

Abstract
Chinese ginseng (Panax ginseng Meyer) is a medicinally important herb and plays crucial roles in traditional Chinese medicine. Pharmacological analyses identified diverse bioactive components from Chinese ginseng. However, basic biological attributes including domestication and selection of the ginseng plant remain under-investigated. Here, we presented a genome-wide view of the domestication and selection of cultivated ginseng based on the whole genome data. A total of 8,660 protein-coding genes were selected for genome-wide scanning of the 30 wild and cultivated ginseng accessions. In complement, the 45s rDNA, chloroplast and mitochondrial genomes were included to perform phylogenetic and population genetic analyses. The observed spatial genetic structure between northern cultivated ginseng (NCG) and southern cultivated ginseng (SCG) accessions suggested multiple independent origins of cultivated ginseng. Genome-wide scanning further demonstrated that NCG and SCG have undergone distinct selection pressures during the domestication process, with more genes identified in the NCG (97 genes) than in the SCG group (5 genes). Functional analyses revealed that these genes are involved in diverse pathways, including DNA methylation, lignin biosynthesis, and cell differentiation. These findings suggested that the SCG and NCG groups have distinct demographic histories. Candidate genes identified are useful for future molecular breeding of cultivated ginseng.
Keywords: artificial selection, domestication, ginsenoside, nucleotide diversity, Panax ginseng
Introduction
The rise of agriculture ∼10,000 years ago has profoundly reshaped human history, fueling the shift of human societies from hunting and gathering to permanent settlement that ultimately gave raise to civilization (Diamond 2002; Doebley etal. 2006; Ross-Ibarra etal. 2007). In the case of East Asia, (Larson etal. 2014), the ancient Chinese have identified 11,146 medicinal species from 383 families, and more than 400 of which are widely used throughout the world (Drašar and Moravcova 2004; Chen etal. 2010). The long-term (>2,500 years) practice of these medicinal plants has laid the foundation of traditional Chinese medicine (TCM) which constitutes a crucial part of Chinese culture (Wang etal. 2005).
Chinese ginseng (Panax ginseng Meyer) is one of the most important medicinal plants of TCM (Wen and Zimmer 1996; Li etal. 2015). As a symbol of the TCM, Chinese ginseng is known as “the lord or king of herbs” and widely used in Chinese medicine to cope with fatigue and physical stress for thousands of years (Wen and Zimmer 1996). Pharmacological investigations demonstrated that Chinese ginseng extracts contain numerous pharmacological bioactive ingredients such as ginsenosides, polysaccharides, pepties, and flavonoids (Kim etal. 2005; Jeong etal. 2006). Among these biomacromolecules, ginsenosides are the major pharmacological active components belonging to steroid glycosides and triterpene saponins (Okazaki etal. 2006). Recent genetic studies based on transcriptome data identified a series of ginsenoside biosynthesis genes in Chinese ginseng and closely related species (Mathur etal. 1999; Le Tran etal. 2002; Wang etal. 2006; King and Murphy 2007; Liang and Zhao 2008; Luo etal. 2011).
To gain a better understanding of the evolutionary trajectory of Chinese ginseng, Shi etal. (2015) investigated the polyploidization process of genus Panax based on multiple chloroplast and nuclear genes. As an allotetraploid species, Chinese ginseng has experienced both of the ancient and recent whole genome duplications. Although several diploid species are extantly distributed in East Asia and North America, the diploid progenitor species of Chinese ginseng were most likely extinct now. Genetic diversity of Chinese ginseng has been investigated using random amplified polymorphic DNA (Ma etal. 1999), amplified fragment length polymorphism (Ma etal. 2000), inter-simple sequence repeat (Li etal. 2011), microsatellites (Jiang etal. 2016), and single copy nuclear genes (Li etal. 2013). These studies demonstrated that cultivated ginseng still maintains a high level of genetic diversity. Origin and domestication history of cultivated ginseng were also investigated based on multiple types of molecular markers, with microsatellite and methylation insensitive amplification polymorphism (MISP) data supporting single origin but nuclear genes indicating independent domestications (Li etal. 2015). However, observations from these studies were mainly based on a few molecular markers, genome-wide view of the domestication, and artificial selection of Chinese ginseng remained unexplored.
In this study, we investigated the population genetic structure of 30 wild and cultivated ginseng accessions based on whole genome data. As a non-model species with large genome size (∼3.1 Gb), no assembled reference genome is currently available for Chinese ginseng. In our previous study, 2,492 BAC-end sequences (∼1.5 Mb in length) were employed as reference to address the origin and domestication of Chinese ginseng (Li etal. 2015). Here, we evaluated the nucleotide variation pattern of wild and cultivated ginseng with three different types of reference. To obtain a genome-wide view of genetic diversity at genic region, we selected 8,660 protein-coding genes (∼18 Mb in length) from the transcriptome data of Chinese ginseng. Nucleotide variation pattern of the ginsenoside biosynthesis pathway genes was also assessed for wild and cultivated ginseng separately. In complement, the available 45s rDNA, chloroplast, and mitochondrial genomes were also applied to infer the origin and domestication of Chinese ginseng. Our aims of this study were to (1) infer the domestication history of Chinese ginseng at both of the nuclear and cytoplasmic genome levels; (2) evaluate how artificial selection and local adaptation have shaped the cultivated ginseng genome.
Materials and Methods
Plant Materials and DNA Extraction
The plant materials used in this study included 11 wild and 19 cultivated ginseng accessions covering their current geographic distribution ranges in northeastern Asia (supplementary fig. S1, Supplementary Material online). In our previous study, a total of 45 wild ginseng accessions were collected representing its current geographic distribution range (Li etal. 2015). Here, two additional wild accessions were collected from the northern distribution region (supplementary fig. S1, Supplementary Material online). Of these wild ginseng samples, 11 accessions were selected for whole genome sequencing. Similarly, nine new cultivated ginseng accessions of the landrace COMMON ginseng were obtained in this study. The other ten accessions of four different ginseng landraces were obtained from previous studies (Choi etal. 2014; Li etal. 2015). Genomic DNA was extracted from the root tissue of each sample using TianGen Plant Kit (TianGen, Beijing, China) following the manufacturer’s instructions.
Reference Assembly and Quality Control
As a non-model tetraploid species with large genome size (∼3.1 Gb), there is no assembled reference genome for Chinese ginseng. We investigated the genome-wide nucleotide variation pattern of 30 ginseng accessions based on three different types of reference. To address how domestication and local adaptation shaped the nucleotide variation pattern at genic region, We performed de novo transcriptome assembly for leaf, flower, fruit, fiber, and main root tissues of P. ginseng (GenBank accession numbers: SRR2952879, SRR2952873, SRR2952876, SRR2952870, SRR2952867) using Trinity (Grabherr etal. 2011). To examine the quality of assembled transcripts, we predicted the open read frame (ORF) for all transcripts and translated them into protein sequences using the program TRAPID (http://bioinformatics.psb.ugent.be/webtools/trapid). To further eliminate the data bias caused by low quality transcripts, only the transcripts that are more than 300 amino acids in length were used as references for subsequent data analyses. In addition, full-length sequences of the 45s rDNA and 15 nuclear genes involving in the ginsenoside biosynthesis pathway were downloaded from GenBank according to published references (Liang and Zhao 2008; Chen etal. 2011; Han etal. 2011, 2012). As a complementary analysis of Chinese ginseng origin, both of the chloroplast (156,355 bp) and mitochondrial (464,680 bp) genomes were downloaded from GenBank (accessions numbers: KF735063 and KF431956 for mitochondrial and chloroplast genomes, respectively).
Genome Sequencing and Variant Calling
DNA libraries of 11 new ginseng samples (two wild and nine cultivated ginseng accessions) were constructed with an insert size of 350 bp and sequenced with the Illumina X10 platform (Illumina, California, USA). Whole genome data of the other 19 wild and cultivated accessions were obtained from previous studies (Choi etal. 2014; Li etal. 2015). Raw reads of the 30 wild and cultivated ginseng accessions were assessed using FastQC (Andrews 2010). All clean short reads (base quality >20) were mapped onto the reference sequences using BWA (Li and Durbin 2010) with the parameter set as “bwa aln -n 0.05”. Single nucleotide polymorphisms (SNPs) and insertion/deletion (INDELs) were detected using SAMtools (Li etal. 2009) with the parameter “mpileup -Dsugf -C 50 -q 30 -Q 20” and “bcftools view -Ncvg”. The raw variants were further filtered using Perl script with the threshold “mapping quality (MQ) > 30, read depth (RD) > 3”. Missing data of each reported locus was detected by outputting the unmapped positions for each transcript using SAMtools (Li etal. 2009) with the command “mpileup -f”. All filtered variants were subjected for subsequent population genetic inferences. In addition, Kim etal. (2015) have sequenced the chloroplast genome of 11 South Korean ginseng (SKG) accessions. To infer the correlations between Chinese and SKG accessions, we combined our chloroplast dataset with available SNP matrix from Kim et al (2015).
Phylogenetic and Population Genetic Analyses
To address the phylogenetic relationships of the 30 wild and cultivated ginseng accessions, only the filtered SNPs (MQ > 30, RD > 3) of chloroplast and mitochondrial genomes were converted from variant call format (VCF) into FASTA alignment using Perl scripts. To further eliminate the systematic bias from our dataset, the SNPs with missing data in any of the 30 ginseng accessions were not included in subsequent analyses. Statistical parsimony network of the chloroplast and mitochondrial haplotypes were constructed using TCS 2.1 (Clement etal. 2000) with a 95% connection limit, respectively. The unprocessed networks produced by TCS were redrawn with the software Adobe Illustrator CS5 (Adobe Systems Inc., San Jose, California). For the population genetic inferences, all filtered SNPs and INDELs of the assembled transcripts were converted from VCF file into Plink format using VCFtools (Danecek etal. 2011) with the command “vcftools –plink”. Then, we employed the program Plink (Purcell etal. 2007) to generated ADMIXTURE input file with the command “Plink –file”. All reported variants from assembled transcripts were merged as a single dataset. The program ADMIXTURE (Alexander etal. 2009) was applied to infer the optimum number of genetic assignments for combined dataset of transcriptome reference. The best genetic assignment was determined by comparing the cross-validation of each K value. Ancestry populations of wild and cultivated ginseng were inferred from 1 to 10 with three independent iterations. As K = 1 exhibited the lowest cross-validation value, we presented the population genetic structure of the 30 wild and cultivated ginseng accessions with K values from 2 to 4. Barplots of the genetic assignments of each ginseng accession were illustrated using R script.
Nucleotide Variation Pattern
Phylogenetic and population genetic analyses revealed spatial genetic structure between the northern and southern cultivated ginseng (SCG) accessions. To this end, we divided the 19 cultivated ginseng accessions into SCG and northern cultivated ginseng (NCG) groups (supplementary fig. S1, Supplementary Material online). For the assembled transcripts, we calculated the nucleotide diversity (π), number of segregating sites (N), Tajima’s D, and genetic differentiation (FST) at the whole genome level and for each transcript, respectively. To further illuminate the systematic bias, the genes with lesser than three SNPs across all ginseng accessions were removed. The candidate genes that showed differentially low nucleotide diversity (πwild/πcultivar > 2) in cultivated ginseng and high genetic differentiation (FST > 0.05) relative to wild ginseng were supposed to have undergone selection during the domestication and local adaptation processes. As the NCG group conserved relatively lower nucleotide diversity and higher genetic differentiation, we employed a stricter cutoff (πwild/πcultivar > 4; FST > 0.10) to identify candidate genes. Distributions and barplots of these candidate transcripts were illustrated using R scripts. The private and shared variants (including SNPs and INDELs) between wild and the two cultivated ginseng groups were calculated using VCFtools (Danecek etal. 2011). Venn diagram of these pair-wise comparisons was illustrated using online service (http://bioinformatics.psb.ugent.be/webtools/Venn). Functions of these candidate genes were annotated using BLASTX (Altschul etal. 1997) with an E-value <10−7. Nucleotide diversity (θ) and number of segregating sites of the ginsenoside biosynthesis genes were calculated for wild ginseng and the two cultivated ginseng groups using VCFtools (Danecek etal. 2011), respectively. Decrease in nucleotide diversity in the SCG and NCG groups compared to wild ginseng group suggested that selection might have acted on these genes during domestication process. As a complement, we also calculated the nucleotide diversity and number of segregating sites at the 45s rDNA, chloroplast and mitochondrial genomes for the wild and cultivated ginseng separately.
Species Distribution Modeling
Potential habitat distribution of P. ginseng in northeastern Asia was predicted using the program Maxent (Phillips etal. 2004, 2006; Phillips and Dudik 2008; Elith etal. 2011). Presence-only data for the P. ginseng were obtained from published literatures and our sampling records (supplementary table S1, Supplementary Material online). Nineteen bioclimatic variables and layer of altitude were downloaded from WorldClim 1.4 database at a resolution of 30 arc-seconds (Hijmans etal. 2005). The following settings were applied to perform the ecological niche modeling: training (75%) for model building and testing (25%), 10 replicates with bootstrap support, 500 maximum iterations and convergence threshold of 0.00001 with Jack-knife option turned on. Area under the receiver operating characteristic curve (AUC) was used as a threshold to measure the reliable of predicted model. AUC score close to 1 is considered as good model. Relative contribution of the environmental variables to predicted ecological model was measured using the Jackknife test (Phillips etal. 2006). Warm and cold colors in the species distribution model indicate high and low predicted probability of suitable conditions for P. ginseng, respectively.
Results
Transcriptome Assembly and Quality Control
For the transcriptome assembly, a total of 149,937 transcripts corresponding to 64,126 unigenes were obtained from the 5 Chinese ginseng tissues. Of these assembled transcripts, the average transcript and ORF length are 781 and 463 bp, respectively. A total of 143,967 transcripts that contained ORF were successfully translated into protein sequences. We then blasted these protein sequences against the TRAPID protein database (including 13 eudicot species). Of these protein sequences, 91,178 (60.8%) are highly similar to available protein sequences of the database; 24,830 (27.2%) of which showed the best similarity hit to Vitis vinifera. Of these protein-coding transcripts, 23,259 (16.2%) possess a protein sequence with more than 300 amino acids in length. To further improve the quality of reference transcripts, only the longest transcript of each unigene was retained, which yielded a total of 8,660 protein-coding genes. Length of these protein-coding genes varied from 904 to 11,072 bp, with an average of 2,138 bp. Similarly, all of the 15 ginsenoside biosynthesis genes contained ORF and can be successfully translated into protein. The 45s rDNA, chloroplast, and mitochondrial genomes were well annotated in previous studies (Kim etal. 2015; Zhao etal. 2015). These attributes suggested that these references are suitable for subsequent population genetic inferences.
Sequence Polymorphism and Variation Distribution Pattern
The 8,660 protein-coding genes yielded a total of 239,735 high-quality variants (including SNPs and INDELs) across the 30 ginseng accessions, with only 0.2% of them being missing data in the variant matrix. Of these variants, 229,512 and 196,543 were found in cultivated and wild ginseng, respectively. Within the cultivated ginseng, the SCG group (N = 219,631) contained obviously more variants than that of the NCG group (N = 171,253) (table 1). Similarly, the statistic of nucleotide diversity (π) also revealed that cultivated ginseng (π = 3.94 × 10−3) possessed slightly higher level of nucleotide diversity compared with wild ginseng (π = 3.89 × 10−3) at the whole genome level. In particular, the SCG group (π = 4.36 × 10−3) harbored the highest nucleotide diversity compared to the other groups (fig. 1A and table 1). We also assessed the nucleotide diversity of the 45s rDNA where cultivated ginseng (N = 50) conserved more segregating sites compared with wild ginseng (N = 37); and the NCG group (N = 26) possessed the smallest number of variants. Compared with the nuclear genome, we found that the number of variants at the cytoplasmic genomes in the wild ginseng (N = 161 for mtDNA; N =10 for cpDNA) was obviously higher than that of cultivated ginseng (N = 124 for mtDNA; N = 4 for cpDNA) (table 1). The decreasing in DNA polymorphisms was more pronounced in the NCG group (N = 46 for mtDNA; N = 1 for cpDNA). Similarly, the estimation of nucleotide diversity also confirmed that wild ginseng (π = 7.00 × 10−5 for mtDNA; π = 12.3× 10−6 for cpDNA) harbored higher level of nucleotide diversity than that of cultivated ginseng (π = 6.49× 10−5 for mtDNA; π = 4.73× 10−6 for cpDNA), and NCG group (π = 3.21 × 10−5 for mtDNA; π = 2.34 × 10−6 for cpDNA) showed the lowest level of nucleotide diversity.
Table 1.
Nucleotide Diversity of Nuclear and Cytoplasmic Genomes for Wild and Cultivated Ginseng, Respectively
| Wild Ginseng |
Cultivated Ginsengb |
SCG |
NCG |
|||||
|---|---|---|---|---|---|---|---|---|
| π | N | π | N | π | N | π | N | |
| Nuclear genesa | 3.89×10−3 | 196,543 | 3.94×10−3 | 229,512 | 4.36×10−3 | 219,631 | 3.49×10−3 | 171,253 |
| 45s rDNA | 1.59×10−3 | 37 | 1.44×10−3 | 50 | 1.64×10−3 | 45 | 1.09×10−3 | 26 |
| Chloroplast | 12.3×10−6 | 10 | 4.73×10−6 | 4 | 6.73×10−6 | 4 | 2.34×10−6 | 1 |
| Mitochondrial | 7.00×10−5 | 161 | 6.49×10−5 | 124 | 6.99×10−5 | 102 | 3.21×10−5 | 46 |
Note.—N, number of segregating sites; SCG, southern cultivated ginseng; NCG, northern cultivated ginseng.
The 8,660 protein-coding genes were combined as a single matrix.
All cultivated ginseng accessions are combined.
Fig. 1.
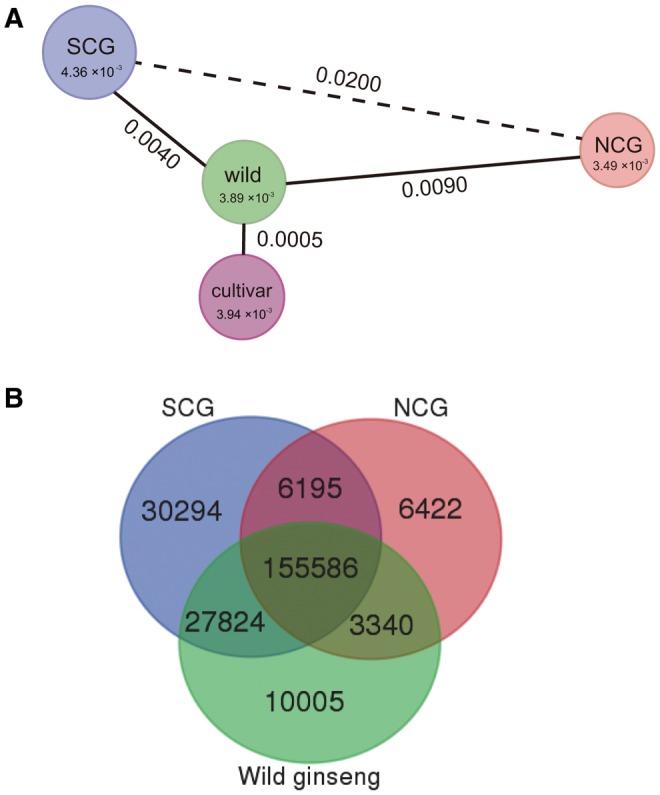
Nucleotide variation patterns of wild and cultivated ginseng. (A) Nucleotide diversity (π) and genetic differentiation (FST) between wild and cultivated ginseng. The size of cycle represents the level of nucleotide diversity. Numbers within each cycle and on each line are the exact nucleotide diversity and genetic differentiation, respectively. (B) Numbers of shared and private variants between wild and cultivated ginseng. SCG, southern cultivated ginseng group; NCG, northern cultivated ginseng group. Numbers within overlapped and non-overlapped areas are shared and private variants, respectively.
To further examine the distribution patterns of these variants within and between different ginseng groups, we calculated shared and private variants (including SNP and INDEL) of the 8,660 genes. Our Venn analysis showed that 155,586 variants (64.9%) were shared among the three ginseng groups (fig. 1B). Of the pair-wise comparisons, the NCG shared smaller number of variants with wild ginseng (66.3%) and SCG group (67.5%), respectively; whereas high proportion of shared variants were found between the wild ginseng and SCG group (76.5%). Notably, the SCG group not only harbored the highest level of genetic diversity, but also possessed high proportion (12.6%) of private variants relative to the wild ginseng (4.2%) and NCG (2.7%) group.
Spatial Genetic Structure and Ecological Niche Modeling
Maximum likelihood estimation of individual ancestries of wild and cultivated ginseng was performed for combined transcriptome data. Genetic assignments of wild and cultivated ginseng revealed spatial structure between the southern and northern accessions (fig. 2). For example, inferred ancestries of the NCG group showed genetic similarity to three wild ginseng accessions collecting from northern distribution range; while the SCG group showed evidence of shared ancestries with the southern wild ginseng accessions (fig. 2). Additional patterns are also apparent, including the differences in the degree of genetic heterogeneity of the SCG and NCG groups. The NCG accessions are highly homogeneous, with inferred ancestries almost entirely assigned to a single genetic cluster (>80%). In contrast, the SCG group accessions are genetically more heterogeneous, with most of accessions showing mixed genetic assignments. Spatial genetic structure was also observed in the resulting parsimony network of mitochondrial haplotypes where all NCG group accessions fall into the same clade (fig. 3A, Clade A), and almost all accessions of the SCG group as well as the SKG accession formed a distinct clade (fig. 3A, Clade B). In contrast, the chloroplast genome does not show spatial genetic structure between the SCG and NCG groups. Instead, one major haplotype (fig. 3A, Hap A) exists in wild ginseng and the three cultivated ginseng groups (SCG, NCG, and SKG) (fig. 3B). Notably, three of the five haplotypes were identified in the 12 SKG accessions only, suggesting the possibility of independent origin of SKG group.
Fig. 2.
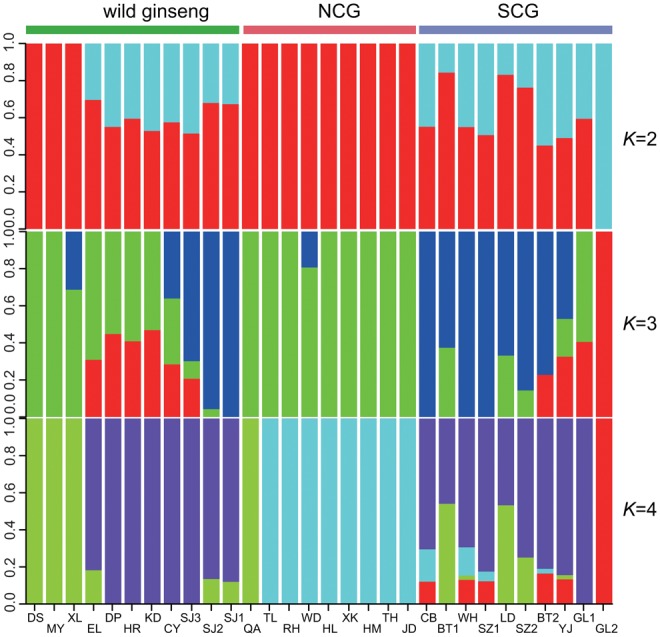
Individual ancestries of the 30 wild and cultivated ginseng accessions. The bars at the top indicate wild and cultivated ginseng groups. Each vertical bar represents one accession and its name is showed on the bottom. NCG, northern cultivated ginseng group. SCG, southern cultivated ginseng group.
Fig. 3.

Parsimony network analyses of the chloroplast and mitochondrial genomes. Relationships of mitochondrial (A) and chloroplast (B) haplotypes of the 30 wild and cultivated ginseng accessions. Size of the cycle indicates number of haplotypes. WG, wild ginseng. NCG, northern cultivated ginseng. SCG, southern cultivated ginseng. SKG, South Korean ginseng.
To further examine the spatial genetic structure between southern and northern ginseng accessions, we calculated pair-wise genetic differentiation between the wild and cultivated ginseng groups. Our results revealed that while the wild ginseng showed low level of genetic differentiation with either of the SCG (FST = 0.0036) or NCG (FST = 0. 0094) groups, obviously higher genetic differentiation was found between the SCG and NCG groups (FST = 0.0200) at the whole genome level (fig. 1A). Similar phenomena were also observed at the statistic of individual gene where most of the 8,660 protein coding genes exhibited low genetic differentiation between wild and cultivated ginseng (supplementary fig. S2A, Supplementary Material online), whereas increase in genetic differentiation was observed at the pair-wise comparisons of three ginseng groups (supplementary fig. S2B and C, Supplementary Material online), especially for those between the NCG and SCG groups (supplementary fig. S2D, Supplementary Material online). We also calculated the Tajima’s D for each gene within the wild and each of the cultivated ginseng groups separately. A large number of these protein-coding genes showed positive value across the wild, SCG, and NCG groups (fig. 4). This trend is more pronounced when all cultivated ginseng accessions were combined (supplementary fig. S3, Supplementary Material online), suggesting the population structure within cultivated ginseng. These observations together suggested spatial differentiation in genetic structure between the southern and northern ginseng accessions.
Fig. 4.

Identification of candidate genes that showed relatively lower nucleotide diversity (π) in SCG and NCG groups and high genetic differentiation (FST) relative to wild ginseng. Red solid lines are the cutoff for SCG (πwild/SCG>2; FST>0.05) (A) and NCG (πwild/SCG>4; FST>0.10) (B), respectively. Each green dot represents a single protein-coding gene. Barplot distributions of Tajima’s D values are showed for wild ginseng, SCG and NCG groups separately. X- and Y-axis are Tajima’s D value and number of genes, respectively.
The ROC of the predicted model demonstrated that average AUC value (0.960±0.006) for the best-fitting model is obviously higher than that of the random distribution model (AUC = 0.500), suggesting stability and good support of the ecological niche model of P. ginseng (supplementary fig. S4A, Supplementary Material online). Similar phenomenon was also observed in the Jackknife test that all the 20 variables generated positive test gain, confirming that the simulated model is better than random model for predicting the distribution of P. ginseng. Of the 20 bioclimatic variables, BIO1 (annual mean temperature), BIO4 (temperature seasonality), and BIO11 (mean temperature of coldest quarter) were the three most effective variables for predicting the suitability value of P. ginseng (supplementary fig. S4B, Supplementary Material online). In contrast, the environmental variable BIO15 (precipitation seasonality) decreased the gain of the predicted model mostly when it was omitted (supplementary fig. S4C, Supplementary Material online). When all 20 bioclimatic variables were pooled together, the predicted distribution model revealed that suitable patches were predicted to be highly probable in the southern ginseng cultivation area (supplementary fig. S1, Supplementary Material online).
Selection on Cultivated Ginseng Genome
The main aim of this study was to assess if selection has acted on specific genes of cultivated ginseng during the domestication and local adaptation processes. In theory, the target genes that have undergone selection would be expected to show low nucleotide diversity with high genetic differentiation relative to neutral genes. To this end, we calculated nucleotide diversity (θ and π) and genetic differentiation for each of the 8,660 protein-coding genes (fig. 4 and supplementary fig. S3, Supplementary Material online). To further examine if allele frequency has observable effect on the estimation of nucleotide diversity, we compared the π and θ ratios between wild and cultivated ginseng for the 8,660 genes. Our comparisons revealed that the genes with high π ratio values also showed high θ ratio values, suggesting that the allele frequency would not result in obvious statistical bias (supplementary fig. S5, Supplementary Material online). Of these comparisons, only 15 candidate genes (0.3%) showed differentially low nucleotide diversity (πwild/πcultivar > 2) in all cultivated ginseng accessions and high genetic differentiation (FST > 0.05) relative to wild ginseng (supplementary table S2, Supplementary Material online). Of the two cultivated ginseng groups, 5 (0.06%) and 97 (1.1%) candidate genes were identified in the SCG (πwild/πcultivar > 2 and FST > 0.05) and NCG (πwild/πcultivar > 4 and FST > 0.10) groups, respectively. Among these candidate genes, only three of them were shared among the three comparisons. In contrast, 5, 2, and 87 of these candidate genes were specific to all cultivated ginseng, SCG, and NCG groups, respectively. Functional analyses further revealed that these genes are associated with diverse biological pathways, including DNA methylation, chromatin structure, and lignin biosynthesis (supplementary table S2, Supplementary Material online). Specifically, some of these genes that exhibited reduced nucleotide diversity in the NCG group only were involved in the pathways of chloroplast and mitochondrial genomes, such as ADP metabolism and cytochrome P450 system (supplementary table S2, Supplementary Material online).
Of the genes involved in the ginsenoside biosynthesis pathway, nucleotide diversity (θ) and segregating site (N) were evaluated for wild ginseng, SCG, and NCG groups separately (fig. 5 and supplementary table S3, Supplementary Material online). Our results revealed that although the nucleotide diversity and segregating site varied dramatically (t-test, P < 0.01) across these genes, a great majority of these genes showed relatively higher nucleotide diversity in cultivated ginseng compared with wild ginseng. Between the cultivated ginseng groups, the SCG group contained the highest nucleotide diversity at most of these genes, which are consistent with above genome-wide scanning of the 8,660 protein-coding genes. We noted that while a portion of these genes exhibited high genetic diversity across the three ginseng groups, decrease in nucleotide diversity was observed at some specific genes in either of the wild ginseng and NCG group. Specifically, three and five of these genes showed no sequence polymorphism in the wild ginseng and NCG group, respectively. These observations indicated that these ginsenoside synthesis pathway genes might have played important roles during ginseng domestication and local adaptation processes.
Fig. 5.
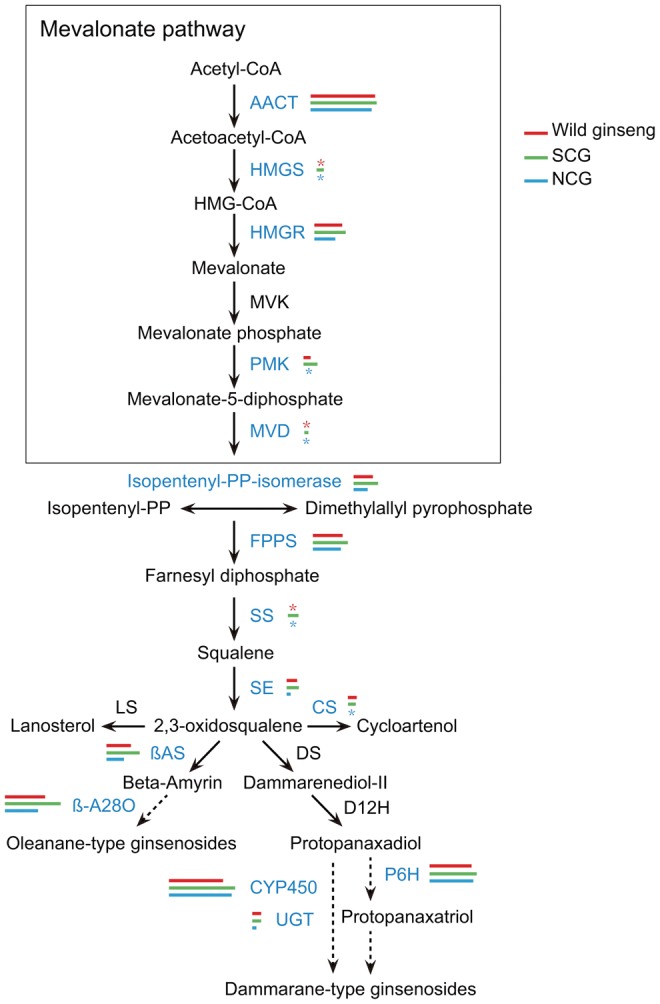
Ginsenoside biosynthesis pathway and DNA polymorphisms for the 15 genes. Genes used in this study are showed in blue color. Lengths of the red, green and blue bars represent the number of SNPs in wild ginseng, SCG, and NCG groups, respectively. Star symbol indicates no DNA polymorphism.
Discussion
Genome-Wide View of Domestication History of Cultivated Ginseng
Chinese ginseng is a remarkable herbal plant with multiple clinical and pharmacological components related to anticancer, antidiabetic, and stress reduction (Xie etal. 2005; Ganesan et al 2015). Ginseng root is highly valuable and has been widely used as a traditional herbal medicine in East Asia for thousands of years (Liu and Xiao 1992; Coon and Ernst 2002). Identifications of the medicinal bioactivities and their effects on human health have been investigated extensively in previous studies (Coon and Ernst 2002; Park etal. 2012). However, domestication history and genomic composition of the Chinese ginseng remained largely uninvestigated.
Recent phylogenetic and population genetic analyses illustrated that all cultivated ginseng landraces were domesticated from their wild progenitor P. ginseng (Wen and Zimmer 1996; Lee and Wen 2004; Li etal. 2015). Then, artificial selection and local adaptation acting on specific genes have resulted in the morphological divergence among different ginseng landraces (Li etal. 2015). With regard to the exact domestication routes, however, it is still unclear on how and where the cultivated ginseng landraces were originated, and opposing views have been proposed based on different molecular markers. For example, while both of the microsatellite and MISP data suggested a single origin of Chinese ginseng landraces, genome-wide BAC-end sequences suggested a multiple origin model in which the various cultivated ginseng landraces were domesticated from different wild ginseng populations independently (Li etal. 2015). In this study, we inferred the origin and domestication history of all the representative cultivated ginseng landraces with evidence from both nuclear and cytoplasmic genomes. Consistent with our previous observation based on the BAC-end sequences, both the nuclear and mitochondrial genomes revealed genetic structure differentiation between the northern and southern ginseng accessions, supporting the multiple origins of current cultivated ginseng landraces. For NCG group, all nine accessions showed high-genetic homogeneity and relatively lower nucleotide diversity compared to the wild ginseng and SCG groups. Given that the northern geographic area has very short ginseng cultivation history and all of these accessions are belonged to the COMMON ginseng landrace, we deemed that all NCG accessions were originated from the same domestication center and then have undergone a severe genetic bottleneck during the domestication process. In contrast, the SCG group contained four ginseng landraces, namely COMMON, BIANTIAO, SHIZHU, and GAOLI. As expected, high genetic heterogeneity and nucleotide diversity were observed in the SCG group, supporting previous hypothesis of the multiple origins of cultivated ginseng (Li etal. 2015). Specifically, the SKG accession(s) showed distinct genetic background at both the nuclear and chloroplast genome levels, confirming the independent origin of South Korean landrace (Kim etal. 2015; Li etal. 2015).
Multiple independent origins suggested distinct demographic histories between the northern and southern ginseng accessions. Indeed, increase in genetic differentiation was observed between the SCG and NCG groups at both the whole genome and individual gene levels. Similar phenomenon was also observed at the nucleotide variation pattern where the NCG group shared much fewer variants with the wild ginseng and SCG groups. These observations can be partially explained by the single origin of the NCG group, and then selection accompanying genetic drift during the domestication process further promoted the genetic divergence. In addition, multiple origins of the SCG group associated with artificial selection might have further driven the genetic differentiation between SCG and NCG groups. We noted that a large number of private variants existed in the wild and cultivated ginseng, respectively. Wild ginseng grows in natural environment and possesses much more bioactive components both qualitatively and quantitatively compared with cultivated ginseng. Morphological differences between the wild and cultivated ginseng are not significant, and most of which are often affected by environmental and developmental factors (Zhao etal. 2015). Previous studies have attempted to isolate molecular markers from the chloroplast genome for authentication of wild ginseng (Kim etal. 2015; Zhao etal. 2015). Our study based on genome-wide scanning identified 10,005 private variants from the 11 wild ginseng accessions which might be useful for future identification of wild ginseng.
Local Adaptation and Selection Shaping the Cultivated Ginseng Genome
The observed spatial genetic structure and phenotypic differences among landraces indicated selection might have acted on the cultivated ginseng genome during the local adaptation and domestication processes. We scanned the nuclear genome of wild and cultivated ginseng by comparing their nucleotide diversity and genetic differentiation. Our results demonstrated that only 15 (0.2%) of the 8,660 protein coding genes were under selection in all cultivated ginseng accessions. In the case of Asian rice, genome-wide scanning identified a total of 1,322 (2.4%) and 1,265 (2.3%) genes that were supposed to have undergone artificial selection in japonica and indica, respectively (Xu etal. 2012). Similarly, 122 (1.2%) and 15 (0.2%) of the 9,768 contigs were associated with the domestication and improvement of cultivated sunflower (Baute etal. 2015). Unlike the other economically important crops, Chinese ginseng has very short domestication history (<1,500 years) and no obvious domestication syndrome was observed across ginseng landraces (Li etal. 2015). Under this reasoning, our genome-wide scanning confirmed that very weak selection pressure has been imposed on the cultivated ginseng genome during the domestication process.
Given that the NCG and SCG groups show spatial genetic structure and inhabit distinct environmental conditions, we therefore detected selected genes for the two cultivated ginseng groups separately. Our comparisons showed that only five genes (0.06%) showed signal of selection across the 8,660 protein-coding genes in the SCG group. It has been proposed in our previous study that the four ginseng landraces (COMMON, BIANTIAO, SHUZHU, and GAOLI) were domesticated from wild ginseng through multiple times (Li etal. 2015). The observed phenotypic differences among these landraces were possibly due to the selection acting on specific traits during the domestication process (Li etal. 2015). In this study, ecological niche modeling further demonstrated that suitable patches are mainly localized in the southern ginseng cultivation area, suggesting that the SCG group might have undergone relatively lower selection pressure compared to the NCG group. Multiple origins together with weak selection pressure resulted in the observation that only a very small part of the SCG genome was the target of selection during the domestication process. In contrast, the NCG group inhabits the ecological niche that is relatively lower predicted probability of suitable conditions. Genome-wide scanning of the nuclear genome revealed that a total of 97 genes (1.1%) were supposed to have undergone selection in the NCG group. It is of note that 87 (85.3%) of the 97 candidate genes are present in the NCG group only. Difference in genomic regions showing selective sweep was also observed between the two Asian rice varieties japonica and indica (Huang etal. 2012). Independent domestication of these varieties was thought to be the main determinant that has led to the observed distinct genomic constitution (He etal. 2011; Huang etal. 2012; Civáň etal. 2015; Choi etal. 2017). Taken together, our genome-wide scanning demonstrated that distinct domestication trajectories and ecological niches have led to the different genomic constitutions between the SCG and NCG groups.
Notably, functional analyses of these candidate genes revealed that ten of them are localized at the nuclear genome but which are subsequently involved in diverse chloroplast and mitochondrial pathways. Meanwhile, our genome-wide screening revealed that, although the cultivated ginseng maintained relatively higher nucleotide diversity at the nuclear genome, clear decrease in nucleotide diversity was found at the chloroplast and mitochondrial genomes. Specifically, only one SNP was identified at the chloroplast genome of the NCG group. The chloroplast genome plays a crucial role in plant cell functions, such as photosynthesis and carbon fixation (Neuhaus and Emes 2000; Jin and Daniell 2015). Chinese ginseng is a shaded perennial plant with very slow growth rate (Hwang etal. 2001; Kim and Lee 2004). Compared with the wild ginseng, cultivated ginseng grows much faster and possesses a shorter lifespan, especially for the landrace COMMON ginseng (Su etal. 2008; Li etal. 2011). These attributes together allowed us to propose that the cytoplasmic genomes as well as their related pathways might be the targets of selection during the domestication and local adaptation processes. In addition, low nucleotide variation level at the plastid genomes can also be explained by the outcross mating system and mixed cultivation of different ginseng landraces (Su etal. 2008). These attributes might together increase genetic variation in nuclear genome leaving lower variation at the plastid genomes.
In addition to the genome-wide screening of the 8,660 protein coding genes, we also investigated the nucleotide diversity of 15 ginsenoside biosynthesis genes. The plant species developed diverse defense mechanisms to prevent insect and pathogenic attacks (Aerts and Mordue 1997; Casanova etal. 2002). In the case of Chinese ginseng, it is grown under shaded area with 7–13 years of cultivation time (Liu and Xiao 1992; Su etal. 2008), which gives favorable growth environments for pathogen infections (Wang etal. 2012). Naturally occurring ginsenosides are antimicrobial and antifungal which confer the Chinese ginseng abilities to cope with biotic stresses (Nicol etal. 2002; Bernards etal. 2006; Sung and Lee 2008). On the other hand, ginsenosides are also the major bioactive components of Chinese ginseng and involved in modulating multiple physiological activities (Leung and Wong 2010). Genetic assessment of these ginsenoside biosynthesis genes is high valuable for future molecular improvement of cultivated ginseng. Consistent with the genome-wide scanning of the 8,660 genes, the NCG group possessed relatively lower level of nucleotide diversity at most of these ginsenoside biosynthesis genes compared to wild ginseng and SCG group. For example, the UDP-glycosyltransferase gene catalyzes the conversion and modification of dammarendiolor β-amyrin to various ginsenosides (e.g., Rb and Rg) (Chen etal. 2011; Wang etal. 2012). While five SNPs were identified in wild ginseng and SCG group, respectively, the NCG group conserved only one SNP across the nine accessions. Differences in effective medicinal ingredients (e.g., ginsenosides) were reported among different ginseng landraces (Woo etal. 1999; Kang etal. 2008). Decrease in nucleotide diversity at these ginsenoside biosynthesis genes might be correlated with the differences in type and level of ginsenosides in the NCG group.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgment
We thank Dr Hongxing Xiao for providing the wild ginseng accessions. This study was supported by The National Natural Science Foundation of China (31470010 and 31670382).
Literature Cited
- Aerts RJ, Mordue AJ.. 1997. Feeding deterrence and toxicity of neem triterpenoids. J Chem Ecol. 23(9):2117–2132. [Google Scholar]
- Alexander DH, Novembre J, Lange K.. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19(9):1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17):3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: A quality control tool for high throughput sequence data. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Baute GJ, Kane NC, Grassa CJ, Lai Z, Rieseberg LH.. 2015. Genome scans reveal candidate domestication and improvement genes in cultivated sunflower, as well as post-domestication introgression with wild relatives. New Phytol. 206(2):830–838. [DOI] [PubMed] [Google Scholar]
- Bernards MA, Yousef LF, Nicol RW.. 2006. The allelopathic potential of ginsenosides In: Inderjit, Mukerji KG, eds. Allelochemicals: biological control of plant pathogens and diseases. Dordrecht: Springer Press; p. 157–175. [Google Scholar]
- Casanova H, et al. 2002. Insecticide formulations based on nicotine oleate stabilized by sodium caseinate. J Agr Food Chem. 50(22):6389–6394. [DOI] [PubMed] [Google Scholar]
- Chen S, et al. 2011. 454 EST analysis detects genes putatively involved in ginsenoside biosynthesis in Panax ginseng. Plant Cell Rep. 30(9):1593–1601. [DOI] [PubMed] [Google Scholar]
- Chen S, et al. 2010. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One 5(1):e8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HI, et al. 2014. Major repeat components covering one-third of the ginseng (Panax ginseng C.A. Meyer) genome and evidence for allotetraploidy. Plant J. 77(6):906–916. [DOI] [PubMed] [Google Scholar]
- Choi JY, et al. 2017. The rice paradox: multiple origins but single domestication in Asian rice. Mol Biol Evol. 34:969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civáň P, Craig H, Cox CJ, Brown TA.. 2015. Three geographically separate domestications of Asian rice. Nat Plants 1(11):15164.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement M, Posada D, Crandall KA.. 2000. TCS: a computer program to estimate gene genealogies. Mol Ecol. 9(10):1657–1659. [DOI] [PubMed] [Google Scholar]
- Coon JT, Ernst E.. 2002. Panax ginseng. Drug Saf. 25(5):323–344. [DOI] [PubMed] [Google Scholar]
- Danecek P, et al. 2011. The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond J. 2002. Evolution, consequences and future of plant and animal domestication. Nature 418(6898):700–707. [DOI] [PubMed] [Google Scholar]
- Doebley JF, Gaut BS, Smith BD.. 2006. The molecular genetics of crop domestication. Cell 127(7):1309–1321. [DOI] [PubMed] [Google Scholar]
- Drašar P, Moravcova J.. 2004. Recent advances in analysis of Chinese medical plants and traditional medicines. J Chromatogr B. 812(1–2):3–21. [DOI] [PubMed] [Google Scholar]
- Elith J, et al. 2011. A statistical explanation of MaxEnt for ecologists. Divers Distrib. 17(1):43–57. [Google Scholar]
- Ganesan P, Ko HM, Kim IS, Choi DK.. 2015. Recent trends of nano bioactive compounds from ginseng for its possible preventive role in chronic disease models. RSC Adv. 5(119):98634–98642. [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JY, Hwang HS, Choi SW, Kim HJ, Choi YE.. 2012. Cytochrome P450 CYP716A53v2 catalyzes the formation of protopanaxatriol from protopanaxadiol during ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol. 53(9):1535–1545. [DOI] [PubMed] [Google Scholar]
- Han JY, Kim HJ, Kwon YS, Choi YE.. 2011. The Cyt P450 enzyme CYP716A47 catalyzes the formation of protopanaxadiol from dammarenediol-II during ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol. 52(12):2062–2073. [DOI] [PubMed] [Google Scholar]
- He Z, et al. 2011. Two evolutionary histories in the genome of rice: the roles of domestication genes. PLoS Genet. 7(6):e1002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A.. 2005. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 25(15):1965–1978. [Google Scholar]
- Huang X, et al. 2012. A map of rice genome variation reveals the origin of cultivated rice. Nature 490(7421):497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HJ, Kim EH, Cho YD.. 2001. Isolation and properties of arginase from a shade plant, ginseng (Panax ginseng C.A. Meyer) roots. Phytochemistry 58(7):1015–1024. [DOI] [PubMed] [Google Scholar]
- Jeong CS, Chakrabarty D, Hahn EJ, Lee HL, Paek KY.. 2006. Effects of oxygen, carbon dioxide and ethylene on growth and bioactive compound production in bioreactor culture of ginseng adventitious roots. Biochem Eng J. 27(3):252–263. [Google Scholar]
- Jiang P, Shi FX, Li YL, Liu B, Li LF.. 2016. Development of highly transferable microsatellites for Panax ginseng (Araliaceae) using whole-genome data. Appl Plant Sci. 4(11):1600075.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Daniell H.. 2015. The engineered chloroplast genome just got smarter. Trends Plant Sci. 20(10):622–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, et al. 2008. NMR-based metabolomics approach for the differentiation of ginseng (Panax ginseng) roots from different origins. Arch Pharm Res. 31(3):330–336. [DOI] [PubMed] [Google Scholar]
- Kim JH, Chang EJ, Oh HI.. 2005. Saponin production in submerged adventitious root culture of Panax ginseng as affected by culture conditions and elicitors. Asia Pacific J Mol Biol Biotechnol. 13:87–91. [Google Scholar]
- Kim K, et al. 2015. Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax ginseng species. PLoS One 10(6):e0117159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Lee HL.. 2004. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11(4):247–261. [DOI] [PubMed] [Google Scholar]
- King ML, Murphy LL.. 2007. American ginseng (Panax quinquefolius L.) extract alters mitogen-activated protein kinase cell signaling and inhibits proliferation of MCF-7 cells. J Exp Ther Oncol. 6:147–155. [PubMed] [Google Scholar]
- Larson G, et al. 2014. Current perspectives and the future of domestication studies. Proc Natl Acad Sci U S A. 111(17):6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tran Q, et al. 2002. Hepatoprotective effect of majonoside R2, the major saponin from Vietnamese ginseng (Panax vietnamensis). Planta Med. 68(5):402–406. [DOI] [PubMed] [Google Scholar]
- Lee C, Wen J.. 2004. Phylogeny of Panax using chloroplast trnC-trnD intergenic region and the utility of trnC-trnD in interspecific studies of plants. Mol Phylogenet Evol. 31(3):894–903. [DOI] [PubMed] [Google Scholar]
- Leung KW, Wong AST.. 2010. Pharmacology of ginsenosides: a literature review. Chin Med. 5:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MR, et al. 2013. A simple strategy for development of single nucleotide polymorphisms from non-model species and its application in Panax. Int J Mol Sci. 14: 24581–24591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MR, et al. 2015. Genetic and epigenetic diversities shed light on domestication of cultivated ginseng (Panax ginseng). Mol Plant. 8(11):1612–1622. [DOI] [PubMed] [Google Scholar]
- Li S, Li J, Yang XL, Cheng Z, Zhang WJ.. 2011. Genetic diversity and differentiation of cultivated ginseng (Panax ginseng C. A. Meyer) populations in Northeast China revealed by inter-simple sequence repeat (ISSR) markers. Genet Resour Crop Evol. 58(6):815–824. [Google Scholar]
- Liang Y, Zhao S.. 2008. Progress in understanding of ginsenoside biosynthesis. Plant Biol. 10(4):415–421. [DOI] [PubMed] [Google Scholar]
- Liu CX, Xiao PG.. 1992. Recent advances on ginseng research in China. J Ethnopharmacol. 36(1):27–38. [DOI] [PubMed] [Google Scholar]
- Luo H, et al. 2011. Analysis of the transcriptome of Panax notoginseng root uncovers putative triterpene saponin-biosynthetic genes and genetic markers. BMC Genomics 12(Suppl 5):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Wang X, Xiao P, Hong D.. 2000. A study on AFLP fingerprinting of land races of Panax ginseng L. Chin J Chin Mater Med. 25(12):707–710. [PubMed] [Google Scholar]
- Ma X, Wang X, Xu Z, Xiao P, Hong D.. 1999. RAPD variation within and among populations of ginseng cultivars. Acta Bot Sin 42:587–590. [Google Scholar]
- Mathur A, Mathur AK, Pal M, Uniyal GC.. 1999. Comparison of qualitative and quantitative invitro ginsenoside production in callus cultures of three Panax species. Planta Med. 65(5):484–486. [DOI] [PubMed] [Google Scholar]
- Neuhaus H, Emes M.. 2000. Nonphotosynthetic metabolism in plastids. Annu Rev Plant Biol. 51:111–140. [DOI] [PubMed] [Google Scholar]
- Nicol R, Traquair J, Bernards M.. 2002. Ginsenosides as host resistance factors in American ginseng (Panax quinquefolius). Can J Bot. 80(5):557–562. [Google Scholar]
- Okazaki H, et al. 2006. Increased cholesterol biosynthesis and hypercholesterolemia in mice overexpressing squalene synthase in the liver. J Lipid Res. 47(9):1950–1958. [DOI] [PubMed] [Google Scholar]
- Park HM, Kim SJ, Kim JS, Kang HS.. 2012. Reactive oxygen species mediated ginsenoside Rg3-and Rh2-induced apoptosis in hepatoma cells through mitochondrial signaling pathways. Food Chem Toxicol. 50(8):2736–2741. [DOI] [PubMed] [Google Scholar]
- Phillips SJ, Anderson RP, Schapire RE.. 2006. Maximum entropy modeling of species geographic distributions. Ecol Model. 190(3–4):231–259. [Google Scholar]
- Phillips SJ, Dudík M.. 2008. Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography 31(2):161–175. [Google Scholar]
- Phillips SJ, Dudík M, Schapire RE.. 2004. A maximum entropy approach to species distribution modeling In Proceedings of the twenty-first international conference on Machine learning, p. 83. [Google Scholar]
- Purcell S, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Ibarra J, Morrell PL, Gaut BS.. 2007. Plant domestication, a unique opportunity to identify the genetic basis of adaptation. Proc Natl Acad Sci U S A. 104:8641–8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi FX, et al. 2015. The impacts of polyploidy, geographic and ecological isolations on the diversification of Panax (Araliaceae). BMC Plant Biol. 15(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su WL, Lv XM, Su YX.. 2008. Planting styles of main types of strains of changbai mountains ginseng and its classification of commodities. Renshen Yanjiu 20:34–39. [Google Scholar]
- Sung WS, Lee DG.. 2008. In vitro candidacidal action of Korean red ginseng saponins against Candida albicans. Biol Pharmaceut Bull. 31(1):139–142. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. 2012. Advances in study of ginsenoside biosynthesis pathway in Panax ginseng C.A. Meyer. Acta Physiol Plant. 34(2):397–403. [Google Scholar]
- Wang M, et al. 2005. Metabolomics in the context of systems biology: bridging traditional Chinese medicine and molecular pharmacology. Phytother Res. 19(3):173–182. [DOI] [PubMed] [Google Scholar]
- Wang W, Zhao ZJ, Xu Y, Qian X, Zhong JJ.. 2006. Efficient induction of ginsenoside biosynthesis and alteration of ginsenoside heterogeneity in cell cultures of Panax notoginseng by using chemically synthesized 2-hydroxyethyl jasmonate. Appl Microbiol Biot. 70(3):298–307. [DOI] [PubMed] [Google Scholar]
- Wen J, Zimmer EA.. 1996. Phylogeny and biogeography of Panax L. (the ginseng genus, araliaceae): inferences from ITS sequences of nuclear ribosomal DNA. Mol Phylogenet Evol. 6(2):167–177. [DOI] [PubMed] [Google Scholar]
- Woo YA, Kim HJ, Chung H.. 1999. Classification of cultivation area of ginseng radix with NIR and Raman spectroscopy. Analyst 124(8):1223–1226. [Google Scholar]
- Xie JT, Mehendale S, Yuan CS.. 2005. Ginseng and diabetes. Am J Chin Med. 33(3):397–404. [DOI] [PubMed] [Google Scholar]
- Xu X, et al. 2012. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol. 30:105–111. [DOI] [PubMed] [Google Scholar]
- Zhao Y, et al. 2015. The complete chloroplast genome provides insight into the evolution and polymorphism of Panax ginseng. Frontiers Plant Sci. 5:696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.