

Abstract
Neuroinflammation is one of the hallmarks of Alzheimer's disease pathology. Amyloid β has a central role in microglia activation and the subsequent secretion of inflammatory mediators that are associated with neuronal toxicity. The recognition of amyloid β by microglia depends on the expression of several receptors implicated in the clearance of amyloid and in cell activation. CD36 receptor expressed on microglia interacts with fibrils of amyloid inducing the release of pro-inflammatory cytokines and amyloid internalization. The interruption of the interaction CD36-amyloid β compromises the activation of microglia cells. We have developed and validated a new colorimetric assay to identify potential inhibitors of the binding of amyloid β to CD36. We have found 7 molecules, structural analogues of the Trichodermamide family of natural products that interfere with the interaction CD36-amyloid β. Molecular docking simulations have suggested the large luminal hydrophobic tunnel, present in the extracellular domain of CD36, as a target of these compounds. These molecules also inhibited the production of TNF-α, IL-6 and IL-1β by peritoneal macrophages stimulated with fibrils of amyloid β. This work serves as a platform for the identification of new potential anti-inflammatory agents for the treatment of Alzheimer's disease.
Keywords: Alzheimer's disease, amyloid β, CD36, Trichodermamide, microglia, inflammation
Introduction
Alzheimer's disease (AD) is the most common form of dementia among the elderly and has become a main public health problem worldwide. This neurodegenerative disorder is characterized by the presence of amyloid beta (Aβ) aggregates, neurofibrillary tangles, and inflammation. The inflammatory response present in AD pathology shows the existence of activated glial cells, cytokines production and complement activation 1-3. Fibrillar Aβ (fAβ) recognition by microglia is believed to be one of the first steps in the production of pro-inflammatory mediators that contributes to AD pathogenesis. Several receptors expressed on microglia have been implicated in the response to Aβ 4,5. These receptors cooperate in the recognition, internalization, and clearance of Aβ and in cell activation.
The scavenger receptor CD36 is expressed by microglia and has been reported as a key element in Aβ recognition in AD pathogenesis 6, 7. This receptor appears to have a dual role in AD as it is involved in both the uptake of Aβ and microglia activation, with the consequent production of pro-inflammatory mediators 7, 8 .The interaction of Aβ with CD36 triggers a cascade of intracellular events that, when interrupted, leads to inhibition of macrophages response in vitro and microglia recruitment in vivo 6. It has been demonstrated that the inflammatory response induced by CD36 after Aβ engagement depends on the assembly of a complex composed of CD36, TLR4 and TLR6 9. This complex appears to be also involved in NLRP3 activation and in the release of IL-1β induced by Aβ 9, 10. Previous studies have identified, by means of a cell-based assay, that ursolic acid is an inhibitor of the Aβ-CD36 interaction and reactive oxygen species (ROS) production in N9 microglia cells 11. A hexapeptide member of the growth hormone-releasing peptides (GHRPs), hexarelin, has been identified as a ligand for CD36 12 and was later demonstrated to interfere with the response of microglia cells to Aβ 13 . Thus, the discovery of small molecules for pharmacological inhibition of the interaction of CD36 and Aβ may be important both to gain understanding of the role of CD36 in pathogenesis of AD and to develop new potential therapeutic agents for this disease.
Many efforts are underway to develop new drugs for stopping or slowing AD progress. The current approved drugs are only a palliative for the management of symptoms and not to cure the disease. Most studies for AD drug discovery are based on the clearance of Aβ by using different approaches, which so far have shown limited success. Recent studies have generated a specific Aβ antibody, aducanumab, which reduced brain Aβ in patients with prodromal or mild AD 14. However, the search for new molecules that may lead to the development of new treatments is still urgent. Existing data shows the need to explore immunomodulation as a new promising target for AD treatment.
Here, we report the development of a colorimetric assay for the screening of molecules that interfere with the CD36-Aβ interaction. We then choose to test a subclass of nitroxy compounds for their capacity to inhibit this interaction. Nitroxy compounds have recently emerged as a privileged scaffold for applications in drug discovery 15, 16. In particular, 1,2-oxazines represent a new structural framework with significant potential for structure diversification. We focused on the Trichodermamide family of natural products that share the 1,2-oxazadecaline unit 17. We choose Trichodermamides A (1) and B (2) (Figure 1) as well as a focused library of structural analogues (Table 1 and S1). From the 24 compounds tested, 7 were able to interfere with the binding of Aβ to CD36 with IC50 values ranging from 11 to 43 μM. Molecular docking simulations supported the strong interaction of Trichodermamide analogues with CD36. All the identified active compounds target a large luminal hydrophobic tunnel present in the extracellular domain of CD36. These molecules also inhibited the production of inflammatory mediators induced by Aβ in macrophages. The assay described herein will allow for rapid identification and further characterization of new molecules as potential agents for the development of novel AD therapeutics. This work adds to the efforts for the identification and characterization of new immunomodulatory agents that may help to stop or delay the progression of the disease.
Figure 1. Trichodermamides A (1) and B (2).
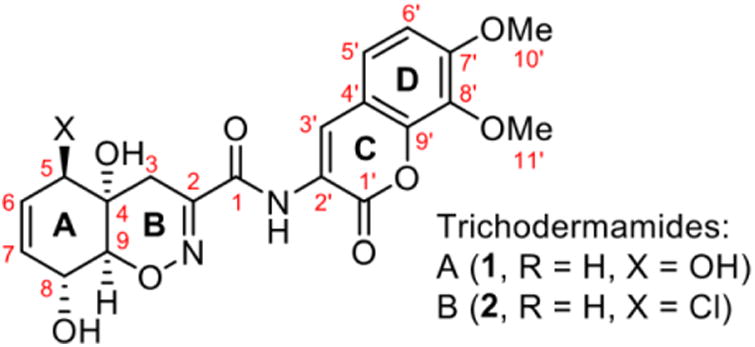
Table 1.
Trichodermamide analogues inhibit the interaction CD36-Aβ.
| Compound Code | 2D Structure | IC50 (μM) | ΔGa (kcal/mol) |
|---|---|---|---|
| 3 |
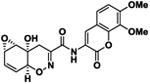
|
20.3 ± 1.2 | -8.2 |
| 4 |
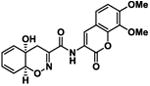
|
16.7 ± 6.3 | -8.6 |
| 5 |
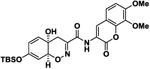
|
11.1 ± 1.3 | -8.6 |
| 6 |
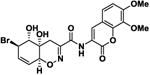
|
40.5 ± 10.5 | -7.9 |
| 7 |
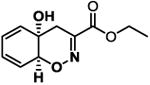
|
11.5 ± 0.86 | -6.9 |
| 8 |
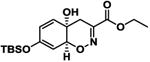
|
11.0 ± 3.6 | -9.7 |
| 9 |
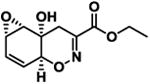
|
43.1 ± 4.5 | -7.9 |
IC50 values are mean ± standard deviation of three independent experiments.
Binding free energy value of each inhibitor calculated using the Autodock Vina scoring function. The control drug was ursolic acid (IC50 = 98.14 μM, ΔG = -6.0 kcal/mol)
Results
Recombinant expression of the extracellular domain of the human CD36 protein
The genetic construct pET30CD36 was assembled to place the open reading frame coding for the extracellular portion of CD36 under the transcriptional control of the bacteriophage T7 promoter (Figure 2A). This construct is also designed to produce the CD36 fused to an N-terminal peptide containing a 6×His tail to aid in the purification of the protein. When transformed into the E. coli strain BL21 (DE3), the expression plasmid is able to drive an IPTG inducible, high level expression of the CD36 fusion protein at the expected molecular weight of ∼52.3 kDa (Figure 2B). The recombinant CD36 was associated with the insoluble fraction of the lysate (data not shown), allowing a fast purification pipeline which includes inclusion body isolation, solubilization in denaturing conditions and affinity purification through affinity Ni-NTA chromatography (Figure 2C). After elution of the denatured protein, refolding was achieved by rapid dilution into PBS and further concentration using ultrafiltration. The final protein preparation was evaluated by SDS PAGE and protein concentration estimation, showing a single major band at the correct size with more than 90% purity, according to gel densitometry (Figure 2C).
Figure 2. Expression of recombinant extracellular human CD36 in Escherichia coli.
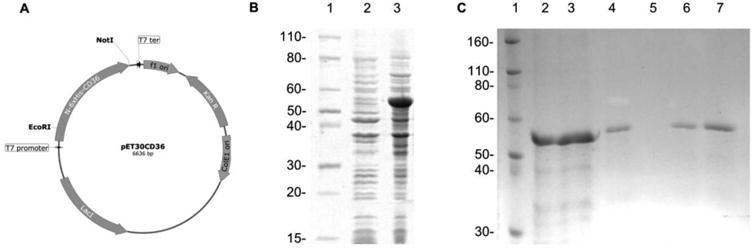
(A) Expression plasmid for IPTG inducible expression. (B) Coomassie stained SDS-PAGE of uninduced (2) and induced culture (3). (C) Coomassie stained SDS-PAGE of purification steps showing cleared lysate (2), column flow-through (3), first wash (4), second wash (5), first elution (6) and second elution (7). Molecular weight markers are indicated in kilodalton, lanes B1and C1.
Development of a colorimetric assay for the detection of inhibitors of CD36-Aβ interaction
The rCD36 immobilized in a 96-well plate was used for the development of the colorimetric assay. It was first established the optimum concentrations of rCD36 and fibrils of human Aβ 1-42 (fAβ) by using checkerboard titrations 18. Results showed a dose response behavior in the binding of rCD36 to fAβ (Figure 3A and B). Concentrations of 15 μg/ml and 0.8 μM for rCD36 and fAβ respectively were selected for further experiments. The primary and secondary antibodies concentrations were similarly titrated to optimize the assay conditions. Results indicated optimum concentration at 0.3 μg/ml for the anti-Aβ and 1:4000 for the HRP-conjugated secondary antibody (Figure 3C and D). The substitution of human fAβ for mouse fAβ in the assay did not alter the results (data not shown).
Figure 3. Immobilized human rCD63 is able to bind fibrillar Aβ in an in vitro binding assay.
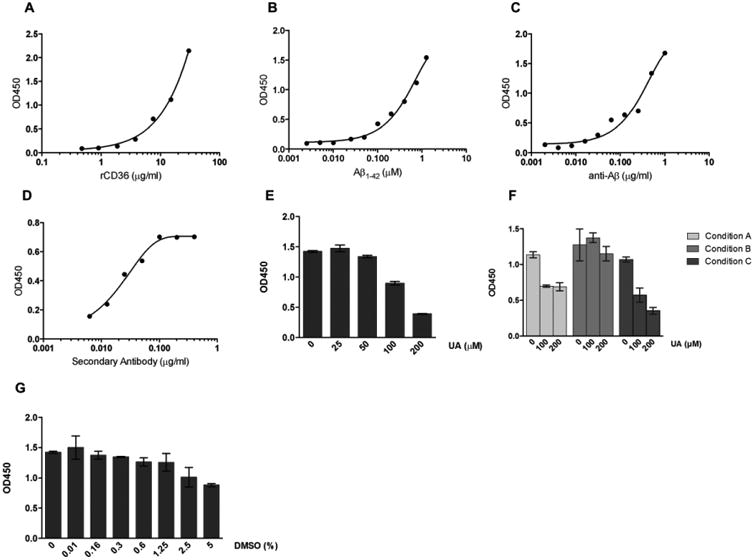
Assay parameters were optimized to achieve best possible signal to noise ratio. (A-D) Curves from checkerboard titrations to determine optimal concentrations for rCD36 (A), fibrillar Aβ (B), anti-Aβ primary antibody (C) and secondary anti-species conjugated antibody (D). (E) Dose response graph of the Ursolic acid (UA) positive control. (F) Optimization of time of addition of inhibitors in the assay. Condition A: the addition of UA and fAβ 1-42 at the same time; Condition B: UA added 30 minutes before and removed just before adding the fAβ 1-42 and; Condition C: UA added 30 minutes before fAβ 1-42 addition. (G) Sensitivity of the assay to DMSO. Results represent means ± S.D. from samples assayed in duplicate.
Ursolic acid (UA) has been previously described and characterized by a cell-based assay as an inhibitor of CD36-Aβ interaction 11. These authors demonstrated also that this compound inhibits the production of ROS induced by Aβ in N9 microglia cells. Therefore, we decided to use UA as positive control of inhibition in our assay. This compound inhibits the interaction of CD36 with Aβ in a dose dependent manner, with an IC50 value of 98.14 μM (Figure 3E and F). Based on this finding we used UA at 200 μM in further experiments. Additionally, we tested several experimental conditions by time-of-addition of UA in the assay. Three options were tested, including (A) the addition of UA and fAβ1-42 at the same time (B) UA added 30 minutes before and removed just before adding the fAβ1-42 and (C) UA added 30 minutes before fAβ1-42 addition (Figure 3G). The condition selected for next assays was the option (C). We confirmed that UA is a competitive inhibitor of the interaction CD36-Aβ based on the dose response profile of the inhibitory response (Figure 3E-G).
As DMSO is the most used solvent in natural or synthetic compounds, we evaluated the sensitivity of the assay to its presence. Several concentrations of DMSO were tested in the assay for 30 minutes before the addition of fAβ1-42. Concentrations of DMSO below 1.25% did not interfere with the binding of CD36 and fAβ1-42 (Figure 3H).
Screening of synthetic compounds
We tested 24 compounds belonging to the Trichodermamide analogues family described above. Trichodermamides A (1) and B (2) belong to a class of 1,2-oxazadecaline secondary metabolites 17 that are produced by marine and terrestrial fungi. Both secondary metabolites were isolated in minute quantities 19, thus necessitating development of a synthetic route to Trichodermamides to enable an in depth investigation of their bioactivity profile. The synthetic approach to Trichodermamides 17 also provided access to a variety of unnatural structural analogues that can have an improved activity and serve as a basis set for a structure-activity relationship study (Supplementary material).
The effect of compounds on the interaction of fAβ 1-42 with CD36 was first evaluated at 100 μM. Those compounds with a statistically significant inhibitory effect compared to CD36-fAβ 1-42 binding control were considered as hits and were tested in four- points dose response. Seven compounds (29%) exhibited a capacity to interfere with the interaction of CD36 to fAβ 1-42 (Table 1 and S1). The IC50 values for these compounds ranged from 11 to 43 μM, lower than the IC50 for ursolic acid (98 μM) (Table 1 and Figure S1). Compounds 5, 7, and 8 presented the smallest IC50 values emerging as promising hits. Dose-response curves suggested these compounds as competitive inhibitors of CD36-Aβ interaction (Figure S2).
Computational studies of the putative binding mode of the hCD36: Inhibitor complexes
In order to investigate the binding mode of the active compounds to CD36 we carried out molecular docking studies. A tridimensional (3D) model of the human CD36 (hCD36) extracellular domain structure was retrieved from the Swiss-Model repository 20 to perform our docking calculations. This model was calculated based on the lysosomal domain structure of human LIMP-2 (PDB code: 4fb7, R = 3.0 Å), a scavenger receptor that shares 35 % of sequence identity (ID) with the human CD36 21. The analysis of the 3D model of hCD36 by the Castp server confirms the presence of interconnected cavities that form a large luminal tunnel (solvent accessible volume around 1181 A3) through the entire protein (Figure 4A). This predominant hydrophobic tunnel is similar to the one described previously in the LIMP-2 structure and it is thought to be important for ligand transference to the membrane 21. We targeted this hydrophobic tunnel as well as other smaller cavities of the extracellular domain of hCD36 to predict the putative binding mode of the 7 active compounds. Notably, the binding free energy of hCD36: Inhibitor complexes with the hydrophobic tunnel displayed a better agreement with experimental results. We further considered it the interaction region of compounds. The analysis of the 3D structures of these complexes suggested that most of the inhibitors established more hydrophobic interactions than hydrogen bond interactions with human CD36. The blockers were anchored in a pocket containing the residues V42, L44, V50, N53, W54, T57, G58, T59, V214, N216, G217, V224, A225, S253, F379, and K426. Compounds 4, 5, 6 and 8 established more interactions with the binding pocket than compounds 3, 7 and 9 (Figure 4B-H). Although the binding free energies predicted for the human CD36: Inhibitor complexes were in agreement with the μM range of experimental IC50 values it was not possible to establish a quantitative structure-activity relationship for these compounds (Table 1). Thus, considering the competitive inhibition profile obtained experimentally, we suggest that residues of the luminal hydrophobic tunnel are involved in the binding between CD36 and the Aβ fibril.
Figure 4. Putative binding mode of human CD36:Inhibitor complexes.
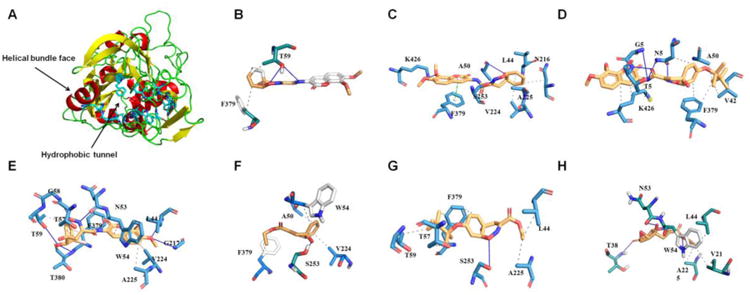
(A) Top view representation of the 3D structure of human CD36. As stick are highlighted the residues comprising the ligand binding site. The black arrows indicated the hydrophobic tunnel and the helical bundle face. (B) Compound 3, (C) 4, (D) 5, (E) 6, (F) 7, (G) 8, and (H) 9. All plots were generated using the PLIP web server. The hydrophobic interactions are displayed by gray dotted lines, hydrogen bonds by the blue line, and π-stacking perpendicular by the green dotted lines.
Inhibitory compounds reduced the cytokine response of macrophages to Aβ
We tested if the compounds that block the CD36-Aβ interaction were capable to inhibit the inflammatory response induced by Aβ in macrophages. Murine peritoneal macrophages were incubated with fibrils of murine Aβ 1-42 for 16 hours in the presence or absence of compounds. Peritoneal macrophages constitutively expressed CD36 on cell membrane (Figure 5A). Fibrillar Aβ1-42 induced the release of IL-6 and TNF-α likely due to the interaction with CD36 receptor (Figure 5B and C). This induction was reduced or blocked by some of the compounds (Figure 5B and C). Similar results were observed for murine primary microglia and SIM-A9 microglia cell line (data not shown). Compounds 5, 8 and 9 abrogated the production of IL-6 by peritoneal macrophages whereas compounds 3, 4, 6, and 7 partially inhibited the production of this cytokine (Figure 5B). The compounds showed different inhibitory profile on TNF-α production, where all compounds, except compound 7, completely impaired the production of this cytokine induced by Aβ (Figure 5C). The molecular basis of this differential effect of the compounds on cytokines secretion warrants further investigation. The inhibitory effect was not due to cell death, since the compounds were not cytotoxic at the inhibitory concentrations (Figure S3).
Figure 5. Compounds inhibiting CD36-Aβ interaction also impair the pro inflammatory activation of macrophages.
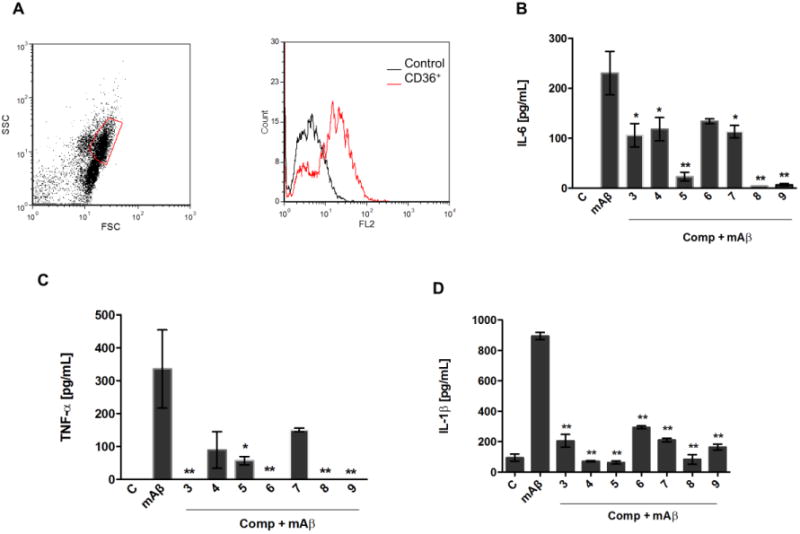
Peritoneal macrophages from C57Bl/6 mice were collected for FACS analysis of CD36 expression. Representative dot plot and histogram of CD36 expressing cells are shown from two different experiments (A). Macrophages were stimulated with fibrils of mouse Aβ1-42 (30 μM) in the presence or absence of compounds (30 μM). Supernatants were harvested 16 h later and concentrations of IL- 6 (B) and TNF-α (C) were measured by the ELISA method. (D) LPS-primed macrophages were stimulated for 6 hours with fibrils of mouse Aβ 1-42 (30 μM) with or without compounds. IL-1β concentration was detected by ELISA. Results represent means ± S.E.M. from stimuli performed in duplicates and are representative of two different experiments. *, P < 0.05; **, P < 0.01, compared with mAβ1-42 stimulus alone.
In order to rule out potential unspecific inhibitory effect of the compounds we evaluated whether the compounds interfered with the response of macrophages to LPS. High levels of TNF-α and IL-6 were detected in the supernatant of peritoneal macrophages cultures stimulated with LPS (Figure S4). In general, no inhibition to LPS response was observed in the presence of the compounds. Although not statistically significant, compounds 7 and 9 showed some reduction in the induction of TNF-α (Figure S4). No decrease in viability was observed along the treatment of cells with LPS and compounds (Figure S4).
It has been reported that CD36 is involved in the activation of NLRP3 induced by Aβ leading to the secretion of mature IL-1β 10. Hence, we tested if the compounds interfere with the production of IL-1β by macrophages primed with LPS and stimulated with the fibrils of mAβ. Our results show that all of the compounds significantly inhibited the secretion of IL-1β (Figure 5D). Taken together these data indicate that the compounds affect the response of macrophages to Aβ, probably by interfering with CD36-Aβ interaction.
Discussion
Inflammation in AD is mediated by the accumulation of microglia surrounding Aβ deposits and by the production of proinflammatory cytokines, chemokines and reactive oxygen and nitrogen species 22,23. The persistent production of inflammatory mediators during the course of AD generates a self-sustained neurotoxic cycle that results in neuronal death and a progressive loss of cognitive and behavioral abilities. The activation of microglia cells by Aβ appears to be one of the first steps in triggering the inflammation in AD. Multiple pieces of evidence implicate microglia receptors in the recognition and internalization of Aβ 24. CD36 receptor has been identified as essential in the Aβ-induced microglia activation and several studies point it out as a suitable target for drug development 7, 25. Engagement of Aβ to CD36 leads to the secretion of TNF-α, IL-1β, MCP-1, among other proinflammatory mediators 6, 7 and the inhibition of this interaction impair the response of cells to Aβ 6,11,13. However, current immunotherapy options for AD are not yet considering modulators of the Aβ-CD36 interaction to block or reduce the subsequent inflammatory reaction. Considering the relevance of CD36 in AD pathology, we have developed a colorimetric assay for the systematical search of molecules that interfere with the interaction of CD36 and fAβ as potential anti-inflammatory agents for AD treatment. The assay has been optimized to be rapid and convenient, and may be used in medium – high throughput screening programs. By screening 24 members of a Trichodermamide analogues family we have identified 7 new molecules, which inhibit CD36-fAβ binding and macrophages inflammatory response to fibrillar Aβ 1-42.
CD36 is a 88-kD glycosylated transmembrane protein, which contains a large extracellular domain and two small transmembrane domains at the N-terminus and the C-terminus of the protein 26. The extracellular domain of the CD36 receptor mediates its interaction with several ligands. Hence, we generated a recombinant protein corresponding to the extracellular domain of the human CD36 (Gly30 - Asn439) expressed on E. coli as a His-tagged protein (rCD36). As previously reported, the high level expression of this protein in bacteria led to accumulation in inclusion bodies, allowing rapid isolation, solubilization and purification under denaturing conditions 27. A rapid, simple refolding protocol yields a protein with high purity that retains the ability to bind fibrillar Aβ, as judged from the binding capability on the described colorimetric assay. The specific binding was demonstrated including ursolic acid, a known disruptor of the CD36-Aβ interaction, which acted in a dose response manner to show a reduction in the generation of color. The inhibition caused by the UA appears to be competitive, as it is able to displace the binding of Aβ in a dose dependent manner, as also reported previously in a cell based system 11. Although the assay was configured to probe the interaction between human CD36 and human Aβ, the same response in color production could be observed when the same protein was assayed with mouse fibrillar Aβ (data not shown). This was expected as hAβ and mAβ share high homology, with amino acid changes only at positions 5, 10 and 13. Two of these changes, at positions 10 (Y → F) and 13 (H → R) involve amino acids with similar chemical properties, with hydrophobic and hydrophilic side chains respectively. The same cross species interaction was observed with human fibrillar Aβ on mouse N9 microglia cell line 11. Although not rigorously tested, our data and those from others also suggest that the N-glycosylation of the CD36, at least in the extracellular portion, is not required to retain the binding capacity to Aβ 27. Interestingly, the N-glycosylation of the CD36 ectodomain was not found to be important for the surface expression or the binding activity to modified low-density lipoproteins 28.
Multiple parameters were optimized for the colorimetric assay development, including concentration of rCD36 binding to solid phase, concentration of Aβ, dilution of primary (anti-Aβ), and secondary antibodies, the interfering compound binding conditions and sensitivity to DMSO, as small compounds to be tested at the screening phase are usually dissolved in this solvent. The assay produced highly reproducible results and presented a high confidence for identifying compounds interfering with CD36-fAβ interaction, with a Z′ value of 0.6, suggesting that the assay is suitable for screening under this experimental setup.
The screening of structurally related molecules also demonstrated the confidence of the assay, since it was able to discriminate among active and non-active compounds with small structural differences (Table 1 and S1). In addition, the assay provided valuable information on the structure-activity relationships within the Trichodermamide family. Interestingly, while the naturally-occurring Trichodermamides A (1) and B (2) lacked activity, the structurally related analogues 3-6 showed inhibition of the CD36-Aβ interaction (Table 1). Our results further show that the aminocoumarin moiety (rings C and D, Figure 1) is not important for the inhibitory activity, since aminocoumarin-containing trichodermamide analogues 3-6 had comparable or lower activity than ester analogues 7-9. This finding significantly simplifies synthetic access to active CD36-fAβ interaction inhibitors. Further, the structure variation within the A ring of the 1,2-oxazadecaline core of analogues 3-6 indicates that increased flattening of the A ring via replacement of sp3 carbons with sp2 carbons in C4-C8 positions leads to the increase of the inhibitory activity. This effect is also evident in compounds 3 and 6, where the more rigid C5-C6-epoxide system in compound 3 affords higher activity than the more flexible bromohydrin unit in analogue 6.
Molecular docking simulations supported the stronger interaction of 7 compounds of the Trichodermamide family with CD36 than that of UA. The predicted binding free energies of the 7 active compounds and the UA explained its IC50 values in the μM range. All the blockers were anchored in a large hydrophobic luminal pocket observed in the 3D structure of the extracellular domain of CD36. This luminal pocket has been described as important for the ligand transference to cell membrane in LIMP-2, a scavenger receptor closely related to CD36 21. The analysis of the 3D structures of the CD36-inhibitor complexes revealed the interactions of the inhibitors with the following residues of CD36: V42, L44, V50, N53, W54, T57, G58, T59, V214, N216, G217, V224, A225, S253, F379, and K426. We proposed that the luminal hydrophobic pocket is implicated in CD36 interaction with Aβ fibrills based on these predictions and the competitive inhibition profile obtained for the ursolic acid and these 7 compounds. Further experiments will be needed, including site-directed mutagenesis, to confirm the role of the CD36 luminal hydrophobic pocket as a putative interacting site of the Aβ fibrils. Also, free energy calculations with end-point methods such as LIE and MM-PB (GB)SA approaches 29-31 will be needed to further understand the structure-activity relationship of the described active compounds.
It has been previously demonstrated that the inhibition of the interaction of CD36 with Aβ leads to the reduction of pro-inflammatory cytokines production and reactive oxygen species generation 11,13. We then evaluated the relevance of the effect of the Trichodermamide analogues identified in the colorimetric assay in cells treated with Aβ. Our results demonstrated that these compounds effectively impair the secretion of TNF-α, IL-6 and IL-1β in macrophages, further validating the usefulness of the described binding assay to identify active molecules. The inhibitory effect of compounds on peritoneal macrophages cytokines secretion was also observed in thioglycolate elicited macrophages and murine SIM-A9 microglia cell line. As have been revealed previously, phenotypic, functional and gene expression differences have been shown for macrophages obtained from different compartments 32, 33 . Further studies directed to characterize the biological effect in primary microglia cells should be performed.
The active compounds did not affect the response of macrophages to LPS. The engagement of LPS to TLR4 triggers a signaling cascade that culminates in the expression and secretion of inflammatory mediators 34. Since TLR4 is involved in signal transduction induced by Aβ through CD36 9 these results suggest that the compounds' effect depends on the impairment of the CD36-Aβ interaction and not on the eventual inhibition of the TLR4 signaling pathway. Other compounds structurally related to the Trichodermamide analogues inhibit the production of TNF induced by LPS in macrophages through a mechanism involving IκB degradation and the subsequent activation of NFκB 35. The biological characterization of the interaction of the compounds identified herein with CD36 in cells should be further investigated.
This work describes the development and validation of a new colorimetric assay for the search of molecules that interfere with an important step in the pathogenesis of Alzheimer's disease. The search for new agents with the potential to modify this pathology is essential en route to drugs that can stop or delay the course of the disease. Here, we have identified 7 new molecules that reduce the inflammatory response of microglia presumably by interfering with the interaction between CD36 and Aβ. This work serves as a platform for the screening of compounds as new potential agents for the treatment of AD.
Materials
Mice
Female and male C57Bl/6 mice, 8 weeks old, were obtained from the mice facility of INDICASAT AIP. The animals were kept at constant temperature (25°C) with free access to chow and water in a room with a 12 h light/dark cycle. The experiments were performed in accordance with the recommendations of the Institutional Animal Welfare Committee guidelines. The protocol was approved by the Institutional Animal Care and Use Committee of INDICASAT AIP (IACUC-14-003).
Materials
Escherichia coli BL21 (DE3) and the expression vector pET30a(+) were purchased from Novagen (Darmstadt, Germany). Endonucleases (EcoRI and NotI) were obtained from New England Biolabs (Ipswich, MA). Cloning vector pGEM-T and Pfu DNA polymerase were obtained from Promega (Madison, WI). Kanamycin and isopropyl-β-D-1-thiogalactopyranoside (IPTG) were from Sigma (St. Louis, MO). The human and murine amyloid beta 1-42 peptides (hAβ 1-42 and mAβ 1-42) were purchased from GenScript (Piscataway, NJ). Lipopolysaccharide (LPS) from E. coli 0111:B4 was purchased by InvivoGen (San Diego, CA). Medium RPMI and fetal bovine serum (FBS) for macrophage culture were obtained from Gibco (Grand Island, NY), while dimethyl sulfoxide (DMSO) was obtained from Sigma-Aldrich (St. Louis, MO).
Cloning and expression in E. coli of the extracellular domain of human recombinant CD36 protein
Total RNA was extracted from THP-1 cells using Trizol reagent (Invitrogen, USA), resuspended in diethyl pyrocarbonate (DEPC) treated water and quantified using Nanodrop. The reverse transcription was performed by reverse transcription system from Promega using random primers following manufacturer's instructions. The cDNA was then amplified by PCR using the primers, forward 5′-AAGAATTCGGAGACCTGCTTATCCAGAAG-3′ and reverse 5′-AGCGGCCGCTTAGTTTATTTTTCCAGTTACTTGAC-3′, containing the sequences of recognition sites of the restriction enzymes EcoRI and NotI, underlined 27. The PCR product was sequence verified, digested and subcloned into corresponding sites of expression vector pET30a(+). The resultant plasmid (pET30CD36) expresses the extracellular portion of the CD36 with and N-terminal fusion peptide including a 6×His tail.
The pET30CD36 plasmid was transformed into E. coli BL21 (DE3) strain. The bacteria were grown at 200 rpm, 37°C in Luria-Bertani (LB) media supplemented with 50 μg/ml of kanamycin until the OD600 reached 0.4 - 0.6. Then IPTG was added to final concentration of 1 mM and further incubated at 37°C for 4 hours. The bacterial pellet was collected by centrifugation at 8 000 × g for 10 min at 4 °C and kept frozen at -20°C.
CD36 purification by immobilized metal affinity chromatography
The cell paste obtained from 300 ml of induced culture was thawed on ice, suspended in 10 ml of lysis buffer (10 mM Tris-Cl pH 7.5, 5 mM EDTA, 100 mM NaCl, 0.3 mg/ml lysozyme, 2 mM PMSF) and lysed by sonication with a Sonic Ruptor 400 (OMNI international, Kennesaw, GA). After verification of the presence of the protein associated to the insoluble fraction of the rupture, the inclusion bodies were collected by centrifugation at 15 000 × g for 15 min at 4°C, resuspended in 5 ml of buffer B (8 M urea, 100 mM NaH2P04, 100 mM Tris-Cl pH 8) and solubilized for one hour at room temperature in shaker. The clear lysate was passed through a 1 ml Ni-NTA superflow column (Qiagen, USA) pre-equilibrated in same buffer and subsequently washed with 10 ml of buffer C (8 M urea, 100 mM NaH2P04, 100 mM Tris-Cl pH 6.3). The protein was eluted in buffer E (8 M urea, 100 mM NaH2P04, 100 mM Tris-Cl pH 4.5). Fractions containing the recombinant CD36 as revealed by SDS-PAGE, were pooled and the protein was refolded by 50-fold dilution with PBS buffer (50 mM NaH2PO4, 150 mM NaCl, pH 7.2) and concentrated using ultra-15 centrifugal devices 3K cutoff (Amicon, USA). Final preparation of the recombinant protein was evaluated by SDS PAGE and the Coomassie stained gel was subjected to densitometry using ImageJ v.1.50 as recommended by the software developer 36.
Amyloid beta fibril (fAβ) formation
Human or mouse Aβ 1–42 peptides were dissolved at 1 mg/ml with sterile water and incubated for 48 hours at 37°C. Before use, the fibrils were vigorously mixed and verified using Thioflavin-T (ThT). Briefly, samples with 10 μM of Aβ 1–42 and 20 μM of ThT in a final volume of 200 μl were incubated at 37°C for 30 min in a black 96-well plate. The ThT fluorescence intensity of each sample was recorded using a microplate reader (synergy HT BioTek) with a wavelength of 450 nm for excitation and 485 nm for emission.
Colorimetric assay for inhibitors of Aβ – CD36 interaction
Ninety-six well plates were coated with 100 μl of 15 μg/mL of recombinant CD36 (rCD36) in PBS and incubated overnight at 4°C. Wells were washed three times with PBS-0.05% Tween 20, and blocked for 2 h with 3% skim milk in PBS at room temperature. After three similar washings, compounds were added at different concentrations in PBS and incubated for 30 min. Then, fibrillar hAβ 1-42 (0.75 μM) diluted in PBS was added. After 2 h of incubation plates were washed three times and incubated for 2 h with a rabbit anti human amyloid β polyclonal antibody (GenScript) (Source, final concentration 0.3 μg/mL). Plates were then washed and incubated for 2 h with a horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1/4000). After a final wash, TMB substrate was added and allowed to react for 15 min at room temperature. The reaction was stopped and absorbance was measured at 450 nm by using a Synergy HT multireader (Biotek, USA). All samples were assayed in duplicate and in the presence of 0.5% DMSO. The ursolic acid was included as positive control of inhibition.
CD36 Sequence Analysis
We analyzed 2 mammalian amino acid sequences homologous to the human CD36 (CD36_HUMAN) (Uniprot code: CD36_RAT (Rat), CD36_MOUSE (Mouse)). These sequences share a sequence identity to human CD36 ranging from 83 to 88%. The multiple sequence alignment of mammalian CD36 was performed using the ClustalW software 37. The multiple alignments were manually parsed by analyzing the gaps, conserved amino acid regions and the secondary structure information using Seaview software 38.
Molecular docking studies
2D structures of the 7 active compounds against CD36 were used to create 3D models from the corresponding SDF files. The building of 3D structures, and conversion to pdbqt format were performed using the Open Babel software 39 Docking simulations were performed for each compound with the Autodock Vina software 40 using as target the coordinates of the 3D model of the extracellular portion of human CD36 protein (Uniprot code: CD36_HUMAN) retrieved from the Swiss-Model repository (http://swissmodel.expasy.org/repository). The ursolic acid was included as positive control of the docking simulations. The putative binding site on human CD36 was identified using the Castp web server (http://sts.bioengr-uic.edu/castp). The ten poses generated for each compound were ranked using the Autodock Vina scoring function 40. Tridimensional representations of the interactions of the best-ranked pose of each hCD36: Inhibitor complex were obtained using default parameters of the PLIP web server (https://projects.biotec.tu-dresden.de/plip-web/plip/index).
Compounds synthesis and purification
Anhydrous dichloromethane, tetrahydrofuran, toluene and diethyl ether were collected under argon from an LC Technologies solvent purification system, having been passed through two columns packed with molecular sieves. N,N-Dimethylformamide, acetonitrile, sym-collidine, pyridine, cyclohexane, dimethyl sulfoxide, benzene and trifluorotoluene were dried over 3 Å molecular sieves. 2,2,6,6-Tetramethylpiperidine was freshly distilled from calcium hydride before use. Benzoquinone was sublimed and stored at −20 °C. N,O-Bis(trimethylsilyl)acetamide (BTSA) was freshly distilled before use. Phenylselenol 41, iodosobenzene 42, tetrakis(triphenylphosphine)palladium(0) 43, ethyl (4aS*,8aS*)-4a-hydroxy-7-oxo-4a,7,8,8a-tetrahydro-4H-benzo[e][1,2]oxazine-3-carboxylate 17, (4aS*,7R*,8aS*)-ethyl 7-((methoxycarbonyl)oxy)-4a-hydroxy-4a,7,8,8a-tetrahydro-4H-benzo[e][1,2]oxazine-3-carboxylate 17, 3-amino-7,8-methoxy-2H-chromen-2-one 17 were prepared according to the literature procedures. Porcine kidney acylase (1100 U/mg) was purchased from Calzyme. All other chemicals were used as commercially available. All reactions were conducted with continuous magnetic stirring under an atmosphere of argon in oven-dried glassware. Reactions were monitored by 1H NMR or by TLC on silica gel-coated glass plates (Merck Kieselgel 60 F254) until deemed complete. Plates were visualized under ultraviolet light (254 nm) and by staining with ceric ammonium molybdate (CAM) or potassium permanganate. Column chromatography was performed using CombiFlash Rf-200 (Teledyne-Isco) automated flash chromatography system with hand-packed RediSep columns.
Compounds Characterization
1H, 13C, NMR spectra were recorded at 500 MHz (1H), 125 MHz (13C) on the Agilent Inova 500 instrument in CDCl3 solutions with and without tetramethylsilane (TMS) as an internal standard unless specified otherwise. Chemical shifts (δ) are reported in parts per million (ppm) from the residual solvent peak and coupling constants (J) in Hz. Proton multiplicity is assigned using the following abbreviations: singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint.), septet (sept.), multiplet (m), broad (br).
Infrared measurements were carried out neat on a Bruker Vector 22 FT-IR spectrometer fitted with a Specac diamond attenuated total reflectance (ATR) module.
Cell culture
Peritoneal macrophages from C57Bl/6 mice were obtained by peritoneal lavage with cold RPMI. Cells were seeded in RPMI with 10% FCS at a density of 2×105/well in 96-well plates and cultured for 2 h at 37°C in an atmosphere of 5% CO2. Non-adherent cells were removed by washing and adherent cells were used for stimulation. For IL-1β induction cells were primed with 100 ng/ml of LPS for 3 h. Then, LPS was removed and mAβ 1-42 (30 μM) was added with or without compounds (30 μM), and cells were incubated for 6 h. For TNF-α and IL-6 induction cells were stimulated with 30 μM of mAβ 1-42 in the presence or absence of compounds (30 μM) and incubated for 16 h. Supernatants were then harvested and cytokines concentrations were determined by the ELISA method. Both, negative controls and stimuli were performed in the presence of 0.5% of DMSO as the compounds are solubilized in this solvent.
Cytokine measurements
The concentrations of TNF-α, IL-6 and IL-1β were determined by the ELISA method (DuoSet kit, R&D System, Inc. Minneapolis, MN), according to the manufacturer's protocol.
Flow cytometry
Peritoneal macrophages from C57Bl/6 were plate at 37°C in 5% atmosphere of CO2. After 2 h non-adherent cells were removed and adherent cells were harvested, passed to a 1.5 ml tube and centrifuged at 150 × g. The cells were washed with phosphate-buffered saline (PBS), and blocked with 200 μl 1 % BSA in PBS for 15 min. The cells were washed and then incubated with (2 μg/ml) PE anti-mouse CD36 antibody (BioLegend, San Diego, CA) diluted in 1% BSA in PBS, for 30 min at 4°C. After several washes, the cells were resuspended in PBS and analyzed by flow cytometry. Event acquisition was performed with a Partec CyFlow® cytometer and the data were analyzed using FCS Express 4 Flow Cytometry (De Novo software, Los Angeles, CA).
Cell viability assay
Cell viability was evaluated by resazurin method, which consists in the measurement of metabolic activity of living cells through its ability to reduce resazurin to resorufin 44. Briefly, after the supernatant was collected from the 96 well plate, 100 μl/well of fresh medium and 20 μl of resazurin solution (0.15 mg/ml in PBS) were added to each well. The plate was incubated for 4 hours at 37°C. The fluorescence was recorded using 560 nm excitation/ 590 nm emission in the synergy HT multireader by Biotek.
Statistical analysis
Data are presented as mean ± S.E.M or mean ± S.D. Results were analyzed using a statistical software package (GraphPad Prism 6). Statistical analysis was performed by one-way ANOVAs followed by post-hoc Tukey test. A difference between groups was considered to be significant if P < 0.05 (*, P < 0.05; **, P < 0.01). Half maximal inhibitory concentration (IC50) values were calculated adjusting a variable slope sigmoidal dose-response curve following GraphPad Prism 6 procedure.
Supplementary Material
Acknowledgments
Financial support by Secretaria Nacional de Ciencia Tecnología e Innovación of the Republic of Panama and the Banco Interamericano de Desarrollo (IND-03-12), the Sistema Nacional de Investigación (SNI34-2014), the Welch Foundation (AX-1788), NIGMS (SC3GM105579), UTSA, and the NSF (CHE- 1455061) is gratefully acknowledged. Mass spectroscopic analysis was supported by a grant from the NIMHD (G12MD007591). We thank Rita M. Giovanni for her assistance with graphical table of contents.
Abbreviations
- AD
Alzheimer's disease
- Aβ
amyloid-β
- SDS
sodium dodecyl sulfate
- fAβ
Fibrillar Aβ
- TNF-α
tumor necrosis factor alpha
- IL-6
Interleukin 6
- IL-1β
Interleukin 1 beta
- TLR4
toll like receptor 4
- TLR6
toll like receptor 6
- NLRP3
NACHT, LRR and PYD domains-containing protein 3
- ROS
reactive oxygen species
- IPTG
isopropyl-β-D-1-thiogalactopyranoside
- GHRPs
growth hormone-releasing peptides
- IC50
half maximal inhibitory concentration
- LIMP-2
lysosome membrane protein 2
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- LPS
lipopolysaccharide
- RPMI
Roswell Park Memorial Institute medium
- BSA
Bovine serum albumin
- UA
Ursolic acid
- MCP-1
Monocyte chemoattractant protein-1
- FBS
fetal bovine serum
- ATR
attenuated total reflectance
- ELISA
Enzyme-linked immunosorbent assay
- DMSO
dimethyl sulfoxide
- ppm
parts per million
- DEPC
diethyl pyrocarbonate
- TMS
tetramethylsilane
- LB
Luria-Bertani
- ThT
Thioflavin-T
- PBS
phosphate-buffered saline
- TMB
3,3′,5,5′-Tetramethylbenzidine
- BTSA
N, O-Bis(trimethylsilyl)acetamide
- CAM
ceric ammonium molybdate
- NMR
nuclear magnetic resonance spectroscopy
- Uniprot
Universal Protein Resource
- PLIP
Protein-Ligand Interaction Profiler
Footnotes
Supporting Information The detailed experimental procedure for Trichodermamide analogs synthesis, the 1H and 13C NMR Spectroscopic Data for the active compounds, additional table with structure of non-active compounds, the IC50 sigmoidal curves and the dose response graphs for the active compounds, the cytotoxicity graphs and compounds' effect on macrophages-LPS response.
Author Contributions: D.D. performed experiments, analyzed data and contributed in writing the manuscript.
P.A.V. performed the bioinformatics analysis and contributed in writing the manuscript.
A.M.M, A.X.T. and V.T.N. prepared the Trichodermamide analogues.
A.T., L.C., M.Q. performed experiments.
O.L. and G.P. directed the work on the Trichodermamide analogues, analyzed data and contributed in writing the manuscript.
P.L.F and R.L. Conceived and designed the work, analyzed data and wrote the manuscript.
Conflict of interests: The authors do not have any competing interests.
References
- 1.Jana M, Palencia CA, Pahan K. Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer's disease. J Immunol. 2008;181:7254–7262. doi: 10.4049/jimmunol.181.10.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crehan H, Hardy J, Pocock J. Microglia, Alzheimer's disease, and complement. Int J Alzheimers Dis. 2012;2012:983640. doi: 10.1155/2012/983640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology. 2011;77:212–218. doi: 10.1212/WNL.0b013e318225ae07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilkinson K, El Khoury J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer's disease. Int J Alzheimers Dis. 2012;2012:489456. doi: 10.1155/2012/489456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doens D, Fernandez PL. Microglia receptors and their implications in the response to amyloid beta for Alzheimer's disease pathogenesis. J Neuroinflammation. 2014;11:48. doi: 10.1186/1742-2094-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002;277:47373–47379. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- 7.El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14:812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson K, Boyd JD, Glicksman M, Moore KJ, El Khoury J. A high content drug screen identifies ursolic acid as an inhibitor of amyloid beta protein interactions with its receptor CD36. J Biol Chem. 2011;286:34914–34922. doi: 10.1074/jbc.M111.232116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demers A, McNicoll N, Febbraio M, Servant M, Marleau S, Silverstein R, Ong H. Identification of the growth hormone-releasing peptide binding site in CD36: a photoaffinity cross-linking study. Biochem J. 2004;382:417–424. doi: 10.1042/BJ20040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulgarelli I, Tamiazzo L, Bresciani E, Rapetti D, Caporali S, Lattuada D, Locatelli V, Torsello A. Desacyl-ghrelin and synthetic GH-secretagogues modulate the production of inflammatory cytokines in mouse microglia cells stimulated by beta-amyloid fibrils. Journal of neuroscience research. 2009;87:2718–2727. doi: 10.1002/jnr.22088. [DOI] [PubMed] [Google Scholar]
- 14.Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O'Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 15.Mfuh AM, Larionov OV. Heterocyclic N-Oxides - An Emerging Class of Therapeutic Agents. Current medicinal chemistry. 2015;22:2819–2857. doi: 10.2174/0929867322666150619104007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrera L, Stephens DE, D'Avila A, George KA, Arman HD, Zhang Y, Perry G, Lleonart R, Larionov OV, Fernández PL. Insights into the Structural Patterns of the Antileishmanial Activity of Bi- and Tricyclic N-Heterocycles. Org Biomol Chem. 2016 doi: 10.1039/c6ob01149g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mfuh AM, Zhang Y, Stephens DE, Vo AX, Arman HD, Larionov OV. Concise Total Synthesis of Trichodermamides A, B, and C Enabled by an Efficient Construction of the 1,2-Oxazadecaline Core. J Am Chem Soc. 2015;137:8050–8053. doi: 10.1021/jacs.5b05205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crowther J. The ELISA guidebook. 2nd. Humana Press Inc; 2009. [Google Scholar]
- 19.Garo E, Starks CM, Jensen PR, Fenical W, Lobkovsky E, Clardy J. Trichodermamides A and B, cytotoxic modified dipeptides from the marine-derived fungus Trichoderma virens. J Nat Prod. 2003;66:423–426. doi: 10.1021/np0204390. [DOI] [PubMed] [Google Scholar]
- 20.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic acids research. 2009;37:D387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, Neculai M, Plumb J, Loppnau P, Pizarro JC, Seitova A, Trimble WS, Saftig P, Grinstein S, Dhe-Paganon S. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature. 2013;504:172–176. doi: 10.1038/nature12684. [DOI] [PubMed] [Google Scholar]
- 22.Zaheer A, Zaheer S, Thangavel R, Wu Y, Sahu SK, Yang B. Glia maturation factor modulates beta-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 2008;1208:192–203. doi: 10.1016/j.brainres.2008.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez PL, Britton GB, Rao KS. Potential immunotargets for Alzheimer's disease treatment strategies. J Alzheimers Dis. 2013;33:297–312. doi: 10.3233/JAD-2012-121222. [DOI] [PubMed] [Google Scholar]
- 24.Yu Y, Ye RD. Microglial Abeta receptors in Alzheimer's disease. Cellular and molecular neurobiology. 2015;35:71–83. doi: 10.1007/s10571-014-0101-6. [DOI] [PubMed] [Google Scholar]
- 25.Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol. 2001;158:63–73. doi: 10.1016/s0002-9440(10)63945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruarin P, Thorne RF, Dorahy DJ, Burns GF, Sitia R, Alessio M. CD36 is a ditopic glycoprotein with the N-terminal domain implicated in intracellular transport. Biochemical and biophysical research communications. 2000;275:446–454. doi: 10.1006/bbrc.2000.3333. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Bao Y, Yang Y, Wu Y, Chen X, Si S, Hong B. Discovery of antagonists for human scavenger receptor CD36 via an ELISA-like high-throughput screening assay. J Biomol Screen. 2010;15:239–250. doi: 10.1177/1087057109359686. [DOI] [PubMed] [Google Scholar]
- 28.Hoosdally SJ, Andress EJ, Wooding C, Martin CA, Linton KJ. The Human Scavenger Receptor CD36: glycosylation status and its role in trafficking and function. The Journal of biological chemistry. 2009;284:16277–16288. doi: 10.1074/jbc.M109.007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 30.Miller BR, 3rd, McGee TD, Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J Chem Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 31.Miranda WE, Noskov SY, Valiente PA. Improving the LIE Method for Binding Free Energy Calculations of Protein-Ligand Complexes. J Chem Inf Model. 2015;55:1867–1877. doi: 10.1021/acs.jcim.5b00012. [DOI] [PubMed] [Google Scholar]
- 32.Turchyn LR, Baginski TJ, Renkiewicz RR, Lesch CA, Mobley JL. Phenotypic and functional analysis of murine resident and induced peritoneal macrophages. Comp Med. 2007;57:574–580. [PubMed] [Google Scholar]
- 33.Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nature reviews Immunology. 2011;11:775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature immunology. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 35.Rether J, Serwe A, Anke T, Erkel G. Inhibition of inducible tumor necrosis factor-alpha expression by the fungal epipolythiodiketopiperazine gliovirin. Biological chemistry. 2007;388:627–637. doi: 10.1515/BC.2007.066. [DOI] [PubMed] [Google Scholar]
- 36.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic acids research. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 39.O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santi C, Santoro S, Testaferri L, Tiecco M. A simple zinc-mediated preparation of selenols. Synlett. 2008;2008:1471–1474. [Google Scholar]
- 42.Saltzman H, Sharefkin JG. Iodosobenzene. Organic Syntheses. 1963;43:60. [Google Scholar]
- 43.Coulson DR, Satek LC, OG S. Tetrakis(triphenylphosphine)palladium(0) In: A., C F, editor. Inorganic Syntheses. 1972. [Google Scholar]
- 44.O'Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. European journal of biochemistry. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.