


Abstract
Base lesions in DNA can stall the replication machinery or induce mutations if bypassed. Consequently, lesions must be repaired before replication or in a post-replicative process to maintain genomic stability. Base excision repair (BER) is the main pathway for repair of base lesions and is known to be associated with DNA replication, but how BER is organized during replication is unclear. Here we coupled the iPOND (isolation of proteins on nascent DNA) technique with targeted mass-spectrometry analysis, which enabled us to detect all proteins required for BER on nascent DNA and to monitor their spatiotemporal orchestration at replication forks. We demonstrate that XRCC1 and other BER/single-strand break repair (SSBR) proteins are enriched in replisomes in unstressed cells, supporting a cellular capacity of post-replicative BER/SSBR. Importantly, we identify for the first time the DNA glycosylases MYH, UNG2, MPG, NTH1, NEIL1, 2 and 3 on nascent DNA. Our findings suggest that a broad spectrum of DNA base lesions are recognized and repaired by BER in a post-replicative process.
INTRODUCTION
Base lesions are continuously generated in the genome by exposure to exogenous sources as well as endogenously via metabolic processes. The majority of these lesions are repaired by the base excision repair (BER) pathway. In BER, damaged bases are excised by DNA glycosylases resulting in apurinic (AP) sites, which are then cleaved by apurinic endonuclease 1 (APE1) or an AP-lyase activity, leading to single strand breaks (SSBs). At this step, BER converges with the single-strand break repair (SSBR) pathway and can further take two directions depending on several factors such as type of lesion and cell cycle state (reviewed in (1)). Single nucleotide insertion by DNA polymerase (POL) β and nick sealing by DNA ligase (LIG) 3 are referred to as short-patch BER (SP-BER) and is organized by X-ray cross complementing 1 protein (XRCC1). Long-patch BER (LP-BER) encompasses removal of a longer fragment of DNA, which requires several DNA replication factors including proliferating cell nuclear antigen (PCNA), flap endonuclease 1 (FEN1), POLδ/ϵ and LIG1 (reviewed in (2)). SSBs also arise independently of BER by cleavage of the sugar phosphate. SSBs are detected by poly(ADP)-ribose) polymerases (PARP1 or PARP2) and one of the end-processing enzymes APE1, polynucleotide kinase 3′-phosphatase (PNKP) or aprataxin (APTX) cleans up the break before repair is completed by nucleotide insertion and ligation (reviewed in (3)).
BER must recognize and repair DNA lesions at all cell cycle phases, from stages of tightly conserved chromatin to less compact structures such as during transcription and DNA replication. However, it remains unclear how BER operates in these different contexts (reviewed in (4)). There are several reports on association between DNA replication and BER. For instance, post-replicative removal of misincorporated uracil was demonstrated to be executed by uracil-DNA glycosylase 2 (UNG2), the first BER-protein to be identified in replication foci where it directly interacts with PCNA (5). UNG2 expression is cell cycle regulated and reaches maximum in S-phase (6–8). Cell cycle regulated expression and association with PCNA have also been proposed for other DNA glycosylases including Nei-like 1 (NEIL1) (9,10), MutY homolog (MYH) (11,12), Nth homolog 1 (NTH1) (13,14) and 3-methylpurine DNA glycosylase (MPG) (15,16). Of these, UNG2, MYH and MPG (inverted) contain the PCNA interacting peptide (PIP)-box. Like UNG2, MYH is also involved in post-replicative BER, where it removes dAMP misincorporated opposite 8-oxoG lesions (11). Interestingly, the other DNA glycosylases have not been suggested to contribute in post-replicative BER.
Because UNG2 directly interacts with PCNA in replication foci, it was initially suggested that removal of misincorporated uracil occurred through LP-BER (5). Later, it was demonstrated that XRCC1 colocalizes and interacts with both PCNA (17) and UNG2 in replication foci (18), and direct UNG2–XRCC1 interaction suggests that these proteins are part of a common S-phase complex. However, UNG2 and XRCC1 were also shown to be part of functionally distinct S-phase complexes. XRCC1 complexes containing low levels of UNG2 performed efficient BER, while UNG2 complexes containing low levels of XRCC1 were inefficient in the ligation step, even though they contained high levels of LIG1 (18). These results led to proposal of a two-step model of post-replicative repair of uracil: (i) UNG2 attached to PCNA is rapidly removing misincorporated dUMPs and (ii) the remaining AP-site is later repaired by a XRCC1 multiprotein complex (18). However, out of all the proteins present in the XRCC1 and UNG2 complexes, only UNG2 has been verified to act post-replicatively (5). Here we verify the presence of UNG2, XRCC1 and other BER proteins behind active replication forks using the high resolution technique isolation of proteins on nascent DNA (iPOND). Interestingly, the DNA glycosylases MPG, NEIL1-3, MYH and NTH1 were also found on nascent DNA, suggesting that a broad spectrum of base lesions are repaired in a post-replicative process.
MATERIALS AND METHODS
Cell lines and culture
HEK293 (Thermo Fischer, Flp-IN™ T-Rex™ 293, R780-07), HeLa (ATCC CCL-2) and U2OS (ATCC HTB-96) cells were cultured in Dulbecco's modified Eagles medium supplemented with amphotericin B (2.5 μg/ml, Sigma-Aldrich), gentamicin (0.1 mg/ml, Life Technologies), L-glutamine (2 mM, Sigma-Aldrich), fetal bovine serum (10% v/v, Sigma-Aldrich) at 37°C and 5% CO2. For HEK293 cells, culture medium was also supplemented with blasticidin (15 μg/ml, Life Technologies) and hygromycin B (50 μg/ml, Life Technologies). MPG knock out (KO) cell lines were generated using CRISPR technology. Briefly, HEK293 cells were transfected with guide RNA and the one vector system lentiCRISPRv2 developed by Feng Zhang lab (Addgene plasmid #52961) (19) using XtremeGENE HP transfection reagent. The cells were grown for 7 days in medium containing puromycin and single cell cloned. Guide RNAs were designed using the CRISPR Design Tool (20) and had the following 5′ to 3′ sequence GTTGGAGTTCTTCGACCAGC targeting exon 3 in the MPG gene.
iPOND
iPOND was performed essentially as previously described (21,22). Briefly, HEK293 cells (3–4·108 cells per sample) were pulsed with medium containing 5-ethynyl-2′-deoxyuridine (EdU) (10 μM, Life Technologies) for 5–15 min (pulse samples). For pulse-chase experiments, medium containing excess thymidine were added for 30 or 90 min after the EdU pulse (10 μM, Sigma-Aldrich). As a negative control medium containing dimethyl sulfoxide (DMSO) (0.1%) was used. Subsequently, cells were crosslinked for 20 min in phosphate buffered saline (PBS) containing formaldehyde (FA) (0.13, 0.25 and 1%), quenched using glycine (0.125 M) and washed in cold PBS. The following steps were performed as previously described in (22). The eluted proteins were analyzed by immunoblotting or mass spectrometry (MS). For MS we also included a negative control where CuSO4, which catalyses the click-reaction between biotin and streptavidin, was omitted. Prior to MS, proteins captured via iPOND were separated onto a NuPAGE 4–12% Bis-Tris gel (Invitrogen) for 10 min. Protein gel lanes were excised and cut into three pieces and the gel pieces were submitted to trypsin digestions as described below.
Immunoblotting
Proteins were transferred to PVDF-membranes (Immobilon, Millipore) and the membranes were blocked in 5% dry milk in PBS-T before incubation with primary antibodies in 5% dry milk. Antibodies against POLβ (ab3181), POLδ (ab10362, Abcam), XRCC1 (ab1838), LIG1 (ab615), LIG3 (ab587), H3 (ab1791), PCNA (SC-56, Santa-Cruz Biotechnology), FEN1 (A300-256, Bethyl Laboratories) and α-UNG2 (TA503755, OriGene) were used. Membranes were incubated in swine α-rabbit and rabbit α-mouse secondary antibodies (Dako Cytomation) diluted 1:5000 in 1% dry milk and visualized in KODAK Image Station 4000R.
In-gel tryptic digestion
Protein gel lanes were excised and cut into five pieces. Gel pieces were washed in pure H2O followed by washes in NH4HCO3 (25 mM, Sigma-Aldrich) and CH3CN (50%, Thermo Fisher Scientific). Further, gel pieces were dehydrated in CH3CN (100%) before incubation (56°C, 45 min) in NH4HCO3 (25 mM) and DTT (10 mM, Sigma-Aldrich). Subsequently, samples were incubated (RT, 45 min) in NH4HCO3 (25 mM) containing iodoacetamide (55 mM, Sigma-Aldrich), followed by washing (RT, 5 min) in NH4HCO3 (25 mM) and CH3CN (50%) and dehydration in CH3CN (100%). Then, each piece was incubated (ice, 30 min) in 15 μl of trypsin (12.5 ng/ml, Thermo Fisher Scientific) and NH4HCO3 (50 mM). The excess of trypsin was removed and gel pieces were covered with NH4HCO3 (50 mM) and incubated over night at 37°C. The tryptic peptides were extracted the following day by incubating twice in HCOOH (5%, Thermo Fisher Scientific) and CH3CN (50%) followed by incubation in CH3CN (100%). Peptides from gel pieces that originated from the same sample were merged and completely dried in a speed vac. The dried peptides were resuspended in 40 μl of HCOOH (0.1%) prior to MS analysis and equal volumes (10 μl) were injected in the mass spectrometer for shotgun and targeted MS analysis.
Tandem mass spectrometry analysis (Shotgun MS)
Shotgun LC-MS/MS analysis was carried on an EASY-nLC 1000 UHPLC system (Thermo Fisher Scientific) coupled to an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Peptides were injected onto an Acclaim PepMap100 C18 column (75 μm i.d. × 2 cm nanoviper, 3 μm particle size, 100 Å pore size) (Thermo Fisher Scientific) and further separated on an Acclaim PepMap100 C18 analytical column (75 μm i.d. × 50 cm nanoviper, 2 μm particle size, 100 Å pore size) (Thermo Fisher Scientific). The peptides were eluted using a 120 min method that consisted of a 250 nl/min flow rate, starting with 98% Buffer A (0.1% HCOOH in H2O) with an increase to 5% Buffer B (0.1% HCOOH in CH3CN) in 2 min, followed by an increase to 35% Buffer B over 98 min and a rapid increase to 100% Buffer B in 6 min, where it was held for 14 min. The peptides eluting from the column were ionized by a nanospray ESI ion source (Thermo Scientific) operating at 2.0 kV. Precursor ions (m/z 400–2000) were measured in the Orbitrap at a resolution of 120 000 FWHM (at 400 m/z). Up to 20 of the most intense peptides were selected from each MS scan for fragmentation and detected in the linear ion trap at Rapid scan rate using collision induced dissociation with normalized collision energy (NCE) of 35.
Raw data files from the mass spectrometer were processed in Proteome Discoverer version 1.4.0.288 software through the SEQUEST database-search algorithm for samples with various FA concentrations. Raw files from six experiments with 0.25% FA were analyzed by label free quantification (LFQ) using Max Quant v 1.5.3.30 (23) which maps the spectra over the Human proteome, downloaded from Uniprot (24) in March 2016. The following search parameters were used: enzyme specified as trypsin with maximum two missed cleavages allowed; precursor mass tolerance was set to 10 ppm and fragment mass tolerance as 0.5 Da; Cysteine Carbamidomethylation as fixed, while Deamidation of Asparagine/Glutamine, Oxidation of Methionine, Protein N-terminal acetylation and Methylation of Lysine/Arginine as variable modifications. False discovery rate was set to 0.01 (high confidence) and minimum of two peptides with high confidence were used for final protein identification. LFQ algorithm (25) was used to estimate the protein amounts in the sample. These LFQ values were log transformed with base 2. Euclidean distance was calculated between proteins over these values which were further used for hierarchical clustering using average linkage procedure. Proteins quantified in less than four out of six biological replicates were discarded. The remaining missing values were imputed with a normal distribution reflecting the lower end of the overall distribution of values (mean downshifted 1.8 and standard deviation scaled with a factor of 0.3) (26) in order to perform two-tailed Student's t-test between P_90C and 15P (replisome) along with one-way ANOVA to capture differentially expressed proteins over all conditions. Gene enrichment analysis was performed on proteins significantly enriched in replisome using Kyoto Encyclopedia of Genes and Genomes (KEGG) (27) and the Gene Ontology Consortium (GO) (28) resources. The false discovery rate (reported as q-value) was estimated using a permutation-based method (26). Perseus plugins (https://github.com/JurgenCox/perseus-plugins) were employed to carry out these steps.
The MS data from six biological replicates (two batches) have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (29) with the identifiers PXD004288 and PDX004494.
Targeted mass spectrometry analysis
Label-free parallel reaction monitoring (PRM)-based targeted MS was employed for relative quantification of selected proteins. All PRM methods were designed, analyzed and processed using Skyline software version 3.6.0.10493 (30). In silico selection of proteotypic peptides was performed via Skyline using the Homo sapiens reference proteome available at www.uniprot.org to exclude non-unique peptides. Furthermore, frequently modified peptides, such as those containing methionine, and peptides containing consecutive lysines or arginines (e.g. KR, RK, KK or RR) were avoided. The standard peptides—namely, synthetic unlabeled purified peptides (Thermo Fisher Scientific) and tryptic digests from purified recombinant proteins—were first analyzed in FullMS-ddMS2 mode (LC-MS system described below) and the raw data were submitted to protein identification using Proteome Discoverer version 1.4.0.288 through SEQUEST and Mascot database-search algorithms. This data were imported into Skyline and used to build a library where the top ionizing peptides (2+ and 3+ charge states) for each protein were selected for PRM analysis.
For both standard peptides and peptides from iPOND samples, PRM LC-MS/MS analysis (t-MS2 mode) was carried on an EASY-nLC 1000 UHPLC system (Thermo Fisher Scientific) coupled to a Q Exactive mass spectrometer (Thermo Fisher Scientific). Peptides were injected onto an Acclaim PepMap100 C18 column (75 μm i.d. × 2 cm nanoviper, 3 μm particle size, 100 Å pore size) (Thermo Fisher Scientific) and further separated on an Acclaim PepMap100 C18 analytical column (75 μm i.d. × 50 cm nanoviper, 2 μm particle size, 100 Å pore size) (Thermo Fisher Scientific). The peptides were eluted using a 115 min method that consisted of a 250 nl/min flow rate, starting with 98% Buffer A (0.1% HCOOH in H2O) with an increase to 5% Buffer B (0.1% HCOOH in CH3CN) in 2 min, followed by an increase to 35% Buffer B over 98 min and a rapid increase to 100% Buffer B in 6 min, where it was held for 9 min. The peptides eluting from the column were ionized by a nanospray ESI ion source (Thermo Scientific) operating at 1.9 kV and analyzed in positive-ion mode using HCD fragmentation. Each MS/MS scan was acquired at a resolution of 35 000 FWHM (at 200 m/z), NCE 28, automatic gain control target value of 2 × 105, maximum injection time of 120 ms and isolation window 2 m/z.
The standard peptides were analyzed first and information on retention time (RT) and fragmentation pattern of the top 2–6 ionizing tryptic standard peptides (2+ or 3+ charge states) for each protein were used to build a scheduled method. Because this is a label-free strategy, wide RT windows of up to 15 min were employed to avoid missing data from peptides showing RT drifts. The scheduled method was then used to detect the corresponding endogenous peptides in the iPOND samples. The raw data from five biological replicates have been deposited to the the ProteomeXchange Consortium via the PeptideAtlas repository (31) with the identifier PASS01008 (http://www.peptideatlas.org/PASS/PASS01008). For relative quantification of a protein in the different samples, we summed the total peak areas for all detected peptides of that protein in each sample. Further, the data from the different experiments were normalized by dividing the summed peptide area (i) with a factor k for each protein:
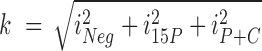 |
Finally, the average normalized peak area and sample standard deviation was calculated based on at least three independent replicates. A two-sided paired Student's t-test was used to identify significant differences between levels in pulse and pulse-chase samples.
Fluorescence protein expression, immunofluorescence staining and imaging
HeLa and U2OS were fixed in paraformaldehyde (2%) and permeabilized in ice-cold methanol. The cells were blocked in bovine serum albumin (BSA)/PBS (2%) prior to incubation with primary antibodies against PCNA (ab18197) and XRCC1 (ab1838) overnight at 4°C. Samples were washed in PBS and stained with Alexa fluor 647 goat α-rabbit and Alexa fluor 532 goat α-mouse (Life Technologies) diluted 1:1000 in BSA/PBS (2%) for 1 h at RT. Samples were washed and maintained in PBS. Images were captured on a Leica SP8 STED 3× confocal microscope using a 100×/1.4 oil immersion objective and the 660 and 775 nm depletion lasers. Cloning of expression construct pECFP-PCNA is previously described (32) and pMPG-EYFP was a gift from Dr Luisa Luna (Department of Microbiology, Oslo University Hospital, Rikshospitalet, Norway). HEK293 cells were transiently transfected with pECFP-PCNA and pMPG-EYFP using XtremeGENE HP transfection reagent and analyzed by live-cell imaging. The images were captured using a Zeiss LSM 510 Meta laser scanning microscope equipped with a 63×/1.4 oil immersion objective. cyano fluorescent protein (CFP) and yellow fluorescent protein (YFP) were excited and detected as previously described (22).
Cell cycle analysis
HEK293 wild-type (WT) and MPG KO cells were treated for 1 h with methyl methanesulfonate (MMS), before they were released into fresh medium and harvested after 6, 12, 24 and 48 h. Cells were fixed in ice-cold methanol, washed with PBS, RNAseA-treated (100 μg/ml in PBS, 37°C, 30 min) and DNA stained with propidium iodide (50 μg/ml in PBS). DNA staining was quantified using a FACS Canto flow cytometer (BD-Life Science) and FlowJo software.
RESULTS
iPOND data suggest post-replicative BER and SSBR
Post-replicative processes are complex and involve many different proteins that are dynamically loaded onto nascent DNA. In order to verify the post-replicative presence of BER proteins we applied the high resolution technique iPOND, developed by Sirbu et al. (21,33). This strategy enabled the isolation of DNA fragments containing newly incorporated EdU and associated proteins from cells pulsed with EdU for various lengths (0–15 min). To identify changes in protein assembly when the EdU segments are distanced from the forks, a chase sample where the EdU pulse was followed by thymidine was included in the analysis. Western blot analysis revealed a clear enrichment of POLδ, LIG1, LIG3, XRCC1, POLβ, FEN1 and UNG2 in EdU pulse samples compared to the chase sample (Figure 1). PCNA was used as a positive control for proteins associated with the replisome and increased as expected in pulse samples with time. Histone H3, known to load further from the replication fork (33), was further enriched in the chase sample. The recruitment pattern of the BER proteins was consistent with the positive control, PCNA.
Figure 1.

Immunoblot of BER proteins captured by iPOND. HEK293 cells were pulsed with EdU for the indicated lengths and the negative control was mock-treated with DMSO. One sample was followed by a thymidine chase for additional 30 min after the EdU-pulse. The cells were crosslinked in formaldehyde (FA) (1%) before harvesting. The input samples contain protein of the cell lysate (100 μg, 0.13%). The capture samples contain protein from nascent DNA. All data are derived from the same biological replicate and from one gel.
These results demonstrate that BER proteins are present at unperturbed replication forks. The presence of XRCC1 in replication foci in absence of PCNA overexpression and replicative stress has been discussed (34,35). EdU labeling has also been reported to stress cells after extended incubation times (36). In order to examine if endogenous XRCC1 and PCNA colocalize in unstressed U2OS and HeLa cells we used high resolution fluorescence microscopy. Indeed, XRCC1 colocalize in PCNA spots resembling replication foci in untreated cells (Figure 2). White spots in the merged pictures appear only when the fluorescence from both antibodies have equal intensity, however similar staining patterns are more pronounced than clear white spots. Colocalization of PCNA and XRCC1 in replication foci is in accordance with our iPOND data and previous data using overexpressed proteins (17,18,37,38). Collectively, these results support that post-replicative BER/SSBR organized by XRCC1 occurs during normal, unperturbed DNA replication.
Figure 2.

Immunofluorescence of endogenous PCNA and XRCC1. U2OS (upper row) and HeLa (lower row) cells were immunostained with PCNA (left panel) and XRCC1 (mid panel) specific antibodies and analyzed by stimulated emission depletion (STED) microscopy. Merged pictures are shown in the right panel. Zoomed regions are shown in inserted boxes. Bars = 5 μm.
iPOND-MS detects additional BER/SSBR proteins on nascent DNA
Immunoblotting requires specific and high affinity antibodies and is a semi-quantitative low resolution technique compared to MS. To verify the results from immunoblotting and potentially detect additional BER/SSBR proteins, we coupled iPOND with high resolution shotgun orbitrap tandem MS (shotgun MS), further referred to as iPOND-MS. Excessive FA crosslinking might interfere with protein detection on MS and could also result in capture of proteins not directly bound to the EdU segment. We therefore tested the effect of lower FA concentrations during iPOND. By decreasing the FA concentration from 1 to 0.25 or 0.13%, the detection of proteins of interest by shotgun MS was markedly improved (Supplementary Figure S1). For further experiments 0.25% FA was used. However, considerable variation in protein score was observed in biological replicates using the same FA concentration. These variations could be due to differences in the replicative potential of the cells, the number of cells used in the experiments, degree of sonication, efficacy of crosslinking and reversal, efficacy of trypsin digestion and MS-performance. We therefore performed LFQ which defines sample specific coefficients for normalization using a protein-abundance profile across samples. Hence, this method is more robust to address variations across samples in terms of quantifiable peptide amounts (25).
When analyzing the proteins enriched in replisome, the BER pathway came up as one of the most significant pathways using the KEGG database or the GO resource (Figure 3A). Many proteins involved in both replication and BER, including POLδ, POLϵ, LIG1, FEN1 and the two BER/SSBR proteins LIG3 and XRCC1 were found to be enriched in replisome in our experiments (Figure 3A, see also Supplementary Table S1). In addition, APE1, MPG, PNKP, PARP1 and PARP2 were detected on nascent DNA, but not significantly enriched in replisome (Figure 3B, see also Supplementary Table S2). However, the trends in the distribution of selected replication and BER/SSBR proteins illustrated in Figure 3C indicated that PARP2 and PNKP were more abundant in pulse samples compared to chase samples. PARP1, MPG and APE1 had increased abundance in the chase samples (Figure 3C). A considerable part of the replisome enriched proteins found in our study overlapped with replisome proteins detected in previous iPOND-MS studies (39–41) (Supplementary Figure S2A and B) and were classified within the same pathways (Supplementary Figure S2C). However, XRCC1 and LIG3 were only detected in replisomes in one of these studies (41). XRCC1, LIG3, APE1 and UNG have been detected in pulse samples in two or three replica out of four replicates by a fourth iPOND-MS study performed on mouse embryonic stem cells, but no statistical analysis was reported (42).
Figure 3.

BER proteins monitored at nascent DNA by shotgun MS. (A) Pathways or processes from KEGG or GO resources overlapping with proteins enriched in 15 min pulse sample compared to 90 min chase sample (replisome) using Students t-test (P < 0.05). The log transformed FDR q-value of the most significant KEGG pathways are presented and the corresponding GO biological process is plotted in comparison. (B) Heat map and average linkage hierarchical clustering of protein levels in iPOND samples based on log2 transformed LFQ intensities. Histones and proteins related to BER in the KEGG and GO database were included in the map. The sample name is indicated on top, where Neg (Cu) = negative control without copper (no EdU-biotin click reaction), Neg = negative control (DMSO), 5–15P = 5–15 min of EdU pulse, P+30-90C = 15 min of EdU pulse + 30–90 min of thymidine chase. Each column represents one biological replicate and a total of six replicates are presented per sample. (C) Median LFQ intensities for selected histones, replication and BER/SSBR proteins Z-scaled across negative, pulse and chase samples, which illustrates the trends in distribution of the proteins on nascent DNA close and distant from the replication fork.
Targeted proteomics identifies the DNA glycosylases MYH, NEIL1-3 and NTH1 on nascent DNA
The proteins UNG2 and POLβ were not detected by shotgun MS, even though they were clearly visible in the immunoblots (Figure 1) and previous functional studies have verified UNG2 to have a post-replicative role (5). Since the detection limit of shotgun LC-MS is dependent on peptide abundance, relative abundance among co-eluting peptides and peptide ionizability, which is defined by factors such as charge, degree of hydrophobicity and length of peptide; we decided to employ a highly selective and sensitive MS approach named PRM. In this targeted strategy, the RT and fragmentation pattern of synthetic standard peptides is used to identify the corresponding peptides in the iPOND samples. Whereas in the shotgun approach, the instrument is set to detect the maximum number of peptides on a given sample with a bias toward high abundance species, the targeted strategy allows the exclusive detection of pre-selected peptides belonging to a protein of interest (reviewed in (43)). Due to the high selectivity and sensitivity of this approach, the detection of low abundance peptides is markedly improved compared to shotgun MS (44). In addition to the core BER/SSBR proteins, we included additional DNA glycosylases in our search. Because there were no considerable differences between 30 and 90 min chase samples in iPOND-MS analysis we analyzed only the 30 min chase sample in the targeted approach, named iPOND-PRM. Peptide peak areas were used for relative quantification of proteins in the different samples (a list of peptides used for quantification and corresponding peak area values is available in Supplementary Table S3).
iPOND-PRM verified that PCNA, POLδ, POLϵ, FEN1, LIG1, LIG3 and XRCC1 are enriched in replisome, while APE1 was significantly enriched in chase samples (Figure 4). MPG and NEIL1 were detected in pulse and chase samples, but they were not specifically enriched in replisomes (Figure 4), i.e. there is no significant difference in protein level in pulse and chase samples. NTH1 was the only glycosylase significantly enriched in the chase sample. NEIL3 and MYH were significantly enriched in pulse samples, suggesting association with the replisome. Interestingly, we found that UNG2, POLβ and NEIL2 were enriched in pulse samples, although only in two experiments (Figure 4). As mentioned before, UNG2 and POLβ were not detected by iPOND-MS although they were visibly enriched in pulse samples by immunoblotting (Figure 1), which indicate that these proteins are of low abundance compared to the rest of the protein pool, but also that these peptides could carry modifications that interfered with their detection. Nevertheless, the iPOND-PRM data verified that UNG2 and POLβ are associated with the replisome. No peptides were detected for OGG1, suggesting that it is not present on nascent DNA. Notably, MPG, NTH1 and NEIL1, which were not enriched in replisomes, were detected in samples pulsed with EdU for 5 min (5P), suggesting that they are loaded rapidly after DNA replication (data not shown). However, because some of the proteins were detected in only three replicates, proper quantification of the proteins was hampered in the 5P samples and the data were therefore not included.
Figure 4.

Replication and BER/SSBR proteins detected by iPOND-PRM. Bars represent normalized peptide peak areas of replisome enriched (light gray), chromatin enriched (dark gray) and proteins with no enrichment (mid gray). Mean ± SD are shown for PCNA, POLδ, POLϵ, FEN1, LIG1, LIG3, XRCC1, APE1, MPG, NEIL1, NTH1 (all n = 5), NEIL3 (n = 4) and MYH (n = 3). For UNG2, POLβ and NEIL2, each biological replicate is illustrated. Statistical significance was calculated using a two-sided paired Student's t-test, where *: P < 0.05, **: P < 0.01, n.s: not significant. The FA concentration used for iPOND was 0.25%.
Altogether, immunoblotting, iPOND-MS and iPOND-PRM demonstrate that XRCC1 and core BER/SSBR proteins, are enriched on unperturbed replication forks, likely performing post-replicative repair when required. Importantly, the DNA glycosylases MYH, NEIL1-3, NTH1, UNG2 and MPG were detected on nascent DNA, suggesting a role in post-replicative repair.
MPG colocalizes with PCNA and is important for S-phase progression after DNA damage
Identification of MPG on nascent DNA prompted us to further investigate its role in S-phase. In absence of specific antibodies, we overexpressed MPG-EYFP and ECFP-PCNA in HeLa cells in order to examine intracellular localization of MPG. We observed that MPG colocalized with PCNA in foci resembling replication foci supporting a role during replication (Figure 5A), which is in agreement with the suggested MPG–PCNA interaction (16). However, MPG-EYFP was also detected in other foci/regions with no or low levels of PCNA. The exact biological nature of these foci, if not an artefact of overexpression, is not known. Next, we generated MPG KO HEK293 cells (Figure 5B) and examined if lack of MPG affected the cells progression through S-phase. In absence of DNA damage, these cells grow similarly to the WT cells (Figure 5C, upper panel). However, when the cells are treated with MMS, the KO cells were delayed in entering S-phase compared to the WT cells (Figure 5C, lower panel, 6 and 12 h). Additionally, the KO cells used twice as long time from early S to late S compared to the WT cells (see 6–12 h for WT and 12–24 h for KO). Together these results support the presence of MPG at sites of replication and a role for MPG for repair of alkylation damage in S-phase. Whether its function is both in front of the fork and post-replicative, as suggested by the iPOND data, remains to be elucidated.
Figure 5.

Localization of MPG-EYFP in replication foci and the effect of MPG on S-phase progression upon DNA alkylation damage. (A) Colocalization of MPG-EYFP and ECFP-PCNA in HeLa cells. Bars = 5 μm. (B) Immunoblot validation of MPG knock out cells generated by CRISPR technology. (C) Cell cycle distributions of HEK293 cells, WT and MPG KO cells. For treated samples, cells were incubated with MMS (1 mM) for 1 h and released into fresh medium. Cells were harvested 6, 12, 24 and 48 h after treatment and stained with PI before flow cytometric analysis. Each cell cycle histograms illustrate the relative cell number at various levels of DNA stain.
DISCUSSION
Several lines of evidence support the existence of XRCC1 multiprotein complexes, and XRCC1 has previously been demonstrated to interact with several of the BER proteins that we identified on nascent DNA in this study (reviewed in (45)). APE1 was the only protein essential for BER that was not enriched at replication forks. However, substantial amounts of APE1 were present in samples pulsed with EdU for only 5 min suggesting that APE1 is rapidly recruited to replisomes for post-replicative BER. Indeed, APE1 has previously been detected in XRCC1 and UNG2 complexes isolated from S-phase cells (18).
Enhanced expression of XRCC1 is found in S/G2 (46), and previous data have demonstrated colocalization of PNKP, POLβ, XRCC1 and PCNA in replication foci (37). Data from iPOND and immunofluorescence experiments presented here, together with previous data of SP-BER competent XRCC1 complexes isolated from S-phase cells support a model where post-replicative XRCC1 complexes are present, independent of DNA damage, ready to act whenever an AP-site or SSB is generated (18). In accordance with this, POLβ was recently identified on chromatin undergoing replication (47). XRCC1 has previously been linked to replication associated repair in the presence of DNA damage, via interactions with the C-terminal domain of REV1 (48) and via interaction with the p58 subunit of the POL α-primase complex (49).
Various SSB/BER intermediates are continuously generated endogenously, and several proteins that sense strand breaks and modify SSB termini interact with XRCC1 (45). In support of a post-replicative XRCC1 SP–BER complex, the XRCC1 interacting end-processor, PNKP, as well as the SSB-sensors PARP1 and PARP2 were detected on nascent DNA. PARP1 has been suggested to regulate fork speed after DNA-damage (50), a function also required for coping with endogenous damage. Both PARP1 and PARP2 contain the AlkB homolog 2 PCNA-interacting motif (51), thus their presence in replisome could also be due to a direct interaction with PCNA.
Interestingly, we detected several DNA glycosylases on nascent DNA, which suggest a role for these glycosylases in post-replicative processes, including BER. UNG2 and MYH are known to remove misincorporated dUTP and dATP, respectively, in a post-replicative process (5,11), which is in agreement with the data presented here. Several findings have also previously linked both NEIL1 and NEIL3 to replication-associated repair (52–55). NEIL1 has been suggested to bind oxidative lesions in single stranded DNA/RPA pre-replicatively (52) and similarly to UNG2, NEIL1 is present in multiprotein complexes containing various BER and replication proteins (53). NEIL3 expression is upregulated in S-phase (54) and recombinant NEIL3 colocalizes with RPA in replication foci (55). NEIL3 is important for cell proliferation and is the main activity removing the oxidative lesions spiroiminodihydantoin (Sp) and guanidinohydantoin (Gh) in single stranded DNA (56,57). Our data support a role for NEIL1 and NEIL3 in replication-associated repair and their presence on nascent DNA suggest that oxidized lesions can be repaired post-replicatively. Presence of NTH1 on nascent DNA further supports this. Notably, NEIL3 is the only one of these that is enriched in replisomes, suggesting it is mainly acting close to the replication fork. Surprisingly, in two experiments we also detected NEIL2 in replisomes. NEIL2 has been reported to have a role in transcription-coupled repair rather than replication-associated repair (58). Thus, NEIL2s presence and function in replisomes need to be verified by additional experimental approaches. In contrast, OGG1 was not detected on nascent DNA, which is in agreement with its cell cycle independent expression (11). NEIL1, NTH1 and MPG seem to bind to nascent DNA, but do not dissociate which could indicate that their function does not require association with the replisome. Nevertheless, they are already detected in 5P samples suggesting that they are loaded early after DNA replication and despite specific enrichments; they could potentially participate in post-replicative repair.
MPG is the major enzyme for removing hypoxanthine (59), which can be found in the DNA due to misincorporation of dITP by DNA polymerases (60). Excision of hypoxanthine by MPG is enhanced by interaction with PCNA and APE1 (16), hence, it is likely that MPG is involved in post-replicative removal of misincorporated dITP. Additionally, MPG as well as the other DNA glycosylases could be involved in repair of base lesions bypassed by translesion synthesis. Here we demonstrate that MPG KO HEK293 cells are delayed in S-phase after MMS treatment, suggesting that base lesions induced by MMS are bypassed, but MPG is required for efficient post-replicative repair of these bases. It could be argued that DNA glycosylases are recruited to replication forks because EdU is recognized as a base lesion. However, it is unlikely that this applies for all the detected DNA glycosylases since they differ in specificity. Also, for several glycosylases, we found specific enrichments in pulse or chase samples, which argue against EdU-mediated recruitment.
In summary, our data provide new insight into the dynamics of BER proteins on nascent DNA and this is to our knowledge the first time DNA-glycosylases have been identified in replisomes. Importantly, we identify core BER/SSBR proteins on nascent DNA, verifying SP-BER is involved in post-replicative repair. We also demonstrate that iPOND coupled with targeted MS greatly improved the identification and quantification of low abundance proteins as compared to iPOND coupled with shotgun MS.
ACCESSION NUMBERS
The shotgun MS data from six biological replicates (two batches) have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the identifiers PXD004288 and PDX004494. The targeted MS data have been deposited to the the ProteomeXchange Consortium via the PeptideAtlas repository with the identifier PASS01008 (http://www.peptideatlas.org/PASS/PASS01008).
Supplementary Material
ACKNOWLEDGEMENTS
The microscopy and MS analyses were provided by the Cellular and Molecular Imaging Core Facility (CMIC) and the Proteomic and Metabolomics Core Facility (PROMEC), NTNU, respectively. CMIC and PROMEC are funded by the Faculty of Medicine and Health Science, NTNU and Central Norway Regional Health Authority. We also would like to thank Nina-Beate Liabakk for technical assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Norwegian University of Science and Technology (NTNU); Trondheim, Norway; Cancer Fund at St Olavs Hospital, Trondheim, Norway; Joint Research Committee between St. Olavs Hospital and Faculty of Medicine and Health Science, NTNU. Funding for open access charge: Joint Research Committee between St. Olavs Hospital and Faculty of Medicine and Health Science, NTNU.
Conflict of interest statement. None declared.
REFERENCES
- 1. Fortini P., Dogliotti E.. Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair (Amst). 2007; 6:398–409. [DOI] [PubMed] [Google Scholar]
- 2. Krokan H.E., Bjoras M.. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013; 5:a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caldecott K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008; 9:619–631. [DOI] [PubMed] [Google Scholar]
- 4. Branzei D., Foiani M.. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008; 9:297–308. [DOI] [PubMed] [Google Scholar]
- 5. Otterlei M., Warbrick E., Nagelhus T.A., Haug T., Slupphaug G., Akbari M., Aas P.A., Steinsbekk K., Bakke O., Krokan H.E.. Post-replicative base excision repair in replication foci. EMBO J. 1999; 18:3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haug T., Skorpen F., Aas P.A., Malm V., Skjelbred C., Krokan H.E.. Regulation of expression of nuclear and mitochondrial forms of human uracil-DNA glycosylase. Nucleic Acids Res. 1998; 26:1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Slupphaug G., Olsen L.C., Helland D., Aasland R., Krokan H.E.. Cell cycle regulation and in vitro hybrid arrest analysis of the major human uracil-DNA glycosylase. Nucleic Acids Res. 1991; 19:5131–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hagen L., Kavli B., Sousa M.M., Torseth K., Liabakk N.B., Sundheim O., Pena-Diaz J., Otterlei M., Horning O., Jensen O.N. et al. . Cell cycle-specific UNG2 phosphorylations regulate protein turnover, activity and association with RPA. EMBO J. 2008; 27:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hazra T.K., Izumi T., Boldogh I., Imhoff B., Kow Y.W., Jaruga P., Dizdaroglu M., Mitra S.. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dou H., Theriot C.A., Das A., Hegde M.L., Matsumoto Y., Boldogh I., Hazra T.K., Bhakat K.K., Mitra S.. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008; 283:3130–3140. [DOI] [PubMed] [Google Scholar]
- 11. Boldogh I., Milligan D., Lee M.S., Bassett H., Lloyd R.S., McCullough A.K.. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 2001; 29:2802–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parker A., Gu Y., Mahoney W., Lee S.H., Singh K.K., Lu A.L.. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem. 2001; 276:5547–5555. [DOI] [PubMed] [Google Scholar]
- 13. Luna L., Bjoras M., Hoff E., Rognes T., Seeberg E.. Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res. 2000; 460:95–104. [DOI] [PubMed] [Google Scholar]
- 14. Oyama M., Wakasugi M., Hama T., Hashidume H., Iwakami Y., Imai R., Hoshino S., Morioka H., Ishigaki Y., Nikaido O. et al. . Human NTH1 physically interacts with p53 and proliferating cell nuclear antigen. Biochem. Biophys. Res. Commun. 2004; 321:183–191. [DOI] [PubMed] [Google Scholar]
- 15. Bouziane M., Miao F., Bates S.E., Somsouk L., Sang B.C., Denissenko M., O’Connor T.R.. Promoter structure and cell cycle dependent expression of the human methylpurine-DNA glycosylase gene. Mutat. Res. 2000; 461:15–29. [DOI] [PubMed] [Google Scholar]
- 16. Xia L., Zheng L., Lee H.W., Bates S.E., Federico L., Shen B., O’Connor T.R.. Human 3-methyladenine-DNA glycosylase: effect of sequence context on excision, association with PCNA, and stimulation by AP endonuclease. J. Mol. Biol. 2005; 346:1259–1274. [DOI] [PubMed] [Google Scholar]
- 17. Fan J., Otterlei M., Wong H.K., Tomkinson A.E., Wilson D.M. 3rd. XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 2004; 32:2193–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Akbari M., Solvang-Garten K., Hanssen-Bauer A., Lieske N.V., Pettersen H.S., Pettersen G.K., Wilson D.M. 3rd, Krokan H.E., Otterlei M.. Direct interaction between XRCC1 and UNG2 facilitates rapid repair of uracil in DNA by XRCC1 complexes. DNA Repair (Amst). 2010; 9:785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanjana N.E., Shalem O., Zhang F.. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014; 11:783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. et al. . DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013; 31:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sirbu B.M., Couch F.B., Cortez D.. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 2012; 7:594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gilljam K.M., Muller R., Liabakk N.B., Otterlei M.. Nucleotide excision repair is associated with the replisome and its efficiency depends on a direct interaction between XPA and PCNA. PLoS One. 2012; 7:e49199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 24. Boutet E., Lieberherr D., Tognolli M., Schneider M., Bansal P., Bridge A.J., Poux S., Bougueleret L., Xenarios I.. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: how to use the entry view. Methods Mol. Biol. 2016; 1374:23–54. [DOI] [PubMed] [Google Scholar]
- 25. Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M.. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014; 13:2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deeb S.J., D'Souza R.C., Cox J., Schmidt-Supprian M., Mann M.. Super-SILAC allows classification of diffuse large B-cell lymphoma subtypes by their protein expression profiles. Mol. Cell. Proteomics. 2012; 11:77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanehisa M., Goto S.. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gene Ontology Consortium Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015; 43:D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vizcaino J.A., Csordas A., del-Toro N., Dianes J.A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T. et al. . 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016; 44:D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. MacLean B., Tomazela D.M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D.L., Liebler D.C., MacCoss M.J.. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010; 26:966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farrah T., Deutsch E.W., Kreisberg R., Sun Z., Campbell D.S., Mendoza L., Kusebauch U., Brusniak M.Y., Huttenhain R., Schiess R. et al. . PASSEL: the PeptideAtlas SRMexperiment library. Proteomics. 2012; 12:1170–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aas P.A., Otterlei M., Falnes P.O., Vagbo C.B., Skorpen F., Akbari M., Sundheim O., Bjoras M., Slupphaug G., Seeberg E. et al. . Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003; 421:859–863. [DOI] [PubMed] [Google Scholar]
- 33. Sirbu B.M., Couch F.B., Feigerle J.T., Bhaskara S., Hiebert S.W., Cortez D.. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011; 25:1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Breslin C., Hornyak P., Ridley A., Rulten S.L., Hanzlikova H., Oliver A.W., Caldecott K.W.. The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 2015; 43:6934–6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ying S., Chen Z., Medhurst A.L., Neal J.A., Bao Z., Mortusewicz O., McGouran J., Song X., Shen H., Hamdy F.C. et al. . DNA-PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 2016; 76:1078–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kohlmeier F., Maya-Mendoza A., Jackson D.A.. EdU induces DNA damage response and cell death in mESC in culture. Chromosome Res. 2013; 21:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanssen-Bauer A., Solvang-Garten K., Sundheim O., Pena-Diaz J., Andersen S., Slupphaug G., Krokan H.E., Wilson D.M. III, Akbari M., Otterlei M.. XRCC1 coordinates disparate responses and multiprotein repair complexes depending on the nature and context of the DNA damage. Environ. Mol. Mutagen. 2011; 52:623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hanssen-Bauer A., Solvang-Garten K., Gilljam K.M., Torseth K., Wilson D.M. 3rd, Akbari M., Otterlei M.. The region of XRCC1 which harbours the three most common nonsynonymous polymorphic variants, is essential for the scaffolding function of XRCC1. DNA Repair (Amst). 2012; 11:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lopez-Contreras A.J., Ruppen I., Nieto-Soler M., Murga M., Rodriguez-Acebes S., Remeseiro S., Rodrigo-Perez S., Rojas A.M., Mendez J., Munoz J. et al. . A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 2013; 3:1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sirbu B.M., McDonald W.H., Dungrawala H., Badu-Nkansah A., Kavanaugh G.M., Chen Y., Tabb D.L., Cortez D.. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 2013; 288:31458–31467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dungrawala H., Rose K.L., Bhat K.P., Mohni K.N., Glick G.G., Couch F.B., Cortez D.. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell. 2015; 59:998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aranda S., Rutishauser D., Ernfors P.. Identification of a large protein network involved in epigenetic transmission in replicating DNA of embryonic stem cells. Nucleic Acids Res. 2014; 42:6972–6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rauniyar N. Parallel reaction monitoring: a targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int. J. Mol. Sci. 2015; 16:28566–28581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peterson A.C., Russell J.D., Bailey D.J., Westphall M.S., Coon J.J.. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics. 2012; 11:1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hanssen-Bauer A., Solvang-Garten K., Akbari M., Otterlei M.. X-ray repair cross complementing protein 1 in base excision repair. Int. J. Mol. Sci. 2012; 13:17210–17229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mjelle R., Hegre S.A., Aas P.A., Slupphaug G., Drablos F., Saetrom P., Krokan H.E.. Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair (Amst). 2015; 30:53–67. [DOI] [PubMed] [Google Scholar]
- 47. Raschle M., Smeenk G., Hansen R.K., Temu T., Oka Y., Hein M.Y., Nagaraj N., Long D.T., Walter J.C., Hofmann K. et al. . DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science. 2015; 348:1253671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gabel S.A., DeRose E.F., London R.E.. XRCC1 interaction with the REV1 C-terminal domain suggests a role in post replication repair. DNA Repair (Amst). 2013; 12:1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Levy N., Oehlmann M., Delalande F., Nasheuer H.P., Van Dorsselaer A., Schreiber V., de Murcia G., Menissier-de Murcia J., Maiorano D., Bresson A.. XRCC1 interacts with the p58 subunit of DNA Pol alpha-primase and may coordinate DNA repair and replication during S phase. Nucleic Acids Res. 2009; 37:3177–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sugimura K., Takebayashi S., Taguchi H., Takeda S., Okumura K.. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008; 183:1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gilljam K.M., Feyzi E., Aas P.A., Sousa M.M., Muller R., Vagbo C.B., Catterall T.C., Liabakk N.B., Slupphaug G., Drablos F. et al. . Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009; 186:645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hegde M.L., Hegde P.M., Bellot L.J., Mandal S.M., Hazra T.K., Li G.M., Boldogh I., Tomkinson A.E., Mitra S.. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E3090–E3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hegde P.M., Dutta A., Sengupta S., Mitra J., Adhikari S., Tomkinson A.E., Li G.M., Boldogh I., Hazra T.K., Mitra S. et al. . The C-terminal domain (CTD) of human DNA glycosylase NEIL1 is required for forming BERosome repair complex with DNA replication proteins at the replicating genome: DOMINANT NEGATIVE FUNCTION OF THE CTD. J. Biol. Chem. 2015; 290:20919–20933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neurauter C.G., Luna L., Bjoras M.. Release from quiescence stimulates the expression of human NEIL3 under the control of the Ras dependent ERK-MAP kinase pathway. DNA Repair (Amst). 2012; 11:401–409. [DOI] [PubMed] [Google Scholar]
- 55. Morland I., Rolseth V., Luna L., Rognes T., Bjoras M., Seeberg E.. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002; 30:4926–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rolseth V., Krokeide S.Z., Kunke D., Neurauter C.G., Suganthan R., Sejersted Y., Hildrestrand G.A., Bjoras M., Luna L.. Loss of Neil3, the major DNA glycosylase activity for removal of hydantoins in single stranded DNA, reduces cellular proliferation and sensitizes cells to genotoxic stress. Biochim. Biophys. Acta. 2013; 1833:1157–1164. [DOI] [PubMed] [Google Scholar]
- 57. Krokeide S.Z., Laerdahl J.k., Salah M., Luna L., Cederkvist F.H., Fleming A.M., Burrows C.J., Dalhus B., Bjoras M.. Human NEIL3 is mainly a monofunctional DNA glycosylase removing spiroimindiohydantoin and guanidinohydantoin. DNA Repair (Amst). 2013; 12:1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Banerjee D., Mandal S.M., Das A., Hegde M.L., Das S., Bhakat K.K., Boldogh I., Sarkar P.S., Mitra S., Hazra T.K.. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 2011; 286:6006–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Engelward B.P., Weeda G., Wyatt M.D., Broekhof J.L., de Wit J., Donker I., Allan J.M., Gold B., Hoeijmakers J.H., Samson L.D.. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:13087–13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Myrnes B., Guddal P.H., Krokan H.E.. Metabolism of dITP in HeLa cell extracts, incorporation into DNA by isolated nuclei and release of hypoxanthine from DNA by a hypoxanthine-DNA glycosylase activity. Nucleic Acids Res. 1982; 10:3693–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.