

Key Points
Activation of αIIbβ3 is required for its ancillary site interactions with fibrinogen fragment D lacking the γ-chain dodecapeptide (‘D98’).
EDTA can paradoxically induce normal αIIbβ3 to interact with fibrinogen fragment ‘D98.’
Abstract
Platelet integrin receptor αIIbβ3 supports platelet aggregation by binding fibrinogen. The interaction between the fibrinogen C-terminal γ-chain peptide composed of residues γ-404-411 (GAKQAGDV) and the Arg-Gly-Asp (RGD) binding pocket on αIIbβ3 is required for fibrinogen-mediated platelet aggregation, but data suggest that other ancillary binding sites on both fibrinogen and αIIbβ3 may lead to higher-affinity fibrinogen binding and clot retraction. To identify additional sites, we analyzed the ability of platelets and cells expressing normal and mutant αIIbβ3 to adhere to an immobilized fibrinogen plasmin fragment that lacks intact γ-404-411 (‘D98’). We found the following: (1) Activated, but not unactivated, platelets adhere well to immobilized ‘D98.’ (2) Cells expressing constitutively active αIIbβ3 mutants, but not cells expressing normal αIIbβ3 or αVβ3, adhere well to ‘D98.’ (3) Monoclonal antibodies 10E5 and 7E3 inhibit the adhesion to ‘D98’ of activated platelets and cells expressing constitutively active αIIbβ3, as do small-molecule inhibitors that bind to the RGD pocket. (4) EDTA paradoxically induces normal αIIbβ3 to interact with ‘D98.’ Because molecular modeling and molecular dynamics simulations suggested that the αIIb L151-D159 helix may contribute to the interaction with ‘D98,’ we studied an αIIbβ3 mutant in which the αIIb 148-166 loop was swapped with the corresponding αV loop; it failed to bind to fibrinogen or ‘D98.’ Our data support a model in which conformational changes in αIIbβ3 and/or fibrinogen after platelet activation and the interaction between γ-404-411 and the RGD binding pocket make new ancillary sites available that support higher-affinity fibrinogen binding and clot retraction.
Visual Abstract
Introduction
Platelets play a major role in both thrombosis and hemostasis. αIIbβ3 is a platelet- and megakaryocyte-specific integrin that mediates adhesion of platelets to ligands and is required for platelet aggregation and clot retraction.1,2 Several ligands for αIIbβ3, including von Willebrand factor (VWF), vitronectin, and fibronectin, contain an Arg-Gly-Asp (RGD) motif that interacts with a pocket on the receptor headpiece composed of contributions by both αIIb and β3.2,3 Fibrinogen contributes to platelet aggregation in vitro and thrombus formation in vivo.4,5 It interacts with the RGD pocket on αIIbβ3 through the last 8 residues (γ-404-411) in its unstructured C-terminal γ-chain dodecapeptide (HHLGGAKQAGDV; γ-12) rather than either of its 2 RGD motifs.6-10 Ligand binding to αIIbβ3 initiates a major conformational change in the receptor resulting in the receptor adopting a high-affinity conformation.11
Although the interaction between the fibrinogen γ-chain and the RGD pocket is necessary for fibrinogen binding to αIIbβ3, it may not be sufficient because of the following: (1) Biochemical and biophysical studies show fibrinogen binding is a time-dependent multistep process leading to higher-affinity and lack of reversibility.9,10,12-22 (2) When reversibly dissociated, both αIIb and β3 can bind to immobilized fibrinogen.16 (3) Platelets can adhere to fibrinogen fragments lacking γ-404-411,23,24 but it is unclear whether the platelets need to be activated in order to bind. (4) Mutations of αIIb at a distance from the RGD pocket, in particular at the αIIb cap domain,25,26 impair fibrinogen binding, as do monoclonal antibodies (mAb’s) that bind in that region. For example, mAb 10E5, which binds to the αIIb cap domain,11 is a potent inhibitor of fibrinogen binding27 even though it does not alter the RGD pocket. Similarly, mutations in the β3 specificity determining loop28 can interfere with fibrinogen binding. (5) Binding of fibrinogen to αIIbβ3 results in changes in the conformation of both αIIb and β3 as determined by the binding of mAb’s specific for ligand-induced binding sites (LIBS),29-31 potentially exposing additional sites. (6) Binding of fibrinogen to αIIbβ3 induces changes in the conformation of fibrinogen, thus also potentially exposing new sites.32-34
There may also be ancillary binding sites for the interaction of fibrin with αIIbβ3 because of the following: (1) αIIbβ3 is required for clot retraction, but clot retraction is essentially normal with fibrinogen lacking the γ-408-411 sequence.35,36 (2) EDTA eliminates fibrinogen binding to the RGD pocket in αIIbβ3 but does not impair clot retraction.37 (3) The conversion of fibrinogen to fibrin exposes new epitopes for mAb’s and thus potentially new interaction sites.38 (4) Binding of fibrin to αIIbβ3 has different physicochemical properties than binding to fibrinogen.39
Identifying ancillary binding sites for fibrinogen and/or fibrin on αIIbβ3 would provide a more comprehensive understanding of fibrinogen binding to platelets. Such sites may furnish novel therapeutic targets to prevent platelet thrombus formation. This is important because current small-molecule αIIbβ3 antagonists act as partial agonists and, under certain experimental conditions, can prime the receptor to bind fibrinogen by inducing the β3 subunit to adopt high-affinity ligand-binding conformations.22,40-42 These conformational changes have been hypothesized to contribute to the development of thrombocytopenia in ∼0.5% to 1% of patients as a result of exposing epitopes for which some people have preformed antibodies,43 and they may limit the efficacy of the current agents. Because the ancillary sites on αIIbβ3 may be different for fibrinogen and VWF, it may be possible to develop ligand-specific antagonists with potential therapeutic advantages, if, for example, selectively blocking fibrinogen binding prevents thrombus formation while preserving hemostasis mediated by VWF binding to αIIbβ3.
Regions of fibrinogen in addition to γ-404-411 and regions of αIIbβ3 in addition to the RGD binding pocket have been reported to affect ligand binding,25,38,44,45 but there has been no detailed description of how any ancillary site on fibrinogen interacts with any ancillary region on αIIbβ3. Specifically, potential ancillary sites have been identified in the fibrinogen γ-module (γ-148-411), including γ-316-322 and γ-370-381.44-47 Such sites are difficult to study using intact fibrinogen because of the confounding effect of the interaction of αIIbβ3 with γ-404-411. As a result, to identify additional sites of interaction between fibrinogen and αIIbβ3, we studied the adhesion of platelets and HEK293 cells expressing either normal αIIbβ3 or mutant αIIbβ3 to fibrinogen and plasmin fragments of fibrinogen that either contain or lack intact γ-404-411 (D100 and ‘D98,’ respectively), in the presence or absence of EDTA. The mutations studied were designed to (1) activate the receptor, (2) disrupt the metal ion dependent adhesion site (MIDAS) (β3 D119A), and (3) alter the interaction of ‘D98’ with αIIb based on inferences from a molecular model of the αIIbβ3 headpiece-fibrinogen γ-module complex.
Methods
Fibrinogen, fibrinogen fragments, and mAb’s
Human fibrinogen was obtained from Enzyme Research Labs, and fibrinogen fragments D100 and ‘D98’ were obtained from Haematologic Technologies. The latter were prepared as described in the supplemental Materials and Methods. Because our fragment resembles to a great extent the D98 fragment described by Lishko et al48 (which was the fragment used in the study by Podolnikova et al24), most importantly for the current study in not having an intact γ-404-411 peptide, but may differ in minor aspects, we have chosen to name our fragment ‘D98.’ The mAb’s 10E5 (anti-αIIbβ3),11,27 7E3 (anti-αIIbβ3 + αVβ3, which binds in the region between the specificity loop and α1 helix of β3),49,50 and 7E9 (anti-C-terminal γ-12 peptide)12 were described previously. The anti-LIBS mAb AP551 and the anti-αVβ3 mAb LM60952,53 were generously provided by Peter Newman and David Cheresh, respectively.
Molecular modeling of the αIIbβ3 headpiece-fibrinogen γ-module complex
The crystal structures of the αIIbβ3 headpiece-γ-12 peptide complex (PDB ID: 2VDO), containing the last 7 residues of the bound dodecapeptide (γ-405-411), and the fibrinogen C-terminal γ-module (PDB ID: 1FIC) (γ-144-392) were used to create a model of the complex formed by the C-terminal γ-module of fibrinogen and αIIbβ3 head domain. Molecular docking was performed using ClusPro,54 Haddock,55 and SwarmDock56 using 3 separate sets of restraints as described in supplemental Materials and Methods and supplemental Figure 1. The 55 structures exhibiting at least 6 ligand-receptor contacts between fibrinogen residues γ-316-322 or γ-370-381 and mAb 10E5 binding site residues on αIIb, as well as suitable conformations of the loop linking residues γ-144-392 of the γ-module and γ-405-411 in the αIIbβ3 RGD binding pocket were clustered in 13 groups. Representative structures of these clusters were subjected to molecular dynamics (MD) simulations as described in the supplemental Materials and Methods.
Generation of mutants and stable cell lines
HEK293 cells expressing normal human αIIbβ3 were previously described,57,58 and the αIIbF992A/F993A (FF), β3N339S, β3Δ717, β3D119A, and αIIb(FF)β3D119A mutants, as well as the αV 144-154/αIIb 148-166 loop swap mutant [αIIb(αV)], were generated using the QuikChange XL Site-directed Mutagenesis kit as described in supplemental Materials and Methods.
Platelet and cell adhesion assay
The platelet and HEK293 cell adhesion assays to immobilized fibrinogen and fibrinogen fragments D100 and ‘D98’ were performed as previously described42 with minor modifications described in the supplemental Materials and Methods.
Mass spectrometry (MS) analysis of fibrinogen fragments D100 and ‘D98’
MS of trypsin-treated fibrinogen fragments D100 and ‘D98’ was performed by liquid chromatography (LC)–MS/MS (Ultimate 3000 nano-HPLC system coupled to a Q-Exactive Plus mass spectrometer, Thermo Scientific) with details described in the supplemental Materials and Methods.
Statistics
Repeated measures analysis of variance and paired Student t test were used to calculate the statistical significance and the P values for adhesion.
Results
Fibrinogen fragment ‘D98,’ unlike D100, lacks the C-terminal γ-chain dodecapeptide
The ‘D98’ fragment contains the majority of the D domain, but unlike fragment D100, does not contain an intact C-terminal γ-12 dodecapeptide. Immunoblot analysis using mAb 7E9 indicated the absence of the intact γ-12 peptide in ‘D98’ (Figure 1A). LC-MS/MS analysis of D100 confirmed a possible N terminus of A63 (supplemental Table 2) and the dominance of the C-terminal peptide containing intact γ-12 (Figure 1B), consistent with the report by Lishko et al.48 In sharp contrast, LC-MS/MS analysis of 2 closely separated ‘D98’ bands showed that the most abundant C-terminal peptide detected ended at A405, with the C-terminal peptide containing intact γ-12 being virtually undetectable (Figure 1B). Lishko et al reported that the N terminus of their D98 fragment was S86, whereas we have identified 1 peptide with an N terminus at M89 in our ‘D98,’ and we infer the presence of at least 1 additional peptide with an N terminus somewhere between P76 and K88. (supplemental Table 2).
Figure 1.

Fibrinogen fragment D100, but not fragment ‘D98,’ is recognized by an antibody to the fibrinogen C-terminal γ-chain dodecapeptide (γ-12); the C-termini of D100 and ‘D98’ are primarily γ-411 and γ-405, respectively. (A) Fragment D100 and fragment ‘D98’ were electrophoresed in a 10% acrylamide gel. Separated proteins were then transferred to a nitrocellulose membrane and stained with Ponceau (left). After washing away the stain, the membrane was immunoblotted with mAb 7E9, which reacts with the fibrinogen γ-12 peptide. (B) MS-based quantitation of C-terminal peptides. D100 and ‘D98’ gel bands were treated with propionic anhydride, which modifies primary amines, and then digested with trypsin. Propionic anhydride-modified residues are marked with [+56]. C-terminal peptides of D100 and ‘D98’ were measured by parallel reaction monitoring71 and signals extracted and analyzed by Skyline.72 Signals normalized to the amount of fibrinogen C-terminal γ-chain in the 2 samples are shown for each peptide. For the D100 sample, the most abundant peptide measured was the C-terminal γ-chain peptide γ-392-411 (LTIGEGQQHHLGGAK[+56]QAGDV) containing the intact γ-12 peptide. No signal for this peptide was identified in the ‘D98’ sample; rather, the most abundant signal was assigned to LTIGEGQQHHLGGA, which ends at γ-405. See supplemental Table 2 for additional validation of C termini, assignment of N termini, and peptides used for normalization.
Unactivated and activated platelets adhere to microtiter wells coated with fibrinogen, whereas only activated platelets adhere well to wells coated with ‘D98’
Platelets from healthy donors adhered to microtiter plates coated with 10 µg/mL fibrinogen, and the adhesion was inhibited by EDTA (10 mM), mAb’s 10E5 and 7E3 (both 20 µg/mL), and tirofiban (1 µM) (Figure 2A). Activating the platelets by adding a thrombin receptor activating peptide (TRAP; 10 µM) resulted in ∼35% overall increase in adhesion (Figure 2B). EDTA, the mAb’s 10E5 and 7E3, and tirofiban all inhibited the adhesion, but not to the same extent as with unactivated platelets.
Figure 2.

Both unactivated and activated platelets adhere to immobilized fibrinogen, whereas only activated platelets adhere to fibrinogen fragment ‘D98.’ Calcein-labeled washed platelets (2 × 105/µL; 50 µL) were tested either before (unactivated) or after activation with 10 µM TRAP for adhesion to fibrinogen or ‘D98’ immobilized in microtiter wells by coating at 10 µg/mL. Platelets were incubated for 1 hour at 22°C in the presence or absence of EDTA (10 mM), 10E5 (20 µg/mL), 7E3 (20 µg/mL), or tirofiban (1 µM), followed by washing of unbound platelets and analysis of calcein fluorescence. The adhesion of unactivated platelets to fibrinogen was defined as 100% adhesion, and all values were normalized to this value in each of 8 separate experiments. (A) EDTA, 10E5, 7E3, and tirofiban inhibited adhesion of unactivated platelets to fibrinogen by 73%, 92%, 94%, and 86%, respectively (P < .0001 for all). (B) Activated platelets adhered to immobilized fibrinogen as well or better than unactivated platelets; EDTA, 10E5, 7E3, and tirofiban inhibited adhesion by 75%, 70%, 63%, and 63%, respectively (P < .001 for all). (C) Unactivated platelets adhered poorly to ‘D98’ (80% less adhesion compared with unactivated platelet adhesion to fibrinogen), whereas activated platelets adhered much better (25% more adhesion compared with unactivated platelet adhesion to fibrinogen). Adhesion was inhibited by 10E5, 7E3, and tirofiban by 66%, 63%, and 57%, respectively (P < .01 for all). (D) Adhesion of unactivated platelets to ‘D98’ was increased by treating the platelets with 10 mM EDTA (P = .02). Data reported as mean ± standard deviation (SD).
In sharp contrast, unactivated platelets bound much less well to immobilized ‘D98’ than to fibrinogen, whereas TRAP-activated platelets adhered to ‘D98’ better than unactivated platelets adhered to fibrinogen, and nearly as well as activated platelets bound to fibrinogen (Figure 2C). The mAb’s 10E5 and 7E3 decreased adhesion of activated platelets to ‘D98’ by 66% and 63%, respectively, and tirofiban decreased adhesion by 57% (all P ≤ .001). The combination of adenosine 5′-diphosphate (ADP) + epinephrine as a platelet activator increased platelet adhesion to ‘D98’ (from 944 ± 363 to 3952 ± 1760 arbitrary fluorescence intensity units (AFU); supplemental Figure 2), but the increase was at the borderline of statistical significance (P = .06). Paradoxically, EDTA increased the adhesion of unactivated platelets to immobilized ‘D98’ in all donors, with the variable increases averaging 3.5-fold (P = .02) (Figure 2D).
Cells expressing normal αIIbβ3 adhere to fibrinogen and D100 but do not adhere to immobilized ‘D98’; cells expressing constitutively active αIIbβ3 adhere to ‘D98’
HEK293 cells expressing normal αIIbβ3 (αIIbβ3-HEK) adhered to fibrinogen and D100 but did not adhere to immobilized ‘D98’ (Figure 3A, left panel). In contrast, HEK293 cells expressing αIIbβ3 containing activating mutations in the cytoplasmic domain of αIIb [αIIb(FF)β3-HEK] bound to ‘D98,’ and HEK293 cells expressing αIIbβ3 with other activating mutations (β3N339S and β3Δ717) also supported adhesion to ‘D98’ (Figure 3A, right panel). In contrast, HEK293 cells expressing αVβ3 showed no adhesion to ‘D98.’
Figure 3.

HEK293 cells expressing normal αIIbβ3 adhere to fibrinogen and D100 but do not adhere to immobilized fibrinogen fragment ‘D98,’ whereas adhesion does occur with HEK293 cells expressing constitutively active αIIbβ3 mutants; adhesion of αIIb(FF)β3-HEK cells to immobilized ‘D98’ is inhibited by function blocking antibodies to αIIbβ3, small-molecules inhibitors of αIIbβ3, and ‘D98,’ but not by an antibody to the fibrinogen γ-12 peptide or soluble fibrinogen. (A) HEK293 cells (2 × 103 cells/ µL; 50 µL) expressing normal αIIbβ3 were labeled with calcein and added to microtiter wells precoated with fibrinogen, D100, or ‘D98’ (10 µg/mL coating concentration) for 1 hour at 22°C (left). After washing, the fluorescence of adherent cells was measured. αIIbβ3-HEK cells adhered to immobilized fibrinogen and D100 but did not adhere to ‘D98.’ Data reported as mean ± SD. HEK293 cells expressing normal αIIbβ3, normal αVβ3, or constitutively active αIIbβ3 mutants were allowed to adhere to ‘D98’ (right). Whereas the cells expressing normal αIIbβ3 or αVβ3 did not adhere, HEK293 cells expressing any 1 of the 3 activating mutations [αIIb(FF), β3N339S, or β3Δ717] demonstrated substantial adhesion (n = 6; P < .01 compared with normal αIIbβ3). Data reported as mean ± SD. Net normalized adhesion is adhesion value (AFU) divided by the expression level (minus background) of each cell line. Expression levels were measured by mAb 10E5 binding and expressed as geometric mean fluorescence intensity (GMFI): normal αIIbβ3 69 ± 40 AFU; αIIb(FF)β3 31 ± 16 AFU; αIIbβ3(N339S) 58 ± 17 AFU; αIIbβ3(Δ717) 86 ± 40 AFU; normal αVβ3 (using mAb LM609) 187 ± 24 AFU. (B) Adhesion of HEK293 cells expressing the αIIb(FF)β3 mutant to ‘D98’ was tested in the presence of mAb’s 10E5 (anti-αIIb ; 20 µg/mL), 7E3 (anti-β3; 20 µg/mL), and mAb 7E9 (anti-fibrinogen γ-12: 20 µg/mL). 10E5 and 7E3 both inhibited adhesion (n = 7; P < .01 for each), whereas 7E9 did not (P = .30). (C) Adhesion of αIIb(FF)β3-HEK cells to immobilized ‘D98’ was also inhibited by the small-molecule inhibitors of αIIbβ3 RUC-4 (5 µM), eptifibatide (100 µM), and tirofiban (10 µM) (n = 3; P < .005 for each). (D) Soluble fibrinogen (1.5 mg/mL) did not inhibit the adhesion of αIIb(FF)β3-HEK cells to ‘D98’ (n = 3; P = .50), but soluble ‘D98’ (1.0 mg/mL) did inhibit the adhesion (n = 3; P = .02). Data reported as mean ± SD.
Effect of mAb’s, αIIbβ3 antagonists, and soluble fibrinogen and ‘D98’ on adhesion of αIIb(FF)β3-HEK to ‘D98’
The adhesion of αIIb(FF)β3-HEK to immobilized ‘D98’ was inhibited by mAb’s 10E5 and 7E3 (Figure 3B). The adhesion was not, however, significantly inhibited by mAb 7E9. In addition, cells treated with either of the RGD-based small-molecule inhibitors of αIIbβ3, tirofiban (10 µM) or eptifibatide (100 µM), or the novel inhibitor RUC-4 (5 µM), which does not bind to the MIDAS Mg2+,59 adhered less well compared with untreated cells (Figure 3C). The adhesion of αIIb(FF)β3-HEK to ‘D98’ was inhibited by soluble ‘D98’ (1.0 mg/mL), but not by soluble fibrinogen (1.5 mg/mL) (Figure 3D).
Paradoxical effect of EDTA in enhancing adhesion of αIIbβ3-HEK293 to ‘D98’
EDTA (10 mM) inhibited the adhesion of αIIbβ3-HEK cells to immobilized fibrinogen in the presence of 2 mM Ca2+/1 mM Mg2+ (Figure 4A, left panel). Surprisingly, 10 mM EDTA enhanced the adhesion of αIIbβ3-HEK to immobilized ‘D98’ nearly 25-fold (Figure 4A, right panel). EDTA did not inhibit the adhesion of αIIbβ3-HEK cells to D100 (Figure 4B).
Figure 4.

Paradoxical effect of EDTA in enhancing adhesion of αIIbβ3-HEK cells to fibrinogen fragment ‘D98’. (A) HEK293 cells expressing normal αIIbβ3 (2 × 103/µL; 50 µL) were added to microtiter wells precoated with fibrinogen (10 µg/mL coating concentration) for 1 hour at 22°C in the absence and presence of EDTA (10 mM) (left). EDTA dramatically inhibited adhesion (n = 9; P < .001). Normal αIIbβ3-HEK cells (2 × 103/µL; 50 µL) were added to microtiter wells precoated with ‘D98’ (10 µg/mL coating concentration) for 1 hour at 22°C in the absence and presence of EDTA (10 mM) (right). The EDTA dramatically increased adhesion (n = 9; P = .001). (B) Normal αIIbβ3-HEK cells bound to D100 in the absence and presence of EDTA. Conditions as per panel A with D100 coated at 10 µg/mL (n = 3). (C) The mAb 10E5 (20 µg/mL) inhibited EDTA-induced adhesion of αIIbβ3-HEK cells to ‘D98’ (n = 7; P = .003). (D) Small-molecule inhibitors of αIIbβ3 RUC-2 (10 µM) and RUC-4 (5 µM) inhibited the EDTA-induced adhesion of αIIbβ3-HEK to ‘D98’ when added after the EDTA (n = 5; P < .005). Similar results, not shown, were obtained when RUC-2 or RUC-4 were added before EDTA (n = 5; P < .005). Data reported as mean ± SD.
Preincubating fibrinogen or ‘D98’ with EDTA did not inhibit adhesion of αIIbβ3-HEK cells to fibrinogen or enhance adhesion of αIIbβ3-HEK cells to ‘D98,’ whereas pretreating the αIIbβ3-HEK cells with EDTA inhibited adhesion to fibrinogen and enhanced adhesion to ‘D98’ (supplemental Figure 3).
EDTA (3 mM) was sufficient to block adhesion to fibrinogen, and 3 to 4 mM EDTA was required to enhance adhesion to ‘D98.’ The effect of EDTA was rapid (control = 403 AFU; 0 minutes = 9667 AFU; 5 minutes = 13 112 AFU; 60 minutes = 11 981 AFU; averages of 2 experiments).
EDTA-induced adhesion of αIIbβ3-HEK to ‘D98’ was inhibited by mAb 10E5 (Figure 4C). The effect of mAb 7E3 could not be tested because EDTA dramatically reduces the binding of mAb 7E3. LM609 (anti-αVβ3) did not inhibit EDTA-induced adhesion to ‘D98’ (data not shown). The effects of eptifibatide and tirofiban could not be tested because both antagonists bind to the MIDAS Mg2+ ion. Both RUC-2 and RUC-4 inhibited EDTA-induced adhesion of αIIbβ3-HEK to ‘D98’ (Figure 4D).
Role of MIDAS residue D119A in EDTA-induced adhesion vs FF-activated adhesion to ‘D98’
αIIbβ3-HEK cells containing the β3 D119A mutation did not adhere to fibrinogen, and adding the β3 D119A mutation to αIIb(FF)β3-HEK led to loss of adhesion to both fibrinogen and ‘D98’ (Figure 5A-B). Of note, the D119A mutation did not affect EDTA-induced adhesion to ‘D98’ (Figure 5C).
Figure 5.

Mutating MIDAS residue D119A eliminates binding of normal αIIbβ3-HEK and αIlb(FF)β3-HEK cells to immobilized fibrinogen and binding of αIlb(FF)β3-HEK cells to ‘D98,’ but does affect adhesion of normal αIIbβ3-HEK cells to ‘D98’ in the presence of EDTA. HEK293 cells (2 × 103/µL; 50 µL) expressing normal αIIbβ3, the αIlbβ3 constitutively active mutant αIlb(FF)β3, the αIlbβ3(D119A) MIDAS-disrupting mutant, or the combined αIlb(FF)β3(D119A) mutant were labeled with calcein (10 µM) and then added to microtiter wells precoated with fibrinogen or ‘D98’ (each at 10 µg/mL coating concentration) for 1 hour at 22°C in the absence and presence of EDTA (10 mM). The fluorescent signal of adherent cells was measured after washing away the nonadherent cells. (A) The normal αIlbβ3-HEK and αIlb(FF)β3-HEK cells adhered to fibrinogen, whereas the αIlbβ3(D119A)-HEK MIDAS mutant and the combined αIlb(FF)β3(D119A)-HEK mutant did not adhere to fibrinogen (n = 3; P ≤ .01). (B) αIlb(FF)β3-HEK cells adhered to ‘D98,’ whereas the same cells with the MIDAS-disrupting mutation β3(D119A), did not adhere (n = 3; P = .004). (C) EDTA treatment (10 mM) of αIlbβ3(D119A)-HEK cells increased their adhesion to ‘D98’ to the level of normal αIlbβ3-HEK cells (n = 4; P > .05). Data reported as mean ± SD. Expression levels for each cell line were measured based on mAb 7E3 binding and are expressed as GMFI: normal αIIbβ3 53 ± 26 AFU; αIIbβ3(D119A) 36 ± 11 AFU; αIIb(FF)β3 25 ± 5 AFU; αIIb(FF)β3(D119A) 27 ± 2 AFU.
Cells expressing αVβ3 bind to immobilized fibrinogen, but not immobilized ‘D98,’ even when treated with EDTA
HEK293 cells expressing normal αVβ3 adhered to immobilized fibrinogen at levels similar to those of HEK293 cells expressing normal αIIbβ3, and EDTA nearly eliminated the adhesion (supplemental Figure 4A). αVβ3-HEK cells did not adhere to ‘D98,’ and adding EDTA did not increase their adhesion (supplemental Figure 4B).
Molecular modeling of the αIIbβ3 headpiece-fibrinogen γ-module complex
The representative structure of the most populated cluster (cluster 1) of the αIIbβ3 headpiece-fibrinogen γ-module complexes deviated substantially from the initial conformation during MD simulations, whereas the representative structure of the second largest cluster (cluster 2) remained reasonably stable (supplemental Figure 5). Figure 6 shows the relaxed, representative structure of this cluster at the end of a 230 ns MD simulation. Direct polar interactions that involve 2 residues (E157 and N158) of the αIIb helix segment L151-D159 are seen in this structure. Although only residue E157 was included in the modeling restraints used in series 1, both E157 and N158 were included in the restraints used in series 2 and 3 (see supplemental Materials and Methods). Specifically, these direct polar interactions are between αIIb E157 and fibrinogen K356 side chains, between αIIb E157 backbone and fibrinogen R375 side chain, and between αIIb N158 side chain and fibrinogen R375 backbone. Water-mediated interactions are also seen between αIIb D159 and fibrinogen Q407 side chains, and between αIIb E157 and fibrinogen W376 side chains. Heavy atoms of 4 residues on the αIIb helix, Leu151, Arg153, Ile154, and Asn158, are within 5 Å from 10E5 in the 10E5-αIIbβ3 headpiece crystal structure 2VDO (supplemental Figure 6), suggesting that our model is consistent with competitive binding between fibrinogen/‘D98’ and 10E5. Of note, both R375 and W376 are contained in the γ-370-381 peptide identified by Podolnikova et al,45 and the αIIb helix segment L151-D159 was contained in 1 of the 3 peptides that reacted most strongly with radiolabeled d-dimer reported by Podolnikova et al.38 Three out of the 13 clusters included structures in which residues contained in the γ-316-322 region interact with αIIb, but because there were only 2 structures in each of these clusters, they were not pursued further.
Figure 6.

αIIbβ3 headpiece-fibrinogen γ-module complex showing interactions between residues on the αIIb helix segment L151-D159 and fibrinogen γ-module. (A) Snapshot at 230 ns from MD simulations of a representative structure of cluster 2 showing a relaxed model of fibrinogen binding to αIIbβ3. Fibrinogen γ-module, αIIb, and β3 are colored in gray, blue, and red, respectively. The last 7 amino acids of fibrinogen are colored in yellow. The important contact region identified is highlighted with a green circle. (B) A zoom-in view of the contacts in the 230 ns snapshot is shown from a slightly different perspective and with fibrinogen residue 291-306 removed for clarity. Direct and water-mediated hydrogen bonds are indicated with dotted lines.
Swapping the αIIb 148-166 loop for the comparable αVβ3 loop leads to loss of adhesion to both fibrinogen and ‘D98,’ and EDTA does not enhance adhesion to ‘D98’
To assess the role of the αIIb helix segment L151-D159 in binding to ‘D98,’ we prepared HEK293 cells expressing αIIbβ3 in which the αV 144-154 loop was swapped for the αIIb 148-166 loop [αIIb(αV)β3-HEK] (supplemental Figure 4C). These cells did not adhere to immobilized fibrinogen, but pretreatment with 5 mM dithiothreitol, a reducing agent that activates platelet αIIbβ3,60 dramatically increased their adhesion (supplemental Figure 4A). They also did not adhere to ‘D98’ even in the presence of EDTA, and adding dithiothreitol did not affect the adhesion (supplemental Figure 4B).
Discussion
Our studies indicate the following: (1) Activated, but not unactivated, platelets adhere well to immobilized ‘D98.’ (2) Cells expressing constitutively active αIIbβ3 mutants, but not cells expressing normal αIIbβ3, adhere well to immobilized ‘D98.’ (3) EDTA treatment increases the adhesion of platelets and cells expressing normal αIIbβ3 to ‘D98.’ (4) The mAb’s 10E5 and 7E3 inhibit the adhesion to ‘D98’ of activated platelets and cells expressing a constitutively active αIIbβ3, as do the small-molecule inhibitors that bind to the RGD pocket. (5) The mAb 10E5 and the small-molecule inhibitors RUC-2 and RUC-4, which do not require the presence of a MIDAS metal ion for binding (as do the other small-molecule αIIbβ3 antagonists based on the RGD sequence), inhibited the adhesion of cells expressing normal αIIbβ3 to ‘D98’ in the presence of EDTA. (6) Cells expressing αVβ3 adhere to fibrinogen, but not ‘D98,’ even in the presence of EDTA. (7) Cells expressing an αIIbβ3 mutant in which the αIIb 148-166 loop was swapped with the corresponding αV loop failed to bind to fibrinogen or ‘D98’ in the absence or presence of EDTA. Our finding that cells expressing normal αIIbβ3 could bind to D100 in the absence or presence of EDTA, whereas they only adhered to fibrinogen in the absence of EDTA, and only bound to ‘D98’ in the presence of EDTA, supports there being 2 separate mechanisms of binding, one involving the interaction of γ-404-411 (in fibrinogen and D100) with the αIIbβ3 RGD binding pocket, and another involving a site on D100 and ‘D98’ (but cryptic in fibrinogen) that interacts with a site on αIIbβ3 exposed by receptor activation or induced by EDTA.
These data provide new insights into the mechanism of adhesion to ‘D98,’ with potential implications for localizing ancillary binding sites on αIIbβ3 and fibrinogen. In an elegant series of studies, Zamarrron et al and Polodnikova et al identified the role of the γ-365-383 region of fibrinogen through studies with mAb 2G5, raised against the fibrinogen D-D fragment, and peptides from this region.24,32 They showed that mAb 2G5 (1) binds to γ-373-385, (2) inhibits platelet aggregation and clot retraction, (3) reacts with fibrinogen bound to αIIbβ3 or immobilized on a microtiter well, (4) does not bind soluble fibrinogen, and (5) does not inhibit fibrinogen binding to activated platelets.24,32,45 Moreover, a peptide containing the sequence γ-365-383 (P3) supported αIIbβ3-mediated cell adhesion but did not inhibit the binding of soluble fibrinogen to activated platelets. The adhesion of platelets to P3 was inhibited by mAb 7E3 (anti-αIIbβ3 + anti-αVβ3) and a mAb to α5β1 but not a mAb to αVβ3. They also identified 8 separate negatively charged sites on the αIIb β-propeller as potential binding sites for the positively charged γ-370-381 peptide.38
Podolinikova et al also studied the impact of P3 on platelet adhesion to D98.24 Using gel-filtered, calcein-labeled platelets in the presence of 1 mM MgCl2, 1 mM CaCl2, they found that adhesion of unactivated platelets to D98 was ∼45% of the adhesion to fibrinogen fragment D100, which contains an intact γ-12 peptide. Activating platelets with a combination of 10 μM ADP and epinephrine increased adhesion to D100 by 1.7-fold but had no impact on adhesion to D98. They concluded that adhesion to D98 was not activation dependent. They went on to show that the P3 peptide inhibited adhesion of unactivated platelets to D98. They concluded that the P3 peptide region of fibrinogen is cryptic on soluble fibrinogen and becomes exposed upon fibrinogen binding to platelets or fibrin formation, where it can mediate additional interactions with unactivated αIIbβ3 that are important in platelet aggregation.
In contrast to their results, we found that unactivated platelet adhesion to ‘D98’ was only ∼20% of the adhesion of unactivated platelets to fibrinogen and that activating platelets with TRAP dramatically increased adhesion to ‘D98,’ reaching just modestly less than the adhesion of activated platelets to fibrinogen. To try to reconcile our findings with those of Podolnikova et al, we also tested the combination of ADP + epinephrine (which they used) in our assay and found that although it increased adhesion to ‘D98,’ the effect was not statistically significant (P = .06). Thus, we conclude that adhesion to ‘D98’ requires potent activation of αIIbβ3.
We confirmed the difference between unactivated and activated αIIbβ3 in mediating adhesion to ‘D98’ using HEK293 cells expressing mutations in αIIb or β3 that induce constitutive fibrinogen binding. These data suggest that the ancillary binding site(s) on αIIbβ3 for ‘D98’ is cryptic on unactivated αIIbβ3 and is exposed with activation by strong agonists or activating mutations. As with platelets, this behavior contrasts sharply with adhesion to intact immobilized fibrinogen, which does not require platelet activation.1,27
Despite the absence of intact γ-12 peptide on ‘D98,’ the small-molecule αIIbβ3 antagonists that bind to the RGD pocket inhibited the adhesion of both platelets and HEK293 cells expressing activated αIIbβ3 to ‘D98.’ Similarly, adding the β3 D119A mutation, which is known to alter the MIDAS and eliminate ligand binding to the constitutively active αIIb(FF)β3 receptor, led to loss of binding to ‘D98.’ Collectively, these data suggest either that the RGD pocket can bind both the γ-404-411 peptide and the ancillary fibrinogen site(s) or that altering the RGD pocket allosterically affects the ancillary site(s). Of note, the mAb 10E5, which binds to the αIIb cap domain and inhibits fibrinogen binding and platelet aggregation,11 also inhibited the adhesion to ‘D98,’ raising the possibility that the ancillary site(s) is in the cap domain, which is unique to αIIb.61 Collectively, our data are similar to those of Peerschke and Galanakis who studied the adhesion of platelets to the D60 plasmin degradation product of fibrinogen, which they confirmed lacked the RGD and intact γ-12 sequences.23 They found that adhesion to D60 required platelet activation with either ADP or thrombin and that the adhesion could be inhibited by RGD peptides and mAb 10E5.
Our docking and molecular dynamics studies suggested that the αIIb cap 3 insert, which includes the α-helical residues E157, N158, and D159, participates in interacting with fibrinogen residues K356, R375, W376, and Q407. Of note, R375 and W376 are contained in the P3 peptide, and the αIIb residues E157-D159 are contained in 1 of the 8 regions Podolnikova et al identified as potential binding sites for P3.38 Moreover, 4 residues on the αIIb L151-D159 helix (Leu151, Arg153, Ile154, and Asn158) are within 5 Å heavy atom distance to 10E5, suggesting that this helix is involved in 10E5 binding. In addition, Podolnikova et al reported that a peptide composed of αIIb residues 153-162 inhibited platelet adhesion to D98 by 33% and inhibited clot retraction with a 50% inhibitory concentration (IC50) of 200 µM, but only inhibited platelet adhesion to fibrinogen by 10%.38 They also showed an ∼65% reduction in the adhesion to D98 of HEK293 cells expressing a mutant αIIbβ3 containing αIIb E157A/D159S/W162F.38 The αIIb helix segment L151-D159 is absent in αV, and so we prepared a hybrid receptor in which the αV 144-154 loop was swapped for the αIIb 148-166 loop. This led to the loss of cell adhesion to both ‘D98’ and fibrinogen. Although this is consistent with a role for this region in forming an ancillary binding site, this experiment cannot differentiate between loss of the residues implicated in binding fibrinogen by the molecular docking and MD simulation studies and other changes produced by the swap (eg, shortening of the loop). Of note, Kamata et al did not find a reduction in fibrinogen binding to αIIbβ3 expressed on CHO cells with single alanine substitutions of E157, N158, or D159 when activated by mAb PT25-2.25
The paradoxical effect of EDTA in enhancing the adhesion of unactivated platelets and cells expressing normal αIIbβ3 to ‘D98’ was unexpected because EDTA is commonly used to inhibit integrin-mediated ligand binding, presumably by chelating the MIDAS divalent cation. In fact, one operational definition of “irreversibly” bound fibrinogen is the inability of EDTA to elute fibrinogen from αIIbβ3.62 The EDTA-induced adhesion of cells expressing normal αIIbβ3 to ‘D98’ was also partially inhibited by mAb 10E5 and the small-molecule antagonist RUC-4, both of which inhibit the adhesion of αIIb(FF)β3-HEK cells to ‘D98.’ The MIDAS-disrupting D119A mutation, however, had no effect on EDTA-induced adhesion of cells expressing normal αIIbβ3 to ‘D98,’ unlike its effect on adhesion of αIIb(FF)β3-HEK cells. Thus, it is possible that EDTA treatment exposes a different site(s) for ‘D98’ than the one exposed by receptor activation, or that EDTA treatment exposes the same site and stabilizes it from allosteric alteration by the D119A mutation. Peerschke and Galanakis did not study the effect of EDTA alone on adhesion of platelets to D60, but they reported that EDTA was less effective in inhibiting platelet adhesion to D60 than to fibrinogen.23 Thus, our findings have implications for using EDTA to probe the reversibility of fibrinogen binding.
EDTA has a number of effects on αIIbβ3. At 22°C and neutral pH, it inhibits ligand binding to αIIbβ3 without dissociating the complex, whereas at higher temperature and pH it irreversibly dissociates the complex.37,63-66 It also exposes the epitopes for several LIBS antibodies, including the AP5 epitope on β351 and the PMI-1 epitope on αIIb,67 indicating that it induces conformational changes akin to those produced by ligand binding. Moreover, the platelets of patients with Glanzmann thrombasthenia because of a D119Y mutation bound PMI-1 in the absence of EDTA, linking disruption of the MIDAS with allosteric modifications in αIIb.67,68 Although EDTA prevents fibrinogen binding to αIIbβ3, it does not inhibit αIIbβ3-mediated clot retraction,37 which is consistent with the finding that clot retraction can be supported by fibrinogen molecules lacking γ-408-411 and both RGD sequences.35,36 Other data that potentially link our results to the mechanism of clot retraction include the observations that both the P3 peptide and mAb 10E5 inhibit clot retraction.24,27 Nonetheless, studies with the d-dimer fibrin fragment showed interactions with 16 separate αIIb peptide regions, 10 of which correspond to regions implicated in fibrinogen binding, but only 3 of which correspond to regions implicated in P3 binding.38
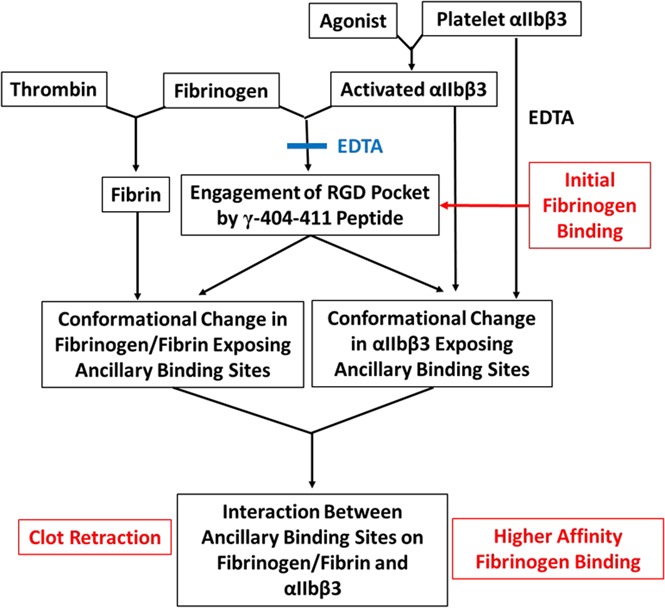
In conclusion, our data are consistent with a model in which activation of platelet αIIbβ3 initially results in fibrinogen binding by γ-404-411 engaging the RGD binding pocket (Figure 7). This binding can be reversed by EDTA or excess fibrinogen.62 Bound fibrinogen undergoes conformational changes as reported by a number of different mAb’s (mAb 2G5 [anti-FG-RIBS1; anti-γ-373-385],32 mAb 9F969 [anti-γ-112-119],33 and mAb 155B16 [anti-Aα-95-98]),33 and 1 or more of these conformational changes could expose ancillary binding sites. The ancillary binding site(s) on fibrinogen and activated αIIbβ3 then interact, resulting in strengthening of the interaction. A similar process may also contribute to platelet-fibrin interactions that support clot retraction, with fibrin formation initiating conformational changes in fibrinogen that can expose new sites that interact with activated αIIbβ3.70 Finally, we found a paradoxical effect of EDTA in inducing a conformational change in αIIbβ3 that supports adhesion to ’D98,’ providing a tool to better understand the conformational changes underlying the exposure of ancillary binding sites for fibrinogen on αIIbβ3 and raising cautions about the use of this reagent to define the reversibility of fibrinogen binding.
Figure 7.

Working model of the multistep process of fibrinogen binding and the platelet-fibrin interaction contributing to clot retraction. Activation of platelet αIIbβ3 with an agonist results in αIIbβ3 being able to bind fibrinogen by the engagement of the γ-404-411 peptide in the αIIbβ3 binding pocket. EDTA can prevent the binding of fibrinogen and reverse the binding if added rapidly after the addition of fibrinogen. The binding of fibrinogen leads to conformational changes in fibrinogen that expose 1 or more ancillary binding sites. Platelet αIIbβ3 activation alone is sufficient to expose ancillary binding site(s), but there may be an additional contribution from fibrinogen engaging the αIIbβ3 binding pocket. The ancillary binding site(s) on fibrinogen and αIIbβ3 then interact, resulting in higher-affinity fibrinogen binding, which under certain experimental conditions may appear irreversible over the time course of the experiments. A similar mechanism may contribute to clot retraction, with fibrin formation initiating conformational changes in fibrinogen that mimic, at least in part, those induced by fibrinogen binding to αIIbβ3. Although data vary, the weight of evidence supports the need for activated platelets to participate in clot retraction. Paradoxically, EDTA treatment appears to initiate conformational changes in αIIbβ3 akin to those produced by activation and ligand binding as judged by the ability of cells expressing normal αIIbβ3 to bind to ‘D98’ in the presence of EDTA.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Ming Cheng who previously produced the αIIbβ3 mutant in which the αIIb and αV loops were swapped; Lorena Buitrago for help with technical issues and valuable suggestions about research design, data analysis, and the written manuscript; and Suzanne Rivera for outstanding administrative support.
This work was supported in part by National Heart, Lung, and Blood Institute, National Institutes of Health (grant HL19278) and National Center for Advancing Translational Sciences, National Institutes of Health Clinical and Translational Science Awards (UL1TR000043), as well as funds from Stony Brook University. Computations were run on resources available through the Scientific Computing Facility at Mount Sinai and the Extreme Science and Engineering Discovery Environment (grant MCB080109N) (M.F.), which is supported by the National Science Foundation (grant OCI-1053575). MS of fragment D100 and ‘D98’ was performed at the Proteomics Resource Center, and the fluorescence intensity measurements for adhesion assays were made using equipment in the High-Throughput and Spectroscopy Resource Center, both at Rockefeller University. The Rockefeller University acknowledges funding from the Leona M. and Harry B. Helmsley Charitable Trust for mass spectrometer instrumentation.
Authorship
Contribution: H.Z. designed and performed research, analyzed data, and wrote the manuscript; Y.S. designed and performed the molecular docking and molecular dynamics studies and wrote the molecular modeling sections; J.L. designed and performed experiments and helped analyze the data; G.A.D. performed the ‘D98’ characterization experiments and analyzed data; J.P.F. and H.M. performed the MS experiment and analyzed the MS data; M.F. designed and supervised the molecular docking and molecular dynamics studies and wrote the molecular modeling sections; and B.S.C. designed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: B.S.C. has royalty interests in abciximab (Centocor) through the Research Foundation of the State University of New York and the VerifyNow Assays (Accumetrics) through the Icahn School of Medicine at Mount Sinai; he is also a consultant to Scholar Rock. The remaining authors declare no competing financial interests.
Correspondence: Barry S. Coller, Allen and Frances Adler Laboratory of Blood and Vascular Biology, The Rockefeller University, 1230 York Ave, New York, NY 10065; e-mail: collerb@rockefeller.edu.
References
- 1.Coller BS. Interaction of normal, thrombasthenic, and Bernard-Soulier platelets with immobilized fibrinogen: defective platelet-fibrinogen interaction in thrombasthenia. Blood. 1980;55(2):169-178. [PubMed] [Google Scholar]
- 2.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104(6):1606-1615. [DOI] [PubMed] [Google Scholar]
- 3.Gralnick HR, Williams SB, Coller BS. Fibrinogen competes with von Willebrand factor for binding to the glycoprotein IIb/IIIa complex when platelets are stimulated with thrombin. Blood. 1984;64(4):797-800. [PubMed] [Google Scholar]
- 4.Ni H, Denis CV, Subbarao S, et al. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest. 2000;106(3):385-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang H, Reheman A, Chen P, et al. Fibrinogen and von Willebrand factor-independent platelet aggregation in vitro and in vivo. J Thromb Haemost. 2006;4(10):2230-2237. [DOI] [PubMed] [Google Scholar]
- 6.Yang Z, Kollman JM, Pandi L, Doolittle RF. Crystal structure of native chicken fibrinogen at 2.7 A resolution. Biochemistry. 2001;40(42):12515-12523. [DOI] [PubMed] [Google Scholar]
- 7.Kloczewiak M, Timmons S, Hawiger J. Recognition site for the platelet receptor is present on the 15-residue carboxy-terminal fragment of the gamma chain of human fibrinogen and is not involved in the fibrin polymerization reaction. Thromb Res. 1983;29(2):249-254. [DOI] [PubMed] [Google Scholar]
- 8.Lam SC, Plow EF, Smith MA, et al. Evidence that arginyl-glycyl-aspartate peptides and fibrinogen gamma chain peptides share a common binding site on platelets. J Biol Chem. 1987;262(3):947-950. [PubMed] [Google Scholar]
- 9.Springer TA, Zhu J, Xiao T. Structural basis for distinctive recognition of fibrinogen gammaC peptide by the platelet integrin alphaIIbbeta3. J Cell Biol. 2008;182(4):791-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farrell DH, Thiagarajan P, Chung DW, Davie EW. Role of fibrinogen alpha and gamma chain sites in platelet aggregation. Proc Natl Acad Sci USA. 1992;89(22):10729-10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432(7013):59-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jirousková M, Smyth SS, Kudryk B, Coller BS. A hamster antibody to the mouse fibrinogen gamma chain inhibits platelet-fibrinogen interactions and FXIIIa-mediated fibrin cross-linking, and facilitates thrombolysis. Thromb Haemost. 2001;86(4):1047-1056. [PubMed] [Google Scholar]
- 13.Hawiger J. Adhesive ends of fibrinogen and its antiadhesive peptides: the end of a saga? Semin Hematol. 1995;32(2):99-109. [PubMed] [Google Scholar]
- 14.Marguerie GA, Edgington TS, Plow EF. Interaction of fibrinogen with its platelet receptor as part of a multistep reaction in ADP-induced platelet aggregation. J Biol Chem. 1980;255(1):154-161. [PubMed] [Google Scholar]
- 15.Müller B, Zerwes HG, Tangemann K, Peter J, Engel J. Two-step binding mechanism of fibrinogen to alpha IIb beta 3 integrin reconstituted into planar lipid bilayers. J Biol Chem. 1993;268(9):6800-6808. [PubMed] [Google Scholar]
- 16.Parise LV, Steiner B, Nannizzi L, Criss AB, Phillips DR. Evidence for novel binding sites on the platelet glycoprotein IIb and IIIa subunits and immobilized fibrinogen. Biochem J. 1993;289(2):445-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peerschke EI. Regulation of platelet aggregation by post-fibrinogen binding events. Insights provided by dithiothreitol-treated platelets. Thromb Haemost. 1995;73(5):862-867. [PubMed] [Google Scholar]
- 18.Huber W, Hurst J, Schlatter D, et al. Determination of kinetic constants for the interaction between the platelet glycoprotein IIb-IIIa and fibrinogen by means of surface plasmon resonance. Eur J Biochem. 1995;227(3):647-656. [DOI] [PubMed] [Google Scholar]
- 19.Litvinov RI, Bennett JS, Weisel JW, Shuman H. Multi-step fibrinogen binding to the integrin (alpha)IIb(beta)3 detected using force spectroscopy. Biophys J. 2005;89(4):2824-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hantgan RR, Stahle MC, Lord ST. Dynamic regulation of fibrinogen: integrin αIIbβ3 binding. Biochemistry. 2010;49(43):9217-9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peerschke EI, Wainer JA. Examination of irreversible platelet-fibrinogen interactions. Am J Physiol. 1985;248(5):C466-C472. [DOI] [PubMed] [Google Scholar]
- 22.Hantgan RR, Stahle MC. Integrin priming dynamics: mechanisms of integrin antagonist-promoted alphaIIbbeta3:PAC-1 molecular recognition. Biochemistry. 2009;48(35):8355-8365. [DOI] [PubMed] [Google Scholar]
- 23.Peerschke EI, Galanakis DK. Platelet adhesion to late fibrinogen degradation products. Blood Coagul Fibrinolysis. 1996;7(3):353-360. [DOI] [PubMed] [Google Scholar]
- 24.Podolnikova NP, Yakubenko VP, Volkov GL, Plow EF, Ugarova TP. Identification of a novel binding site for platelet integrins alpha IIb beta 3 (GPIIbIIIa) and alpha 5 beta 1 in the gamma C-domain of fibrinogen. J Biol Chem. 2003;278(34):32251-32258. [DOI] [PubMed] [Google Scholar]
- 25.Kamata T, Tieu KK, Irie A, Springer TA, Takada Y. Amino acid residues in the alpha IIb subunit that are critical for ligand binding to integrin alpha IIbbeta 3 are clustered in the beta-propeller model. J Biol Chem. 2001;276(47):44275-44283. [DOI] [PubMed] [Google Scholar]
- 26.Kamata T, Irie A, Tokuhira M, Takada Y. Critical residues of integrin alphaIIb subunit for binding of alphaIIbbeta3 (glycoprotein IIb-IIIa) to fibrinogen and ligand-mimetic antibodies (PAC-1, OP-G2, and LJ-CP3). J Biol Chem. 1996;271(31):18610-18615. [DOI] [PubMed] [Google Scholar]
- 27.Coller BS, Peerschke EI, Scudder LE, Sullivan CA. A murine monoclonal antibody that completely blocks the binding of fibrinogen to platelets produces a thrombasthenic-like state in normal platelets and binds to glycoproteins IIb and/or IIIa. J Clin Invest. 1983;72(1):325-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong X, Hudson NE, Lu C, Springer TA. Structural determinants of integrin β-subunit specificity for latent TGF-β. Nat Struct Mol Biol. 2014;21(12):1091-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frelinger AL III, Lam SC, Plow EF, Smith MA, Loftus JC, Ginsberg MH. Occupancy of an adhesive glycoprotein receptor modulates expression of an antigenic site involved in cell adhesion. J Biol Chem. 1988;263(25):12397-12402. [PubMed] [Google Scholar]
- 30.Frelinger AL III, Cohen I, Plow EF, et al. Selective inhibition of integrin function by antibodies specific for ligand-occupied receptor conformers. J Biol Chem. 1990;265(11):6346-6352. [PubMed] [Google Scholar]
- 31.Frelinger AL III, Du XP, Plow EF, Ginsberg MH. Monoclonal antibodies to ligand-occupied conformers of integrin alpha IIb beta 3 (glycoprotein IIb-IIIa) alter receptor affinity, specificity, and function. J Biol Chem. 1991;266(26):17106-17111. [PubMed] [Google Scholar]
- 32.Zamarron C, Ginsberg MH, Plow EF. A receptor-induced binding site in fibrinogen elicited by its interaction with platelet membrane glycoprotein IIb-IIIa. J Biol Chem. 1991;266(24):16193-16199. [PubMed] [Google Scholar]
- 33.Ugarova TP, Budzynski AZ, Shattil SJ, Ruggeri ZM, Ginsberg MH, Plow EF. Conformational changes in fibrinogen elicited by its interaction with platelet membrane glycoprotein GPIIb-IIIa. J Biol Chem. 1993;268(28):21080-21087. [PubMed] [Google Scholar]
- 34.Kouns WC, Wall CD, White MM, Fox CF, Jennings LK. A conformation-dependent epitope of human platelet glycoprotein IIIa. J Biol Chem. 1990;265(33):20594-20601. [PubMed] [Google Scholar]
- 35.Rooney MM, Parise LV, Lord ST. Dissecting clot retraction and platelet aggregation. Clot retraction does not require an intact fibrinogen gamma chain C terminus. J Biol Chem. 1996;271(15):8553-8555. [DOI] [PubMed] [Google Scholar]
- 36.Rooney MM, Farrell DH, van Hemel BM, de Groot PG, Lord ST. The contribution of the three hypothesized integrin-binding sites in fibrinogen to platelet-mediated clot retraction. Blood. 1998;92(7):2374-2381. [PubMed] [Google Scholar]
- 37.White JG. EDTA-induced changes in platelet structure and function: clot retraction. Platelets. 2000;11(1):49-55. [DOI] [PubMed] [Google Scholar]
- 38.Podolnikova NP, Yakovlev S, Yakubenko VP, Wang X, Gorkun OV, Ugarova TP. The interaction of integrin αIIbβ3 with fibrin occurs through multiple binding sites in the αIIb β-propeller domain. J Biol Chem. 2014;289(4):2371-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Litvinov RI, Farrell DH, Weisel JW, Bennett JS. The platelet integrin alphaIIbbeta3 differentially interacts with fibrin versus fibrinogen. J Biol Chem. 2016;291(15):7858-7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bassler N, Loeffler C, Mangin P, et al. A mechanistic model for paradoxical platelet activation by ligand-mimetic alphaIIb beta3 (GPIIb/IIIa) antagonists. Arterioscler Thromb Vasc Biol. 2007;27(3):E9-E15. [DOI] [PubMed] [Google Scholar]
- 41.Du XP, Plow EF, Frelinger AL III, O’Toole TE, Loftus JC, Ginsberg MH. Ligands “activate” integrin alpha IIb beta 3 (platelet GPIIb-IIIa). Cell. 1991;65(3):409-416. [DOI] [PubMed] [Google Scholar]
- 42.Blue R, Murcia M, Karan C, Jirousková M, Coller BS. Application of high-throughput screening to identify a novel alphaIIb-specific small- molecule inhibitor of alphaIIbbeta3-mediated platelet interaction with fibrinogen. Blood. 2008;111(3):1248-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aster RH. Immune thrombocytopenia caused by glycoprotein IIb/IIIa inhibitors. Chest. 2005;127(2, suppl):53S-59S. [DOI] [PubMed] [Google Scholar]
- 44.Hogan KA, Gorkun OV, Lounes KC, et al. Recombinant fibrinogen Vlissingen/Frankfurt IV. The deletion of residues 319 and 320 from the gamma chain of fibrinogen alters calcium binding, fibrin polymerization, cross-linking, and platelet aggregation. J Biol Chem. 2000;275(23):17778-17785. [DOI] [PubMed] [Google Scholar]
- 45.Podolnikova NP, Gorkun OV, Loreth RM, Yee VC, Lord ST, Ugarova TP. A cluster of basic amino acid residues in the gamma370-381 sequence of fibrinogen comprises a binding site for platelet integrin alpha(IIb)beta3 (glycoprotein IIb/IIIa). Biochemistry. 2005;44(51):16920-16930. [DOI] [PubMed] [Google Scholar]
- 46.Remijn JA, IJsseldijk MJ, van Hemel BM, et al. Reduced platelet adhesion in flowing blood to fibrinogen by alterations in segment gamma316-322, part of the fibrin-specific region. Br J Haematol. 2002;117(3):650-657. [DOI] [PubMed] [Google Scholar]
- 47.Lounes KC, Ping L, Gorkun OV, Lord ST. Analysis of engineered fibrinogen variants suggests that an additional site mediates platelet aggregation and that “B-b” interactions have a role in protofibril formation. Biochemistry. 2002;41(16):5291-5299. [DOI] [PubMed] [Google Scholar]
- 48.Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP. Regulated unmasking of the cryptic binding site for integrin alpha M beta 2 in the gamma C-domain of fibrinogen. Biochemistry. 2002;41(43):12942-12951. [DOI] [PubMed] [Google Scholar]
- 49.Coller BS. A new murine monoclonal antibody reports an activation-dependent change in the conformation and/or microenvironment of the platelet glycoprotein IIb/IIIa complex. J Clin Invest. 1985;76(1):101-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Artoni A, Li J, Mitchell B, et al. Integrin β3 regions controlling binding of murine mAb 7E3: implications for the mechanism of integrin alphaIIbbeta3 activation. Proc Natl Acad Sci USA. 2004;101(36):13114-13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Honda S, Tomiyama Y, Pelletier AJ, et al. Topography of ligand-induced binding sites, including a novel cation-sensitive epitope (AP5) at the amino terminus, of the human integrin beta 3 subunit. J Biol Chem. 1995;270(20):11947-11954. [DOI] [PubMed] [Google Scholar]
- 52.Cheresh DA. Human endothelial cells synthesize and express an Arg-Gly-Asp-directed adhesion receptor involved in attachment to fibrinogen and von Willebrand factor. Proc Natl Acad Sci USA. 1987;84(18):6471-6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin αVβ3 for angiogenesis. Science. 1994;264(5158):569-571. [DOI] [PubMed] [Google Scholar]
- 54.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32(suppl 2):W96-W99. [DOI] [PMC free article] [PubMed]
- 55.de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5(5):883-897. [DOI] [PubMed] [Google Scholar]
- 56.Moal IH, Bates PA. SwarmDock and the use of normal modes in protein-protein docking. Int J Mol Sci. 2010;11(10):3623-3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng M, Li J, Negri A, Coller BS. Swing-out of the β3 hybrid domain is required for αIIbβ3 priming and normal cytoskeletal reorganization, but not adhesion to immobilized fibrinogen. PLoS One. 2013;8(12):e81609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buitrago L, Rendon A, Liang Y, et al. ; ThromboGenomics Consortium. αIIbβ3 variants defined by next-generation sequencing: predicting variants likely to cause Glanzmann thrombasthenia. Proc Natl Acad Sci USA. 2015;112(15):E1898-E1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li J, Vootukuri S, Shang Y, et al. RUC-4: a novel αIIbβ3 antagonist for prehospital therapy of myocardial infarction. Arterioscler Thromb Vasc Biol. 2014;34(10):2321-2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zucker MB, Masiello NC. Platelet aggregation caused by dithiothreitol. Thromb Haemost. 1984;51(1):119-124. [PubMed] [Google Scholar]
- 61.Zhu J, Luo BH, Xiao T, Zhang C, Nishida N, Springer TA. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell. 2008;32(6):849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peerschke EI. Reversible and irreversible binding of fibrinogen to platelets. Platelets. 1997;8(5):311-318. [DOI] [PubMed] [Google Scholar]
- 63.Zucker MB, Grant RA. Nonreversible loss of platelet aggregability induced by calcium deprivation. Blood. 1978;52(3):505-513. [PubMed] [Google Scholar]
- 64.Gachet C, Hanau D, Spehner D, et al. Alpha IIb beta 3 integrin dissociation induced by EDTA results in morphological changes of the platelet surface-connected canalicular system with differential location of the two separate subunits. J Cell Biol. 1993;120(4):1021-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pidard D, Didry D, Kunicki TJ, Nurden AT. Temperature-dependent effects of EDTA on the membrane glycoprotein IIb-IIIa complex and platelet aggregability. Blood. 1986;67(3):604-611. [PubMed] [Google Scholar]
- 66.Fitzgerald LA, Phillips DR. Calcium regulation of the platelet membrane glycoprotein IIb-IIIa complex. J Biol Chem. 1985;260(20):11366-11374. [PubMed] [Google Scholar]
- 67.Ginsberg MH, Lightsey A, Kunicki TJ, Kaufmann A, Marguerie G, Plow EF. Divalent cation regulation of the surface orientation of platelet membrane glycoprotein IIb. Correlation with fibrinogen binding function and definition of a novel variant of Glanzmann’s thrombasthenia. J Clin Invest. 1986;78(4):1103-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Loftus JC, O’Toole TE, Plow EF, Glass A, Frelinger AL III, Ginsberg MH. A β3 integrin mutation abolishes ligand binding and alters divalent cation-dependent conformation. Science. 1990;249(4971):915-918. [DOI] [PubMed] [Google Scholar]
- 69.Abrams CS, Ellison N, Budzynski AZ, Shattil SJ. Direct detection of activated platelets and platelet-derived microparticles in humans. Blood. 1990;75(1):128-138. [PubMed] [Google Scholar]
- 70.Hantgan RR, Taylor RG, Lewis JC. Platelets interact with fibrin only after activation. Blood. 1985;65(6):1299-1311. [PubMed] [Google Scholar]
- 71.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 2012;11(11):1475-1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.