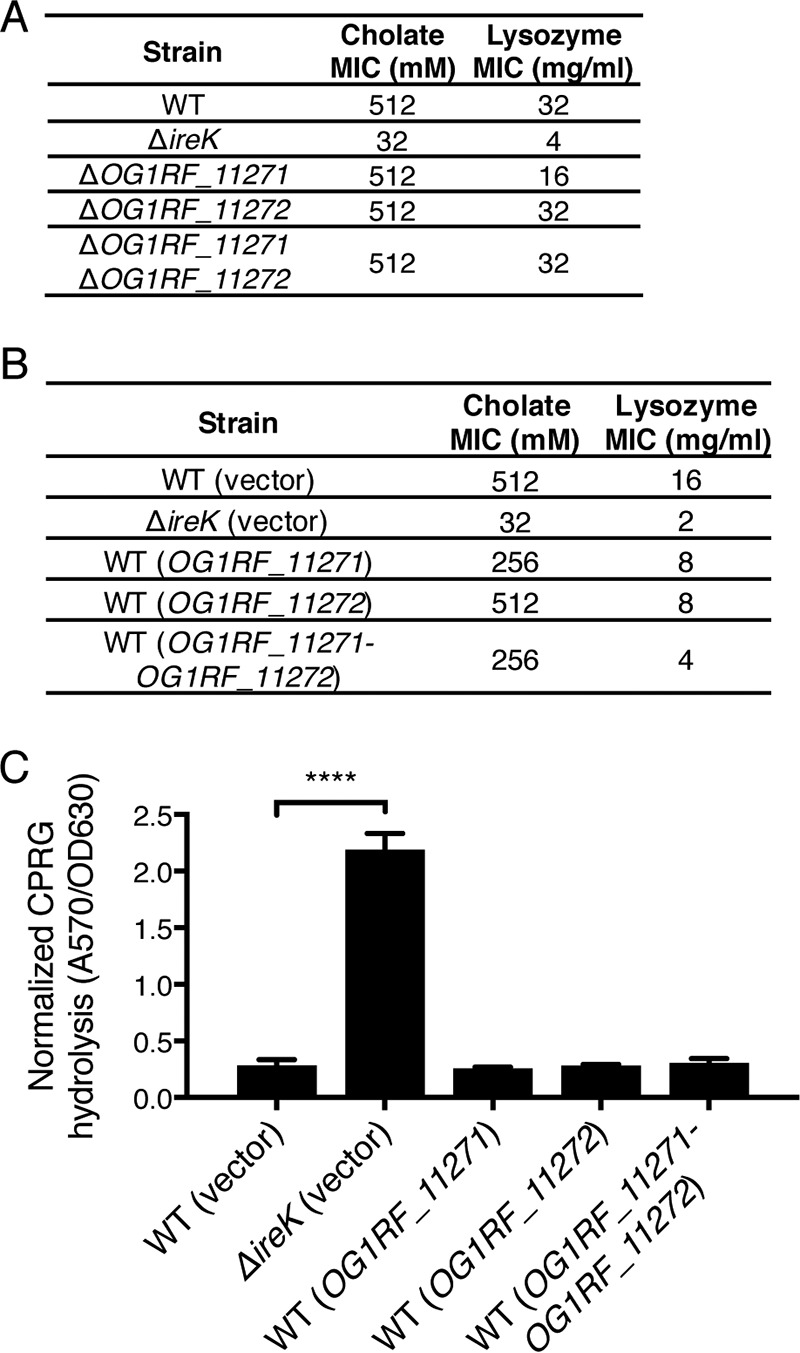
The inhibitory actions of OG1RF_11271 and OG1RF_11272 are limited in the presence of IreK. (A) The cholate and lysozyme resistance of the WT E. faecalis strain (OG1RF) and the ΔireK (CK119), ΔOG1RF_11271 (IB18), ΔOG1RF_11272 (IB19), and ΔOG1RF_11271 ΔOG1RF_11272 (IB20) mutants was determined. The reported MICs represent the median values from three independent biological replicates. (B) The cholate and lysozyme resistance of the WT E. faecalis strain (OG1RF) and the ΔireK mutant (CK119) carrying the empty vector (pJRG9) or expressing wild-type copies of the indicated genes was determined. (C) CPRG hydrolysis was measured for the strains listed in panel B. The reported measurements represent averages from three independent cultures, and error bars represent standard deviations. Statistical significance was evaluated by t test. ****, P < 0.0001.
FIG 7.
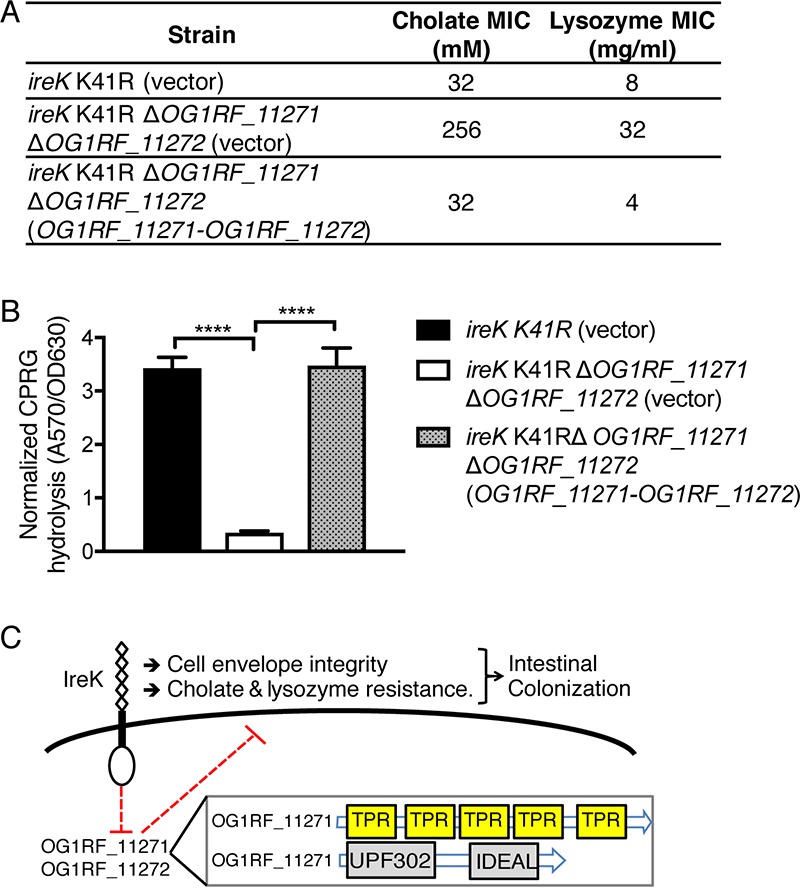
Restriction of OG1RF_11271 and OG1RF_11272 actions require IreK's kinase activity. (A) The cholate and lysozyme resistance of a strain with an ireK K41R mutation (BL102) carrying an empty vector as well as an ireK K41R ΔOG1RF_11271 ΔOG1RF_11272 mutant (IB36) carrying an empty vector or complemented with OG1RF_11271-OG1RF_11272 was determined. The reported MICs represent the median values from three independent biological replicates. (B) CPRG hydrolysis was measured for the strains listed in panel A. The reported measurements represent averages from three independent cultures, and error bars represent standard deviations. Statistical significance was evaluated by t test. ****, P < 0.0001. (C) Proposed model for IreK and OG1RF_11271/OG1RF_11272 modulation of cell envelope integrity, antimicrobial resistance, and colonization. (Inset) A nonscaled depiction of the protein domain architecture for OG1RF_11271 and OG1RF_11272.
To determine whether IreK modulates OG1RF_11271 and OG1RF_11272 expression by regulating their mRNA levels, we assessed the expression of these genes by reverse transcription-quantitative PCR (RT-qPCR). We observed that the expression of these genes does not depend on either IreK or lysozyme stress (Fig. S7). We also found that deletion of ireP (which results in a constitutive activation of IreK [13]) does not alter the levels of OG1RF_11271 or OG1RF_11272 mRNA.
IreK restricts the activity of OG1RF_11271 and OG1RF_11272 via its kinase activity.
To test if the kinase activity of IreK is required for restriction of OG1RF_11271 and OG1RF_11272, we analyzed the enzymatically impaired IreK K41R mutant (13). The K41R mutation impairs the kinase activity of IreK (20) and results in a defect in antimicrobial resistance that phenocopies the ΔireK mutant, although the mutant IreK is expressed and stable. Deletion of OG1RF_11271 and OG1RF_11272 in the IreK K41R background suppressed the defect in cholate and lysozyme resistance, just as was observed in the ΔireK mutant, and this effect could be complemented by expression of OG1RF_11271 and OG1RF_11272 from a plasmid (Fig. 7A). Similar trends were observed when we analyzed cell envelope integrity by the CPRG hydrolysis assay. In the IreK K41R background, deletion of OG1RF_11271-OG1RF_11272 enhanced cell envelope integrity, and this phenotype was reversed by ectopic expression of OG1RF_11271-OG1RF_11272 (Fig. 7B). Collectively, the data indicate that the catalytically impaired IreK K41R mutant is unable to restrict the activity of OG1RF_11271 and OG1RF_11272, highlighting the requirement of IreK's kinase activity for this restriction.
DISCUSSION
An understanding of GIT colonization by enterococci can establish a foundation for the development of new therapies targeting this essential step of pathogenesis. More generally, insights into the mechanisms of enterococcal GIT colonization might reveal principles that help shape the composition of the GIT microbiota. Previous studies showed that the microbiota forms a major barrier to enterococcal GIT colonization. This colonization resistance is achieved directly through competition and indirectly through modulation of innate immune defenses (3, 34, 35), suggesting that nutrient acquisition and resistance to both microbe- and host-produced antimicrobials are important for successful colonization. Indeed, we previously described that E. faecalis can use bacteriocins to compete for a niche in the GIT, confirming the importance of antimicrobials in shaping bacterial communities in the GIT (7).
Genetically distinct clades of E. faecium differ in their ability to colonize the GIT (36), and the ability to colonize the GIT can be transferred among E. faecium strains via conjugation (37), suggesting that specific enterococcal genes modulate GIT colonization. In accordance with this idea, several studies identified specific genes that promote GIT colonization. For example, EpaX, an enzyme involved in the synthesis of the surface polysaccharide Epa was found to be critical for GIT colonization by E. faecalis (4). Both the biofilm transcription regulator EbrB and PtsD, a subunit of an uncharacterized phosphotransferase system, were observed to promote colonization by E. faecium (5, 6). The genes encoding EpaX, EbrB, and PtsD are carried on mobile genetic elements and/or are enriched in infection-causing clinical enterococcal strains relative to commensal strains and are therefore not part of the core enterococcal genome. We speculate that these genes may be especially useful to promote GIT colonization under conditions of dysbiosis, for example, in hospitalized patients undergoing antibiotic therapy. However, enterococci have evolved over millions of years to be GIT commensals; hence, the core enterococcal genome must contain colonization determinants that fundamentally enable enterococci to thrive in the GIT. It is also worth noting that previous studies of enterococcal GIT colonization were performed using antibiotic-treated mice or germfree animals. As a result, we lack an understanding of enterococcal colonization of the unperturbed GIT. Here we exploited our antibiotic-free colonization model to explore the importance of antimicrobial resistance in GIT colonization by E. faecalis.
We found that IreK, a determinant of cell envelope integrity as well as cholate and lysozyme resistance encoded in the core genome of E. faecalis, is required for GIT colonization. However, a defect in neither cholate nor lysozyme resistance is individually sufficient to prevent GIT colonization by E. faecalis. Instead, our results suggest that the colonization defect of the ΔireK mutant is multifactorial, resulting from the loss of some combination of antimicrobial resistance traits and cell envelope integrity. Although it seems likely that the defects in antimicrobial resistance or cell envelope integrity are responsible for the inability of the ΔireK mutant to colonize the GIT, we cannot exclude other as-yet-unknown mechanisms. For example, previous studies implicated PASTA kinases of other bacteria in the regulation of central metabolism (17, 18, 38). Although we do not have evidence that this is the case for IreK, regulation of energy metabolism may be an additional mechanism underlying IreK's contribution to GIT colonization. Nutrient acquisition and energy metabolism are essential for bacterial survival in all environments. At present, the metabolic adaptations that E. faecalis undergoes in the GIT are unknown. Although the ΔireK mutants did not exhibit a growth defect in Mueller-Hinton (MH) broth or brain heart infusion medium (BHI) (the laboratory media used in this study), it remains possible that IreK is involved in metabolic adaptations in the GIT. Future studies will investigate possible links between energy metabolism pathways and IreK.
We found that the intestinal environment can select for ΔireK* suppressor mutants that exhibit enhanced antimicrobial resistance and cell envelope integrity. That recovery of these phenotypes is associated with a recovery in colonization points toward the importance of cell envelope integrity and antimicrobial resistance to GIT colonization. Examination of the ΔireK* suppressor mutants led to the identification OG1RF_11271 and OG1RF_11272, which act antagonistically to IreK. While OG1RF_11272 was sufficient to prevent intestinal colonization and antimicrobial resistance in ΔireK ΔOG1RF_11271 mutants, both OG1RF_11271 and OG1RF_11272 were required to disrupt envelope integrity in ΔireK mutants (Fig. 5). These observations are consistent with a model in which OG1RF_11272 is the primary effector of the processes that disrupt GIT colonization, antimicrobial resistance, and envelope integrity in ΔireK mutants, while OG1RF_11271 enhances the activity of OG1RF_11272. In this model, OG1RF_11272 is sufficient to disrupt antimicrobial resistance and GIT colonization but requires the assistance of OG1RF_11271 to disrupt envelope integrity.
PASTA kinases in bacteria are thought to promote cell envelope homeostasis. For example, some PASTA kinases directly phosphorylate enzymes in the peptidoglycan synthesis pathway (39, 40), and other studies described reduced levels of peptidoglycan precursors and altered expression of peptidoglycan biosynthetic enzymes in mutants lacking PASTA kinases (16, 41). Mutants of E. faecalis lacking IreK or expressing catalytically impaired IreK exhibit a marked loss of cell envelope integrity. Our data suggest that at least part of the role of IreK in maintenance of cell envelope integrity involves restriction of the activity of OG1RF_11271 and OG1RF_11272 in a kinase-dependent manner. Understanding the mechanism of this restriction can therefore help uncover novel ways in which PASTA kinases promote cell wall homeostasis. IreK does not affect the mRNA levels of OG1RF_11271 and OG1RF_11272; therefore, IreK-mediated restriction of OG1RF_11271 and OG1RF_11272 occurs downstream of transcription. IreK might influence the translation or stability of OG1RF_11271 and OG1RF_11272 or even affect their activity by directly phosphorylating them. We also cannot exclude the possibility that IreK and the OG1RF_11271 and OG1RF_11272 pair could modulate cell envelope integrity in opposing ways without interacting in a direct manner.
Homologs of OG1RF_11271 and OG1RF_11272 are conserved in enterococcal species and are found in other Firmicutes, such as Bacillus, Listeria and Streptococcus spp., suggesting that these proteins play an important role in the biology of these organisms. In Bacillus subtilis, YpiB (a homolog of OG1RF_11272) was observed to be induced upon nutrient starvation (42, 43). To our knowledge, no further studies were done to assess the roles of YpiB or YpiA (a homolog of OG1RF_11271) during the starvation response. Our data suggest that OG1RF_11271 and OG1RF_11272 work in concert to interfere with antimicrobial resistance and envelope integrity in E. faecalis mutants lacking IreK (in what is presumably an unregulated state). OG1RF_11272 has two conserved but uncharacterized domains (UPF0302 and IDEAL), while the Simple Modular Architecture Research Tool (SMART) (44) identifies five tetratricopeptide repeats (TPRs; which are known to be involved in protein-protein interactions [45]) in OG1RF_11271. We speculate that these TPRs allow OG1RF_11271 to interact with OG1RF_11272 to execute a common function. OG1RF_11271 and OG1RF_11272 are predicted to be cytoplasmic proteins, as they do not contain transmembrane domains or secretion signal sequences. In E. faecalis, it seems likely that the normal function of OG1RF_11271 and OG1RF_11272 is not to interfere with adaptation to the GIT per se but, rather, that this effect is a consequence of the loss of proper regulation of OG1RF_11271 and OG1RF_11272 that is only relevant in the absence of IreK.
The unregulated activity of OG1RF_11271 and OG1RF_11272 in the ΔireK mutant leads to a cell envelope defect associated with a profound GIT colonization defect in E. faecalis. The mechanisms by which OG1RF_11271 and OG1RF_11272 interfere with envelope integrity and antimicrobial resistance are unknown. Dissecting the molecular details of this phenomenon is of high importance, as its modulation could form the basis of therapies aimed at interfering with E. faecalis colonization.
MATERIALS AND METHODS
Bacterial strains, growth media, and chemicals.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. The oligonucleotides used for plasmid construction and reverse transcription-quantitative PCR (RT-qPCR) were synthesized by Eurofins MWG Operon LLC. All restriction enzymes were purchased from New England BioLabs. Phusion high-fidelity DNA polymerase (Thermo Scientific) was used for all PCRs performed for strain and plasmid construction. The Escherichia coli strains were cultured in LB medium at 30°C with shaking at 200 rpm. The E. faecalis strains were cultured in Mueller-Hinton (MH) broth, prepared per the manufacturer's instruction (Difco). Brain heart infusion medium (BHI) agar (Difco) was prepared as described by the manufacturer. When required, antibiotics were added at the following concentrations: 10 μg/ml (for E. faecalis) or 100 μg/ml (for E. coli) erythromycin (Em), 20 μg/ml (for E. coli) or 10 μg/ml (for E. faecalis) chloramphenicol (Cm), 200 μg/ml Rif, and 500 μg/ml kanamycin (Kan).
Animals.
The Committee for Animal Care and Use at the Medical College of Wisconsin approved all animal-related procedures and experiments. Five-week-old male C57BL/6 mice were obtained from The Jackson Laboratory (RB08 room). Upon arrival, the mice were allowed to adapt to the new environment for 1 week before the start of the experiments. Animals were housed under specific-pathogen-free conditions in the Medical College of Wisconsin vivarium. Experimental sample sizes were determined by appropriate husbandry considerations determined by the Medical College of Wisconsin vivarium, and experiments were repeated as described below. No blinding was performed, and no scheme of randomization was applied when allocating mice for the experiments.
Construction of E. faecalis mutants.
Construction of in-frame deletion mutants of E. faecalis was performed using the previously described markerless allelic exchange strategy (46, 47). Allelic exchange vectors were constructed by amplifying flanking regions of target genes and seamlessly inserting those amplicons into pJH086 (48) or a derivative of pCJK245 (pJH082) by Gibson assembly (49). Deletion alleles retained 2 to 10 codons at the 5′ and 3′ ends to minimize the risk of disrupting the expression of downstream genes by polar effects.
To improve the temperature-sensitive allelic exchange vector for E. faecalis, pCJK245 (47), RepA was analyzed for hydrophobic residues that may be disrupted to generate a more temperature-sensitive phenotype. The method described by Varadarajan and colleagues (50, 51) was used to identify V71G as a candidate substitution in RepA to provide temperature sensitivity. We previously showed that the V71G substitution provides temperature-sensitive replication of a closely related vector (48). Inverse PCR on pCJK245 was used to introduce the V71G substitution into RepA. During sequence confirmation of the V71G substitution, we discovered that the clone of pCJK245 used did not carry the original temperature-sensitive RepA substitutions. Therefore, the resulting plasmid, pJH082, carries a repA allele that encodes only the V71G substitution but otherwise retains the features of pCJK245 (Cmr, lacZ, thyA*). E. coli and E. faecalis carrying pJH082 grew at 30°C but were significantly impaired in growth at 42°C, confirming that the V71G substitution provides temperature sensitivity.
Antibiotic susceptibility determinations.
MICs were determined after 24 h at 37°C with 2-fold serial dilutions of antimicrobial in MH broth. Microtiter plates were inoculated from stationary-phase cultures to a final density of ∼105 CFU/ml, and growth was monitored using a Bioscreen C plate reader. The lowest antibiotic concentration that prevented growth was recorded as the MIC. Plasmid-bearing strains were tested in the presence of chloramphenicol for plasmid maintenance.
Mouse GIT colonization experiments.
Colonization experiments were conducted as previously described (7). Stationary-phase cultures of E. faecalis grown in Rif-supplemented MH broth (Em was also added for plasmid-bearing strains) were washed with sterile water and added to sterile water to a final density of an optical density at 600 nm (OD600) of 0.25; this suspension was fed to mice as drinking water. The persistence of E. faecalis in drinking water was confirmed to remain between 107 and 108 CFU ml−1 over 4 days for all strains tested. E. faecalis-supplemented drinking water was changed every 3 to 4 days to maintain an appropriate inoculum, and mice were allowed to drink ad libitum for 14 days, after which the mice were provided sterile water for the remainder of the experiment. Bacterial loads were determined by plating feces on Rif-supplemented BHI agar (for strain OG1RF and its derivatives) or Kan-supplemented BHI agar (for strain V583 and its derivative) and enumerating viable colonies. Plasmid retention was assessed by enumerating viable colonies growing on Rif- and Em-supplemented BHI agar.
CPRG hydrolysis to assess cell envelope integrity.
Hydrolysis of CPRG was monitored to assess cell envelope integrity, as described previously (26). Bacterial cultures were grown at 37°C to stationary phase in MH broth supplemented with 40 μg/ml CPRG and erythromycin (for the maintenance of pCJK205). CPRG hydrolysis was quantified by measuring the absorbance of cell-free supernatants at 570 nm and normalized to the bacterial density (optical density at 630 nm).
Whole-genome sequencing.
Genomic DNA was obtained from exponential-phase cultures of ΔireK* and OG1RF isolates from colonized mice. Cells were treated with lysozyme (10 mg/ml) and mutanolysin (500 U/ml) at 37°C for 30 min, following which their genomic DNA was isolated using Qiagen genomic tip 100/G columns, according to the manufacturer's instructions. Genome sequencing was performed on an Illumina platform through Genewiz Next Generation Sequencing Services. Sequencing yielded ∼100-fold coverage of the genome. The Illumina paired-end reads were imported into the CLC Genomics Workbench program and mapped to the E. faecalis OG1RF genome (NCBI accession number CP002621). Basic variant detection was performed to identify variants occurring with a ≥90% frequency in read assemblies. Variants were confirmed by Sanger sequencing of PCR amplicons of the corresponding genomic regions derived from the mutants. Three independent ΔireK* suppressor mutant isolates and 12 independent OG1RF isolates recovered from the ileum, cecum, and colon were sequenced.
Phos-tag SDS-PAGE and immunoblotting.
Stationary-phase cultures were diluted in MH broth (supplemented with 10 μg/ml Cm for plasmid maintenance, when needed) and grown to exponential phase at 37°C and 200 rpm (OD600, approximately 0.2). Bacteria were mixed with an equal volume of cold ethanol-acetone (1:1) and collected by centrifugation. Cells were washed with water and normalized on the basis of the OD600 prior to treatment with lysozyme and lysis with SDS Laemmli sample buffer. Without boiling, samples were loaded on 15% acrylamide gels supplemented with or without 30 μM Phos-tag, 60 μM Zn(NO3)2. Gels were run at 200 V in Laemmli's buffer system until the dye front reached the bottom of the gel. After electrophoresis, the gels were soaked in a 5 mM EDTA solution before they were transferred to a nitrocellulose membrane using a Bio-Rad semidry transfer apparatus. RpoA, IreK, and IreB were detected using custom rabbit polyclonal antiserum. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibody was used for detection (Invitrogen). Acrylamide-pendant Phos-tag was from Wako Chemicals.
Gene expression analysis by RT-qPCR.
Stationary-phase cultures were diluted in MH broth and grown to exponential phase at 37°C and 200 rpm (as described above). Cultures were then subjected to either treatment with 50 μg/ml lysozyme or mock treatment for 30 min, after which bacteria were mixed with an equal volume of cold ethanol-acetone (1:1) and collected by centrifugation. RNA was extracted using a total RNeasy kit (Qiagen), and contaminating DNA was degraded with DNase Max (Mo Bio). cDNA was made using an iScript cDNA synthesis kit (Bio-Rad). Quantitative amplification was achieved by using iTaq universal SYBR green supermix (Bio-Rad). Primer efficiencies were determined using serial dilutions of E. faecalis genomic DNA. Calculations of fold changes in gene expression used the Pfaffl method (52) and gyrB as a reference gene.
Accession number(s).
Sequence data have been submitted to GenBank under BioProject no. PRJNA415857.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grant R01 AI081692 and OD006447 from the National Institutes of Health (NIH) to C.J.K. and grant R01 AI116610 to K.L.P., as well as grant R01 GM099526 to N.H.S. This work was also supported by the Children's Research Institute of Children's Hospital of Wisconsin and by the Advancing a Healthier Wisconsin Endowment Research and Education Program to N.H.S. This work was supported in part by American Heart Association Midwest Affiliate Predoctoral Fellowship 16PRE29700011 to I.L.B.
The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00381-17.
REFERENCES
- 1.Shepard BD, Gilmore MS. 2002. Antibiotic-resistant enterococci: the mechanisms and dynamics of drug introduction and resistance. Microbes Infect 4:215–224. doi: 10.1016/S1286-4579(01)01530-1. [DOI] [PubMed] [Google Scholar]
- 2.Donskey CJ, Chowdhry TK, Hecker MT, Hoyen CK, Hanrahan JA, Hujer AM, Hutton-Thomas RA, Whalen CC, Bonomo RA, Rice LB. 2000. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 343:1925–1932. doi: 10.1056/NEJM200012283432604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MRM, Kamboj M, Pamer EG. 2010. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 120:4332–4341. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rigottier-Gois L, Madec C, Navickas A, Matos RC, Akary-Lepage E, Mistou MY, Serror P. 2015. The surface rhamnopolysaccharide Epa of Enterococcus faecalis is a key determinant of intestinal colonization. J Infect Dis 211:62–71. doi: 10.1093/infdis/jiu402. [DOI] [PubMed] [Google Scholar]
- 5.Top J, Paganelli FL, Zhang X, van Schaik W, Leavis HL, van Luit-Asbroek M, van der Poll T, Leendertse M, Bonten MJ, Willems RJ. 2013. The Enterococcus faecium enterococcal biofilm regulator, EbrB, regulates the esp operon and is implicated in biofilm formation and intestinal colonization. PLoS One 8:e65224. doi: 10.1371/journal.pone.0065224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X, Top J, de Been M, Bierschenk D, Rogers M, Leendertse M, Bonten MJ, van der Poll T, Willems RJ, van Schaik W. 2013. Identification of a genetic determinant in clinical Enterococcus faecium strains that contributes to intestinal colonization during antibiotic treatment. J Infect Dis 207:1780–1786. doi: 10.1093/infdis/jit076. [DOI] [PubMed] [Google Scholar]
- 7.Kommineni S, Bretl DJ, Lam V, Chakraborty R, Hayward M, Simpson P, Cao Y, Bousounis P, Kristich CJ, Salzman NH. 2015. Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature 526:719–722. doi: 10.1038/nature15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourgogne A, Garsin DA, Qin X, Singh KV, Sillanpaa J, Yerrapragada S, Ding Y, Dugan-Rocha S, Buhay C, Shen H, Chen G, Williams G, Muzny D, Maadani A, Fox KA, Gioia J, Chen L, Shang Y, Arias CA, Nallapareddy SR, Zhao M, Prakash VP, Chowdhury S, Jiang H, Gibbs RA, Murray BE, Highlander SK, Weinstock GM. 2008. Large scale variation in Enterococcus faecalis illustrated by the genome analysis of strain OG1RF. Genome Biol 9:R110. doi: 10.1186/gb-2008-9-7-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Islam KBMS, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. 2011. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 141:1773–1781. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 10.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. 2010. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, Goodman AL. 2015. Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 347:170–175. doi: 10.1126/science.1260580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kristich CJ, Wells CL, Dunny GM. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc Natl Acad Sci U S A 104:3508–3513. doi: 10.1073/pnas.0608742104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kristich CJ, Little JL, Hall CL, Hoff JS. 2011. Reciprocal regulation of cephalosporin resistance in Enterococcus faecalis. mBio 2:e00199-11. doi: 10.1128/mBio.00199-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pompeo F, Foulquier E, Galinier A. 2016. Impact of serine/threonine protein kinases on the regulation of sporulation in Bacillus subtilis. Front Microbiol 7:568. doi: 10.3389/fmicb.2016.00568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah IM, Laaberki MH, Popham DL, Dworkin J. 2008. A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell 135:486–496. doi: 10.1016/j.cell.2008.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liebeke M, Meyer H, Donat S, Ohlsen K, Lalk M. 2010. A metabolomic view of Staphylococcus aureus and its Ser/Thr kinase and phosphatase deletion mutants: involvement in cell wall biosynthesis. Chem Biol 17:820–830. doi: 10.1016/j.chembiol.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Pietack N, Becher D, Schmidl SR, Saier MH Jr, Hecker M, Commichau FM, Stülke J. 2010. In vitro phosphorylation of key metabolic enzymes from Bacillus subtilis: PrkC phosphorylates enzymes from different branches of basic metabolism. J Mol Microbiol Biotechnol 18:129–140. doi: 10.1159/000308512. [DOI] [PubMed] [Google Scholar]
- 18.Pensinger DA, Boldon KM, Chen GY, Vincent WJ, Sherman K, Xiong M, Schaenzer AJ, Forster ER, Coers J, Striker R, Sauer JD. 2016. The Listeria monocytogenes PASTA kinase PrkA and its substrate YvcK are required for cell wall homeostasis, metabolism, and virulence. PLoS Pathog 12:e1006001. doi: 10.1371/journal.ppat.1006001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira SF, Goss L, Dworkin J. 2011. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol Mol Biol Rev 75:192–212. doi: 10.1128/MMBR.00042-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall CL, Tschannen M, Worthey EA, Kristich CJ. 2013. IreB, a Ser/Thr kinase substrate, influences antimicrobial resistance in Enterococcus faecalis. Antimicrob Agents Chemother 57:6179–6186. doi: 10.1128/AAC.01472-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desbonnet C, Tait-Kamradt A, Garcia-Solache M, Dunman P, Coleman J, Arthur M, Rice LB. 2016. Involvement of the eukaryote-like kinase-phosphatase system and a protein that interacts with penicillin-binding protein 5 in emergence of cephalosporin resistance in cephalosporin-sensitive class A penicillin-binding protein mutants in Enterococcus faecium. mBio 7:e02188-15. doi: 10.1128/mBio.02188-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labbe BD, Kristich CJ. 2017. Growth- and stress-induced PASTA kinase phosphorylation in Enterococcus faecalis. J Bacteriol 199:e00363-17. doi: 10.1128/JB.00363-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bauer TM, Steinbruckner B, Brinkmann FE, Ditzen AK, Schwacha H, Aponte JJ, Pelz K, Kist M, Blum HE. 2001. Small intestinal bacterial overgrowth in patients with cirrhosis: prevalence and relation with spontaneous bacterial peritonitis. Am J Gastroenterol 96:2962–2967. doi: 10.1111/j.1572-0241.2001.04668.x. [DOI] [PubMed] [Google Scholar]
- 24.Lorenzo-Zuniga V, Bartoli R, Planas R, Hofmann AF, Vinado B, Hagey LR, Hernandez JM, Mane J, Alvarez MA, Ausina V, Gassull MA. 2003. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology 37:551–557. doi: 10.1053/jhep.2003.50116. [DOI] [PubMed] [Google Scholar]
- 25.Kristich CJ, Nguyen VT, Le T, Barnes AMT, Grindle S, Dunny GM. 2008. Development and use of an efficient system for random mariner transposon mutagenesis to identify novel genetic determinants of biofilm formation in the core Enterococcus faecalis genome. Appl Environ Microbiol 74:3377–3386. doi: 10.1128/AEM.02665-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Djoric D, Kristich CJ. 2015. Oxidative stress enhances cephalosporin resistance of Enterococcus faecalis through activation of a two-component signaling system. Antimicrob Agents Chemother 59:159–169. doi: 10.1128/AAC.03984-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwata T, Watanabe A, Kusumoto M, Akiba M. 2016. Peptidoglycan acetylation of Campylobacter jejuni is essential for maintaining cell wall integrity and colonization in chicken intestines. Appl Environ Microbiol 82:6284–6290. doi: 10.1128/AEM.02068-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller CA, Autenrieth IB, Peschel A. 2005. Innate defenses of the intestinal epithelial barrier. Cell Mol Life Sci 62:1297–1307. doi: 10.1007/s00018-005-5034-2. [DOI] [PubMed] [Google Scholar]
- 29.Le Jeune A, Torelli R, Sanguinetti M, Giard JC, Hartke A, Auffray Y, Benachour A. 2010. The extracytoplasmic function sigma factor SigV plays a key role in the original model of lysozyme resistance and virulence of Enterococcus faecalis. PLoS One 5:e9658. doi: 10.1371/journal.pone.0009658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varahan S, Iyer VS, Moore WT, Hancock LE. 2013. Eep confers lysozyme resistance to Enterococcus faecalis via the activation of the extracytoplasmic function sigma factor SigV. J Bacteriol 195:3125–3134. doi: 10.1128/JB.00291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comenge Y, Quintiliani R, Li L, Dubost L, Brouard J-P, Hugonnet J-E, Arthur M. 2003. The CroRS two-component regulatory system is required for intrinsic β-lactam resistance in Enterococcus faecalis. J Bacteriol 185:7184–7192. doi: 10.1128/JB.185.24.7184-7192.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hancock L, Perego M. 2002. Two-component signal transduction in Enterococcus faecalis. J Bacteriol 184:5819–5825. doi: 10.1128/JB.184.21.5819-5825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snyder H, Kellogg SL, Skarda LM, Little JL, Kristich CJ. 2014. Nutritional control of antibiotic resistance via an interface between the phosphotransferase system and a two-component signaling system. Antimicrob Agents Chemother 58:957–965. doi: 10.1128/AAC.01919-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, Schnabl B, DeMatteo RP, Pamer EG. 2008. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 455:804–807. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ubeda C, Bucci V, Caballero S, Djukovic A, Toussaint NC, Equinda M, Lipuma L, Ling L, Gobourne A, No D, Taur Y, Jenq RR, van den Brink MRM, Xavier JB, Pamer EG. 2013. Intestinal microbiota containing Barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infect Immun 81:965–973. doi: 10.1128/IAI.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montealegre MC, Singh KV, Murray BE. 2016. Gastrointestinal tract colonization dynamics by different Enterococcus faecium clades. J Infect Dis 213:1914–1922. doi: 10.1093/infdis/jiv597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rice LB, Lakticova V, Carias LL, Rudin S, Hutton R, Marshall SH. 2009. Transferable capacity for gastrointestinal colonization in Enterococcus faecium in a mouse model. J Infect Dis 199:342–349. doi: 10.1086/595986. [DOI] [PubMed] [Google Scholar]
- 38.Ravikumar V, Shi L, Krug K, Derouiche A, Jers C, Cousin C, Kobir A, Mijakovic I, Macek B. 2014. Quantitative phosphoproteome analysis of Bacillus subtilis reveals novel substrates of the kinase PrkC and phosphatase PrpC. Mol Cell Proteomics 13:1965–1978. doi: 10.1074/mcp.M113.035949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Novakova L, Saskova L, Pallova P, Janecek J, Novotna J, Ulrych A, Echenique J, Trombe MC, Branny P. 2005. Characterization of a eukaryotic type serine/threonine protein kinase and protein phosphatase of Streptococcus pneumoniae and identification of kinase substrates. FEBS J 272:1243–1254. doi: 10.1111/j.1742-4658.2005.04560.x. [DOI] [PubMed] [Google Scholar]
- 40.Parikh A, Verma SK, Khan S, Prakash B, Nandicoori VK. 2009. PknB-mediated phosphorylation of a novel substrate, N-acetylglucosamine-1-phosphate uridyltransferase, modulates its acetyltransferase activity. J Mol Biol 386:451–464. doi: 10.1016/j.jmb.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 41.Donat S, Streker K, Schirmeister T, Rakette S, Stehle T, Liebeke M, Lalk M, Ohlsen K. 2009. Transcriptome and functional analysis of the eukaryotic-type serine/threonine kinase PknB in Staphylococcus aureus. J Bacteriol 191:4056–4069. doi: 10.1128/JB.00117-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eymann C, Homuth G, Scharf C, Hecker M. 2002. Bacillus subtilis functional genomics: global characterization of the stringent response by proteome and transcriptome analysis. J Bacteriol 184:2500–2520. doi: 10.1128/JB.184.9.2500-2520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tam LT, Antelmann H, Eymann C, Albrecht D, Bernhardt J, Hecker M. 2006. Proteome signatures for stress and starvation in Bacillus subtilis as revealed by a 2-D gel image color coding approach. Proteomics 6:4565–4585. doi: 10.1002/pmic.200600100. [DOI] [PubMed] [Google Scholar]
- 44.Letunic I, Doerks T, Bork P. 2015. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res 43:D257–D260. doi: 10.1093/nar/gku949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allan RK, Ratajczak T. 2011. Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones 16:353–367. doi: 10.1007/s12192-010-0248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vesić D, Kristich CJ. 2013. A Rex family transcriptional repressor influences H2O2 Accumulation by Enterococcus faecalis. J Bacteriol 195:1815–1824. doi: 10.1128/JB.02135-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kristich CJ, Djorić D, Little JL. 2014. Genetic basis for vancomycin-enhanced cephalosporin susceptibility in vancomycin-resistant enterococci revealed using counterselection with dominant-negative thymidylate synthase. Antimicrob Agents Chemother 58:1556–1564. doi: 10.1128/AAC.02001-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kellogg SL, Little JL, Hoff JS, Kristich CJ. 2017. Requirement of the CroRS two-component system for resistance to cell wall-targeting antimicrobials in Enterococcus faecium. Antimicrob Agents Chemother 61:e02461-16. doi: 10.1128/AAC.02461-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 50.Varadarajan R, Nagarajaram HA, Ramakrishnan C. 1996. A procedure for the prediction of temperature-sensitive mutants of a globular protein based solely on the amino acid sequence. Proc Natl Acad Sci U S A 93:13908–13913. doi: 10.1073/pnas.93.24.13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chakshusmathi G, Mondal K, Lakshmi GS, Singh G, Roy A, Babu Ch R, Madhusudhanan S, Varadarajan R. 2004. Design of temperature-sensitive mutants solely from amino acid sequence. Proc Natl Acad Sci U S A 101:7925–7930. doi: 10.1073/pnas.0402222101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.