

ABSTRACT
The heteromeric acyl coenzyme A (acyl-CoA) dehydrogenase FadE28-FadE29 and the enoyl-CoA hydratase ChsH1-ChsH2, encoded by genes within the intracellular growth (igr) operon of Mycobacterium tuberculosis, catalyze the dehydrogenation of the cholesterol metabolite 3-oxo-4-pregnene-20-carboxyl-CoA (3-OPC-CoA), with a 3-carbon side chain, and subsequent hydration of the product 3-oxo-4,17-pregnadiene-20-carboxyl-CoA (3-OPDC-CoA) to form 17-hydroxy-3-oxo-4-pregnene-20-carboxyl-CoA (17-HOPC-CoA). The gene downstream of chsH2, i.e., ltp2, was expressed in recombinant Rhodococcus jostii RHA1 in combination with other genes within the igr operon. His-tagged Ltp2 copurified with untagged ChsH1-ChsH2, ChsH2, or the C-terminal domain of ChsH2, which contains a domain of unknown function (DUF35). Ltp2 in association with ChsH1-ChsH2 or just the DUF35 domain of ChsH2 was shown to catalyze the retroaldol cleavage of 17-HOPC-CoA to form androst-4-ene-3,17-dione and propionyl-CoA. Steady-state kinetic analysis using the Ltp2-DUF35 complex showed that the aldolase had optimal activity at pH 7.5, with a Km of 6.54 ± 0.90 μM and a kcat of 159 ± 8.50 s−1. ChsH1-ChsH2 could hydrate only about 30% of 3-OPDC-CoA, but this unfavorable equilibrium could be overcome when the aldolase was present to remove the hydrated product, providing a rationale for the close association of the aldolase with the hydratase. Homologs of ChsH1, ChsH2, and Ltp2 are found in steroid-degrading Gram-positive and Gram-negative bacteria, suggesting that side chains of diverse steroids may be cleaved by aldolases in the bacteria.
IMPORTANCE The C-C bond cleavage of the D-ring side chain of cholesterol was shown to be catalyzed by an aldolase. The aldolase associates with the hydratase that catalyzes the preceding reaction in the cholesterol side chain degradation pathway. These enzymes are encoded by genes within the intracellular growth (igr) operon of M. tuberculosis, and the operon was demonstrated previously to be linked to the pathogenicity and persistence of the bacteria in macrophages and in mice.
KEYWORDS: DUF35, M. tuberculosis, steroid degradation, aldolase, cholesterol, hydratase
INTRODUCTION
Mycobacterium tuberculosis primarily invades alveolar macrophages, resulting in the formation of granulomas within the lungs of infected individuals. M. tuberculosis can survive within these granulomas by utilizing host lipids, such as cholesterol, for carbon and energy (1, 2). When the intracellular growth operon (igr) that encodes genes involved in cholesterol degradation was deleted, the resultant strain showed reduced growth within resting macrophages and its virulence in mouse models was attenuated (3, 4). The strain accumulated a cholesterol metabolite containing a 2-propionate side chain on the D-ring when it was grown in cholesterol-containing medium (5). Subsequent studies showed that genes within the igr operon are involved in cholesterol D-ring side chain degradation and encode enzymes analogous to those involved in fatty acid β-oxidation. The fadE28 and fadE29 genes within the igr operon encode a heteromeric acyl coenzyme A (acyl-CoA) dehydrogenase that catalyzes dehydrogenation of the Cα-Cβ carbon bond of the 3-carbon-side-chain cholesterol CoA thioester metabolite 3-oxo-4-pregnene-20-carboxyl-CoA (3-OPC-CoA) (5, 6). The genes chsH1 and chsH2, downstream of fadE29, encode a heteromeric enzyme that catalyzes the subsequent hydration reaction (7).
ChsH1 and ChsH2 share homology with the MaoC family of hydratases (7). ChsH1 contains one MaoC domain, while ChsH2 contains a MaoC domain at the N terminus and a domain of unknown function at the C terminus, designated DUF35 (Pfam accession no. PF01796). ChsH2 with a truncation that removed the DUF35 domain (ChsH2ΔDUF35) could form a functional complex with ChsH1 that retained catalytic activity, indicating that the DUF35 domain is not necessary for hydratase function (7). The crystal structure showed that ChsH1-ChsH2ΔDUF35 adopted an α2β2 quaternary structure (7). ChsH1 has a typical complete hot dog fold, composed of a “bun” with five antiparallel β-sheets and a “hot dog” with a central α-helix, like other MaoC hydratases. The carbon bond of the substrate targeted for hydration was found in apposition to catalytic histidine and aspartate residues in ChsH1. The MaoC domain of ChsH2 has an incomplete hot dog fold with a shortened α-helix, which provides a large cavity for binding of the steroid rings of the substrate. Therefore, the functional unit of the hydratase is a dimer containing one protomer each of ChsH1 and ChsH2.
Although the structure of the DUF35 domain within ChsH2 has not been solved, the crystal structure of a homolog, SSO2064 from Sulfolobus solfataricus P2 (about 25% sequence similarity to the DUF35 domain of ChsH2), has been determined (8). SSO2064 consists of only a DUF35 domain, i.e., it does not have an associated MaoC hydratase domain, and its function in S. solfataricus is unknown. Proteins containing a DUF35 domain have also been found to form complexes with 3-ketothiolases that catalyze the formation of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) from acetyl-CoA/propionyl-CoA in Haloferax mediterranei (9). Interestingly, the last gene in the igr operon of M. tuberculosis, ltp2, is annotated as a lipid transfer protein and shares homology with these thiolases (about 40% sequence similarity to the H. mediterranei thiolases). However, Ltp2 does not contain the catalytic cysteine/histidine residues shared among thiolases. In addition, thiolases act on 3-ketoacyl-CoA substrates, which cannot be produced from the 3-carbon side chain of 3-OPC-CoA, as it would result in the carbon in the steroid D-ring having a valency of 5. Here we show that Ltp2 interacts with the DUF35 domain of ChsH2 and the complex catalyzes cleavage of the 3-carbon side chain on the cholesterol metabolite after hydration by ChsH1-ChsH2, forming androst-4-ene-3,17-dione and propionyl-CoA products (Fig. 1).
FIG 1.

Pathway of 3-oxo-4-pregnene-20-carboxyl-CoA (3-OPC-CoA) degradation catalyzed by enzymes encoded in the igr operon of M. tuberculosis. Metabolites in the pathway include 3-oxo-4,17-pregnadiene-20-carboxyl-CoA (3-OPDC-CoA) and 17-hydroxy-3-oxo-4-pregnene-20-carboxyl-CoA (17-HOPC-CoA). Ltp2 in complex with the DUF35 domain of ChsH2 is proposed to catalyze the retroaldol cleavage of 17-HOPC-CoA.
RESULTS
Ltp2 interacts with the DUF35 domain of ChsH2.
The ChsH1-ChsH2 complex, the ChsH1-ChsH2ΔDUF35 complex, and the DUF35 domain of ChsH2 could be produced and purified from recombinant Escherichia coli. In contrast, the Ltp2 protein could not be produced alone or in combination with ChsH1-ChsH2 in recombinant E. coli, although ltp2 could be expressed in the actinobacterial strain Rhodococcus jostii RHA1 and the encoded protein with an N-terminal His tag could be purified by Ni2+-nitrilotriacetic acid (Ni2+-NTA) chromatography (see Fig. S1 in the supplemental material). The yield of Ltp2 was low (75 μg per liter of culture). Ltp2 could be produced in good yield (at least 2 mg from 1 liter of culture) and in high purity from R. jostii RHA1 when the ltp2 gene was coexpressed with chsH1 and chsH2. Ltp2 (with an N-terminal or C-terminal His tag) produced by this recombinant R. jostii RHA1 strain copurified with untagged ChsH1 and ChsH2 in Ni2+-NTA chromatography, indicating that the three proteins formed a stable complex (Fig. 2, lanes 3 and 4). The identities of untagged ChsH1 and ChsH2 in SDS-PAGE were confirmed through tryptic peptide mass fingerprinting. By using a false discovery rate of <1% (probability of >99%), 15 peptides were matched to ChsH1, with 91% sequence coverage, while 38 peptides were matched to ChsH2, with 85% sequence coverage. Gel filtration on a calibrated column showed that the purified proteins eluted in a single peak corresponding to a molecular mass of 224 kDa, indicating a complex containing two molecules each of ChsH1, ChsH2, and Ltp2.
FIG 2.
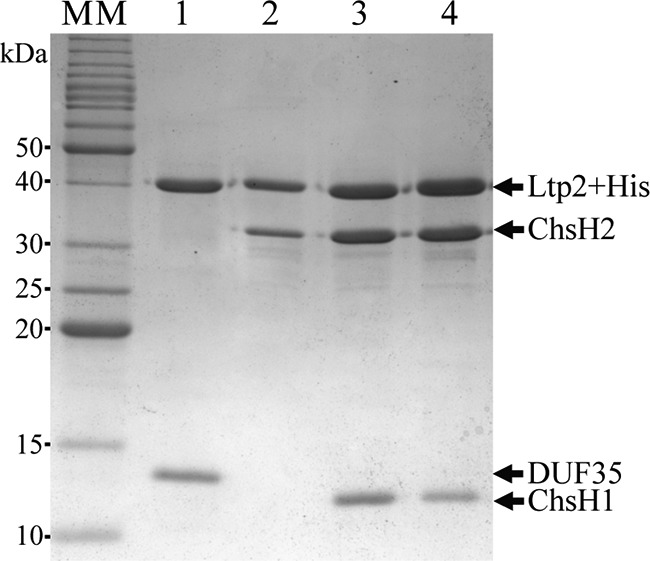
Coomassie blue-stained 12% SDS-PAGE gel of copurified proteins from Ni2+-NTA chromatography. Proteins loaded are N-terminal His-tagged Ltp2 and untagged DUF35 domain of ChsH2 (lane 1), N-terminal His-tagged Ltp2 and untagged ChsH2 (lane 2), C-terminal His-tagged Ltp2 and untagged ChsH1 and ChsH2 (lane 3), or N-terminal His-tagged Ltp2 and untagged ChsH1 and ChsH2 (lane 4). A benchmark protein ladder was used as a molecular mass (MM) marker. Bands corresponding to the expected sizes of His-tagged Ltp2, ChsH1, and ChsH2 and the DUF35 domain of ChsH2 are indicated by arrows.
Combinations of plasmids containing various gene constructs were transformed into R. jostii RHA1 to determine which proteins or domains interacted with Ltp2. The ltp2 gene could be coexpressed with chsH2 and the encoded His-tagged Ltp2 copurified with full-length untagged ChsH2 alone, in the absence of ChsH1, on a Ni2+-NTA column (Fig. 2, lane 2). The 3′ end of chsH2, encoding the DUF35 domain, was amplified by PCR and inserted into a rhodococcal plasmid for coexpression with ltp2 in R. jostii RHA1. The resulting His-tagged Ltp2 copurified with a protein corresponding to the molecular mass of the untagged DUF35 domain of ChsH2 on SDS-PAGE (Fig. 2, lane 1). When subjected to tryptic peptide mass fingerprinting, 28 peptides were matched to DUF35, corresponding to 84% coverage. The purified Ltp2-DUF35 complex was subjected to gel filtration and eluted in a single peak corresponding to a molecular mass of 99 kDa, suggesting that it was a dimer containing two molecules each of Ltp2 and DUF35. Conversely, His-tagged Ltp2 did not copurify with untagged ChsH1-ChsH2ΔDUF35 complex (Fig. S2). Together, the results showed that Ltp2 interacted with the hydratase through the DUF35 domain of ChsH2.
Ltp2 in association with ChsH1-ChsH2 catalyzes a retroaldol reaction.
3-OPC-CoA was incubated with purified acyl-CoA dehydrogenase FadE28-FadE29, ChsH1-ChsH2, or ChsH1-ChsH2-Ltp2, and the products were separated on a C18 column by high-performance liquid chromatography (HPLC). In the presence of Ltp2, two new product peaks were observed, corresponding to the Rf values of authentic propionyl-CoA and androst-4-ene-3,17-dione (Fig. 3). Peak I was subjected to electrospray ionization (ESI)-mass spectrometry (MS) and corresponded to a [M-H]− of 822.1334 (expected mass, 822.1341 Da). The Molecular Formula Generator program assigned an empirical formula of C24H40N7O17P3S, corresponding to propionyl-CoA, with an error of 0.9 ppm and a score of 98. Peak II corresponded to a [M+H]+ of 287.2025 (expected mass, 287.2006 Da). The Molecular Formula Generator program assigned an empirical formula of C19H26O2, corresponding to androst-4-ene-3,17-dione, with an error of 8 ppm and a score of 88.
FIG 3.

HPLC traces of reaction mixtures. Five-hundred-microliter samples were loaded onto a C18 column and eluted with a gradient of 50 mM phosphate buffer (pH 5.3)-acetonitrile mixture (percent acetonitrile is indicated by the dashed lines). CoA esters were detected by absorbance at 254 nm. (A) Androst-4-ene-3,17-dione standard (100 μM). (B) Propionyl-CoA standard (100 μM). (C to E) Reaction mixtures containing 100 μM 3-OPC-CoA and 200 μM ferrocenium hexafluorophosphate in 100 mM TAPS buffer (pH 8.5), with the addition of 0.5 μM (each) FadE28-FadE29 and ChsH1-ChsH2-Ltp2 (C), 0.5 μM (each) FadE28-FadE29 and ChsH1-ChsH2 (D), or 0.5 μM FadE28-FadE29 (E). In the presence of Ltp2, peak III, corresponding to the retention times of 3-OPDC-CoA and 17-HOPC-CoA in chromatograms D and E, was reduced and two new peaks (I and II) appeared, corresponding to the retention times of propionyl-CoA and androst-4-ene-3,17-dione, respectively.
Kinetic analysis of Ltp2-DUF35 activity.
The aldolase activity could be continuously monitored spectrophotometrically, by coupling the formation of propionyl-CoA with NADH oxidation using the aldehyde dehydrogenase from Thermus thermophilus, TTHB247. In this assay, neither DUF35 nor ChsH1-ChsH2 showed any detectable aldolase activity toward 17-hydroxy-3-oxo-4-pregnene-20-carboxyl-CoA (17-HOPC-CoA). The specific aldolase activity of the purified ChsH1-ChsH2-Ltp2 complex in the presence of a saturating concentration of 17-HOPC-CoA (15 μM) was 38.0 ± 1.5 μmol min−1 mg−1. The purified Ltp2-DUF35 complex also possessed aldolase activity, with a specific activity of 125.1 ± 5.7 μmol min−1 mg−1. In contrast, Ltp2 produced alone and partially purified from recombinant R. jostii RHA1 had a low specific activity of (9.7 ± 0.15) × 10−3 μmol min−1 mg−1. The addition of purified DUF35 or ChsH1-ChsH2 in 3-fold molar excess to Ltp2 in assay mixtures enhanced the aldolase activity by only about 3- or 6-fold, respectively. As indicated previously, the expression level of Ltp2 produced alone in R. jostii RHA1 was low. It is possible that, in the absence of DUF35, Ltp2 is not stable and can be misfolded, accounting for the low aldolase activity.
The Ltp2-DUF35 aldolase activity toward 17-HOPC-CoA was determined at a range of pH values (Fig. 4). Activity was determined for a range of substrate concentrations at the optimal pH of 7.5, and the kinetic data could be fitted to a Michaelis-Menten equation (Fig. 5), giving a Km of 6.54 ± 0.90 μM and a kcat of 159 ± 8.50 s−1. The addition of 10 mM sodium EDTA to assay mixtures did not affect the aldolase activity of Ltp2-DUF35, indicating that the aldolase is not metal dependent.
FIG 4.
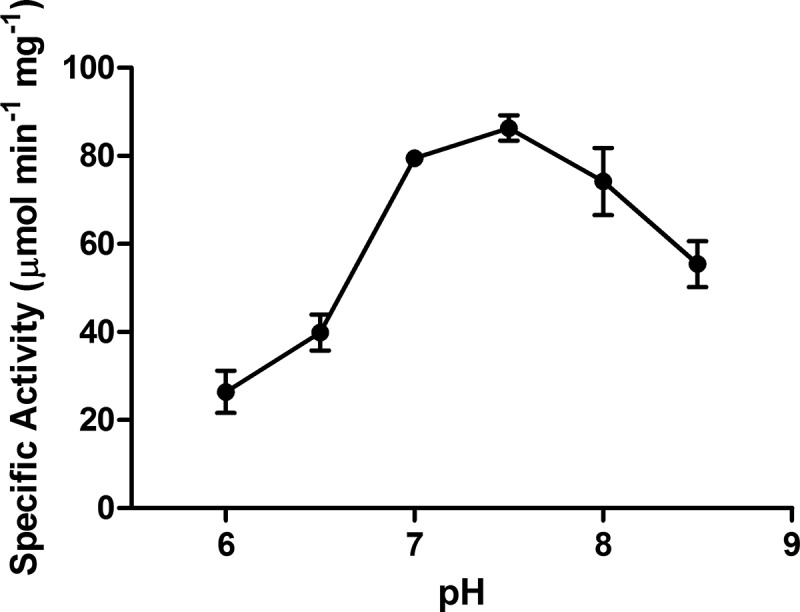
pH dependence of Ltp2-DUF35 aldolase activity. Assay mixtures contained 18 μM 17-HOPC-CoA, 200 μM NADH, and 16 μM TTHB247 in three-component buffer containing 0.1 M Tris, 0.05 M MES, and 0.05 M acetic acid. Error bars indicate standard deviations of triplicate data.
FIG 5.

Plot of initial velocity of the aldolase reaction catalyzed by Ltp2-DUF35 versus the concentration of 17-HOPC-CoA. The assay mixture contained 16 μM TTHB247, 200 μM NADH, and various concentrations of 17-HOPC-CoA in 100 mM sodium HEPES buffer (pH 7.5). The data were fitted to the Michaelis-Menten equation by nonlinear regression with the program GraphPad Prism. Error bars indicate standard deviations of triplicate data.
Equilibrium of hydration reaction catalyzed by ChsH1/ChsH2.
The Ltp2-DUF35 complex had no detectable hydratase activity toward 3-oxo-4,17-pregnadiene-20-carboxyl-CoA (3-OPDC-CoA). In contrast, ChsH1-ChsH2, ChsH1-ChsH2-Ltp2, and ChsH1-ChsH2ΔDUF35 complexes were able to catalyze the hydration of 3-OPDC-CoA. However, an endpoint assay revealed that, in comparison with the ChsH1-ChsH2-Ltp2 complex, ChsH1-ChsH2 and ChsH1-ChsH2ΔDUF35 could hydrate only about 30% of the 3-OPDC-CoA substrate, regardless of the amount of enzyme added to the assay mixture (14 μM 3-OPDC-CoA in 100 mM sodium HEPES buffer [pH 7.5] at 25°C). Therefore, the hydration reaction catalyzed by ChsH1-ChsH2 or ChsH1-ChsH2ΔDUF35 appeared to have reached equilibrium (estimated Keq of 7 × 10−3 M−1) in the absence of the aldolase, with 3-OPDC-CoA being more thermodynamically stable than the hydrated product, 17-HOPC-CoA. When the Ltp2-DUF35 complex was added to a reaction mixture containing ChsH1-ChsH2ΔDUF35, complete hydration of 3-OPDC-CoA was observed, indicating that removal of the hydrated compound, 17-HOPC-CoA, by the aldolase enabled complete turnover of 3-OPDC-CoA.
Occurrence of homologs of Ltp2 and DUF35-containing proteins within steroid-degrading bacteria.
Homologs of chsH1, chsH2, and ltp2 are found in the cholesterol and bile acid degradation gene clusters of other actinobacteria, including R. jostii RHA1 (Fig. 6). Homologs of these genes are also found within the genomes of the Gram-negative bile-acid-degrading bacteria Comamonas testosteroni and Pseudomonas sp. strain Chol1. The arrangement of genes in the bile acid degradation gene clusters differ from the igr operon in M. tuberculosis, in that chsH1 homologs are separated from chsH2 and ltp2 homologs by an acyl-CoA dehydrogenase gene.
FIG 6.

Genes in bacterial steroid degradation gene clusters that contain homologs of ltp2. The names of genes from the respective annotated genomes are written above each gene. R. jostii RHA1 has separate gene clusters for cholesterol and bile acid side chain degradation, which are indicated accordingly. The genes in the igr operon encode a heteromeric acyl-CoA dehydrogenase containing a catalytic subunit (FadE29) (orange) and a noncatalytic subunit (FadE28) (yellow), a heteromeric hydratase containing a catalytic subunit (ChsH1) with a MaoC domain (blue) and a noncatalytic subunit (ChsH2) with a MaoC domain at the N terminus (blue) and a DUF35 domain at the C terminus (gray), and Ltp2 (purple). White, linker. The same colors are used to depict the corresponding homologous genes found in cholesterol or bile acid degradation gene clusters of R. jostii RHA1, Pseudomonas sp. strain Chol1, and C. testosteroni KF1.
Rv3521 and Ro04685 are additional genes containing DUF35; they are found within the cholesterol gene clusters of M. tuberculosis and R. jostii RHA1, respectively. These genes encode proteins containing two tandem DUF35 domains. Downstream of these genes are two genes (designated ltp3 and ltp4 in M. tuberculosis) that are homologous to ltp2. The steroid degradation gene clusters of C. testosteroni and Pseudomonas sp. strain Chol1 also contain genes that encode a protein containing an N-terminal DUF35 domain and a C-terminal MaoC hydratase domain (C211_11482; also known as shy), followed by a gene homologous to ltp2 (C211_11487; also known as sal). These enzymes are proposed to catalyze a hydration and aldolase reaction involving the 5-carbon side chain of cholyl-CoA in Pseudomonas sp. strain Chol1, based on accumulated metabolites in gene knockout studies (10).
DISCUSSION
Aerobic bacteria degrade steroid side chains through a series of reactions analogous to fatty acid β-oxidation. In M. tuberculosis, the thiolase FadA5 was proposed to catalyze the C-C bond cleavage of the side chain in the first two rounds of β-oxidation of the 8-carbon cholesterol side chain, releasing one molecule each of propionyl-CoA and acetyl-CoA and forming the steroid metabolite 3-OPC-CoA, with a 3-carbon side chain (11, 12). In this study, we showed that cleavage of the 3-carbon side chain of this steroid metabolite during the last round of β-oxidation is catalyzed by an aldolase, involving Ltp2, instead of a thiolase. Ltp2 associates with DUF35, a domain at the C terminus of ChsH2. ChsH2 is one of two protomers of enoyl-CoA hydratase that catalyze the preceding step in the metabolism of 3-OPC-CoA. The findings that the ChsH1-ChsH2ΔDUF35 complex retained hydratase activity and Ltp2 in complex with just the DUF35 domain of ChsH2 was sufficient for robust aldolase activity suggest that the hydratase and aldolase activities are catalyzed by distinct active sites in the ChsH1-ChsH2-Ltp2 complex. The close proximity of two enzymatic activities within a protein complex may increase the efficiency of the hydration reaction and aid in overcoming the limitations of the unfavorable hydration equilibrium.
Ltp2 is not related in sequence to aldolases from other metabolic pathways. Rather, it is related to thiolases and acetoacetyl-CoA synthases. Interestingly, some of those homologs of Ltp2 have been known to associate with and to require a protein containing a DUF35 domain for activity (9). DUF35 domains have also been found fused to proteins that also contain thiolase, 3-hydroxy-acyl-CoA dehydrogenase, crotonase, NTF2, or sterol carrier protein domains (8), but the roles of DUF35 in those proteins have not been elucidated. The structure of a stand-alone DUF35 domain protein, SSO2064, from S. solfataricus contains a rubredoxin-like zinc finger motif and an oligonucleotide/oligosaccharide binding (OB) fold. There is a zinc atom bound within the zinc ribbon by residues Cys52, Cys49, Cys63, and Cys66 of SSO2064, but sequence alignment with the DUF domain of ChsH2 indicates that the last three cysteines are replaced by leucine, valine, and aspartate, respectively, while Cys52 corresponds to a gap. These residues are not conducive to metal ion binding. The reaction catalyzed by the Ltp2-DUF35 complex of M. tuberculosis was unaffected by the presence of the metal chelator EDTA (10 mM), providing further evidence that metal ions are not necessary for aldolase activity. The DUF35 domains of some ChsH2 homologs, such as those from the bile acid degradation clusters of R. jostii RHA1, C. testosteroni KF1, and Pseudomonas sp. strain Chol1, contain these cysteine residues, and the functional significance of a potential zinc binding site in these proteins is currently not known. The OB fold found in SSO2064, however, is an architecture shared by a variety of different proteins that are involved in interacting with oligosaccharides, metal ions, nucleic acids, and proteins. A hydrophobic groove formed along the OB fold of SSO2064 is proposed to serve as an acyl-CoA binding site, given that many proteins associated with DUF35 domains act on substrates that are CoA thioesters (8). Although it is possible that the DUF35 domain assists in substrate binding within the Ltp2-DUF35 complex, the possibility that it is an allosteric activator of Ltp2 aldolase activity or functions as a conduit for channeling of the product from the hydratase to Ltp2 requires further investigation.
Homologs of Ltp2 and hydratases containing DUF35 domains have been found encoded in steroid degradation gene clusters of bacteria. It is likely that many of these homologs are also involved in cleavage of the isopropyl side chain from the D-ring of various sterols and bile acids in the last round of β-oxidation, similar to the cholesterol degradation pathway of M. tuberculosis. Interestingly, in Mycobacterium and Rhodococcus, the steroid degradation gene cluster also contained a protein with two tandem DUF35 domains followed by two genes homologous to ltp2 (designated ltp3 and ltp4). Single-gene deletions of ltp3 or ltp4 in R. rhodochrous resulted in mutants that were capable of cholesterol side chain degradation but were deficient in β-sitosterol or campesterol side chain degradation (13). Both campesterol and β-sitosterol contain branch side chains. The typical β-oxidation reaction, in which the alcohol produced by hydration is further oxidized by 3-hydroxy-acyl-CoA dehydrogenase, could not occur for these Cβ branched sterol side chains during the first β-oxidation cycle. Instead, the evidence suggests that Ltp3 and Ltp4 catalyze the retroaldol cleavage in the first β-oxidation cycle of β-sitosterol or campesterol side chain degradation, possibly in association with the DUF35 domain-containing protein encoded upstream of ltp3 and ltp4. It is unclear, however, why two DUF35 domains and Ltps are required to catalyze this single reaction.
In the Gram-negative bile-acid-degrading Pseudomonas sp. strain Chol1, formation, by aldolytic cleavage, of an aldehyde from a cholate metabolite with a 5-carbon side chain that does not contain a Cβ branch has been proposed. This aldehyde was not produced in a mutant with a deletion in an ltp2 homolog named skt or sal (10). Interestingly, sal is adjacent to a gene that encodes a hydratase containing an N-terminal DUF35 domain and a C-terminal MaoC hydratase domain; this hydratase was proposed, based on gene knockout studies, to catalyze the preceding step of hydration of the cholate metabolite with a 5-carbon side chain. This finding suggests that, in some bacteria, C-C bond cleavage of unbranched steroid side chains may proceed through a retroaldol reaction instead of a retro-Claisen reaction, which typically is catalyzed by thiolases in fatty acid β-oxidation. However, aldol cleavage of an unbranched side chain would be energetically less favorable than a reaction catalyzed by thiolases, since the aldehyde produced in the former would need to be oxidized and CoA esterified to enable the subsequent round(s) of β-oxidation to continue.
In summary, we have successfully purified and biochemically characterized an enzyme responsible for the cleavage of the side chain from the D-ring of steroids. This will pave the way for future structure-function studies of this novel aldolase and may facilitate the development of new antibiotics against M. tuberculosis that target the cholesterol degradation pathway.
MATERIALS AND METHODS
Chemicals.
Propionyl-CoA, flavin adenine dinucleotide (FAD), and ferrocenium hexafluorophosphate were from Sigma-Aldrich (Oakville, Canada). 3-OPC was from Steraloids Inc. (Newport, RI). Coenzyme A was from BioShop Canada Inc. (Burlington, Canada). Androst-4-ene-3,17-dione was from Toronto Research Chemicals (Toronto, Canada). Restriction enzymes and Pfu polymerase were from Thermo Scientific (Ottawa, Canada). T4 DNA ligase was from New England BioLabs (Pickering, Canada). Ni2+-NTA Superflow resin was from Qiagen (Mississauga, Canada). All other chemicals were obtained from Thermo Scientific (Nepean, Canada), unless otherwise stated.
Bacterial strains and plasmids.
R. jostii RHA1 was obtained from Lindsay Eltis (Department of Microbiology and Immunology, University of British Columbia). The M. tuberculosis H37Rv genomic DNA was a gift from Marcel Behr (McGill University). Plasmids used in this study and their sources are indicated in Table 1.
TABLE 1.
Plasmids used in this study
| Plasmid name | Descriptiona | Reference(s) or source |
|---|---|---|
| pTIPQc2 | E. coli-Rhodococcus shuttle plasmid; Apr Cmr; thiostrepton-inducible promoter for gene expression in R. jostii RHA1 | 17 |
| pTIPQcHis | Plasmid derived from pTIPQc2 except that XbaI-HindIII fragment containing His tag and multiple cloning sites was replaced with corresponding fragment from pET28a | 18 |
| pTIPRT2 | E. coli-Rhodococcus shuttle plasmid; Apr Tetr; thiostrepton-inducible promoter for gene expression in R. jostii RHA1 | 17 |
| pET28a | E. coli expression plasmid; Kanr; T7 promoter, encoding N-terminal His tag | Novagen |
| pBTLT7 | E. coli expression plasmid; Tetr; lac promoter and RBS of pBTL-4 were replaced with T7 promoter and RBS of pT7-7 | 19, 20 |
| pTIPQcHis_ltp2 | ltp2 (PCR) inserted into NdeI and HindIII sites of pTIPQcHis; produces N-terminal His-tagged Ltp2 | This work |
| pTIPQc2_chsH1ltp2 | chsH1 and ltp2 (PCR) inserted into NdeI and HindIII sites of pTIPQc2; produces C-terminal His-tagged Ltp2 and untagged ChsH1 | This work |
| pTIPRT2_chsH2chsH1 | chsH2 and chsH1 (PCR) inserted into NdeI and HindIII sites of pTIPRT2 | This work |
| pTIPRT2_chsH2 | chsH2 (PCR) inserted into NdeI and HindIII sites of pTIPRT2 | This work |
| pTIPRT2_chsH2ΔDUF35 | chsH2 without 3′ end encoding DUF35 domain (PCR) inserted into NdeI and HindIII sites of pTIPRT2 | This work |
| pTIPRT2_DUF35 | DNA encoding DUF35 of chsH2 (PCR) inserted into NdeI and HindIII sites of pTIPRT2 | This work |
| pET28a_chsH1 | chsH1 (PCR) inserted into NdeI and HindIII sites of pET28a | This work |
| pBTLT7_chsH2 | chsH2 (PCR) inserted into NdeI and HindIII sites of pBTLT7 | This work |
| pBTLT7_chsH2ΔDUF35 | chsH2 without 3′ end encoding DUF35 domain (PCR) inserted into NdeI and HindIII sites of pBTLT7 | This work |
Apr, ampicillin resistance; Cmr, chloramphenicol resistance; Tetr, tetracycline resistance; Kanr, kanamycin resistance; RBS, ribosome binding site.
DNA manipulation.
DNA was purified, digested, and ligated using standard protocols. The genes ltp2 (Rv3540c), chsH1 (Rv3541c), and chsH2 (Rv3542c), chsH2 lacking the 3′ end that encodes the DUF35 domain (chsH2Δduf35), and DNA encoding the DUF35 domain of chsH2 were PCR amplified from M. tuberculosis H37Rv genomic DNA with the primers listed in Table S1 in the supplemental material. The primers introduced NdeI and HindIII restriction sites, which could be used to insert the amplified DNA into E. coli and Rhodococcus jostii RHA1 expression vectors. Coexpression of genes was facilitated by the use of pairs of plasmids, in E. coli (pET28a and pBTLT7) or R. jostii RHA1 (pTIPQc and pTIPRT2), that are from incompatible groups and have distinct antibiotic resistance genes for selection and maintenance within the recombinant hosts.
Protein expression and purification.
FadE28-FadE29 and TTHB247 were purified according to the methods described previously (6, 14). Recombinant E. coli BL21(λDE3) containing chsH1-chsH2, chsH1-chsH2Δduf35, and duf35 was grown at 37°C in 4 liters of LB medium supplemented with kanamycin (50 μg/ml) and/or tetracycline (15 μg/ml). Expression of the recombinant proteins was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at mid-log phase (optical density at 600 nm [OD600] of 0.4 to 0.6). Cells were grown for an additional 20 h at 15°C after induction. Recombinant R. jostii RHA1 was grown at 30°C in 4 liters of LB medium supplemented with chloramphenicol (25 μg/ml) or chloramphenicol and tetracycline (8 μg/ml). Expression of the recombinant proteins was induced with 1 μg/ml thiostrepton at mid-log phase (OD600 of 0.4 to 0.6), and the cells were grown for an additional 24 h at 30°C. All cells were harvested by centrifugation at 9,605 × g for 10 min.
Cell pellets were resuspended in 20 mM HEPES buffer (pH 7.5). E. coli and R. jostii RHA1 cells were lysed by passage through a French press at 15,000 lb/in2 and 20,000 lb/in2, respectively. E. coli lysates were centrifuged at 39,191 × g for 30 min, while R. jostii lysates were centrifuged twice at 39,191 × g for 15 min each time, with the removal of cellular debris at each step. Cell extracts were filtered through a 0.45-μm filter and incubated for 1 h at 4°C with Ni2+-NTA resin in buffer (50 mM sodium phosphate, 300 mM sodium chloride [pH 8.0]) containing 20 mM imidazole. The mixture was poured into a gravity column and washed with the same buffer. The His-tagged proteins were eluted with buffer containing 150 mM imidazole (pH 8.0). The buffer was exchanged for 20 mM HEPES (pH 7.5) by dilution in a stirred cell equipped with a YM10 filter (Amicon). Purified enzymes were stored at −80°C.
Determination of protein concentrations, purities, and molecular masses.
Concentrations of proteins were determined by the Bradford assay using bovine serum albumin as the standard (15). Coomassie blue-stained SDS-PAGE gel was used to assess the purity of the enzymes. Liquid chromatography-MS analyses of tryptic fragments of various proteins were performed on an Agilent UHD 6530-Q-TOF mass spectrometer at the Mass Spectrometry Facility of the Advanced Analysis Centre, University of Guelph. Samples were loaded onto a C18 column (Agilent Advance Bio Peptide Map; 100 mm by 2.1 mm by 2.7 μm). Elution buffers used were buffer A, containing 0.1% formic acid in water, and buffer B, containing 0.1% formic acid in acetonitrile. Peptides were eluted from the column using a gradient of 2% buffer B to 45% buffer B over 8 ml. The mass-to-charge ratio was scanned across the m/z range of 300 to 2,000 at 4 GHz. Precursor ions per cycle were selected for fragmentation. The native molecular masses of purified proteins were estimated using gel filtration on a HiLoad 26/60 Superdex 200 column (GE Healthcare), using a previously described protocol (6).
Substrate synthesis.
3-OPC-CoA was synthesized using acyl-CoA synthetase CasI, as described previously (6). 3-OPDC-CoA was synthesized from 200 μM 3-OPC-CoA, in a 30-ml volume, by the addition of 500 nM FadE28-FadE29 and 400 μM ferrocenium hexafluorophosphate, while an equilibrium mixture of 3-OPDC-CoA and 17-HOPC-CoA was synthesized similarly, with the addition of 1 μM ChsH1-ChsH2 to the reaction mixture. Reaction mixtures were incubated at room temperature for 2.5 h, the reactions were stopped by acidification using HCl, and the reaction mixtures were filtered using 0.2-μm filters and YM10 filters. The filtrates were passed through a 3-ml HyperSep disposable C18 column (500-mg bed weight; Thermo Scientific) that had been preequilibrated with 6% acetonitrile in 50 mM sodium phosphate buffer (pH 5.3). The compounds were eluted using 8 ml of 48% acetonitrile in 50 mM sodium phosphate buffer (pH 5.3). 17-HOPC-CoA preparations contained 3-OPDC-CoA due to the unfavorable hydration equilibrium. Analysis by HPLC with a C18 column showed that the preparation contained 57% 3-OPDC-CoA and 43% 17-HOPC-CoA, based on peak ultraviolet (UV) light absorbance (254 nm). Minor peaks constituted <1% of the total peak absorbance of these two compounds. The compounds were dried by lyophilization and stored at −20°C.
Analysis of Ltp2 reaction products by HPLC and MS.
Reactions mixtures containing 100 μM 3-OPC-CoA and 200 μM ferrocenium hexafluorophosphate in 1 ml of 100 mM [tris(hydroxymethyl)methylamino]propanesulfonic acid (TAPS) (sodium-potassium salt) buffer (pH 8.5) were incubated at 25°C for 30 min. FadE28-FadE29, ChsH1-ChsH2, and ChsH1-H2-Ltp2 complexes, when included, were added at concentrations of 0.5 μM. Reactions were stopped by the addition of 70 mM HCl, and precipitated proteins and ferrocenium hexafluorophosphate were removed by centrifugation at 21,000 × g for 3 min. Five hundred microliters of reaction products were separated by HPLC with a HyPURITY C18 column (150 by 4.6 mm; Thermo Scientific), using isocratic elution at 6% acetonitrile-50 mM sodium phosphate (pH 5.3) followed by a gradient of 6% to 80% acetonitrile over 20 column volumes, with UV detection (254 nm). The same volumes of 100 μM authentic androst-4-ene-3,17-dione and propionyl-CoA were similarly separated by HPLC as standards. Fractions were collected and analyzed with an Agilent 1200 HPLC system (Agilent Poroshell 120 EC-C18 column, 50 mm by 3.0 mm, 2.7 μm) interfaced with an Agilent UHD 6530 Q-TOF mass spectrometer, using a gradient of 0.1% formic acid and acetonitrile, at the University of Guelph Advanced Analysis Centre. The mass spectrometer electrospray capillary voltage was maintained at 4.0 kV and the drying gas temperature at 250°C, with a flow rate of 8 liters/min. The nebulizer pressure was 30 lb/in2, and the fragmentor was set to 160. Nozzle, skimmer, and octapole radiofrequency (RF) voltages were set at 1,000, 65, and 750 V, respectively. Nitrogen (purity, >98%) was used as the nebulizing, drying, and collision gas. The mass-to-charge ratio was scanned across the m/z range of 50 to 1,500 at 4 GHz (extended dynamic range), in positive- and negative-ion modes. Androst-4-ene-3,17-dione was observed only in negative-ion mode. Data were collected by target- or data-dependent MS/MS acquisition with MS and MS/MS scan rates of 1.41 spectra/s. The acquisition rate was set at 2 spectra/s. The mass axis was calibrated using Agilent tuning mixture HP0321, prepared in acetonitrile. Mass spectrometer control, data acquisition, and data analysis were performed with MassHunter Workstation software (B.04.00).
Steady-state kinetic assays.
All assays were performed at least in triplicate, in a total volume of 1 ml, at 25°C, using a Varian Cary 3 spectrophotometer equipped with a temperature-controlled cuvette holder. Hydratase activity was determined by measuring the decrease in absorbance at 263 nm due to hydration of the Cα-Cβ double bond of the substrate (ε263 of 6,700 M−1 cm−1) (16). Enzyme-catalyzed retroaldol cleavage of 17-HOPC-CoA was measured by coupling the formation of the propionyl-CoA product to NADH utilization catalyzed by the purified Thermus thermophilus aldehyde dehydrogenase TTHB247 (14), which could be followed spectrophotometrically at 340 nm (ε340 of 6,220 M−1 cm−1). 17-HOPC-CoA preparations used for kinetic analysis of aldolase activity contained 3-OPDC-CoA, due to the equilibrium of the two species. The precise concentration of 17-HOPC-CoA in the mixture was determined using an endpoint assay containing Ltp2-DUF35, TTHB247, and NADH and assuming a stoichiometry of 1:1 for 17-HOPC-CoA present and NADH oxidized. Within the time scale of each aldolase kinetic assay with the Ltp2-DUF35 complex (5 to 15 min), there was no detectable spontaneous conversion of 3-OPDC-CoA to 17-HOPC-CoA, as monitored spectrophotometrically at 263 nm. 3-OPDC CoA was verified to be noninhibitory to aldolase activity up to a concentration of 50 μM. The concentrations of TTHB247 and NADH were adjusted to ensure that these reagents are not limiting in the kinetic assays. The pH dependence of aldolase activity was determined using 18 μM 17-HOPC-CoA, 200 μM NADH, and 16 μM TTHB247, in constant-ionic-strength three-component buffers containing 0.1 M Tris, 0.05 M 2-(N-morpholino)ethanesulfonic acid (MES), and 0.05 M acetic acid. Kinetic parameters for the Ltp2-DUF35 complex were determined using 100 mM sodium HEPES buffer (pH 7.5), 16 μM TTHB247, 200 μM NADH, and concentrations of 17-HOPC-CoA ranging from 2.5 μM to 30 μM. Data were fitted to the Michaelis-Menten equation by nonlinear regression using GraphPad Prism software.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant 2015-05366 from the Natural Science and Engineering Research Council of Canada.
We thank Steven Lam and Amy Stenhouse for cloning of chsH1 and chsH2. Dyanne Brewer and Armen Charchoglyan from the University of Guelph Advanced Analysis Centre are thanked for the acquisition of mass spectra.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00512-17.
REFERENCES
- 1.Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A 105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD. 2007. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A 104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, Sherman DR. 2009. igr genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol 191:5232–5239. doi: 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang JC, Harik NS, Liao RP, Sherman DR. 2007. Identification of mycobacterial genes that alter growth and pathology in macrophages and in mice. J Infect Dis 196:788–795. doi: 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- 5.Thomas ST, VanderVen BC, Sherman DR, Russell DG, Sampson NS. 2011. Pathway profiling in Mycobacterium tuberculosis: elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism. J Biol Chem 286:43668–43678. doi: 10.1074/jbc.M111.313643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruprecht A, Maddox J, Stirling A, Visaggio N, Seah SYK. 2015. Characterization of novel acyl coenzyme A dehydrogenases involved in bacterial steroid degradation. J Bacteriol 197:1360–1367. doi: 10.1128/JB.02420-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang M, Guja KE, Thomas ST, Garcia-Diaz M, Sampson NS. 2014. A distinct MaoC-like enoyl-CoA hydratase architecture mediates cholesterol catabolism in Mycobacterium tuberculosis. ACS Chem Biol 9:2632–2645. doi: 10.1021/cb500232h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishna SS, Aravind L, Bakolitsa C, Caruthers J, Carlton D, Miller MD, Abdubek P, Astakhova T, Axelrod HL, Chiu HJ, Clayton T, Deller MC, Duan L, Feuerhelm J, Grant JC, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kumar A, Marciano D, McMullan D, Morse AT, Nigoghossian E, Okach L, Reyes R, Rife CL, Van Den Bedem H, Weekes D, Xu Q, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wilson IA. 2010. The structure of SSO2064, the first representative of Pfam family PF01796, reveals a novel two-domain zinc-ribbon OB-fold architecture with a potential acyl-CoA-binding role. Acta Crystallogr Sect F Struct Biol Cryst Commun 66:1160–1166. doi: 10.1107/S1744309110002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hou J, Feng B, Han J, Liu H, Zhao D, Zhou J, Xiang H. 2013. Haloarchaeal-type β-ketothiolases involved in poly(3-hydroxybutyrate-co-3-hydroxyvalerate) synthesis in Haloferax mediterranei. Appl Environ Microbiol 79:5104–5111. doi: 10.1128/AEM.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holert J, Kulić Ž, Yücel O, Suvekbala V, Suter MJF, Möller HM, Philipp B. 2013. Degradation of the acyl side chain of the steroid compound cholate in Pseudomonas sp. strain chol1 proceeds via an aldehyde intermediate. J Bacteriol 195:585–595. doi: 10.1128/JB.01961-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaefer CM, Lu R, Nesbitt NM, Schiebel J, Sampson NS, Kisker C. 2015. FadA5, a thiolase from Mycobacterium tuberculosis: a steroid-binding pocket reveals the potential for drug development against tuberculosis. Structure 23:21–33. doi: 10.1016/j.str.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nesbitt NM, Yang X, Fontán P, Kolesnikova I, Smith I, Sampson NS, Dubnau E. 2010. A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect Immun 78:275–282. doi: 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilbrink MH, van der Geize R, Dijkhuizen L. 2012. Molecular characterization of ltp3 and ltp4, essential for C24-branched chain sterol-side-chain degradation in Rhodococcus rhodochrous DSM 43269. Microbiology 158:3054–3062. doi: 10.1099/mic.0.059501-0. [DOI] [PubMed] [Google Scholar]
- 14.Baker P, Hillis C, Carere J, Seah SYK. 2012. Protein-protein interactions and substrate channeling in orthologous and chimeric aldolase-dehydrogenase complexes. Biochemistry 51:1942–1952. doi: 10.1021/bi201832a. [DOI] [PubMed] [Google Scholar]
- 15.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 16.Binstock JF, Schulz H. 1981. Fatty acid oxidation complex from Escherichia coli. Methods Enzymol 71:403–411. doi: 10.1016/0076-6879(81)71051-6. [DOI] [PubMed] [Google Scholar]
- 17.Nakashima N, Tamura T. 2004. Isolation and characterization of a rolling circle type plasmid from Rhodococcus erythropolis and application of the plasmid to multiple recombinant protein expression. Appl Environ Microbiol 70:5557–5568. doi: 10.1128/AEM.70.9.5557-5568.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carere J, McKenna SE, Kimber MS, Seah SYK. 2013. Characterization of an aldolase-dehydrogenase complex from the cholesterol degradation pathway of Mycobacterium tuberculosis. Biochemistry 52:3502–3511. doi: 10.1021/bi400351h. [DOI] [PubMed] [Google Scholar]
- 19.Lynch MD, Gill RT. 2006. Broad host range vectors for stable genomic library construction. Biotechnol Bioeng 94:151–158. doi: 10.1002/bit.20836. [DOI] [PubMed] [Google Scholar]
- 20.Baker P, Pan D, Carere J, Rossi A, Wang W, Seah SYK. 2009. Characterization of an aldolase-dehydrogenase complex that exhibits substrate channeling in the polychlorinated biphenyls degradation pathway. Biochemistry 48:6551–6558. doi: 10.1021/bi9006644. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.