


Abstract
Toxin–antitoxin (TA) loci are widespread in bacteria including important pathogenic species. Recent studies suggest that TA systems play a key role in persister formation. However, the persistence phenotype shows only weak dependence on the number of TA systems, i.e. they are functionally redundant. We use a mathematical model to investigate the interaction of multiple TA systems in the switching between growth and persistence. We explore two scenarios: (i) TA systems are bistable and each TA system experiences its own noise and (ii) the noise in the level of common stress signal (e.g. (p)ppGpp) coordinates all TA systems simultaneously. We find that in the first scenario the exit from the persister state strongly depends on the number of TA systems. However in the second case, we could reproduce the weak dependence. The duration of the high (p)ppGpp state was found to be the key parameter for persistence. The (p)ppGpp-driven synchronized transition of all TA systems results in the redundancy.
INTRODUCTION
In 1944, Joseph Bigger reported that a small fraction of Staphylococcus population was insensitive to a transient penicillin treatment and referred those cells as persisters (1). The descendants of persisters were as sensitive to penicillin as normal cells. It was then proposed that the persisters are a subpopulation of cells that happened to be in a phase in which they are insusceptible to the action of penicillin at the time of exposure. It was later revealed that persistence is a universal phenomenon among many bacteria and archaea species and for many antibiotics (2).
The molecular mechanisms of persistence is currently under active research, but various research indicates the involvement of type II toxin–antitoxin (TA) loci in persister formation ((3,4) for review). A type II TA locus encodes a long-lived toxin protein that inhibits cell growth and an unstable antitoxin protein that neutralizes the toxin’s activity by forming a tight complex with the toxin (5,6). In Escherichia coli, mutations in a TA locus hipBA gene were found to severely increase the level of persister formation (7–9). In single cell experiments, persisters are found to be in a dormant state that provides antibiotics tolerance, but can stochastically switch back to growth state (4,9). It was hypothesized that the persisters are a dormant subpopulation that is more tolerant to stress, and TA systems enable switching between growth and dormancy.
TA loci are surprisingly abundant in free-living prokaryotes (10), and are very often redundant. Previous research has identified at least 11 type II TA loci in E. coli (3) and 88 putative TA loci in Mycobacterium tuberculosis (11). Among them, toxins of 10 type II TA loci in E. coli were found to be mRNases (3). Interestingly, deletion of a mRNase-encoding TA locus had only minor effect on the frequency of persister cells (3,12,13). This is rather puzzling, since one TA system can easily cause visible dormancy if its antitoxin gene is inactivated (14).
The antitoxin-toxin battle of a single TA system was well-studied by mathematical models. A TA system is often modeled as a bistable system, where the antitoxin-dominant stable state represents cell growth, while the toxin-dominant stable state represents dormancy, serving as a possible explanation for persistence. Molecular noises, especially the intrinsic noises within the TA system, can facilitate cells to switch between growth and dormancy (15–18).
However, great difficulties arise when one extends a single bistable TA system with intrinsic noise to coupled TA systems and attempts to explain the observed redundancy. Fasani et al. proposed a model by assuming that translation inhibition by toxins requires activation of all TA modules present in the cell (18), but the assumption of such cooperativity lacks experimental support. Alternatively, if one employs the intuitive assumption that action of a given toxin is independent of other TA systems, then the persister formation rate and/or waking up time is expected to be very sensitive to the number of TA systems (19). The reason is that if one kind of toxin sufficiently accumulates, it will help other TA systems to be toxin-dominant by preventing translation and by slowing down the cell growth. As the existing antitoxin proteins are degraded, long-lived toxin proteins become free and exert their growth inhibitory effect. The cross-activation of TA systems was indeed observed experimentally (20). If all of TA systems are in the toxin-dominant state, any TA system fluctuating to the high antitoxin state will be pulled back to the high toxin state due to other free toxins (Supplementary Figure S1). A successful exit of dormancy requires a synchronous switching of all TA systems, and the chance of this process decreases exponentially with the number of TA systems (21). In other words, persistence with this mechanism is expected to be very sensitive to the number of TA systems (see also the result section for the model analysis).
Interestingly, guanosine tetra- and pentaphosphate [(p)ppGpp], the alarmone molecule for stringent response (22–24), was identified as a regulator for TA systems (4). All ten mRNase-encoding TA systems in E. coli were found to be up-regulated in response to amino-acid starvation, a signal for (p)ppGpp production (25). Overproduction of (p)ppGpp also increased the frequency of persister cells (4), likely due to the promotion of antitoxin degradation (26). This suggests an alternative scenario for the growth-dormancy transition. The fluctuation of the master regulator (p)ppGpp will act as a common source of noise for all the TA systems to switch between being antitoxin-dominant and toxin-dominant. If (p)ppGpp fluctuation is the main governing factor of the transition between growth and dormancy, then all TA systems will switch between the toxin and antitoxin dominant states synchronously according to the (p)ppGpp fluctuation, and the growth-dormancy transition is expected to show a very weak dependence on the number of TA systems.
Indeed, experimental findings support the strong fluctuation of (p)ppGpp (4). This can be created by transcription bursts (refer to (27)) of (p)ppGpp-related enzymes, such as the synthetase RelA and the synthetase/hydrolase SpoT. Positive feedbacks, which can amplify the fluctuation, are also involved in (p)ppGpp regulation. For example, Amato et al. proposed that (p)ppGpp and SpoT form a bistable system, allowing (p)ppGpp to switch between two characteristic levels (28). Also, the hipBA module, which was the first TA locus that was suggested to form a bistable system (15), can induce amino-acid starvation and hence (p)ppGpp synthesis by inhibiting the Glu-tRNA-synthetase, GltX (29,30).
In this work, we explore the relationship between (p)ppGpp fluctuation and redundancy of TA systems by using mathematical models. We first present a model of one TA system that can show bistability between growth and dormancy. We update our previous model (16) based on our experimental data. We then build a model for coupled TA systems, and show that without external common fluctuation, the growth-dormancy transition rates tend to be very sensitive to the number of TA systems, contrary to the experimental observation. We then introduce explicit (p)ppGpp fluctuation to the model as the driver for transition, and show that it successfully reproduces the weak dependence of the persistence on the number of TA systems. In particular, we identify the duration of high (p)ppGpp state as a key quantity, governing the properties of persister cells including frequency of persister cells and waking up times.
MATERIALS AND METHODS
Experimental procedure
Plasmid construction
Plasmid pSEM3187 was constructed in two steps. First, the XbaI–PsiI fragment of plasmid pTYB1 (NEB) containing the lacI gene and the pMB1 replication origin together with the rop gene was ligated with the XbaI–PvuII fragment of plasmid pSEM2027 containing a strong LacI controlled synthetic promoter (Figure 4 top, (31)). Next, a synthetic ribosome binding sequence and the relE open reading frame were inserted downstream, of the LacI controlled promoter, between the PstI and BamHI sites. The sequence of the plasmid is provided in the Supplementary Data.
Figure 4.

(p)ppGpp fluctuation synchronizes the activity of 10 Toxin-Antitoxin systems. (A) A sample trajectory of the model with 10 TA systems. The total amount of free toxins is plotted as a function of time. Light green region represents low (p)ppGpp level and dark green region represents high (p)ppGpp level. The black line represents the threshold level of free toxins for dormancy, which corresponds to a 5-hour cell doubling time when (p)ppGpp level is low. (B) Duration of high-(p)ppGpp state determines the entry of dormancy. The top row represents a sample trajectory of antitoxin (first column) and toxin (second column) for each TA system, and total free toxin (third column) level with the duration of the high (p)ppGpp state being 24 hours. The bottom row represents a sample trajectory when the duration is 1 h.
Plasmids pSEM4049 was constructed by inserting a synthetic fragment encoding a translational fusion of mCherry and venus YFP between the KpnI and SalI sites of plasmid pBAD33 (32). Plasmid pSEM4063 was made in a similar way except that the mCherry and venus YFP open reading frames are separated by the sequence responsible for translational coupling of the E. coli MG1655 relB and relE genes. The sequences of the synthetic fragments are shown in the Supplementary Data.
Fluorescence measurement
SC34/pSEM3187/pBAD33, SC34/pSEM3187/pSEM4049 and SC34/pSEM3187/pSEM4063 cells were grown overnight in LB medium containing zeocin (70 μg/ml) and chloramphenicol (30 μg/ml) before they were diluted 1:1000 in M9 fructose (0.2%) medium containing zeocin (70 μg/ml), chloramphenicol (30 μg/ml), and supplemented with 0.1 mg/ml amino acids (excluding Phe, Tyr and Trp to limit background fluorescence). Cells were grown at 37°C in a FLUOstar Omega Microplate Reader (BMG Labtech). OD (600 nm), mCherry and YFP fluorescence was recorded at regular intervals. The fluorescence data was corrected with the fluorescence of SC34/pSEM3187/pBAD33 cells (not carrying mCherry/YFP).
Model for one TA system
We construct a mathematical model for one TA system (Figure 2A) based on our previous work (16). We assume antitoxin proteins rapidly form tight dimers upon translation (A2,f) and model the interaction between antitoxin and toxins with
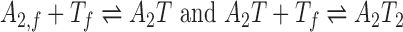 |
(1) |
where Tf represents isolated toxin molecules (free toxins). We assume that the reactions reach equilibrium with association constant KT = [A2T]/[A2, f][Tf] = [A2T2]/[A2T][Tf]. For convenience, we define the total level of antitoxins as [A] = 2[A2, f] + 2[A2T] + 2[A2T2] and the total level of toxins as [T] = [Tf] + [A2T] + 2[A2T2].
Figure 2.

(p)ppGpp level modulates steady state behaviors of a TA system. (A) Schematic description of the model. A toxin–antitoxin locus encodes an antitoxin protein (blue) and a toxin protein (red). Antitoxin proteins rapidly form tight dimers A2,f and may bind toxins to form trimers A2T and tetramers A2T2. Isolated toxin proteins deplete mRNA and inhibit cell growth, and trimers A2T repress promoter activity. (p)ppGpp promotes the degradation of antitoxins and inhibit cell growth. (B) The effect of (p)ppGpp level on the steady states of the model. We increase (p)ppGpp levels by reducing antitoxin half lives and by increasing cell doubling times. The top row describes the steady states (black cross) and nullclines (blue for d[A]/dt = 0 and red for d[T]/dt = 0). The bottom row describes the steady states in the ball-in-potential-well interpretation where a system (brown ball) stochastically switches between steady states. The antitoxin half-life of the four columns are 40, 26.7, 22.8 and 20 min, respectively; the cell doubling times in absence of translation inhibition are 40, 100, 160 min and no growth. (C) The effect of translation inhibition on the steady states of the model. We construct two mathematical models by choosing the values of βA to satisfy βA/βT = 0.5 (blue) and βA/βT = 1 (red). All other parameter values remain identical to Supplementary Table S1. We choose the cell doubling time of growth state to be 40 min and compute the steady states of the two models (plotted in free toxin levels) as a function of antitoxin half lives.
We describe the antitoxin concentration [A] and the antitoxin concentration [T] with the following dynamical equations:
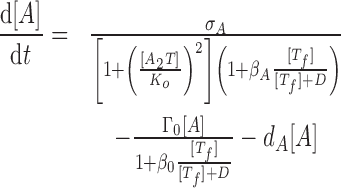 |
(2) |
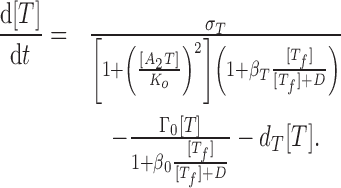 |
(3) |
The first lines of Eqs. (2) and (3) describe protein synthesis, where σA and σT represent the maximal synthesis rates of antitoxins and toxins, respectively. Protein synthesis can be inhibited both transcriptionally and translationally. Transcriptional inhibition is mainly mediated by cooperative binding of two trimers A2T to the promoter (33), which we describe by the term 1 + ([A2T]/Ko)2 where Ko is the dissociation constant between A2T and promoter. We exclude the inhibitory effect of A2 from the model due to its relatively weak strength (refer to (16)), and tetramers A2T2 were shown to de-repress transcription, a phenomenon called ‘conditional cooperativity’ (33,34). The mRNase activities of free toxins Tf mediate translation inhibition, which we model by the Michaelis–Menten form 1 + β[Tf]/([Tf] + D) where β describes the maximal fold-reduction of translation and D controls the saturation of toxins’ activity.
The second lines of Eqs. (2) and (3) describe the reduction of protein levels due to cell growth (first term) and degradation (second term). We assume that the cell growth is also regulated by free toxins (refer to (16)). Since cell division requires coordination of multiple pathways and all such pathways are subject to disruption by free toxins, we assume that toxins have a stronger inhibitory effect on cell growth than expression of individual genes. Consequently, we choose the maximal reduction in growth rate (β0) to be much greater than βA and βT.
To control the complexity of the model, we do not model the explicit levels of (p)ppGpp and only describe its effect on TA systems by modulating parameter values in Eq. (2) and (3). Here, we consider two major effects of a high (p)ppGpp level: repression of cell growth rate (35) and promotion of antitoxin degradation (3,4). The first one is captured by decreasing cell dilution rate (Γ0) and the second one is captured by increasing antitoxin degradation rate dA. We exclude other regulation by (p)ppGpp from the model.
The model is implemented with parameter values listed in Supplementary Table S1. The steady states and the nullclines are computed by solving the equations analytically. Since the dependence of dA and Γ0 on (p)ppGpp levels has not been fully quantified, we manually determine the values of these two parameters such that the model reaches one of four characteristic cases. We do not argue whether the values are biologically feasible or not.
Model for coupled TA systems
We consider the coupling of multiple TA systems with identical kinetic properties. Coupling among equations comes from the free toxin activity. We assume that all free toxins cleave mRNAs independently, so the toxin activity is dependent on the sum of all free toxin concentrations (19). The resulting model of coupled TA systems is described by the equations
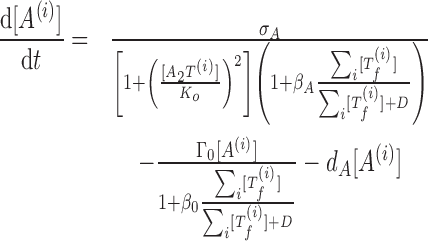 |
(4) |
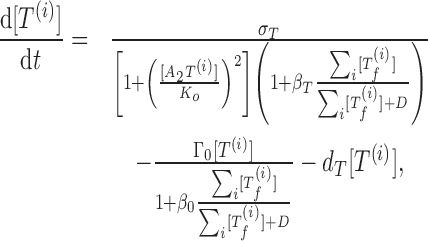 |
(5) |
where the superscript i represents the ith TA system.
We allow (p)ppGpp to stochastically jump between two levels, with a transition rate from the low level ([Pl]) to the high level ([Ph]) to be r+ and the reverse rate to be r−. We choose the half-life of antitoxins and cell doubling time to be both 40 min under low (p)ppGpp level, and choose a 20-min antitoxin half-life and no cell growth under high level (Supplementary Table S1). We do not include negative feedback mediated by toxins to (p)ppGpp fluctuation (refer to (36)) for simplicity.
We implement the model with a hybrid Gillespie algorithm (37). For simplicity, we describe the amount of antitoxin in dimer concentrations ([A2]) during simulation, though we present the results in monomer levels ([A]). The amount of toxins is always described in monomer levels ([T]). We keep the total amount of toxins ([T]) and antitoxin dimers ([A2]) as integers. For each toxin (monomer) and antitoxin (dimer) species, we construct two reactions, one for synthesis and one for degradation/dilution. At each time step, the values of [A2, f], [A2T], [A2T2] and [Tf] are calculated by solving Eq. (1) in equilibrium. We allow these values to be non-integer. We then calculate the reaction rates and update the time and state of the model following Gillespie algorithm. We record simulation data around every 30 min. We define that cells enter dormancy state if toxins induce a growth-reduction of >7.5-fold, corresponding to a cell doubling time of more than 5 h in absence of (p)ppGpp, which is necessary to survive a persister assay with 5 h exposure to antibiotics. We define cells exit dormancy state if toxin-induced growth reduction is <3-fold. We compute the fraction of persister cells by averaging the transition rates from growth to dormancy states in generation time (9) and compute the waking up time from dormancy by averaging the time spent in dormancy state. The amount of accumulated free toxins is defined as the maximal amount of free toxins during dormancy.
When we study the coupling without (p)ppGpp fluctuation, we remove (p)ppGpp from the model and choose the value of dA to be 5.1 × 10−4s−1 (1.75-fold higher than dA,low). All other parameter values remain unchanged. We implement the model with the hybrid Gillespie algorithm with initial state [A] = 4000, [T] = 3000, which allows the system to reach the dormancy stable steady state rapidly upon simulation. We perform 10 independent simulation for each number of TA systems and examine the timescale in which cells return to the growth state.
RESULTS
Toxins attack their own mRNA more than antitoxin mRNA
The toxin and antitoxin modules of a TA locus typically locate in the same operon. Among the 10 known mRNase-encoding TA loci in E. coli, seven loci, including the well-studied relBE locus, encode the modules in the sequence of antitoxin and toxin (hicBA, mqsRA and higBA encode in the reverse order) (38–41). Since the toxin module contains no ribosome binding sites, translation of toxins can only be initiated after a ribosome occasionally passes through the switching-over region after finishing the translation of the upstream antitoxins. Consequently, this sequential order ensures that the toxin module is always less translated than the antitoxin module.
The mRNase activity of toxins is hypothesized to induce global translation inhibition by depleting mRNA at various locations, including its own mRNA (16). Since translation of a toxin module relies on a successful translation of its antitoxin counterpart, we expect that translation inhibition affects toxins and antitoxins to different extents. As depicted in Figure 1A, a complete mRNA contains information of both antitoxin (blue) and toxin (red) and can be translated into both proteins (top line). If the mRNA is cleaved within the antitoxin module (second line), no protein can be synthesized. Meanwhile, antitoxin proteins can still be synthesized if the cutting site locates within the toxin module (third line), as the information of antitoxin is complete. Therefore, translation inhibition results in a higher synthesis rate ratio between antitoxins and toxins. This effect acts as a negative feedback to the toxin activity and can possibly affect the growth-dormancy transition significantly.
Figure 1.

Translation inhibition of Toxin-Antitoxin mRNA. (A) Schematic description of translation inhibition by toxins. A full mRNA consists of an antitoxin region (blue) and a toxin region (red), and can be translated to both antitoxin and toxin proteins. If mRNA is cut within the antitoxin region, no proteins can be synthesized. If mRNA is cut within the toxin region, the cleaved mRNA contains complete information of antitoxins and antitoxins can be produced. (B-D) Effect of RelE expression on the production of translationally coupled fluorescent proteins. Cells carrying the reporter constructs (B) were grown in a temperature controlled plate reader. Cell growth and expression of the fluorescent proteins were monitored in the presence (D) and absence (C) of RelE. RelE expression was induced by adding 2 mM IPTG at 130 min (D). Expression of the fluorescent proteins was induced by 0.2% arabinose added at 0 min (C) or 280 min (D).
To quantify this negative feedback, we made two reporter constructs (Figure 1B). These constructs express the mCherry and Venus YFP fluorescent proteins, which are easy to distinguish based on their excitation and emission spectra. The construct pSEM4049 carried a translational fusion of the two proteins, i.e. it encodes a single mCherry-YFP polypeptide. In the construct pSEM4063, the coding sequences of the two proteins are separated by the sequence responsible for translational coupling of RelB and RelE. In this case the two proteins are translated separately, but only the ribosome that have already translated mCherry can translate YFP.
These constructs were used to study the effect of RelE expression on translationally coupled genes. Two reporter constructs were introduced into E. coli SC34 cells (ΔrelBEF) (42) carrying pSEM3187. In these cells, the expression of RelE and therefore cell growth rate can be controlled by IPTG (Supplementary Figure S2). Cells were grown in a temperature controlled plate reader as described in Materials and Methods. To induce the expression of fluorescence proteins, 0.2% arabinose was added at time zero (Figure 1C) or 150 min after the addition of 2 mM IPTG inducing RelE expression (Figure 1D). Cell density and the fluorescence level of YFP and mCherry were measured at regular intervals. Determined from the YFP/mCherry fluorescence ratios for the two constructs in absence of RelE expression, the efficiency of translation coupling was ∼30% (Table 1). Introduction of a TAG stop codon into pSEM4063, terminating mCherry translation 30 nt upstream from the translational start of YFP, abolished YFP translation (<3% coupling efficiency), confirming the translational coupling. When RelE is induced by IPTG, the efficiency of translational coupling decreased to 16% (Table 1). This result suggests that translation inhibition towards the toxin module is twice stronger than towards antitoxin. We include this finding into our mathematical model.
Table 1. Expression of translationally fused and translationally coupled fluorescence proteins.
| Translational fusion | Translational coupling | |||
|---|---|---|---|---|
| RelE induction | − | + | − | + |
| Growth rate (doublings/h) | 0.45 | 0.135 | 0.44 | 0.16±0.01 |
| Rate of YFP expression (a.u.) | 1583 | 674 | 210 | 122 ± 10 |
| Rate of mCherry expression (a.u.) | 101 | 37 | 44 | 41 ± 1 |
| YFP/mCherry expression | 15.7 | 18.2 | 4.8 | 2.9 ± 0.2 |
The standard errors of mean for translational coupling are calculated based on two independent measurements.
(p)ppGpp level regulates steady state behaviors of TA systems
To understand how (p)ppGpp and the bias of translation inhibition control the switching between growth and dormancy, we construct a mathematical model for a typical TA system (for example, relBE) illustrated in Figure 2A (details in Materials and Methods). We assume that antitoxin proteins rapidly form tight dimers upon translation and may reversibly bind free toxin molecules to form trimers A2T and tetramers A2T2. We include ‘conditional cooperativity’ into the model (refer to (16,33)) by only allowing trimers A2T to repress promoter activities. Free toxin proteins may cleave mRNA and inhibit cell growth, and we choose the effects of free toxins on the toxin production to be twice as high as on antitoxin production (βA:βT = 1:2) to account for the bias of translation inhibition. Two effects of (p)ppGpp are accounted into this model: repression of cell growth rate (35) and promotion of antitoxin degradation through activation of proteases (3,4). We therefore represent an up-shift in (p)ppGpp level by modulating the two relevant parameters in the model. We fit the parameter values to literature data (Supplementary Table S1) and analyze the model in the deterministic limit.
We compute the nullclines and the steady states of the model under four characteristic (p)ppGpp levels. As shown in Figure 2B, the model with the lowest (p)ppGpp level exhibits a monostable steady state where the antitoxin level exceeds toxin (interpreted as growth state). The model becomes bistable with a higher (p)ppGpp level where growth state ([A] > [T]) and dormancy state ([A] < [T]) co-exist and cells may stochastically switch between these two states facilitated by molecular noises. The growth state is more stable, suggesting that only a minority of cells enters dormancy. By further increasing (p)ppGpp level, one observes that the model maintains bistability but the dormancy state dominates. Finally, at the highest (p)ppGpp level, model exhibits the dormancy state as the monostable steady state.
Our analysis suggests two possible mechanisms for growth-dormancy transition. Firstly, cells may contain a constant intermediate level of (p)ppGpp and TA systems exhibit bistability (second and third columns of Figure 2B). Transition between growth and dormancy is solely facilitated by the molecular noises within the TA systems, without a possible role for (p)ppGpp. This mechanism is consistent with various modeling works (15,16,18). Alternatively, (p)ppGpp may fluctuate between the lowest (first column) and the highest (fourth column) levels and TA systems exhibit monostability in both cases. Growth-dormancy transition is then controlled by the (p)ppGpp fluctuation and less contributed by the molecular noises within TA systems.
To evaluate the effects of translation inhibition bias, we construct two mathematical models with identical equations and parameter values except that free toxin proteins inhibit toxin translation twice stronger than antitoxin (βA:βT = 1:2) in the first model (blue curve in Figure 2c, parameter values listed in Supplementary Table S1) and free toxins equally inhibit translation of antitoxin and toxin (βA:βT = 1:1) in the second model (red curve in Figure 2c). We compute the steady states of the two models as a function of antitoxin half lives and plot the bifurcation diagrams in Figure 2C. The bistable region for model without bias ranges over 30 min of antitoxin half lives, while the presence of bias reduces the range to ∼10 min. This is because the high-toxin state was de-stabilized due to the bias that inhibits the toxins more strongly than antitoxin.
Bistable model of coupled TA systems without common noise gives strong dependence on the number of TA systems
To examine the redundancy of TA systems, we construct a mathematical model for coupled TA systems (details in Materials and Methods). For simplicity, we model all TA systems with the same equations and parameter values. We allow all free toxin proteins to deplete mRNA with the same rate, thus translation inhibition is dependent on the sum of free toxin levels from all TA systems. All TA systems are coupled through the mRNase activity of toxins, as free toxin proteins from one TA system may deplete other TA’s mRNA as well as induce growth repression. Finally, we allow (p)ppGpp to equally regulate the antitoxin degradation rates of all TA systems as well as to modulate the cell growth rate.
We first use this model to demonstrate that the bistable model of coupled TA systems without common fluctuation shows sensitivity to the number of TA systems. We assume that (p)ppGpp level is constant and choose the parameter values such that the coupled TA systems have two stable steady states: a growth state where all TA systems contain a higher antitoxin level than toxin, and a dormancy state where all TA systems contain a lower antitoxin level. Transition between the two states can be facilitated by the molecular noises within the TA systems, which is realized by simulating the model with a hybrid Gillespie algorithm (details in Materials and Methods). The switching rate from growth state to dormancy state is proportional to the fraction of persister cells in exponentially growth phase, while the duration of dormancy is quantified as waking-up time (9).
While some parameter values result in a weak dependence of persister fractions on the number of TA systems, the average waking-up times often scale strongly. Figure 3 demonstrates one sample simulation where the model is around the dormancy state at time zero and is simulated until returning to the growth state. For model with one TA system, the average waking up time is around 2 × 104 h. Meanwhile, for model with two TA systems, we perform 10 independent simulations for ∼106 h and none of the simulations shows a successful wake up. Similar results were obtained previously (19). This observation is consistent with our analysis in the Introduction, suggesting that we can not reproduce the redundancy of TA systems with bistable mechanism within reasonable parameter regions.
Figure 3.

Growth-dormancy transition of coupled TA systems with bistability-based mechanism. We assume that (p)ppGpp level is constant and simulate the model with one to two TA systems, and plot the sample trajectories as a function of time. For clarity, we only plot the levels of one TA system. The simulation starts at a dormancy state, and the time required to reach growth state indicates the waking up times. Our simulation shows that addition of one TA system increases the waking up times by at least two orders of magnitudes. For clarity of the figure, we record data points every 50 h from simulation.
(p)ppGpp fluctuation mediates synchronized behaviors of TA systems
We next examine whether the (p)ppGpp fluctuation mechanism is possible to reproduce the redundancy of TA systems. For simplicity, we assume that (p)ppGpp stochastically jumps from(to) a low level to(from) a high level with a constant rate r+ (r−). We choose the parameter values such that with the low (p)ppGpp level, the model exhibits a monostable steady state where the levels of all antitoxins are greater than the levels of their toxin counterparts. Similarly, with the high (p)ppGpp level, the model is monostable and all toxins are of higher concentrations than antitoxins at steady state (Supplementary Figure S4). We ignore the mild negative feedback of mRNase toxins to (p)ppGpp level (36) in the model for simplicity.
We consider the coupling of 10 TA systems and simulate the model with the hybrid Gillespie algorithm. Figure 4 and S5 plot sample time courses of antitoxin, toxin and free toxin levels. Models are in growth state with a low amount of free toxin proteins with a low (p)ppGpp level (light green region, Figure 4A and Supplementary Figure S5). Upon an up-shift of (p)ppGpp level (dark green region), cells rapidly accumulates free toxins proteins. The levels of antitoxins and toxins increase as well due to de-repression of promoters. TA systems return to the growth state after (p)ppGpp jumps back to the low level. Throughout the entire time courses, all 10 TA systems share similar levels and respond to (p)ppGpp fluctuation in the same timescale (Figure 4B).
Duration of high-(p)ppGpp state determines persister fraction and waking-up times
To quantify the properties of growth-dormancy transition, we define that a cell enters dormancy if free toxin proteins induce a growth-reduction of >7.5-fold and the threshold amount of free toxin proteins is plotted as a black line in Figure 4A. A cell is considered exiting dormancy if the growth-reduction is <3-fold (details in Materials and Methods). Simulation shows that if (p)ppGpp stays at the high level for a sufficiently long duration, cells accumulate a large quantity of free toxins and remain in dormancy for hours (top, Figure 4B). Meanwhile, cells remain in growth if the duration of high-(p)ppGpp state is too short (bottom, Figure 4B). This observation suggests that the duration of the high (p)ppGpp state is a key quantity governing whether cells exhibit persistence.
To further explore the relationship between the duration of the high (p)ppGpp state and persistence, we sample this duration by over 2 orders of magnitudes and compute the maximal amount of free toxin proteins during dormancy (Figure 5A) and waking-up times (Figure 5B). We find that the duration of high-(p)ppGpp state can be partitioned into three regions: a growth region (orange) where cells are unable to enter dormancy, an exponential region (green) where cells accumulate free toxin proteins exponentially in time and a saturated region (purple) where the amount of free toxins converges its maximal value as cells reach the steady state with high (p)ppGpp level. The waking-up time is not well-defined for the growth region, but exhibits good linear relationship with the duration of the high (p)ppGpp state for both exponential and saturated regions despite different slopes.
Figure 5.

Duration of the high (p)ppGpp state determines properties of persisters. We simulate a model with 10 TA systems with variable durations of the high (p)ppGpp state. (A) The amount of accumulated free toxins and (B) waking up times from dormancy are plotted. We discover that the duration can be partitioned into three regions: the growth region (orange) where (p)ppGpp fluctuation is unable to induce dormancy, the exponential region (green) where cell accumulates free toxins exponentially in time, and the saturated region (purple) where cells reach steady state when (p)ppGpp level is high.
Figure 5A can be explained by analyzing the model in the high toxin deterministic limit. Cells exhibit a high amount of free toxin proteins if the duration of the high (p)ppGpp state falls in the exponential or saturated region. In the high toxin limit, when (p)ppGpp level is high, the dominant form of antitoxin proteins is tetramer A2T2 and the amount of trimers A2T is negligible. Consequently, the promoters of TA systems are usually de-repressed and the terms for transcription inhibition vanish. Translation inhibition also reaches its maximal strength under high toxin limit, so the corresponding terms can be simplified as well (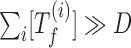 ). As a result, synthesis rates of antitoxins and toxins are approximately constant (σA/βA and σT/βT) and both proteins degrade/dilute with first order kinetics (dA[A] and dT[T]). The solution to this simplified model is that the toxin and antitoxin proteins reach their steady state values with high (p)ppGpp level exponentially in time. The amount of free toxins, characterized by the difference between toxin levels and antitoxin levels, scales exponentially in time as well. With the same argument, cells lose free toxins exponentially in time when (p)ppGpp switches back to the low level.
). As a result, synthesis rates of antitoxins and toxins are approximately constant (σA/βA and σT/βT) and both proteins degrade/dilute with first order kinetics (dA[A] and dT[T]). The solution to this simplified model is that the toxin and antitoxin proteins reach their steady state values with high (p)ppGpp level exponentially in time. The amount of free toxins, characterized by the difference between toxin levels and antitoxin levels, scales exponentially in time as well. With the same argument, cells lose free toxins exponentially in time when (p)ppGpp switches back to the low level.
An operational definition of waking-up times is the duration of a cell in dormancy, and this quantity consists of two periods: a high (p)ppGpp period (within dark green regions) where cells enter dormancy but keep accumulating free toxins and a low (p)ppGpp period (within light green regions) where cells lose free toxin proteins but still remain in dormancy. In the high toxin limit, the length of high (p)ppGpp period approximately equals to the duration of high-(p)ppGpp state. If the duration falls in the exponential region, the levels of free toxins change exponentially in time for both periods. Since the amount of toxins to accumulate during high (p)ppGpp period equals to the amount to lose during the low period, the lengths of the two periods should be proportional to each other. The waking-up time of the cell is then proportional to the duration of high (p)ppGpp state. Meanwhile, if the duration of high (p)ppGpp state falls in the saturated region, the majority of dormancy is spent around the steady state with the high (p)ppGpp level. The time spent for other activities, such as accumulation/losing free toxin proteins, is negligible. Therefore, the waking-up time can be approximated by the length of high (p)ppGpp period, which equals to the duration of the high (p)ppGpp state. Since the mechanisms causing linear relations are different for the two regions, their slopes are distinct.
Persister fraction and waking up times are weakly correlated with the number of Toxin-Antitoxin systems
Finally, we calculate the dependency between the number of TA systems and the properties of persister cells in our model. We consider the coupling of 1–10 TA systems and simulate the mathematical model with Gillespie algorithm. We calculate the average dormancy entry rate and waking-up times and plot the values as a function of the number of TA systems (Figure 6). The dormancy entry rate, which can be interpreted as the fractions of persister cells (9), differs by 2-fold between wt (10TA) and Δ9 (1TA) (Figure 6a). Meanwhile, the average waking-up times of all models are ∼18 h, which is consistent with previous measurements (Figure 6B) (9).
Figure 6.

Persister fraction and waking up time are weakly correlated with the number of TA systems. We perform simulation for models with varying number of TA systems (1–10). (A) Dormancy entry rates and (B) average waking up times are plotted as a function of the number of TA systems. We estimate each data point with at least 50 simulated persister samples. The error bars represent the standard error of the means.
DISCUSSION
In this work, we investigated a possible explanation of the redundancy of TA systems in bacterial persistence.
We first updated our previous model of one TA system by taking into account the experimental results that mRNase toxin inhibits the toxin module more than the antitoxin module. Our experiments using fluorescent reporter genes gave a 2-fold stronger inhibition of toxins. However, it should be noted that the mRNase toxin cleavage activity is known to have some sequence specificity (43,44). This suggests that the TA sequences can evolve to have more or less inhibition by mRNase toxins. Our model showed that the more biased to the toxin cleavage is, the narrower the bistability parameter range (Figure 2), making dormancy to growth transition easier to be driven by (p)ppGpp fluctuation.
We showed that, persister formation in cells with multiple coupled bistable TA systems without common noise is very sensitive to the number of TA systems, while the (p)ppGpp-driven switching mechanism of TA systems reproduces the experimentally observed redundancy of TA systems. We identify the duration of (p)ppGpp state as a key quantity governing persister properties including persister fractions, waking up times and amount of accumulated free toxins. This would suggest that a short pulse of (p)ppGpp, such as a sudden amino-acid starvation, would not drastically increase the persister fraction (Figure 5A, orange region). Also, the present model predicts that TA systems turn on and off in synchrony. It was indeed shown that upon isoleucin starvation all 10 TA systems were activated (25). The model further predicts that the waking-up time correlates positively with the duration of the high (p)ppGpp state (Figure 5B), which can be tested experimentally if (p)ppGpp levels and waking-up times are measured simultaneously at single cell level.
In this paper, we assumed an extreme case that the steady state in the high (p)ppGpp state is the high toxin state for all TA systems. The realistic growth-dormancy transition mechanism can be a combination of bistability and (p)ppGpp fluctuation. For example, (p)ppGpp may fluctuate between a low level giving only growth steady state (first column in Figure 2B) and a high level giving bistability (second or third columns). The entry of dormancy then requires both a high (p)ppGpp level and intrinsic noises in TA expression. Meanwhile, the exit process remains only dependent on (p)ppGpp fluctuation, indicating that the redundancy regarding to waking up times (Figure 6B) will be maintained. In this case, even if the cells are kept in the high (p)ppGpp state for long time, the fraction of persisters will not reach 100%. In addition, we assumed that all the TAs are identical, which is also a simplification of reality, though the lack of strong phenotype in cells having single TA loci deleted suggests that none of them are dominant.
For simplicity, our model for coupled TA systems assumes that all 10 TA systems in E. coli share the same properties with relBE. Meanwhile, recent experimental evidences have shown that TA systems have distinct features and regulations. For example, the model uses identical antitoxin and toxin production rates and sets the ratio of these two rates to be 6:1 (Supplementary Table S1). In reality, this ratio was measured to vary from 2:1 to 7:1 (38), suggesting that TA systems possibly have different activation thresholds and induce distinct levels of translation inhibition. In addition, TA systems may have different configurations of complexes (Ref. (45) suggests that MazE/MazF shows a 2:1 ratio in solution and a 1:1 ratio when binding to DNA) and not all systems have been shown to exhibit conditional cooperativity (46). A final example is that hicBA encodes the toxin in the first gene and the antitoxin in the second gene. However, the hicBA module is transcribed from two separate promoters that are controlled by different factors, suggesting that activation of this TA module requires a specific signal (47). Furthermore, the conclusion from Figure 1 that free toxins inhibit toxin expression more than antitoxin expression relies on the gene order and it does not hold for the TA systems coded in the reversed order.
It should also be noted that the effect of the known feedbacks from TA systems to (p)ppGpp were not taken into account in the present model. The hipBA TA system (7–9) is likely to act as a positive feedback via (p)ppGpp (4,48). This may help high-(p)ppGpp state to maintain long enough time to induce the persistence. On the other hand, mRNase toxins constitute a negative feedback via (p)ppGpp because cleavage of mRNAs reduce the (p)ppGpp production (36). This may shorten the waking-up time from dormancy. However, further experimental analyses are needed to understand the roles of these feedback loops in the entry and exit processes.
Finally, though we used (p)ppGpp as an example, our model only required the existence of a common fluctuation signal to drive the simultaneous activation/inactivation of TA systems. Other persister-induction mechanisms which generate a similar common signal also fit into our modeling framework. For example, Radzikowski et al. showed that metabolic fluxes form a bistable system, where cells maintain homeostasis in one stable steady state and exhibit antibiotic persistence in the other stable steady state. TA systems were proposed to be a downstream component of metabolic bistability (49). The switching between two metabolic states can be regarded as the common fluctuation signal and our model can be adjusted to test this mechanism as well.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Kim Sneppen and Kenn Gerdes for fruitful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Danish National Research Foundation through the Center for Models of Life (to C.T., S.S., N.M.); Center for Bacterial Stress Response and Persistence [DNRF120 to S.S., N.M.]. Funding for open access charge: Danish National Research Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bigger J.W. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet. 1944; 244:497–500. [Google Scholar]
- 2. Lewis K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 2008; 322:107–131. [DOI] [PubMed] [Google Scholar]
- 3. Gerdes K., Maisonneuve E.. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012; 66:103–123. [DOI] [PubMed] [Google Scholar]
- 4. Maisonneuve E., Gerdes K.. Molecular mechanisms underlying bacterial persisters. Cell. 2014; 157:539–548. [DOI] [PubMed] [Google Scholar]
- 5. Galvani C., Terry J., Ishiguro E.E.. Purification of the RelB and RelE Proteins ofEscherichia coli: RelE Binds to RelB and to Ribosomes. J. Bacteriol. 2001; 183:2700–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pedersen K., Christensen S.K., Gerdes K.. Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol. Microbiol. 2002; 45:501–510. [DOI] [PubMed] [Google Scholar]
- 7. Moyed H.S., Bertrand K.P.. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983; 155:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Keren I., Kaldalu N., Spoering A., Wang Y., Lewis K.. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004; 230:13–18. [DOI] [PubMed] [Google Scholar]
- 9. Balaban N.Q., Merrin J., Chait R., Kowalik L., Leibler S.. Bacterial persistence as a phenotypic switch. Science. 2004; 305:1622–1625. [DOI] [PubMed] [Google Scholar]
- 10. Pandey D.P., Gerdes K.. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005; 33:966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramage H.R., Connolly L.E., Cox J.S.. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009; 5:e1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Keren I., Shah D., Spoering A., Kaldalu N., Lewis K.. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 2004; 186:8172–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim Y., Wood T.K.. Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli. Biochem. Biophys. Res. Commun. 2010; 391:209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rotem E., Loinger A., Ronin I., Levin-Reisman I., Gabay C., Shoresh N., Biham O., Balaban N.Q.. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:12541–12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lou C., Li Z., Ouyang Q.. A molecular model for persister in E. coli. J. Theor. Biol. 2008; 255:205–209. [DOI] [PubMed] [Google Scholar]
- 16. Cataudella I., Sneppen K., Gerdes K., Mitarai N.. Conditional cooperativity of toxin-antitoxin regulation can mediate bistability between growth and dormancy. PLoS Comput. Biol. 2013; 9:e1003174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gelens L., Hill L., Vandervelde A., Danckaert J., Loris R.. A general model for toxin-antitoxin module dynamics can explain persister cell formation in E. coli. PLoS Comput. Biol. 2013; 9:e1003190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fasani R.A., Savageau M.A.. Molecular mechanisms of multiple toxin–antitoxin systems are coordinated to govern the persister phenotype. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E2528–E2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Catuadella I. Toxin-Antitoxin Battle in Bacteria. 2013; Copenhagen: University of Copenhagen, PhD thesis. [Google Scholar]
- 20. Kasari V., Mets T., Tenson T., Kaldalu N.. Transcriptional cross-activation between toxin-antitoxin systems of Escherichia coli. BMC Microbiol. 2013; 13:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tian C., Mitarai N.. Bifurcation of transition paths induced by coupled bistable systems. J. Chem. Phys. 2016; 144:215102. [DOI] [PubMed] [Google Scholar]
- 22. Gallant J.A. Stringent control in E. coli. Annu. Rev. Genet. 1979; 13:393–415. [DOI] [PubMed] [Google Scholar]
- 23. Potrykus K., Cashel M.. (p) ppGpp: still magical?. Annu. Rev. Microbiol. 2008; 62:35–51. [DOI] [PubMed] [Google Scholar]
- 24. Dalebroux Z.D., Swanson M.S.. ppGpp: magic beyond RNA polymerase. Nat. Rev. Microbiol. 2012; 10:203–212. [DOI] [PubMed] [Google Scholar]
- 25. Shan Y., Gandt A.B., Rowe S.E., Deisinger J.P., Conlon B.P., Lewis K.. ATP-Dependent persister formation in Escherichia coli. mBio. 2017; 8:e02267–e02316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christensen S.K., Mikkelsen M., Pedersen K., Gerdes K.. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:14328–14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Golding I., Paulsson J., Zawilski S.M., Cox E.C.. Real-time kinetics of gene activity in individual bacteria. Cell. 2005; 123:1025–1036. [DOI] [PubMed] [Google Scholar]
- 28. Amato S.M., Orman M.A., Brynildsen M.P.. Metabolic control of persister formation in Escherichia coli. Mol. Cell. 2013; 50:475–487. [DOI] [PubMed] [Google Scholar]
- 29. Germain E., Castro-Roa D., Zenkin N., Gerdes K.. Molecular mechanism of bacterial persistence by HipA. Mol. Cell. 2013; 52:248–254. [DOI] [PubMed] [Google Scholar]
- 30. Kaspy I., Rotem E., Weiss N., Ronin I., Balaban N.Q., Glaser G.. HipA-mediated antibiotic persistence via phosphorylation of the glutamyl-tRNA-synthetase. Nat. Commun. 2013; 4:3001. [DOI] [PubMed] [Google Scholar]
- 31. Bendtsen K.M., Erdossy J., Csiszovszki Z., Svenningsen S.L., Sneppen K., Krishna S., Semsey S.. Direct and indirect effects in the regulation of overlapping promoters. Nucleic Acids Res. 2011; 39:6879–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guzman L.-M., Belin D., Carson M.J., Beckwith J.. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995; 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Overgaard M., Borch J., Gerdes K.. RelB and RelE of Escherichia coli Form a tight complex that represses transcription via the Ribbon–Helix–Helix Motif in RelB. J. Mol. Biol. 2009; 394:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Overgaard M., Borch J., J⊘rgensen M.G., Gerdes K.. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 2008; 69:841–857. [DOI] [PubMed] [Google Scholar]
- 35. Bremer H., Dennis P.P.. Modulation of chemical composition and other parameters of the cell by growth rate. Escherichia coli and Salmonella. 1996; Washington D.C.: ASM Press; 1553–1569. [Google Scholar]
- 36. Tian C., Roghanian M., Jorgensen M.G., Sneppen K., Sorensen M.A., Gerdes K., Mitarai N.. Rapid curtailing of the stringent response by toxin-antitoxin-encoded mRNases. J.. Bacteriol. 2016; 198:1918–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gillespie D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977; 81:2340–2361. [Google Scholar]
- 38. Li G.-W., Burkhardt D., Gross C., Weissman J.S.. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014; 157:624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jorgensen M.G., Pandey D.P., Jaskolska M., Gerdes K.. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 2009; 191:1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brown B.L., Grigoriu S., Kim Y., Arruda J.M., Davenport A., Wood T.K., Peti W., Page R.. Three dimensional structure of the MqsR:MqsA complex: a novel TA pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog. 2009; 5:e1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Christensen Dalsgaard M., Gerdes K.. Two higBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol. Microbiol. 2006; 62:397–411. [DOI] [PubMed] [Google Scholar]
- 42. Christensen S.K., Pedersen K., Hansen F.G., Gerdes K.. Toxin–antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J. Mol. Biol. 2003; 332:809–819. [DOI] [PubMed] [Google Scholar]
- 43. Pedersen K., Zavialov A.V., Pavlov M.Y., Elf J., Gerdes K., Ehrenberg M.. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell. 2003; 112:131–140. [DOI] [PubMed] [Google Scholar]
- 44. Zhang Y., Zhang J., Hoeflich K.P., Ikura M., Qing G., Inouye M.. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell. 2003; 12:913–923. [DOI] [PubMed] [Google Scholar]
- 45. Kamada K., Hanaoka F., Burley S.K.. Crystal structure of the MazE/MazF complex: molecular bases of antidote-toxin recognition. Mol. Cell. 2003; 11:875–884. [DOI] [PubMed] [Google Scholar]
- 46. Page R., Peti W.. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016; 12:208–214. [DOI] [PubMed] [Google Scholar]
- 47. Turnbull K.J., Gerdes K.. HicA toxin of Escherichia coli derepresses hicAB transcription to selectively produce HicB antitoxin. Mol. Microbiol. 2017; 104:781–792. [DOI] [PubMed] [Google Scholar]
- 48. Germain E., Roghanian M., Gerdes K., Maisonneuve E.. Stochastic induction of persister cells by HipA through (p) ppGpp-mediated activation of mRNA endonucleases. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:5171–5176. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49. Radzikowski J.L., Vedelaar S., Siegel D., Ortega A.D., Schmidt A., Heinemann M.. Bacterial persistence is an active sigma-s stress response to metabolic flux limitation. Mol. Syst. Biol. 2016; 12:882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.