

Bruton’s tyrosine kinase (BTK) is a pivotal protein in the B-cell antigen receptor pathway.1 Investigations in murine hematopoietic cells demonstrated that BTK is expressed in B lymphocytes but not in T cells. This signal transducing protein is present in hematopoietic stem cells and expressed throughout the development of B cells, including in mature B-cells. However, compared with B cells in the bone marrow, peripheral blood B-lymphocytes express lower total amount of BTK protein. These observations suggest that expression of BTK protein is context dependent.2 Furthermore, activation of the B-cell antigen receptor pathway resulted in a dramatic increase in the expression of BTK in murine B cells in vitro and in intact animal model systems.3 Mechanistically, this upregulation was at the protein, not mRNA level, and was mediated through the PI3Kdelta axis. Consequently, a pan-PI3K inhibitor decreased B-cell antigen receptor-mediated increase in BTK protein level.
Recently, there has been a renewed interest in BTK because of the emergence of a small molecule inhibitor (ibrutinib) that irreversibly binds to cysteine-481 residue of the protein. Importantly, the drug showed remarkable overall response rates and improved survival in previously treated4 and untreated5 patients with chronic lymphocytic leukemia (CLL), a mature B-cell neoplasm. In fact, because of the high response rates and prolongation of survival observed without much toxicity, the drug is now FDA-approved for patients with CLL. Pharmacodynamic correlative studies, conducted in the context of a phase I trial of this oral drug, demonstrated almost complete (>95%) occupancy of cellular BTK protein at doses of ≥ 2.5 mg/kg/day.6 This high occupancy of BTK by irreversible binding of the drug was a requirement for clinical response. This underscores the importance of BTK in B-cell CLL in general and levels of total protein for pharmacodynamic inhibition in particular. A unique and key feature observed in clinical trials is an egress of leukemia cells from lymph nodes to the peripheral blood resulting in a dramatic increase in the circulating lymphocyte count paralleled by a decrease in lymphadenopathy.
A role for SDF1/CXCL12 and its cognate receptor CXCR4 in tumor cell trafficking has been identified in several tumor types, including CLL.7 Recently, Chen et al.8 mimicked the CLL lymphocytosis phenomenon in a TCL1 adoptive transfer mouse model and identified, in systematically and comprehensively performed experiments, that CXCR4 and CXCL12 are determinants of this mobilization of CLL cells from the tissues to the blood. Ibrutinib’s action on cell trafficking was due to a decline in CXCR4 phosphorylation, which is mediated by kinases such as Pim1, PKCμ and GTK6. Interestingly, in parallel with these changes, ibrutinib therapy in diseased mice resulted in a decrease in phospho- as well as total BTK protein in CLL lymphocytes8 which is consistent with the prior observation in murine normal B-lymphocytes.3
We postulated that B-cell antigen receptor pathway-induced BTK protein observed in normal murine B-lymphocytes3 and ibrutinib-mediated decline in BTK levels in CLL B-cells in TCL1 mice8 should also occur during ibrutinib therapy of human CLL. To test this hypothesis, we obtained circulating CLL cells from patients on ibrutinib. The present study was carried out in lymphocytes isolated from peripheral blood samples of patients with CLL who received 420 mg/day of ibrutinib on phase II and III clinical trials. All participants signed written informed consent forms in accordance with the Declaration of Helsinki, and the laboratory protocols were approved by the Institutional Review Board at the University of Texas MD Anderson Cancer Center. CLL cells were isolated from peripheral blood samples, protein was extracted, and analyzed for quantitation using immunoblots.
Similar to Chen et al.,8 we observed a decline in BTK protein in the circulating leukemia cells in the peripheral blood of patients after 4 weeks of ibrutinib therapy (Figure 1). This was associated with a decrease in phospho- and total-CXCR4. In contrast to the prior report in murine CLL cells,8 where cell surface (membrane) as well as and intracellular expression were evaluated, in the current work, we analyzed only total intracellular CXCR4 protein levels. This decrease in CXCR4 receptor level may be owing to diminished phosphorylation of the receptor, which may be due to three different upstream kinases (Pim1, GRK6 and PKCμ). Total protein levels of all three kinases appear to decrease after 4 weeks of ibrutinib therapy. In the murine Tcl-1 model system, reduction in phospho-PKC and total Pim-1 kinase was reported.8
Figure 1.
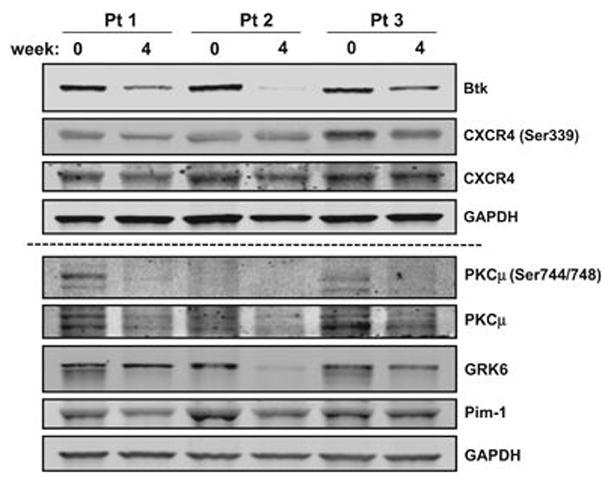
Impact of ibrutinib therapy on BTK- and CXCR4-associated proteins in primary CLL cells. Primary CLL cells from three patients (Pt) were isolated fresh from peripheral blood samples before treatment (baseline, designated as 0 week), and 4 weeks after 420 mg daily oral doses of ibrutinib. Blood samples were collected in green-top tubes and CLL cells were isolated by Ficoll-Hypaque gradient. Cell pellets were prepared for two immunoblots. The first immunoblot was used for total BTK (BD Biosciences 611116), and phospho-(Abcam 74012) and total CXCR4 (Abcam 58176). GAPDH (Novus NB600-502) was blotted for loading control. The second immunoblot (below dashed line) was used to test phospho-(Cell Signaling 2054) and total PKC (Abnova H00005587-B01P), and total GRK6 (Cell Signaling 5878) and Pim-1 (Sigma SAB1404205). GAPDH (Novus NB600-502) served as a protein loading control.
Because a decrease in BTK protein during ibrutinib therapy has clinical and pharmacological consequences, we further investigated changes in the total protein levels in additional patients. It is noteworthy that as a corollary to almost complete occupation of BTK protein by ibrutinib at this dose (420 mg per day), a decline in phospho-BTK9 was observed in circulating CLL lymphocytes after 2, 4 and 12 weeks of continuous therapy. Quantitation of total BTK protein in 2, 5 and 2 patient samples at 2, 4 and 12 weeks, respectively of ibrutinib showed a statistically significant time-dependent decline in the level of total BTK protein compared with the untreated (baseline) sample (Figure 2a). Because the number of samples is low at each time point, comparison of all samples was done to determine overall statistical significance (P=0.0001). This reduction in protein level was also observed at the mRNA transcript level (Figure 2b), albeit to a lesser extent. Compared with the baseline value (0 week), the mean values were 0.7, 0.5 and 0.35 at 2, 4 and 12 weeks, respectively (P=0.0003 for the whole group).
Figure 2.
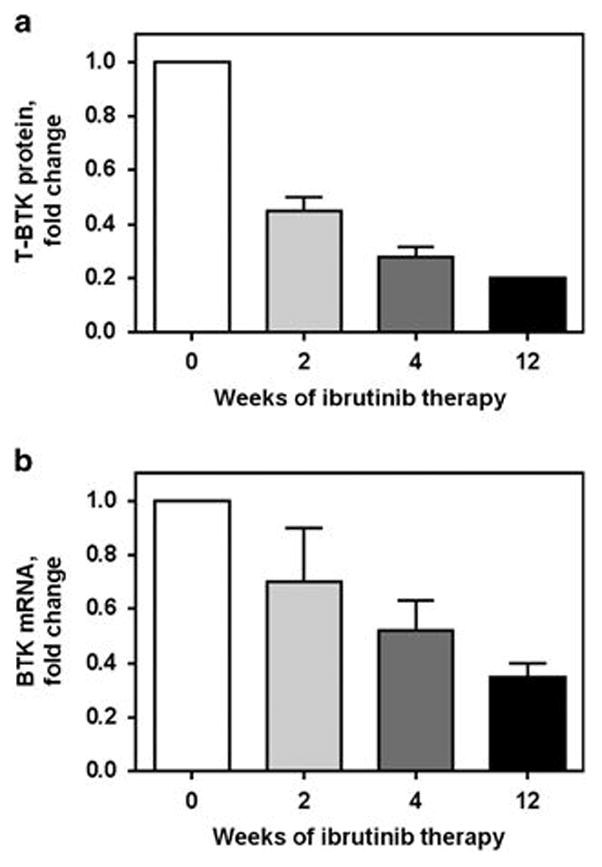
Ibrutinib therapy decreases total protein and mRNA transcript levels of BTK. Primary CLL cells from patients were isolated fresh from peripheral blood samples before treatment (baseline), and at 2, 4 and 12 weeks of 420 mg daily oral dosing of ibrutinib. Blood samples were collected from six patients in green-top tubes and CLL cells were isolated by Ficoll-Hypaque gradient. Cell pellets were used for total BTK protein levels (a) and BTK mRNA levels (b) from six patients with CLL. For the former, cell lysates were prepared and after protein quantitation, immunoblots were run to analyse total BTK protein levels. Immunoblots were quantitated and plotted as a percentage of the control level (baseline sample). For mRNA transcript levels, RT-PCR assay was performed in triplicate from each sample as described previously.8 Student’s t-tests (two-tailed) were performed using the GraphPad Prism6 software (GraphPad Software, Inc., San Diego, CA, USA) to compare values in untreated samples compared with all treated samples and the P-values for the comparisons in a and b are 0.0001 and 0.0003, respectively.
As mentioned above, the mechanism of action of ibrutinib in CLL relies on almost complete and irreversible covalent binding of drug to the target, that is, BTK. Hence, drug (ibrutinib) to substrate (BTK) stoichiometry is important in identifying quantitative relationships and optimal dosing of ibrutinib. On the basis of our current data in human CLL cells during ibrutinib therapy, one could postulate that after a few weeks of ibrutinib, the target protein will decrease, suggesting that a lower dose will suffice to stochiometrically inhibit this kinase. We have designed a pilot protocol to test this hypothesis wherein each patient will receive progressively lower ibrutinib doses over three cycles of therapy.
Acknowledgments
We thank Benjamin Hayes and Mark Nelson for coordinating sample distribution during therapy. This work was supported in part by MD Anderson CLL Moon Shot Program and Sponsored Research Agreement form Pharmacyclics, Inc.
Footnotes
Competing interests
VG has a sponsored research agreement with Pharmacyclics, Inc. Other authors do not have any conflict of interest.
Contributor Information
F Cervantes-Gomez, Department of Experimental Therapeutics, Houston, TX, USA.
V Kumar Patel, Department of Experimental Therapeutics, Houston, TX, USA.
P Bose, Department of Leukemia, The University of Texas M D Anderson Cancer Center, Houston, TX, USA.
M J Keating, Department of Leukemia, The University of Texas M D Anderson Cancer Center, Houston, TX, USA.
V Gandhi, Department of Experimental Therapeutics, Houston, TX, USA. Department of Leukemia, The University of Texas M D Anderson Cancer Center, Houston, TX, USA.
References
- 1.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14:219–232. doi: 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- 2.Rawlings DJ, Witte ON. The Btk subfamily of cytoplasmic tyrosine kinases: structure, regulation and function. Semin Immunol. 1995;7:237–246. doi: 10.1006/smim.1995.0028. [DOI] [PubMed] [Google Scholar]
- 3.Nisitani S, Satterthwaite AB, Akashi K, Weissman IL, Witte ON, Wahl MI. Posttranscriptional regulation of Bruton’s tyrosine kinase expression in antigen receptor-stimulated splenic B cells. Proc Natl Acad Sci USA. 2000;97:2737–2742. doi: 10.1073/pnas.050583597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371:213–223. doi: 10.1056/NEJMoa1400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373:2425–2437. doi: 10.1056/NEJMoa1509388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 8.Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia. 2016;30:833–843. doi: 10.1038/leu.2015.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cervantes-Gomez F, Lamothe B, Woyach JA, Wierda WG, Keating MJ, Balakrishnan K, et al. Pharmacological and protein profiling suggests venetoclax (ABT-199) as optimal partner with ibrutinib in chronic lymphocytic leukemia. Clin Cancer Res. 2015;21:3705–3715. doi: 10.1158/1078-0432.CCR-14-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]