

Abstract
Background
Triple-negative breast cancer (TNBC), lacking expression of hormone and human epidermal growth factor receptor 2 receptors, is an aggressive subtype that frequently metastasizes to the brain and has no FDA-approved systemic therapies. Previous literature demonstrates mitogen-activated protein kinase kinase (MEK) pathway activation in TNBC brain metastases. Thus, we aimed to discover rational combinatorial therapies with MEK inhibition, hypothesizing that co-inhibition using clinically available brain-penetrant inhibitors would improve survival in preclinical models of TNBC brain metastases.
Methods
Using human-derived TNBC cell lines, synthetic lethal small interfering RNA kinase screens were evaluated with brain-penetrant inhibitors against MEK1/2 (selumetinib, AZD6244) or phosphatidylinositol-3 kinase (PI3K; buparlisib, BKM120). Mice bearing intracranial TNBC tumors (SUM149, MDA-MB-231Br, MDA-MB-468, or MDA-MB-436) were treated with MEK, PI3K, or platelet derived growth factor receptor (PDGFR; pazopanib) inhibitors alone or in combination. Tumors were analyzed by western blot and multiplexed kinase inhibitor beads/mass spectrometry to assess treatment effects.
Results
Screens identified MEK+PI3K and MEK+PDGFR inhibitors as tractable, rational combinations. Dual treatment of selumetinib with buparlisib or pazopanib was synergistic in TNBC cells in vitro. Both combinations improved survival in intracranial SUM149 and MDA-MB-231Br, but not MDA-MB-468 or MDA-MB-436. Treatments decreased mitogen-activated protein kinase (MAPK) and PI3K (Akt) signaling in sensitive (SUM149 and 231Br) but not resistant models (MDA-MB-468). Exploratory analysis of kinome reprogramming in SUM149 intracranial tumors after MEK ± PI3K inhibition demonstrates extensive kinome changes with treatment, especially in MAPK pathway members.
Conclusions
Results demonstrate that rational combinations of the clinically available inhibitors selumetinib with buparlisib or pazopanib may prove to be promising therapeutic strategies for the treatment of some TNBC brain metastases. Additionally, effective combination treatments cause widespread alterations in kinase pathways, including targetable potential resistance drivers.
Keywords: brain metastases, MEK inhibitor, PI3K inhibitor, rational combinations, triple-negative breast cancer
Importance of the study.
There are currently no FDA-approved systemic treatments for TNBC brain metastases. This study supports the combined use of brain-penetrant inhibitors against the MEK pathway with either PI3K or PDGFR inhibition to more effectively treat patients with TNBC brain metastases. In sensitive TNBC models, combination treatments proved synergistic in vitro and effective in vivo, nearly doubling or tripling survival compared with controls, depending on the model. Although these combinations did not prove effective in all models tested, collective results demonstrate that a population of patients with TNBC brain metastases may benefit from these treatment strategies. As the inhibitors tested are already clinically available for other indications, this strategy could be quickly translated to the design of TNBC brain metastases trials to address this medically unmet need.
Triple-negative breast cancer (TNBC) is an aggressive subtype which lacks expression of the estrogen and progesterone receptors and human epidermal growth factor receptor 2 (HER2). TNBC is commonly classified as either the basal-like or claudin-low molecular subtype by gene expression analysis, subtypes that have a predilection for brain relapse.1–3 Nearly 50% of TNBC patients with advanced disease will present with CNS recurrence.4,5 Survival after TNBC brain metastases (BM) diagnosis is less than 6 months.4,5 A lack of targeted agents for TNBC BM coupled with the unique biology of TNBC, BM, and the blood–brain barrier (BBB) continue to thwart effective therapeutics.4,6,7
While breast cancer brain metastases (BCBM) have compromised the BBB, they exhibit a wide heterogeneity of permeability and are overall less permeable than extracranial metastases.8,9 Non-brain-penetrant drugs do not reach effective doses in BM,8,9 while liposomal-packaged drugs with increased brain penetration demonstrate increased efficacy.10,11 Thus, brain-penetrant, targeted systemic therapies are needed to effectively treat patients with TNBC BM.
The mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK/ERK) pathway is activated in both TNBC and BM,12,13 thus providing a possible target for therapeutic intervention. Activation of this pathway in BM enhances colonization, survival, and growth.13,14 TNBC specifically is initially sensitive to MEK inhibition in vivo,15,16 but resistance ensues without combinatorial therapy due to kinome reprogramming.17 Dual inhibition strategies involving MEK are needed for increased efficacy.16–19
Two potential compensatory pathways include the phosphatidylinositol-3 kinase (PI3K) and platelet-derived growth factor receptors (PDGFRα/β) pathways. Basal-like tumors exhibit the highest activation of the PI3K and PDGFR pathways among the breast cancer subtypes,12,20–22 and BM exhibit even greater activation of the PI3K pathway than primary breast tumors.13,23,24 PDGFR plays an important role in the epithelial-to-mesenchymal transition–mediated process of TNBC shifting from nonstem to cancer stem cells20 and in the neuro-inflammatory response by astrocytes to BCBM.25
Combination therapy must be achieved with brain-penetrant molecules to effectively treat patients with TNBC BM. We hypothesized that synthetic-lethal hits from a kinome small interfering (si)RNA screen with MEK inhibitors would be effective as combinatorial therapy in intracranial TNBC models. We investigated 3 clinically available brain-penetrant inhibitors against MEK1/2 (selumetinib, AZD6244),26,27 pan-PI3K (buparlisib, BKM120),19 and PDGFRα/β (pazopanib, Votrient),28,29 which have also shown promise in prior preclinical studies of extracranial TNBC. Selumetinib, combined with inhibition of PI3K or PDGFRβ, induces tumor regression and increases survival in primary TNBC mouse models.17,18 Buparlisib alone reduces tumor growth in a genetically engineered mouse TNBC model and patient-derived xenografts.30 In a mouse BCBM model, pazopanib reduces the number and size of brain metastases.14 Our results in 4 intracranial TNBC models provide preclinical data for early-phase clinical trials in TNBC BM patients.
Materials and Methods
Cell Lines
The human-derived TNBC cell lines SUM149 (Asterand; basal-like BRCA1-mutant, PTEN−), MDA-MB-231Br (Dr Toshiyuki Yoneda; claudin-low BRCA1-wildtype, PTEN-wildtype, KRAS-mutant, BRAF-mutant), MDA-MB-468 (American Type Culture Collection [ATCC]; basal-like BRCA1-wildtype, PTEN−), and MDA-MB-436 (ATCC; claudin-low BRCA1-mutant, PTEN−) (Fig. 1A) were transfected with luciferase vector under control of a cytomegalovirus promoter as described,31,32 were confirmed mycoplasma free (September 2015), and were verified by gene expression (September 201031,32). Cell lines were cultured in Invitrogen media with antibiotic-antimycotic additive and maintained at 37°C with 5% CO2: SUM149 in HuMEC + supplements + 5% fetal bovine serum (FBS), MDA-MB-468 in Roswell Park Memorial Institute 1640 medium + 10% FBS with plug seal capped flasks (Corning), MDA-MB-231Br and MDA-MB-436 in high glucose Dulbecco’s modified Eagle’s medium + 10% FBS. See Supplementary material for additional details.
Fig. 1.

Potency, efficacy, and synergy of buparlisib, selumetinib, and pazopanib in four TNBC human-derived cancer cell lines in vitro. (A) Utilized cell lines, with their molecular classifications and relevant known mutational statuses. Drug response curves of SUM149 (149), MDA-MB-231Br (231Br), MDA-MB-468 (468), and MDA-MB-436 (436) TNBC cells after 72 hours with (B) buparlisib, (C) selumetinib, or (D) pazopanib. Synergy fraction affected (FA) versus combination index (CI) curves quantification for combined (E) buparlisib+selumetinib or (F) selumetinib+pazopanib in 149, 231Br, 468, and 436. CI categories: synergistic: <0.1–0.9, additive: 0.9–1.1, antagonistic: >1.1.
Drugs
Buparlisib, selumetinib, and pazopanib (Chemietek) (Supplementary Table S1) were diluted in dimethyl sulfoxide (DMSO) in vitro. Dosing, schedule, and administration routes (Supplementary Table S1) were conducted as published for in vivo studies.14,18 As monotherapy, buparlisib (30 mg/kg/day) and selumetinib (37 mg/kg/day) were delivered in the chow, while pazopanib was given at 200 mg/kg by oral gavage daily. In combination, doses were reduced to 25 mg/kg/day buparlisib + 18 mg/kg/day selumetinib and 125/mg/kg/day pazopanib + 18 mg/kg/day selumetinib.
In Vitro Chemosensitivity Drug and Synergy Studies
Cells were harvested and plated into 384-well, flat-bottom plates (MicroFlo Select, Biotek Instruments) at a density of 600 (SUM149) or 550 (231Br) cells per well. For determinations of half-maximal inhibitory concentration (IC50), buparlisib, selumetinib, or pazopanib was added to cells 24 hours after plating on duplicate plates (Biomek FXP automated system, Beckman Coulter). For synergy studies, DMSO, selumetinib, and/or buparlisib or pazopanib was added to cells 24 hours after plating (Chou–Talalay method).33 Cell viability was assessed via CellTiterGlo (Promega) luminescence (Pherastar FS plate reader, BMG Labtech) at 72 hours after adding drug(s). See Supplementary material for additional details.
High-Throughput siRNA Screens for Synthetic Lethality
A human RNA interference (RNAi) library (Dharmacon)34 containing 4 pooled siRNAs against 720 kinases was applied to SUM149 and MDA-MB-231Br. siRNAs on 384-well plates were resuspended in media and transfection reagent (Dharmafect 2, Dharmacon, or RNAiMax, Invitrogen). After 30 minutes, 600 (SUM149) or 550 (231Br) cells per well were added. After 24 hours, media containing DMSO, buparlisib, or selumetinib were added to triplicate plates at the calculated IC25 concentration. Cell viability was assessed at 72 hours after drug addition. A validation screen with 58 siRNAs (Dharmacon) was then conducted.
In Vivo Efficacy and Pharmacodynamic Studies
In vivo studies were conducted as previously described31 according to the Institutional Animal Care and Use Committee (IACUC) (12.059, 15.038). Briefly, 200000 cells in 5 μL of 5% methylcellulose + culture media or phosphate buffered saline + 0.5% FBS were stereotactically injected into the right caudate nucleus of the basal ganglia using a 27-gauge needle. All 4 TNBC models achieved a >95% intracranial take rate. Drug doses and routes of administration (above, Supplementary Table S1) were based on previously conducted maximum tolerated dose studies.18 Treatment began the day after randomization (day 8 post-injection for 231Br, day 15 for all other models to normalize for differential growth rates between models and maintain treatment initiation at 30%–40% of respective median survivals). For pharmacodynamic studies, mice were sacrificed at 2 weeks posttreatment or due to poor health.31 Mice in the combination 5-2 groups were harvested following a 2-day drug holiday. For survival studies, mice were sacrificed due to poor health or prespecified post-injection date (per IACUC protocol). Tumor burden was assessed weekly following tumor cell implantation by IVIS Lumina Camera (Caliper Life Sciences) imaging of the luciferase-positive cells and quantified with Living Image 4.0 Software (Caliper Life Sciences) as photons/second after correction for background signal.31 Intracranial tumors were immediately dissected and stored at −80°C. For histology, whole brains were fixed in 10% formalin prior to processing for hematoxylin and eosin staining.31
Western Blot Analysis
Tumors were homogenized (Fisher PowerGen Model 125 Homogenizer) for 3 × 10 seconds in lysis buffer consisting of 1/2X phosphate buffered saline + 2.0% NP-40 + 1 mM DL-dithiothreitol (Sigma 43819) + Roche cOmplete Protease Inhibitor Cocktail (11697498001) + Phosphatase Inhibitor Cocktail II (Sigma P-5726) + Phosphatase Inhibitor Cocktail III (Sigma P-0044) per manufacturer’s instructions. Lysates were centrifuged for 12 minutes at 14000 relative centrifugal force at 4°C, and supernatants were stored at −80°C. Bicinchoninic acid assay (Pierce 23225) was used to calculate protein concentrations. Proteins were separated on NuPage Novex Midi 4–12% Bis-Tris sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis gels (ThermoFisher Scientific) prior to transfer to 0.45 micron Hybond-P polyvinylidene difluoride membranes (Amersham RPN303F). The following Cell Signaling (unless otherwise noted) antibodies were used per manufacturer’s instructions: pAkt (Ser473, 9271), total Akt (9272), pERK1/2 (p44/42 MAPK Thr202/Tyr204, 4370), total ERK1/2 (p44/42 MAPK, 4695), pMEK1/2 (Ser217/221, 9154), total MEK1/2 (9122), β-actin (Sigma A5316).
Multiplex Inhibitor Beads/Mass Spectrometry
Intracranial SUM149 tumors treated for 2 weeks (pharmacodynamic tumors) with control, buparlisib, selumetinib, or combination chow were lysed (n = 4/treatment) and processed for global activated kinome profiling as described previously.35 Briefly, lysates were flowed over affinity columns packed with kinase inhibitor resin, bound to kinase-bound inhibitor beads, washed with high- and low-salt buffers followed by 0.1% SDS, and eluted in buffer containing 0.5% SDS by boiling. Samples were purified using methanol chloroform extraction, trypsin digested, and cleaned via ethyl acetate extraction and Pepclean C-18 columns before being analyzed on a Thermo Q-Exactive ESI mass spectrometer. Spectra were searched using MaxQuant v1.5.1.2 software, and protein intensities were quantified as label free. Additional details are in the Supplementary material.
Statistical Analysis
Synthetic lethality screens
Screen cell viability data were quantile normalized and log transformed. Genes were tested between control and treatment with the significance analysis of microarray.36 Genes which decreased viability with treatment were prioritized with a false discovery rate ≤ 0.1. For the 231Br validation screen, a t-test for each gene was performed with P < 0.1 considered further.
IC50 and synergy
IC50, 50% growth inhibition (GI50), maximum inhibition (Imax), and Hill slope values were calculated (GraphPad Prism v6) from 2–5 independent experiments for each cell line. Synergy determinations were conducted according to the Chou–Talalay method.33 See the Supplementary material for additional details.
In vivo efficacy (survival and tumor burden)
Survival data were analyzed using the Kaplan–Meier method (GraphPad Prism). For the SUM149 and 231Br models, the results from 2 independent cohorts were pooled, as no significant differences were evident between cohorts. Additional details are in the Supplementary material.
Western blots
For 14-day treated tumors, ANOVA followed by paired t-tests with Tukey multiple correction was used to compare the ratios of the band intensities of phosphorylated to total protein levels for pERK/ERK, pAkt/Akt, and pMEK/MEK.
Multiplexed kinase inhibitor beads/mass spectrometry
Label-free quantification (LFQ) normalized values were filtered to kinases and ranked. Missing values were imputed via multiple imputation, assuming that the abundances of unobserved values were less than the observed LFQ values within a sample. For each kinase, 20 imputed datasets were generated, collated, and analyzed by Kruskal–Wallis. Results were combined using Rubin’s Rules. Pairwise comparisons between control versus treatment group were run using t-tests followed by false discovery rate. Adjusted P (Q) < 0.1 was considered significant. Kinome tree diagrams of treatment effects relative to controls were reproduced courtesy of Cell Signaling Technology (www.cellsignal.com). See Supplementary material for additional details.
Results
Synthetic Lethality Screens Identify Multiple Rational Co-targets with MEK Inhibition in TNBC
A synthetic enhancement of lethality screen was performed in 2 human-derived TNBC cell lines capable of forming intracranial tumors in mice, SUM149 and MDA-MB-231Br, using PI3K and MEK inhibitors (Fig. 1A). Exposure of SUM149 and MDA-MB-231Br (231Br) cells to the MEK1/2 inhibitor selumetinib or the pan-PI3K inhibitor buparlisib yielded several co-target kinase pathways that enhanced lethality (Table 1, Supplementary Table S2). Knockdown of genes within the same pathway as the individual drug treatment enhanced the effects of the drug. Increased lethality with selumetinib occurred when combined with siRNA-mediated knockdown of other MAPK/MEK pathway genes: BRAF, RAF1, MEK1, and MAP2K2/MEK2. Similarly, knockdown of PI3K pathway genes (AKT1, PIK3CB, AKT2, and RPS6KB1) enhanced the effects of buparlisib.
Table 1.
Synthetically lethal genes in the siRNA screens in SUM149 and MDA-MB-231Br TNBC cells with PI3K or MEK inhibition*
| Drug | Selumetinib | Buparlisib | ||||
|---|---|---|---|---|---|---|
| Cell Line | SUM149 | MDA-MB-231Br | Combined | SUM149 | MDA-MB-231Br | Combined |
| Dose (IC 25) | 200 nM | 5.04 µM | N/A | 810 nM | 850 nM | N/A |
| Gene “Hits” |
BRAF, RAF1 (CRAF), MAP4K3, MAP2K1 (MEK1), MAP2K5, MAPK10, MAPK11
PIK3C2A, PIK3C3, PIK3CB, PIK3R4 PDGFRB CDK2, CDK3, CDK5, CDK6, AURKB, BUB1B EPHA1, EPHA3, EPHA4, EPHA6, EPHB1, EPHB3, EPHB4, EFNB3 INSR, FLT1 (VEGFR1), FGFR4, ERBB4, TGFBR2 |
BRAF, RAF1 (CRAF), MAP4K1, MAP4K3, MAP3K5, MAP3K7,
MAP3K12, MAP2K2 (MEK2), MAP2K7, MAPK1 (ERK2), MAPK4 (ERK4), MAPK7, MAPK10, RPS6KA2 (RSK3), RPS6KA4 (MSK2), RPS6KA5 (MSK1) PIK3C2B, AKT2, RPS6KB1 (S6K1) PDGFRB CDK2, CDK3, CDK4, CDK5, CDK5R1, CDK7, CDK9, CDK11, CHEK2 EPHA1, EPHA3, EPHA4, EPHA10, EPHB1, EPHB3, EPHB4 INSR, EGFR, FLT1 (VEGFR3), TGFBR2 |
BRAF, RAF1 (CRAF), MAP4K1, MAP4K3, MAP3K1, MAP3K3, MAP3K5, MAP3K6, MAP3K7, MAP3K10, MAP3K12,
MAP2K1 (MEK1), MAP2K2 (MEK2), MAP2K4, MAP2K5, MAP2K6, MAP2K7, MAPK1 (ERK2), MAPK10, MAPK11, RPS6KA1 (RSK1), RPS6KA5 (MSK1), DUSP21 PIK3CB, PIK3C2B, PIK3C3, PIK3CD, PIK3R4 PDGFRA, PDGFRB CDK2, CDK3, CDK5, CDK5R1, CDK6, CDK9, CDK11, AURKB, PLK3, DDR1, CHEK2 EPHA1, EPHA3, EPHA4, EPHA6, EPHB1, EPHB3, EPHB4 INSR, INSRR, FLT1 (VEGFR1), FGFR4, TGFBR2 |
MAP2K1 (MEK1) AURKA, BUB1 |
BRAF, RAF1 (CRAF), TAO1 (TAOK1),
MAP4K3, MAP3K5, MAP3K13, MAP3K14, MAP2K7, MAPK6 (ERK3), RPS6KA1 (RSK1), RPS6KA3 (RSK2) AKT2, RPS6KB1 (S6K1) AURKC, BUB1, BUB1B, PLK3, DDR2 EPHB3 TGFBR1 |
BRAF, RAF1 (CRAF), TAO1 (TAOK1), MAP4K3, MAP3K5,
MAP3K14, MAP2K1 (MEK1), MAP2K2 (MEK2), MAP2K5, MAP2K7, MAPK1 (ERK2), MAPK10, RPS6KA1 (RSK1), RPS6KA3 (RSK2), RPS6KA4 (MSK2), DUSP21 PIK3CB, AKT2 AURKA, AURKC, BUB1, BUB1B, DDR2 EFNA3, EFNA5, EFNB3, EPHB1, EPHB4 ERBB3, TGFBR1 |
*Genes with common alternative names are in parentheses, and those related to the PI3K, MEK, or PDGFR pathways are in bold
Several promising genes emerged as synthetically lethal with MEK inhibitor treatment: PI3K subunit genes (PIK3C2B/C3/CB/CD), cell cycle regulators (CDK2/3/5/6, AURKB), and extracellular receptors (PDGFRα/β, EPHA1/3, EPHB1/3, INSR, VEGFR1) (Table 1, Supplementary Table S2). With PI3K inhibition, the MAPK cascade (TAO1, MEK1/MEK2, ERK2, RSK1/2, DUSP21), cell cycle regulation (AURKA), and extracellular receptors (EFNA3/B3, EPHB1, TGFβR1) (Table 1, Supplementary Table S2) were synthetically lethal. Both selumetinib and buparlisib treatment yielded cross-validation of the MEK+PI3K combination as being synthetically lethal in 2 of our 4 TNBC cell lines, and identified MEK+PDGFR as another possible combination.
Small Molecule PI3K, MEK, or PDGFR Inhibitors Reduce Cell Growth in Human-Derived TNBC Cell Lines
The effects of PI3K, MEK, or PDGFR inhibitors were evaluated in 4 TNBC cell lines (SUM149, 231Br, MDA-MB-436, and MDA-MB-468) representing both heterogeneous molecular subtypes and mutational backgrounds (Fig. 1A). Exposure to buparlisib (PI3Ki), selumetinib (MEKi), and pazopanib (PDGFRi) in vitro demonstrated differential patterns of drug responses based on cell viability. For buparlisib, all 4 models exhibited similar IC50 responses (1.3–1.9 µM) but variable GI50 (1.4–11.7 µM) and Imax (92%–49%) values (Fig. 1B, Supplementary Table S3). Selumetinib had a higher potency in the SUM149 and 231Br models (IC50: 0.8–19.3 µM) relative to the 468 and 436 models (IC50: 97.0–220.7 µM) (Fig. 1C, Supplementary Table S3). Pazopanib yielded similar IC50 (4.1–10 µM) but was relatively ineffective (GI50: never reached; Imax: 13%–30%; Fig. 1D, Supplementary Table S3).
Synergistic Combination of PI3K+MEK and PDGFR+MEK
In comparing the combination index (CI) to fraction affected (FA), dual buparlisib+selumetinib exposure was synergistic. Synergism occurs in all 4 cell lines (Fig. 1E). This synergy was generally greater (lower CI) in the SUM149 and 231Br lines compared with the 468 and 436 lines and occurred at lower absolute drug concentrations, due to the relatively lower IC50 concentrations in the SUM149 and 231Br lines (Fig. 1E). Similar results were obtained with the selumetinib+pazopanib combination. In the SUM149 and 231Br models, dual selumetinib+pazopanib treatment was synergistic at or below the IC50, in contrast to the 468 and 436 models (Fig. 1F). Thus, both the PI3K+MEK and MEK+PDGFR combinations are synergistic at potentially physiologically relevant concentrations in SUM149 and 231Br, but not 468 and 436 cell lines.
PI3K+MEK Inhibition Improves Survival and Reduces Tumor Burden in Some Intracranial TNBC Models
We tested the survival and efficacy of buparlisib ± selumetinib in mice with intracranial TNBC (Fig. 2A). In the SUM149 model, survival significantly improved from controls (45 days) with buparlisib (52 days; P = 0.003) and selumetinib (72 days, P = 0.004; Fig. 2B, C). A similar effect on tumor burden was observed in response to single agents, with buparlisib slightly reducing and selumetinib greatly reducing tumor burden relative to controls (Supplementary Fig. S1A, B). Daily buparlisib+selumetinib modestly improved survival (50 days, P = 0.023), but with continued daily weight loss. Thus, we tested a 5 days on/2 days off schedule (5-2), which significantly improved survival while avoiding toxicity (87 days; P of 5-2 vs control: <0.0001; buparlisib: 0.0077; selumetinib: 0.17; Fig. 2B, C). Further, a third (4/12) of the mice on 5-2 combination treatment were alive at the end of the study compared with 0/11 mice in the controls, 1/13 with buparlisib, and 3/13 with selumetinib. Reduction in tumor burden was most profound in the daily combination treatment group (Supplementary Figs S1B, S2A–F).
Fig. 2.

In vivo efficacy of PI3K ± MEK inhibition in established intracranial TNBC. (A) Experimental schedule. (B) The median survival (MS) and number of mice alive at the end of the study for mice with intracranial SUM149, MDA-MB-231Br, MDA-MB-468, or MDA-MB-436 tumors. Median survival (MS) for treatment groups statistically different from control (*) or buparlisib (^). Kaplan–Meier curves by treatment group for the (C) SUM149, (D) MDA-MB-231Br, (E) MDA-MB-468, and (F) MDA-MB-436 models. Vertical dotted lines indicate the study’s end.
The MDA-MB-231Br model demonstrated a similar sensitivity to the buparlisib+selumetinib combination. Buparlisib alone did not improve survival (29 days) relative to the control group (30 days; P = 0.39), but selumetinib did (37 days; P = 0.014; Fig. 2B, D). Again, toxicity from daily buparlisib+selumetinib precluded improved survival (31 days; P = 0.31), but administration on a 5-2 schedule increased survival (55 days; P of 5-2 vs controls: <0.0001; buparlisib: 0.0004; selumetinib: 0.058; overall P < 0.0001). Treatment with selumetinib, alone or combined with buparlisib, reduced tumor burden relative to controls (Supplementary Figs S1C, S2G–L).
In contrast, the MDA-MB-468 and MDA-MB-436 intracranial TNBC murine models did not show significant improvements in survival or reduction in tumor burden with buparlisib and/or selumetinib (overall P 468: 0.12; 436: 0.35; Fig. 2B, E, F; Supplementary Figs S1D–E, S2M–X).
Buparlisib and Selumetinib Differentially Inhibit Downstream Targets in Intracranial TNBC Tumors
Target inhibition was compared in 231Br (responsive) and 468 (resistant) intracranial tumors after 14 days of treatment. The ratio of phosphorylated to total protein of ERK/MEK and Akt were used as markers of MEK and PI3K pathway inhibition, respectively. Intracranial 231Br tumors treated with selumetinib ± buparlisib daily had significantly reduced pERK/ERK ratios compared with controls (overall P = 0.025) (Fig. 3A, B). Similarly, daily exposure to buparlisib and/or selumetinib significantly reduced pAkt/Akt ratios in 231Br intracranial tumors (overall P = 0.0011) (Fig. 3A, C). Finally, selumetinib alone or combined with buparlisib on a daily or 5-2 schedule maintained MEK inhibition in the 231Br model (overall P = 0.013) (Fig. 3A, D). In contrast, phosphorylated and total levels of ERK or Akt in the 468 model were similar across treatment groups (Supplementary Fig. S3A–C), while pMEK/MEK ratios increased with selumetinib treatment (overall P = 0.017; Supplementary Fig. S3D). Thus, the responsive 231Br demonstrates target inhibition and improved survival, while the resistant 468 model maintains MEK and PI3K signaling despite ongoing treatment.
Fig. 3.

Western blot analysis of MEK ± PI3K inhibitor treatment in MDA-MB-231Br. (A) Immunoblots of phosphorylated (p-) and total ERK, Akt, and MEK in control, selumetinib, buparlisib, combination (“combo”) daily, and combo 5-2 treated tumors, n = 4 per group. Quantitation of (B) pERK to total ERK, (C) pAkt to Akt, and (D) pMEK to MEK levels by densitometry of immunoblot bands from (A). Overall P shown; *pairwise vs control P ≤ 0.05.
Widespread Reprogramming of the Kinome Follows PI3K and/or MEK Treatment in Intracranial SUM149 Tumors
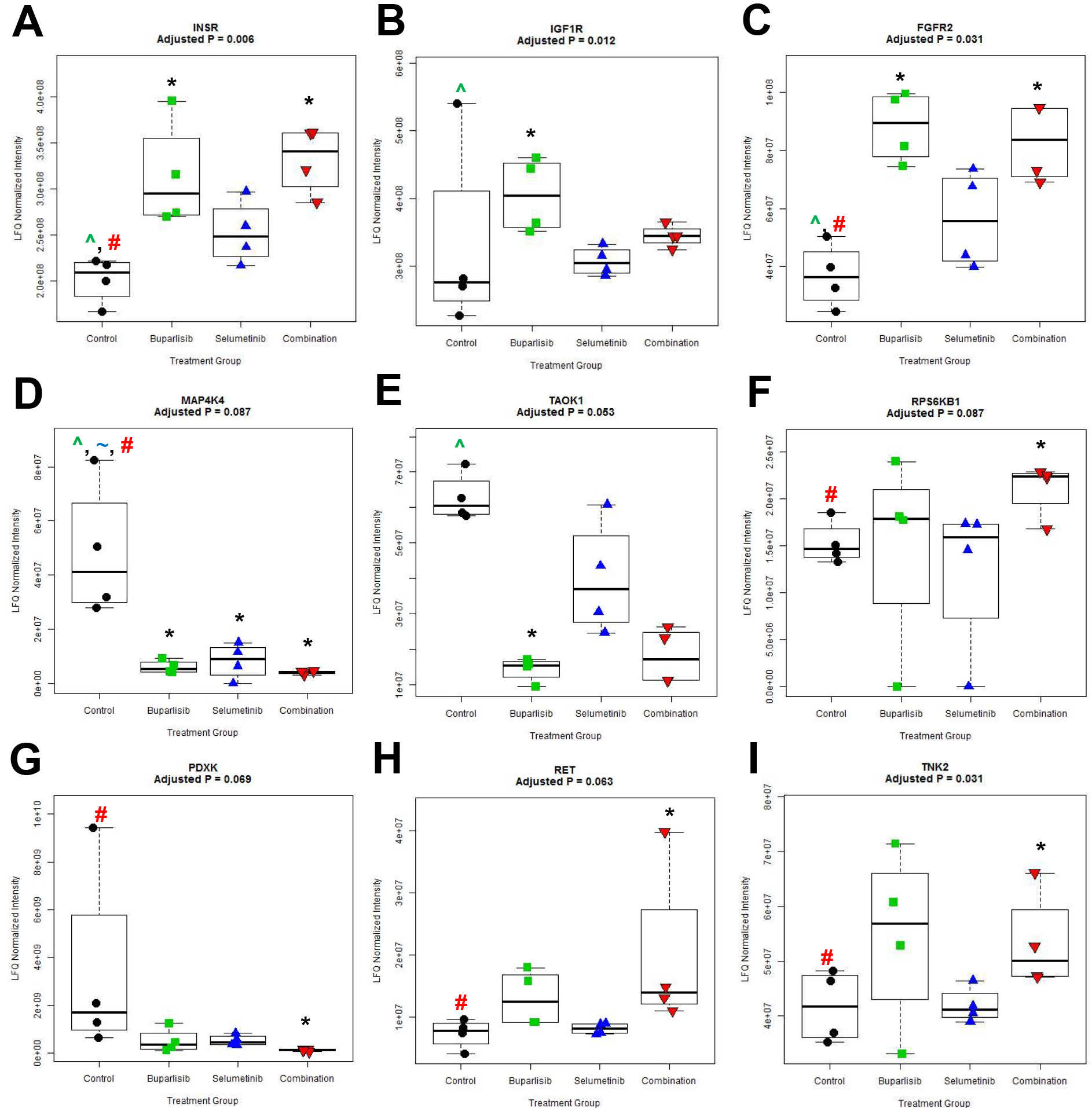
An affinity chromatography technique using multiplexed inhibitor beads (MIB) coupled with mass spectrometry (MS) was used to assess the functional kinome in response to drug treatment. In intracranial SUM149 tumors, widespread alterations in kinase binding to the beads occurred, but with varying kinome families activated with each treatment (Fig. 4). Forty-eight of 251 kinases captured were significantly different across treatments (Q < 0.1, Supplementary Table S4). Of these, 9/48 kinases overlapped in at least one pairwise comparison between control and treatment. Relative to control-treated tumors, buparlisib significantly altered the levels of 5 kinases: insulin receptor (INSR), insulin-like growth factor 1 receptor (IGF1R), fibroblast growth factor receptor 2 (FGFR2), mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4), and thousand-and-one amino acid kinase 1 (TAOK1) (Supplementary Fig. S4A–E, Supplementary Table S5). Selumetinib treatment caused a reduction only in MAP4K4 (Supplementary Table S6). In contrast, buparlisib+selumetinib significantly altered binding of the most responsive kinases across all treatment groups (n = 7/9) relative to controls: INSR, FGFR2, MAP4K4, ribosomal protein S6 kinase B1 (RPS6KB1), pyridoxine, vitamin B6 kinase (PDXK), the “rearranged during transfection” gene (RET), and tyrosine nonreceptor kinase 2 (TNK2) (Supplementary Fig. S4A, C–D, F–I, Supplementary Table S7).
Fig. 4.

Kinome alteration following 2 weeks of treatment with buparlisib, selumetinib, or combination in intracranial SUM149 TNBC tumors. (A) Fold change of kinases comparing buparlisib (green), selumetinib (blue), or combination (red) relative to controls. Only kinases with a Log2 fold change of >0.5 or <−0.5 in any treatment group relative to controls are shown. B‒D. Kinome tree diagrams of altered kinases following (B) buparlisib, (C) selumetinib, and (D) combination, colored by fold change relative to controls: ≥2x (red), 1.5x–2x (pink), ≤0.5x (blue), unchanged (black), detected only in treatment (gray).
PDGFR+MEK Inhibition as a Tractable Therapeutic Strategy in Intracranial TNBC
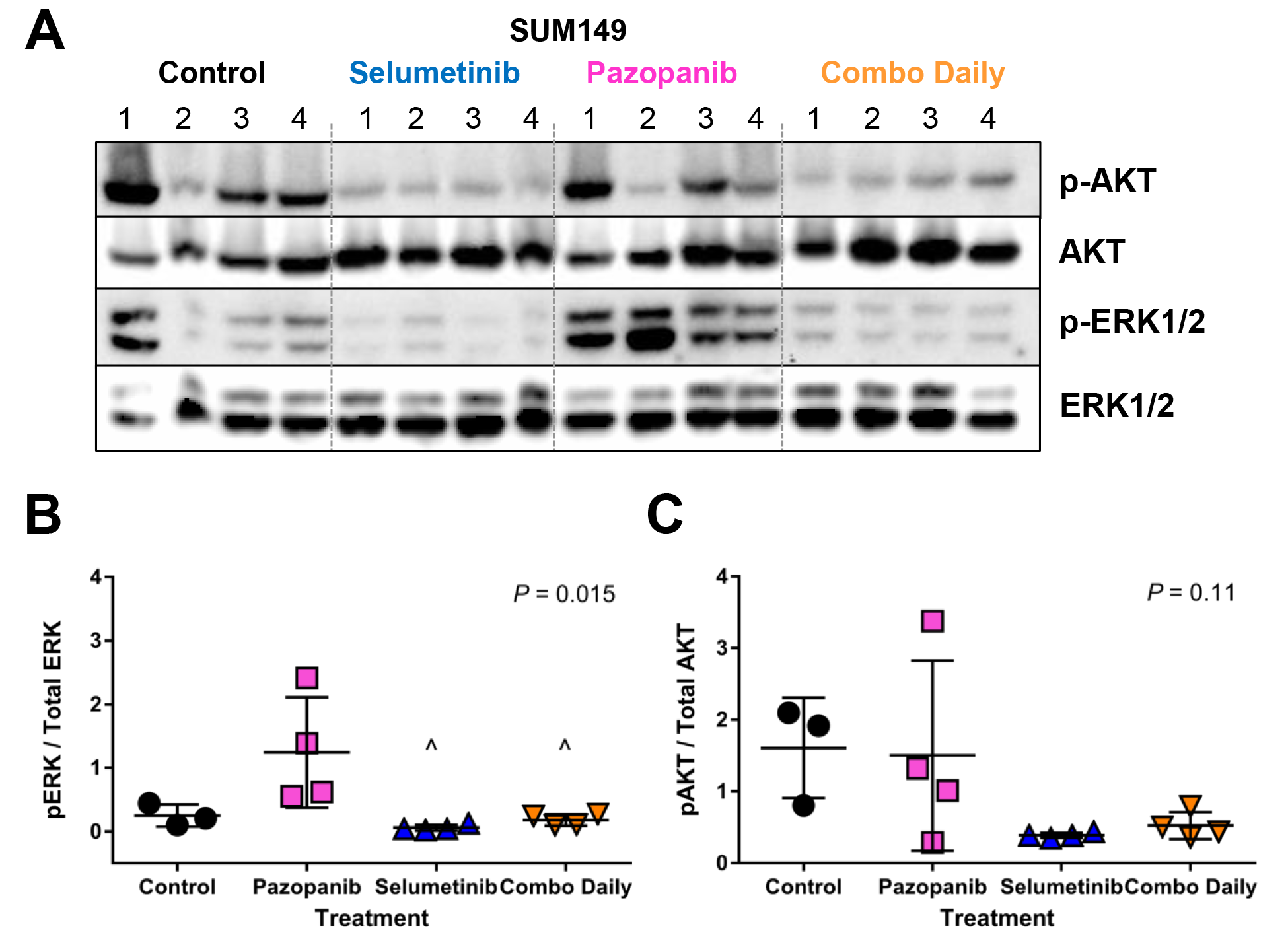
As a result of the siRNA kinome screen and in vitro synergy studies, we also evaluated PDGFR+MEK in SUM149 and 231Br. In the SUM149 model, pazopanib (PDGFRi) did not improve median survival (37.5 vs 34 days in controls; overall P < 0.0001; pairwise P = 0.66), whereas selumetinib demonstrated efficacy (63 days; P < 0.0001) (Fig. 5A‒B). Combined daily pazopanib+selumetinib significantly improved survival (92.5 days; P < 0.0001) and drastically reduced tumor burden (Fig. 5C). Western blot analysis showed reduction of pERK/ERK with 2 weeks of selumetinib or combination treatment (P = 0.015), but no change in pAkt/Akt (Supplementary Fig. 5C).
Fig. 5.

Efficacy of PDGFR+MEK inhibition. (A) Median survival (MS) and number of mice alive at the study’s end for intracranial SUM149 and MDA-MB-231Br tumors, statistically different from control (*), pazopanib (^), selumetinib (~). SUM149 (B) MS and (C) in vivo intracranial tumor burden by treatment group. MDA-MB-231Br (D) MS and (E) tumor burden by treatment group. Vertical dotted lines indicate study’s end per IACUC-approved protocols. N/D = not determined.
Results were more modest in the 231Br model. Single agent pazopanib did not alter median survival (24 days vs 28 days in controls; overall P = 0.0072; pairwise P = 0.94) (Fig. 5A, D). Pazopanib+selumetinib modestly improved survival (38 days; P = 0.016) and reduced tumor burden (Fig. 5E).
Discussion
Herein, we demonstrate hypothesis-driven discovery of 2 rational combinatorial approaches for the treatment of intracranial triple-negative breast cancer (TNBC). Examining 4 intracranial TNBC models, the combination of MEK+PI3K and MEK+PDGFR inhibitors increased survival and decreased tumor burden in 2 models and induced kinome-wide alterations in 1 model. We demonstrate effective intracranial tumor penetration of and a durable response to the combination of these 3 clinically approved therapies in 2 models.
This work supports growing appreciation that combination therapy must be utilized to effectively target cancer. Due to dynamic resistance mechanisms in breast cancer, monotherapy is not an effective strategy for treating TNBC. Concurrent inhibition of MEK+PI3K or MEK+PDGFRα/β has been suggested as a rational partnering strategy in primary breast cancer.17,18 This study is the first, to our knowledge, to demonstrate efficacy of rational MEK inhibitor combination strategies in multiple orthotopic models of TNBC brain metastases, established or otherwise.
Our data illustrate potentially targetable resistance mechanisms to kinase inhibitors, including the INSR/IGF1R and FGFR2 pathways. Prior literature has demonstrated heterodimerization of INSR+IGF1R and IGF1R+PDGFR receptors, activating the PI3K and MAPK pathways.37 Interestingly, buparlisib+selumetinib treatment in the SUM149 model also increased 2 kinases, RET and TNK2, associated with both neuronal/synaptic functions and cancer progression.38,39 Alteration of neuronal kinases as resistance mechanisms adds to recent literature suggesting a “breast-to-brain” transition wherein cancer cells express neuronal-like features that confer survival advantages.40,41
The results of the present study should be interpreted in the context of the following limitations. Substantial toxicity of the MEK+PI3K inhibitor treatment, as seen in our data, is an important consideration in the clinical applicability of this approach. Continued weight loss (data not shown) and ultimate demise of the animals (Fig. 2C‒D) despite stable tumor burden (Supplementary Fig. S1B–C) in both the SUM149 and MDA-MB-231Br models occurred on daily combination therapy. As has been reported in the clinical setting, the incorporation of a weekly drug holiday significantly improved survival by abating toxicity.42,43 MEK+PI3K inhibition has dose limiting toxicities of stomatitis, diarrhea, and creatinine kinase elevation in <10% of patients, with many patients (65%) exhibiting grade 3–4 adverse events of those above plus aspartate aminotransferase/alanine aminotransferase elevations and rashes.43 Pazopanib induces similar manageable gastrointestinal events (ie, diarrhea, nausea, vomiting, and anorexia) and fatigue.44,45 Further investigation into strategies that manage toxicity while maintaining efficacy with these treatments are needed. Secondly, our direct intracranial implantation method addresses whether these agents are effective in treating established BM, mimicking the current clinical approach, and not leptomeningeal disease or as a preventative measure blocking TNBC brain metastases. Finally, the use of immunocompromised mice precludes study on the effects of MEK inhibition on the immune system and its involvement in the treatment response, a direction worthy of further study.
The present study demonstrates that models of established intracranial TNBC exhibit differential sensitivity to targeted therapies, highlighting the need to test multiple models when evaluating potential treatment strategies. The differential responses in our models are likely due in part to the molecular heterogeneity of TNBC, which comprises as many as 6 subtypes, each with unique responsiveness to treatment and outcome.46,47 We continue to explore the underlying mechanism of differential sensitivity to MEK inhibition in intracranial TNBC. Identification and validation of biomarkers predictive of sensitivity or resistance to MEK inhibition will foster the most effective translation of our preclinical findings to the design of biomarker-driven, early-phase clinical trials to more effectively treat patients with TNBC BM.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This research was supported by the Breast Cancer Research Foundation-AACR (10-60-26-ANDE, to C.K.A.), NCI (K23157728, to C.K.A.), the Mary Kay Ash Charitable Foundation (to C.K.A.), NCI (CA16086, to C.S.), and the Damon Runyon Cancer Research Foundation (CI-64-12 CKA; CI-45-09, to C.R.M.). The content is solely the responsibility of the authors.
Conflict of interest statement. C.K.A. is an uncompensated consultant/advisory board member for Novartis, Sanofi, to-BBB, GERON, Angiochem, Merrimack, Lily, Genentech, Nektar, and Kadmon and receives unrelated research funding from Novartis, Sanofi, to-BBB, GERON, Angiochem, Merrimack, PUMA, Lily, Merck, and Oncothyreon. The other authors have no potential conflicts of interest to disclose.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank Dr Toshiyuki Yoneda (Osaka University) for providing MDA-MB-231Br cells. Animal studies were performed within the Animal Studies Core Facility, supported in part by an NCI Center Core Support Grant (CA16086) to the Lineberger Comprehensive Cancer Center. We also appreciate the expertise of the Mouse Phase 1 Unit.
References
- 1. Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009;9(Suppl 2):S73–S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. [DOI] [PubMed] [Google Scholar]
- 3. Harrell JC, Prat A, Parker JS, et al. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat. 2012;132(2):523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113(10):2638–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Niwińska A, Murawska M, Pogoda K. Breast cancer brain metastases: differences in survival depending on biological subtype, RPA RTOG prognostic class and systemic treatment after whole-brain radiotherapy (WBRT). Ann Oncol. 2010;21(5):942–948. [DOI] [PubMed] [Google Scholar]
- 6. Siegel MB, Van Swearingen AED, Anders CK. Approaches for optimal drug development and clinical trial design for breast cancer brain metastasis. Oncology. 2014;28(7):579, 584–575. [PMC free article] [PubMed] [Google Scholar]
- 7. Muldoon LL, Soussain C, Jahnke K, et al. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol. 2007;25(16):2295–2305. [DOI] [PubMed] [Google Scholar]
- 8. Lockman PR, Mittapalli RK, Taskar KS, et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res. 2010;16(23):5664–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taskar KS, Rudraraju V, Mittapalli RK, et al. Lapatinib distribution in HER2 overexpressing experimental brain metastases of breast cancer. Pharm Res. 2012;29(3):770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anders CK, Adamo B, Karginova O, et al. Pharmacokinetics and efficacy of PEGylated liposomal doxorubicin in an intracranial model of breast cancer. PLoS One. 2013;8(5):e61359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sambade M, Deal A, Schorzman A, et al. Efficacy and pharmacokinetics of a modified acid-labile docetaxel-PRINT(®) nanoparticle formulation against non-small-cell lung cancer brain metastases. Nanomedicine (Lond). 2016;11(15):1947–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Da Silva L, Simpson PT, Smart CE, et al. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res. 2010;12(4):R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gril B, Palmieri D, Qian Y, et al. Pazopanib reveals a role for tumor cell B-Raf in the prevention of HER2+ breast cancer brain metastasis. Clin Cancer Res. 2011;17(1):142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jing J, Greshock J, Holbrook JD, et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther. 2012;11(3):720–729. [DOI] [PubMed] [Google Scholar]
- 16. Mirzoeva OK, Das D, Heiser LM, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;69(2):565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duncan JS, Whittle MC, Nakamura K, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149(2):307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roberts PJ, Usary JE, Darr DB, et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res. 2012;18(19):5290–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maira SM, Pecchi S, Huang A, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–328. [DOI] [PubMed] [Google Scholar]
- 20. Tam WL, Lu H, Buikhuisen J, et al. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell. 2013;24(3):347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jansson S, Bendahl PO, Grabau DA, et al. The three receptor tyrosine kinases c-KIT, VEGFR2 and PDGFRα, closely spaced at 4q12, show increased protein expression in triple-negative breast cancer. PLoS One. 2014;9(7):e102176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marty B, Maire V, Gravier E, et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008;10(6):R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adamo B, Deal AM, Burrows E, et al. Phosphatidylinositol 3-kinase pathway activation in breast cancer brain metastases. Breast Cancer Res. 2011;13(6):R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salhia B, Kiefer J, Ross JT, et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS One. 2014;9(1):e85448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gril B, Palmieri D, Qian Y, et al. Pazopanib inhibits the activation of PDGFRβ-expressing astrocytes in the brain metastatic microenvironment of breast cancer cells. Am J Pathol. 2013;182(6):2368–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase ½ inhibitor. Clin Cancer Res. 2007;13(5):1576–1583. [DOI] [PubMed] [Google Scholar]
- 27. Chen HY, Yang YM, Han R, Noble M. MEK1/2 inhibition suppresses tamoxifen toxicity on CNS glial progenitor cells. J Neurosci. 2013;33(38):15069–15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwamoto FM, Lamborn KR, Robins HI, et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro Oncol. 2010;12(8):855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amiri-Kordestani L, Tan AR, Swain SM. Pazopanib for the treatment of breast cancer. Expert Opin Investig Drugs. 2012;21(2):217–225. [DOI] [PubMed] [Google Scholar]
- 30. Juvekar A, Burga LN, Hu H, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2(11):1048–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karginova O, Siegel MB, Van Swearingen AE, et al. Efficacy of carboplatin alone and in combination with ABT888 in intracranial murine models of BRCA-mutated and BRCA-wild-type triple-negative breast cancer. Mol Cancer Ther. 2015;14(4):920–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prat A, Karginova O, Parker JS, et al. Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res Treat. 2013;142(2):237–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. [DOI] [PubMed] [Google Scholar]
- 34. Zawistowski JS, Nakamura K, Parker JS, Granger DA, Golitz BT, Johnson GL. MicroRNA 9-3p targets β1 integrin to sensitize claudin-low breast cancer cells to MEK inhibition. Mol Cell Biol. 2013;33(11):2260–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stuhlmiller TJ, Miller SM, Zawistowski JS, et al. Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 2015;11(3):390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. King H, Aleksic T, Haluska P, Macaulay VM. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat Rev. 2014;40(9):1096–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Runeberg-Roos P, Saarma M. Neurotrophic factor receptor RET: structure, cell biology, and inherited diseases. Ann Med. 2007;39(8):572–580. [DOI] [PubMed] [Google Scholar]
- 39. Mahajan K, Mahajan NP. ACK1/TNK2 tyrosine kinase: molecular signaling and evolving role in cancers. Oncogene. 2015;34(32):4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Swearingen AE, Siegel MB, Anders CK. Breast cancer brain metastases: evidence for neuronal-like adaptation in a ‘breast-to-brain’ transition? Breast Cancer Res. 2014;16(3):304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neman J, Termini J, Wilczynski S, et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A. 2014;111(3):984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jokinen E, Koivunen JP. MEK and PI3K inhibition in solid tumors: rationale and evidence to date. Ther Adv Med Oncol. 2015;7(3):170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bedard PL, Tabernero J, Janku F, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21(4):730–738. [DOI] [PubMed] [Google Scholar]
- 44. Santoni M, Conti A, De Giorgi U, et al. Risk of gastrointestinal events with sorafenib, sunitinib and pazopanib in patients with solid tumors: a systematic review and meta-analysis of clinical trials. Int J Cancer. 2014;135(4):763–773. [DOI] [PubMed] [Google Scholar]
- 45. de Jonge MJ, Hamberg P, Verweij J, et al. Phase I and pharmacokinetic study of pazopanib and lapatinib combination therapy in patients with advanced solid tumors. Invest New Drugs. 2013;31(3):751–759. [DOI] [PubMed] [Google Scholar]
- 46. Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lehmann BD, Pietenpol JA. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast. 2015;24(Suppl 2):S36–S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.