

ABSTRACT
Though Vγ9Vδ2-T cells constitute only a small fraction of the total T cell population in human peripheral blood, they play a vital role in tumor defense and are therefore of major interest to explore for cancer immunotherapy. Vγ9Vδ2-T cell-based cancer immunotherapeutic approaches developed so far have been generally well tolerated and were able to induce significant clinical responses. However, overall results were inconsistent, possibly due to the fact that these strategies induced systemic activation of Vγ9Vδ2-T cells without preferential accumulation and targeted activation in the tumor. Here we show that a novel bispecific nanobody-based construct targeting both Vγ9Vδ2-T cells and EGFR induced potent Vγ9Vδ2-T cell activation and subsequent tumor cell lysis both in vitro and in an in vivo mouse xenograft model. Tumor cell lysis was independent of KRAS and BRAF tumor mutation status and common Vγ9Vδ2-T cell receptor sequence variations. In combination with the conserved monomorphic nature of the Vγ9Vδ2-TCR and the facile replacement of the tumor-specific nanobody, this immunotherapeutic approach can be applied to a large group of cancer patients.
KEYWORDS: cancer, EGFR, gamma delta T cells, immunotherapy, nanobody, single-domain antibody fragment, tumor, VHH
Introduction
Although the majority of human T cells expresses an αβ-T cell receptor (TCR), a smaller proportion of T cells expresses a γδ-TCR. The most predominant γδ-T cell subset in human peripheral blood consists of Vγ9Vδ2-T cells that account for approximately 1–5% of all T cells. Vγ9Vδ2-T cells are able to induce apoptosis in a broad spectrum of cancer cells and their reduced frequency and/ or impaired functionality in the peripheral blood is a commonly observed phenomenon in cancer patients.1-8 In melanoma patients, reduced Vγ9Vδ2-T cell levels in the tumor microenvironment are related to more advanced clinical stages and reduced Vγ9Vδ2-T cell levels in the peripheral blood were recently shown to be a negative predictor for response upon treatment with ipilimumab.9,10 These observations clearly point to a vital role for Vγ9Vδ2-T cells in natural and induced immunity to cancer. In contrast to conventional T cells, ligand recognition by Vγ9Vδ2-T cells is independent of MHC-molecule presentation, tumor neo-epitope burden and classical immuno-editing,11,12 This underscores their great potential as anti-tumor effector cells, a potential that has been hitherto largely untapped.
Vγ9Vδ2-T cells become activated by the recognition of non-peptidic phosphoantigens (pAg).13-15 These are upregulated by stressed cells, including malignant cells, as a consequence of an enhanced activity of the mevalonate pathway16 or through the non-mevalonate pathway upon bacterial infection.14,17,18 Furthermore, therapeutic agents such as aminobisphosphonates (NBP) can inhibit the mevalonate pathway and thus lead to intracellular pAg accumulation. Upon elevated intracellular levels of pAg in target cells, the GTPase RhoB translocates from the nucleus to the cytoplasm where it presumably binds to the membrane protein butyrophilin 3A1 (BTN3A1, also known as CD277). This binding might induce a conformational change of BTN3A1 that in turn is sensed by the Vγ9Vδ2-TCR and results in Vγ9Vδ2-T cell activation.19-23 Activation of Vγ9Vδ2-T cells can be enhanced by interactions between the NKG2D receptor expressed on most Vγ9Vδ2-T cells and by stress-related MICA, MICB and ULBP molecules that are upregulated in infected or transformed cells.3,24 This, in combination with enhanced pAg levels, allows Vγ9Vδ2-T cells to distinguish “normal” cells from ”altered-self” or tumor cells.25 Activated Vγ9Vδ2-T cells produce pro-inflammatory cytokines (e.g. IFN-γ, TNF-α, TRAIL and the chemokines MIP-1 and RANTES) in addition to cytolytic mediators (perforin, granzyme B) to induce the specific lysis of target cells, which is regulated through the perforin pathway or through Fas- and TRAIL-induced apoptosis.25,26
Their monomorphic recognition of activating ligands, their effective induction of tumor cell lysis and their rapid effector response provide Vγ9Vδ2-T cells with a unique combination of features that make them of major interest for cancer immunotherapeutic approaches. As a result, several clinical trials have been initiated to evaluate the use of Vγ9Vδ2-T cells in the treatment of both hematological and solid malignancies. Clinically explored approaches have included adoptive transfer of ex vivo expanded Vγ9Vδ2-T cells and the in vivo activation of Vγ9Vδ2-T cells through the administration of NBPs or synthetic pAg, alone or in combination with low-dose IL-2 treatment.27,28 These Vγ9Vδ2-T cell-based therapeutic approaches were well tolerated and capable of inducing clinically relevant anti-tumor responses in several cases. However, the overall results were inconsistent and are possibly related to the fact that these approaches induced a systemic Vγ9Vδ2-T cell activation without necessarily affecting their preferential accumulation and activation in the tumor microenvironment, where these cells should exert their anti-tumor effects.
To date, various bispecific T cell engagers (BiTEs) targeting both CD3 and a tumor antigen through the coupling of single-chain variable fragments (scFv) have been developed and were shown to induce clinical responses.29 However, as CD3 is expressed by all T cells, including immunosuppressive regulatory T cells (Tregs) that actually predominate in the tumor microenvironment and are related to poor prognosis30, antibody-based constructs designed to exclusively trigger immune cells with a pro-inflammatory function, such as Vγ9Vδ2-T cells, might well constitute a more effective approach.31 Recently, we have reported on the generation of a set of Vγ9Vδ2-TCR specific nanobodies with activating properties that could form the basis for a novel therapeutic approach aimed at tumor-specific Vγ9Vδ2-T cell accumulation and activation.32 Nanobodies (or VHHs) are defined by the variable antigen binding regions derived from heavy chain only antibodies, naturally occurring in camelids (i.e. llamas, camels and dromedaries).33,34 Single-domain VHH have several advantages over full-length antibodies or scFv when used for the generation of multivalent and/or multispecific molecules. Due to the absence of light chain domains, pairing issues do not apply, VHHs refold easily and they are provided with increased solubility. Moreover, VHHs can easily be produced by bacteria or yeast allowing time and cost reduction during manufacturing.35,36 Furthermore, VHH domains are low immunogenic because of their high homology with human VH genes and the absence of the Fc-region.29,36 VHHs are ten times smaller than conventional antibodies allowing them to reach clefts in antigen structures and granting them with enhanced tissue penetration as compared with conventional antibodies.37,38
Here, we describe the generation and evaluation of a bispecific VHH-based construct that combines inhibition of the epidermal growth factor receptor (EGFR)-signaling pathway via an antagonistic anti-EGFR VHH with the target-dependent activation of effector Vγ9Vδ2-T cells via an anti-Vγ9Vδ2-TCR VHH. Vγ9Vδ2-T cells activated in this manner produced pro-inflammatory cytokines such as IFN-γ and TNF-α and efficiently lysed EGFR-expressing tumor cell lines both in vitro and in vivo. This therapeutic effect was independent of KRAS or BRAF mutations, which are normally associated with resistance to anti-EGFR monoclonal antibody (mAb) therapy.39,40 Moreover, variations in Vγ9Vδ2-TCR δ2-CDR3 sequence that are known to be associated with reduced Vγ9Vδ2-T cell responses1 to pAg stimulation did not affect cell killing efficacy. This novel bispecific VHH-based immunotherapeutic approach can be applied to many tumor types by simply replacing the tumor-specific VHH and does not require further individualization due to the conserved monomorphic nature of the Vγ9Vδ2-TCR.
Results
Selection of a human Vγ9Vδ2-TCR specific and -activating VHH
Vγ9Vδ2-TCR specific VHHs were generated by immunizing two lama glamas multiple times with human Vγ9Vδ2-T cells pooled from different healthy donors. Through phage display and after screening for Vγ9Vδ2-TCR specific fragments, 20 different Vγ9Vδ2-TCR specific VHHs were identified, either directed to the Vδ2- or to the Vγ9-chain, and either with activating or with non-activating potential as determined using a Vγ9Vδ2-TCR transduced JurMa luciferase reporter cell line. The specificity of these Vγ9Vδ2-TCR specific VHHs and their applicability for flow cytometry, immunocytochemistry, and magnetic activated cell sorting was previously reported.32 The VHHs with activating potential identified in this screen were then tested for their capability to induce activation of human healthy donor-derived Vγ9Vδ2-T cells via cross-linking. For this purpose, Vγ9Vδ2-T cells were cultured with plate-bound VHHs for 24 hrs. Activation of Vγ9Vδ2-T cells was determined by assessing up-regulation of the activation marker CD25, induction of CD107a expression reflecting the release of cytotoxic granules, and the intracellular production of IFN-γ as determined by flow cytometry. As a positive control we used NBP-pretreated HeLa cells in which the endogenous pAg isopentenyl pyrophosphate (IPP) accumulates as a result of NBP`s inhibitory effect on farnesyl pyrophosphate synthase. These screens led to the identification of the anti-Vδ2 VHH 6H4 and the anti-Vγ9 VHH 6H1 as the most consistently activating VHHs, inducing Vγ9Vδ2-T cell activation in all three assays across multiple donors (Fig. 1A-C). Their ability to activate Vγ9Vδ2-T cells was further confirmed by studying the activation of Vγ9Vδ2-T cells from PBMC directly ex-vivo (Supplementary Fig. 1).
Figure 1.

Characteristics of Vγ9Vδ2-T cell activating VHH. (A-C) Vγ9Vδ2-T cells were cultured with individual plate bound (wells coated with 500 nM) anti-Vγ9Vδ2-TCR VHH, control VHH R2, HeLa cells or NBP-pretreated HeLa cells in a 1:1 ratio. After 24 hrs, Vγ9Vδ2-T cell activation was determined by assessing the percentage of Vγ9Vδ2-T cells positive for (A) CD25, (B) CD107a, or (C) intracellular IFN-γ using flow cytometry. Shown are means substracted by background levels ± SEM of n = 3-5 experiments. p-Values were calculated with a one-way ANOVA and Bonferroni's post-hoc test. (* indicates p<0.05 and ** indicates p<0.01). (D) The anti-Vδ2 VHH 6H4 (40 nM) binds to Jurkat-Vγ9Vδ2-TCR cells (thick line), but not to Jurkat cells without TCR expression (filled grey) or Jurkat-Vα24Vβ11-TCR cells (dotted line). (E) The anti-Vδ2 VHH 6H4 (350 nM) binds to healthy donor-derived Vγ9+Vδ2+ (thick line) and Vγ9−Vδ2+ γδ-T cells but not to Vγ9−Vδ2− (filled grey) or Vγ9+Vδ2− γδ-T cells (dotted line). (F) Vγ9Vδ2-T cells were cultured with plate bound or soluble monovalent VHH (filled squares), bivalent VHH (filled triangles) or control VHH R2 (open circles) at the indicated concentrations for 24 hrs. Expression of CD25 was assessed using flow cytometry. Representative figures of n = 3 experiments are shown. Abbreviations: aminobisphosphonates (NBP); Gly4Ser (GS).
Although the vast majority of γδ-T cells in the human peripheral blood consist of Vγ9Vδ2-T cells, γδ-T cells expressing either the Vγ9-chain (i.e. Vγ9+Vδ2− γδ-T cells) or the Vδ2-chain (i.e. Vγ9−Vδ2+ γδ-T cells) exist; these, however, do not respond to pAg stimulation. As the relative frequency of Vγ9−Vδ2+ γδ-T cells is very low and substantially lower than the level of Vγ9+Vδ2− γδ-T cells17,41, we reasoned that a Vδ2-TCR chain specific VHH would more selectively target Vγ9Vδ2-T cells and would therefore be the preferred VHH to be used in a bispecific VHH construct aimed at the specific targeting of Vγ9Vδ2-T cells. For this reason, the Vδ2-TCR specific VHH 6H4 (Fig. 1D and 1E) was selected for further experiments.
To determine whether coupling of two anti-Vγ9Vδ2-TCR VHHs into one bivalent VHH construct could result in an even stronger activation of Vγ9Vδ2-T cells, two 6H4 VHH genes were engineered into one construct and separated by a flexible Gly4Ser-linker (GS) of varying length (5-30 amino acids). First, bivalent VHH 6H4-5GS-6H4 was compared to the monovalent VHH 6H4 with respect to its ability to activate Vγ9Vδ2-T cells in a plate-bound assay. At all tested concentrations, stimulation with the bivalent VHH resulted in a stronger Vγ9Vδ2-T cell activation as compared to the monovalent VHH (Fig. 1F). However, to be optimally effective in a tumor targeting construct it is desirable that the VHH does not induce Vγ9Vδ2-T cell activation either on its own or in the absence of tumor cells. We observed that when Vγ9Vδ2-T cells were cultured with the bivalent VHH 6H4-5GS-6H4 added in solution, a strong Vγ9Vδ2-T cell activation was induced, even at very low concentrations (Fig. 1F). This was independent of the linker length between both 6H4 VHHs (data not shown). In contrast, when monovalent VHH 6H4 was added in solution to Vγ9Vδ2-T cell cultures, Vγ9Vδ2-T cells did not become activated (Fig. 1F). As Vγ9Vδ2-TCR cross-linking by the bivalent VHH constructs would likely result in non-specific systemic activation as opposed to target-specific activation of Vγ9Vδ2-T cells, the monovalent anti-Vγ9Vδ2 TCR VHH 6H4 was selected for incorporation into a bispecific tumor-targeting VHH construct.
Generation and functional evaluation of a bispecific anti-EGFR-anti-Vγ9Vδ2-TCR VHH construct
To generate a bispecific VHH construct, the anti-Vγ9Vδ2-TCR VHH 6H4 was joined to the previously generated and characterized high-affinity anti-EGFR VHH 7D12. This VHH is able to compete with EGF for EGFR binding and inhibits both EGFR phosphorylation and EGFR+ tumor cell proliferation in vitro and in vivo.42,43 To determine the optimal format with respect to binding and functionality, multiple bispecific VHH constructs were created with variations in orientation and spacing between the individual VHHs. First, bispecific 7D12-6H4 and 6H4-7D12 VHH constructs were generated with a flexible Gly4Ser-linker of 10 amino acids to determine whether target binding and affinity of the individual VHHs was maintained in the bispecific format and whether this was dependent on their orientation. Whereas the affinity of 6H4 to Vγ9Vδ2-T cells did not differ by its relative (i.e. N-terminal or C-terminal) position in the bispecific VHH, the anti-EGFR 7D12 VHH clearly bound to EGFR+ A431 tumor cells more efficiently when it was positioned at the N-terminus (Fig. 2A). Therefore, the 7D12-6H4 format was considered optimal and variations in the linker length were made to assess whether this would influence binding efficiency and functionality. As shown in Fig. 2B, linker length variations did not influence the binding of the construct to target cells. Neither did these influence the efficacy of Vγ9Vδ2-T cell activation, degranulation or tumor cell lysis induced by the bispecific VHH construct upon co-culture of Vγ9Vδ2-T cells with EGFR-expressing tumor cells (Fig. 2C). This was also observed when the linker length was replaced to the smallest Gly4Ser-linker consisting of 5 amino acids (data not shown). As a small linker is least prone to proteolysis in vivo, we used the 5 amino acid (5GS) linker for all subsequent experiments.
Figure 2.

The effect of orientation and linker length in the bispecific VHH. (A and B) Vγ9Vδ2-T cells (left) or EGFR-expressing A431 cells (right) were incubated in the presence or absence of the indicated VHHs and bound VHH was assessed by flow cytometry. Mean fluorescence intensity (MF) of bound VHH to the cells is depicted. (C) Vγ9Vδ2-T cells and A431 cells were co-cultured in a 1:1 ratio for 24 hrs in the presence or absence of the indicated bispecific VHH. Both CD25 (left) and CD107a (middle) expression on Vγ9Vδ2-T cells were assessed by flow cytometry. The percentage of lysed A431 cells (right) was determined using 7-AAD staining and flow cytometry. Representative figures of n = 3 experiments are shown. Abbreviations: Gly4Ser (GS).
Vγ9Vδ2-T cell activation and subsequent tumor cell lysis was formally demonstrated to depend on simultaneous binding of the bispecific VHH construct to Vγ9Vδ2-T cells and EGFR-expressing tumor cells by the use of control constructs incorporating the irrelevant VHH R244 (Fig. 3A). Of note, Vγ9Vδ2-T cell activation and degranulation levels induced by the 7D12-5GS-6H4 bispecific VHH construct were equivalent to those observed when Vγ9Vδ2-T cells were co-cultured with NBP-pretreated EGFR+ tumor cells. The maximum level of tumor cell lysis induced by Vγ9Vδ2-T cells was observed at concentrations as low as 10 nM of the 7D12-5GS-6H4 bispecific construct. Importantly, this was at least as effective as when tumor cells were pretreated with 100 µM NBP. Furthermore, when titrating down the 7D12-5GS-6H4 concentration, efficient lysis of EGFR+ tumor cells was observed at concentrations as low as 10 pM in a 1:1 effector:target cell ratio. Importantly, this was not observed when the immortalized human B-cell line JY, lacking EGFR expression, was used as target (Fig. 3B) and herewith demonstrated the specificity of this targeting approach.
Figure 3.
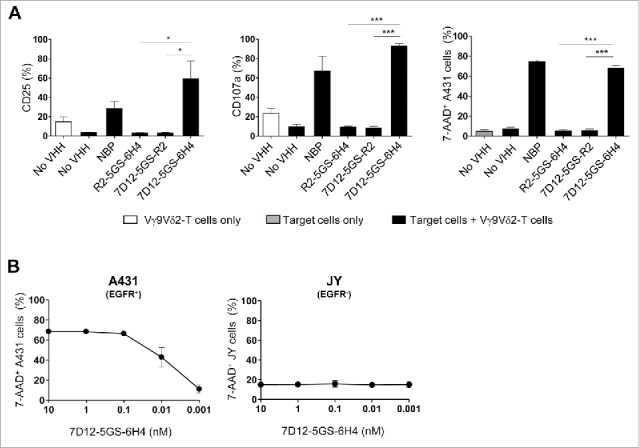
The 7D12-5GS-6H4 bispecific VHH induces Vγ9Vδ2-T cell activation and lysis of EGFR expressing tumor cells. Vγ9Vδ2-T cells were cultured with or without EGFR+ A431 tumor cells (A-B) or EGFR− JY cells (B) in a 1:1 ratio in the presence of the 7D12-5GS-6H4 bispecific VHH or a bispecific control VHH. VHH concentrations: (A) 10 nM; (B) as indicated. For control situations, Vγ9Vδ2-T cells were co-cultured with target cells in the absence of VHH (no VHH; negative control) or with NBP-pretreated target cells (positive control). After 24 hrs, Vγ9Vδ2-T cell activation and degranulation was determined by assessing the percentage of CD25 or CD107a expression, respectively by flow cytometry. The percentage of lysed target cells was determined using 7-AAD staining and flow cytometry. A) White bars represent Vγ9Vδ2-T cell mono-cultures in the absence of VHH, grey bars represent target cell mono-cultures in the absence of VHH and black bars represent Vγ9Vδ2-T cell co-cultures with target cells and indicated VHH. B) Co-cultures of target cells with Vγ9Vδ2-T cells and indicated amount of 7D12-5GS-6H4 bispecific VHH. Shown are mean ± SEM of n = 3-4 experiments. p-Values were calculated with a one-way ANOVA and Bonferroni's post-hoc test (* indicates p<0.05 and *** indicates p<0.001). Abbreviations: aminobisphosphonates (NBP); Gly4Ser (GS).
As treatment with anti-EGFR mAbs such as cetuximab or panitumumab is often accompanied by skin toxicity,45,46 we explored whether primary skin-derived (EGFR+) keratinocytes were lysed by Vγ9Vδ2-T cells in the presence of the 7D12-5GS-6H4 construct. EGFR expression was confirmed by flow cytometry demonstrating that the mean fluorescence index (MFI) of EGFR expression on keratinocytes was 3.2 ± 1.5 (n = 3, mean ± SD) (as a reference, the MFI of EGFR on A431 was 9.4, and the MFI of EGFR on JY was 0.8). Even at high concentrations of the bispecific targeting construct, only minor activation and cytolytic activity of Vγ9Vδ2-T cells was observed in the presence of primary keratinocytes (Supplementary fig. 2).
7D12-5GS-6H4 induces Vγ9Vδ2-T cell-mediated lysis of EGFR+ tumor cells irrespective of KRAS or BRAF mutation status
Of note, the 7D12 VHH retained its capacity to inhibit EGFR signaling in a dose-dependent manner upon incorporation into the bispecific VHH construct. As demonstrated by the analysis of EGFR phosphorylation in EGFR expressing tumor cells upon their exposure to EGF, this inhibitory activity was equivalent to that of the monovalent 7D12 VHH (Fig. 4A). To test if the 7D12-5GS-6H4 bispecific VHH exerted antitumor activity even in the presence of activating mutations in the EGFR signaling pathway, Vγ9Vδ2-T cells were co-cultured with EGFR+ human colon cancer cell lines carrying either a mutation in KRAS (i.e. SW480 cells, expressing one of the most common and oncogenic KRAS mutations G12V) or BRAF (i.e. HT-29 cells, expressing the most common BRAF mutation V600E)47,48 in the presence or absence of 7D12-5GS-6H4. As shown in Fig. 4B-I, 7D12-5GS-6H4 induced potent Vγ9Vδ2-T cell activation, degranulation and tumor cell lysis of EGFR+ colon tumor cells, irrespective of their KRAS or BRAF mutation status. Next, the antitumor activity of the 7D12-5GS-6H4 bispecific VHH was assessed using primary tumor material from colon cancer patients. For this purpose, Vγ9Vδ2-T cells were co-cultured for 4 hrs at a 1:1 effector:target ratio with tumor cells of three different colon carcinoma patients (1 patient with a RASwt BRAFwt tumor, 1 patient with a KRASmt tumor, and 1 patient with a BRAFmt tumor). In the presence of the 7D12-5GS-6H4 bispecific VHH efficient lysis of primary colon cancer cells was observed (Fig. 4J). Again, no apparent differences between the wild-type tumor and KRAS or BRAF mutant primary tumors could be noted. EGFR expression was confirmed on all primary colon cancer samples using flow cytometry (Fig. 4K).
Figure 4.

The 7D12-5GS-6H4 bispecific VHH inhibits EGFR signaling but does not depend on this to induce tumor cell lysis. (A) The anti-EGFR 7D12 VHH retains its capacity to inhibit phosphorylation of EGFR when incorporated in a bispecific 7D12-5GS-6H4 VHH format. Her14 cells were incubated with a mixture of 8 nM human EGF and the indicated VHH. Cell lysates were run on SDS-PAGE gel and western blotted for phosphorylated EGFRTyr1068 and β-actin as a loading control. (B-I) Vγ9Vδ2-T cells were cultured with or without the EGFR+ colon tumor cells SW480 KRASG12V (B-E) or HT29 BRAFV600E (F-I) for 24 hours or primary colon cancer cells for 4 hours (J) in a 1:1 ratio in the presence of the 7D12-5GS-6H4 bispecific VHH or a bispecific control VHH. VHH concentrations: B-D, F-H and J) 10 nM; E and I) as indicated. For control conditions, Vγ9Vδ2-T cells were co-cultured with target cells in the absence of VHH (no VHH; negative control) or with NBP-pretreated target cells (positive control). Vγ9Vδ2-T cell activation and degranulation was determined by assessing the percentage of CD25 (B) and (F) or CD107a (C and G) expression, respectively by flow cytometry. The percentage of lysed target cells was determined using 7-AAD staining and flow cytometry (D-E and H-J). B-D and (F-H) White bars represent Vγ9Vδ2-T cell mono-cultures in the absence of VHH, grey bars represent target cell mono-cultures in the absence of VHH and black bars represent Vγ9Vδ2-T cell co-cultures with target cells and indicated VHH. E and (I) Co-cultures of target cells with Vγ9Vδ2-T cells and the indicated amount of 7D12-5GS-6H4 bispecific VHH. J) Lysis of patient derived primary colon cancer cells; tumor 1: mutation in KRAS exon 2, c.38G>A, p.G13D; tumor 2: mutation in BRAF exon 15, c.1799 T>A, p.V600E; tumor 3: RAS and BRAF wild-type. (K) All three colon cancer samples expressed EGFR as determined by flow cytometry. Shown are means ± SEM of n = 3 experiments. p-Values were calculated with a one-way ANOVA and Bonferroni's post-hoc test (* indicates p<0.05, ** indicates p<0.01 and *** indicates p<0.001). Abbreviations: aminobisphosphonate (NBP); Gly4Ser (GS); mean fluorescence index (MFI).
7D12-5GS-6H4 activates Vγ9Vδ2-TCR-G115 with various δ2-CDR3 sequence variations
The sequence and length of the δ2-CDR3 region of the Vγ9Vδ2-TCR varies between individuals and in part determines the TCR affinity and cytolytic capacity upon binding of pAg expressing target cells.1 Since VHH 6H4 specifically binds to the Vδ2-chain of the Vγ9Vδ2-TCR, we determined if common Vδ2-CDR3 variations would influence the binding of the VHH 6H4 to the Vγ9Vδ2-TCR and whether this affected its Vγ9Vδ2-T cell activating capacity. To this end, JurMa cells were transduced to express the wildtype Vγ9Vδ2-TCR-G115 or the Vγ9Vδ2-TCR-G115 with δ2-CDR3 variations in the 110–112 region (98-103 by Kabat numbering). This region was either replaced by i) a single alanine, creating a “short length” mutant (δ2-G115LM1) with complete abolishment of pAg/BTN3A1-reactivity; ii) 9 alanine amino acids, creating an “enlongated length” mutant (in δ2-G115LM9) with approximately 40% reduced pAg/BTN3A1-reactivity compared to wild-type Vγ9Vδ2-TCR-G115; iii) the δ2-CDR3 sequence of the naturally weakly pAg/BTN3A1-reactive cl3 clone (δ2-G115cl3); or iiii) the δ2-CDR3 sequence of the naturally highly pAg/BTN3A1-reactive cl5 clone (δ2-G115cl5) (Supplementary table 1).1 The δ2-G115 length mutants and δ2-G115cl3 showed a slightly reduced binding of VHH 6H4 compared to δ2-G115WT and δ2-G115cl5 (Fig. 5A). However, and more importantly, no significant difference was observed in the ability of VHH 6H4 to trigger activation as determined by CD69 expression on the JurMa cells expressing the modified Vγ9Vδ2-TCRs as compared to the JurMa cells expressing the Vγ9Vδ2-TCR-G115WT (Fig. 5B). Thus, donor sequence variations in δ2-CDR3 that are known to impact pAg recognition, do not substantially affect the capacity of the 7D12-5GS-6H4 bispecific VHH construct to trigger Vγ9Vδ2-T cell activation and function.
Figure 5.
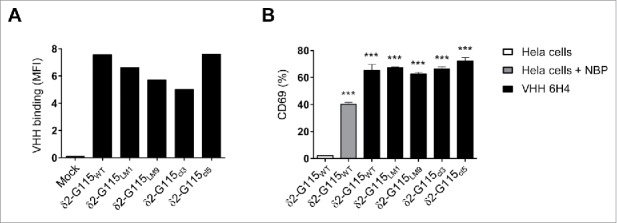
The anti-Vγ9Vδ2-TCR specific VHH 6H4 efficiently activates Vγ9Vδ2-T cells harbouring δ2-CDR variations. (A) Indicated JurMa transductants were incubated with 500 nM VHH 6H4 and bound VHH was assessed by flow cytometry. Mean fluorescence intensity (MF) of VHH bound to the cells is depicted. A representative figure of n = 3 experiments is shown. (B) Indicated JurMa transductants were cultured with HeLa cells (negative control, white), NBP-pretreated HeLa cells (positive control, grey) or plate bound (wells coated with 500 nM) VHH 6H4 (black). After 24 hrs, the activation status of the cells was determined by assessing CD69 expression on the cells by flow cytometry. Indicated significant differences are relative to values of δ2-G115WT cells stimulated with HeLa cells. A representative figure of triplicate samples (mean ± SEM) of n = 3 experiments is shown. p-Values were calculated with a one-way ANOVA and Bonferroni's post-hoc test (*** indicates p<0.001). Abbreviations: aminobisphosphonates (NBP).
7D12-5GS-6H4 enhances the Vγ9Vδ2-T cell-mediated inhibition of tumor outgrowth in vivo
We next assessed the effect of the 7D12-5GS-6H4 VHH on the outgrowth of human EGFR-overexpressing tumors in vivo. Immunodeficient BRGS mice were engrafted with SW480Gluc tumor cells carrying a KRASG12V-mutation and transduced to stably express Gaussia luciferase (Gluc). Expression of Gluc allowed real-time monitoring of viable primary and metastatic tumor cell load and response to treatment using bioluminescence imaging (BLI).49 At days 1, 4, and 7, mice were treated with either PBS, cetuximab, Vγ9Vδ2-T cells, or Vγ9Vδ2-T cells in combination with 7D12-5GS-6H4 (Fig. 6A). At day 35, BLI clearly demonstrated that mice treated with the combination of Vγ9Vδ2-T cells and 7D12-5GS-6H4 had an overall lower tumor burden compared to the mice treated with PBS, cetuximab or Vγ9Vδ2-T cells alone (Fig. 6B and C). Most importantly, and in accordance with the BLI data, mice treated with the combination of Vγ9Vδ2-T cells and 7D12-5GS-6H4 had a significantly improved overall survival compared with mice treated with either PBS, cetuximab or Vγ9Vδ2-T cells alone (Fig. 6D). These results confirm the expected treatment failure with cetuximab of KRAS mutated colorectal cancer cells and demonstrate that EGFR-mediated tumor targeting by 7D12-5GS-6H4 enhances the Vγ9Vδ2-T cell-mediated inhibition of tumor outgrowth in vivo, even for cetuximab resistant (i.e. KRAS mutated) tumors.
Figure 6.

The 7D12-5GS-6H4 bispecific VHH inhibits tumor growth in vivo. Immunodeficient BRGS mice grafted with SW480Gluc cells were treated with PBS (control group; (A), cetuximab (500 µg i.p.; group (B), Vγ9Vδ2-T cells (1*107 i.v.; group (C) or Vγ9Vδ2-T cells and 7D12-5GS-6H4 VHH (1*107and 1 µg, respectively, both i.v.; group (D) at days 1, 4 and 7. IL-2 (10,000 U, i.p.) was administered on days 1, 4, 7, 10, and 14 to the groups receiving Vγ9Vδ2-T cells. A) A schematic overview of the treatment schedule. B and C) Bioluminescence imaging at day 35 of 4 mice per treatment group. B) Heat map indicating the sites and relative level of tumor cell activity in individual mice. Red squares indicate the image field used for quantification analysis. C) Quantified bioluminescence signal measured per mouse expressed as the measured radiance normalized to the number of pixels, time and angle of imaging. Shown are means ± SEM of n = 4 mice per group. p-Values were calculated with a unpaired T-test (* indicates p<0.05). D) Kaplan-Meier analyses of mouse survival, n = 6 mice per group. p-Values were calculated with a Mantel-Cox test (* indicates p<0.05, ** indicates p<0.01 and *** indicates p<0.001). Abbreviations: Gly4Ser (GS).
Discussion
Vγ9Vδ2-T cells have a unique combination of features that make them highly promising for use in cancer immunotherapy, i.e. the recognition of ligands exclusively exposed by stressed or altered cells in an MHC-independent manner, a rapid innate-like response, the ability to induce efficient target cell lysis via multiple routes (Fas/FasL and perforin pathway) against a wide variety of tumor targets, the induction of dendritic cell maturation, and even efficient antigen presentation to αβ-T cells.22,25,26 Several attempts have been made to clinically exploit Vγ9Vδ2-T cell activation in cancer patients but results thus far lack consistency.27,28 This is likely related to the absence of a specific trigger for the activated Vγ9Vδ2-T cells to home to and infiltrate tumor sites.
Here, we explored whether the antitumor activity of Vγ9Vδ2-T cells could be enhanced and directed to the tumor by using a bispecific VHH construct that would allow Vγ9Vδ2-T cell accumulation and activation specifically at the tumor site. As a model tumor antigen we selected EGFR, which is a key factor in epithelial malignancies as its activity enhances tumor growth, invasion, and metastasis.50,51 Agents aimed at EGFR inhibition, such as anti-EGFR mAbs competing for ligand binding (e.g. cetuximab and panitumumab) and EGFR-specific tyrosine kinase inhibitors (TKI; e.g. erlotinib or gefitinib), are currently registered treatments for various advanced-stage epithelial cancers, including non-small-cell lung cancer, colorectal cancer, pancreatic cancer, and head and neck squamous cell carcinoma.52 Treatment with these agents is related to improved progression free and overall survival, though the overall efficacy is generally limited and frequently restricted to certain patient subsets, leaving ample room for improvement.52
From a set of 20 Vγ9Vδ2-TCR specific VHHs generated from immunized llamas and selected by phage display, we selected the Vδ2-specific VHH 6H4 on the grounds that it consistently induced Vγ9Vδ2-T cell activation and since targeting the Vδ2-chain would be more specific for Vγ9Vδ2-T cells than targeting the Vγ9-chain, as in general Vγ9+Vδ2− γδ-T cells are more abundant than Vγ9−Vδ2+ γδ-T cells in the human peripheral blood.17,41 As bivalent formats of the Vδ2-specific VHH 6H4 already induced striking activation of Vγ9Vδ2-T cells in the absence of target cells, probably due to the crosslinking of the TCRs, and this was expected to result in systemic activation of Vγ9Vδ2-T cells when applied therapeutically, we decided to use the monovalent VHH 6H4 for incorporation in the bispecific VHH in order to minimize the chances of non-specific and tumor unrelated activation upon systemic administration. By joining an anti-EGFR VHH (7D12) to the 6H4 VHH, we created a bispecific VHH construct targeting both EGFR and the Vγ9Vδ2-TCR. The generated 7D12-5GS-6H4 bispecific VHH induced strong activation and degranulation of Vγ9Vδ2-T cells resulting in potent lysis of EGFR expressing tumors at picomolar concentrations in an EGFR and Vγ9Vδ2-TCR dependent fashion. Previously, we determined that VHH 7D12 inhibited EGFR phosphorylation and pathway activation by binding to EGFR domain III, thus preventing its conformational change to an active state.42,43 Importantly, integration of anti-EGFR VHH 7D12 into the bispecific format did not alter its ability to inhibit EGF-induced signaling in EGFR overexpressing cancer cells. Moreover, and in contrast to the currently available anti-EGFR mAb therapies which are mainly effective through the inhibition of EGFR signaling52, the 7D12-5GS-6H4 bispecific VHH construct described here also induced efficient Vγ9Vδ2-T cell-mediated lysis of both colon cancer cell lines and primary colon cancer cells carrying common KRAS or BRAF mutations. Mutations such as these in the proto-oncogenes of the RAS family (e.g. KRAS, NRAS, HRAS) and BRAF frequently occur in e.g. colorectal, pancreatic and lung cancers, which together account for a major proportion of cancer cases. This often makes tumors resistant to the currently available anti-EGFR therapies (e.g. mAbs and TKI)39,40 and leads to poor prognosis53,54. Our data demonstrate that 7D12-5GS-6H4 has a dual mechanism of action by combining the inhibition of EGF-induced signaling (involved in tumor survival, growth and metastasis) with the direct induction of tumor cell lysis. The effective and superior anti-tumor effect of the bispecific VHH construct was confirmed using mice xenograft experiments. Mice grafted with human EGFR-overexpressing KRAS-mutated tumor cells that were treated with the bispecific VHH in combination with Vγ9Vδ2-T cells showed significant reduction of tumor outgrowth and improved overall survival. These are promising results that suggest that the bispecific 7D12-5GS-6H4 VHH construct may also inhibit growth of EGFR+ tumors in patients independent of the RAS or BRAF mutation status of the tumor and might thereby allow a more widespread applicability of EGFR-targeted treatments by bypassing the need for RAS and BRAF mutation analyses.55 Moreover, the data presented here provide a proof of concept for Vγ9Vδ2-T cell targeted therapy for a broad range of tumor types, which may be facilitated by simply exchanging the anti-EGFR VHH for VHHs targeting various other tumor antigens.
Interestingly, the ability to overcome the therapeutic barrier posed by RAS mutations was also noted for bispecific T cell engagers (BiTEs) wherein the scFv variable domains of cetuximab or panitumumab were fused to a scFv against CD3 expressed by T cells.56 However, though some BiTEs have induced clinical responses, limitations of this approach include the requirement for continuous infusions of the drug due to its short serum half-life time and the fact that it targets CD3 which is expressed by all T cells including immunosuppressive T cells such as Tregs.29 Tregs predominate in the tumor microenvironment, actively suppress the activation and proliferation of effector T cells and are related to unfavorable prognosis.30 For this reason, antibody-based constructs designed to exclusively trigger immune cells with a pro-inflammatory function, such as Vγ9Vδ2-T cells, may be preferable over the targeting of CD3.31
In a recently published preclinical study the antitumor efficacy of Vγ9Vδ2-T cells was explored through the use of a tribody targeting the γ9-chain of the Vγ9Vδ2-TCR and the tumor antigen Her2/neu. This (Her2)2xVγ9 tribody efficiently lysed Her2/neu overexpressing pancreatic cells in vitro and in mouse xenografts.57 Although this nearly full-sized (∼100kD) antibody approach underscores the potential of tumor-targeted Vγ9Vδ2-T cell-based immunotherapies, more specific targeting of the Vγ9Vδ2-T cell population can be achieved using an antibody (fragment) directed to Vδ2 compared to Vγ9, as in our bispecific VHH. Furthermore, the development and use of whole mouse mAbs has several limitations including the mispairing of heavy and light chains and the risk of developing human-anti-mouse antibodies (HAMA) in patients which leads to antibody neutralization and adverse events in the form of a cytokine release syndrome.29,36 These limitations can be overcome by the use of VHHs. VHH are low immunogenic because they share high homology with human VH genes and are devoid of an Fc-region.29,33,36 Because of the single domain nature of VHHs, pairing issues do not apply. This advantage in combination with their small size and the fact that they do not require post-translational modifications, allow VHHs to be easily produced in bacteria or yeast, which remain the most cost- and time-efficient production systems to date.36,58,59 VHHs are known for their high stability at elevated temperature and pH, providing them with enhanced solubility and making them less prone to aggregation.36,60 The small size of VHHs (∼30 kDa for a bispecific VHH) also facilitates deep tumor tissue penetration compared to larger sized antibody constructs, but like other small antibody fragments (such as BiTEs) this is also associated with a short serum half-life time due to fast renal clearance. This can be circumvented by fusion of the VHH to a serum albumin binding VHH.29,38,42,61
As anti-EGFR mAb therapy can be complicated by (generally well manageable) skin toxicity as a result of mAb binding to EGFR expressed on keratinocytes, it was encouraging to see only minor keratinocyte lysis when keratinocytes were cultured with Vγ9Vδ2-T cells in the presence of the 7D12-5GS-6H4 bispecific VHH. Although preclinical tests such as this do not necessarily predict safety in patients, these results are encouraging with regards to the future clinical exploration of this particular bispecific VHH construct.
In conclusion, we here describe the development of a bispecific VHH construct with a dual mechanism of action, combining ligand deprivation crucial for tumor cell proliferation and survival with the efficient and exclusive lysis of EGFR expressing tumor cells by conserved immune effector Vγ9Vδ2-T cells. Since EGFR is a widely expressed and clinically validated tumor antigen, a large patient group could benefit from this therapy. This group can be even broadened by the fact that, in contrast to currently available anti-EGFR therapies, the effectiveness of this therapy will not be influenced by downstream mutations in e.g. RAS or BRAF and, due to the monomorphic nature of the Vγ9Vδ2-TCR this immunotherapeutic approach requires no further individualization. Furthermore, as recently VHHs directed to various other tumor antigens have been developed62 and continue to be developed, these can easily be exchanged for the anti-EGFR VHH enabling future Vγ9Vδ2-T cell targeted therapy for a broad range of tumor types.
Materials & methods
Cell lines
HeLa, A431, HT29, Colo320 and SW480 cell lines were obtained from ATCC and, as well as Her1463 cultured in DMEM+, i.e. Dulbecco's Modified Eagle's Medium (Lonza, catalog #BE12-614 F) supplemented with 10% (v/v) fetal calf serum (FCS) (HyClone GE Healthcare, catalog #SV30160.03), 100 IU/mL sodium penicillin, 100 μg/mL streptomycin sulfate and 2.0 mM L-glutamine (Life Technologies, catalog #10378-016).
The SW480 cell line was stably transduced with lentivirus carrying the Gaussia Luciferase (Gluc) and Cerulean Fluorescent Protein (CFP) genes (LV-CFP-Gluc)64, kindly provided by Tom Würdinger (VU University medical center (VUmc), Amsterdam, NL), to generate the SW480Gluc cell line. CFP positive SW480Gluc cells were sorted and used for tumor injection in mice when CFP expression was >95% as determined by flow cytometry.
JY cells and Jurkat transductants32 were cultured in IMDM+, i.e. Iscove's modified Dulbecco's medium (Lonza, catalog #BE12-722 F) supplemented with 10% (v/v) FCS, 0.05 mM β-mercaptoethanol, 100 IU/mL sodium penicillin, 100 μg/mL streptomycin sulfate and 2.0 mM L-glutamine. JurMa cell lines transduced with Vγ9Vδ2-TCR-G115WT and δ2-CDR3 variants, were generated as previously described1 and cultured in IMDM+.
Primary cells
Keratinocytes were isolated from human adult skin as described previously.65 In brief, epidermal sheets were separated from dermis by incubation with Dispase II (Roche, catalog #04942078001) overnight at 4 °C. Subsequently, keratinocytes were isolated from the epidermis by a 10 minute 0.125% trypsin incubation (HyClone GE Healthcare, catalog # SH3004201) and seeded per 3*106 cells in keratinocyte culture medium on 9-cm-diameter tissue culture dishes coated with 0.5 μg/cm2 human placental collagen IV (Sigma-Aldrich, catalog #C5533). Keratinocyte culture medium consisted of Dulbecco's Modified Eagle's Medium and Ham's F12 (Invitrogen, catalog #21765-029) in a 3:1 ratio, supplemented with 10% (v/v) FCS, 100 IU/mL sodium penicillin, 100 μg/mL streptomycin sulfate, 2.0 mM L-glutamine, 1 μmol/L hydrocortisone (Sigma-Aldrich, catalog #H0888), 1 μmol/L isoproterenol hydrochloride (Sigma-Aldrich, catalog #I6504), 0.09 μmol/L insulin (Sigma-Aldrich, catalog #I5500), and 2 ng/ml human keratinocyte growth factor (Sigma-Aldrich, catalog #K1757). All cell lines were maintained at 37 °C with 5% CO2 in a humidified atmosphere and tested mycoplasma negative.
Colon cancer tissues from patients participating in trials conducted at the VU University medical center were collected and processed after written informed consent as described previously66. In brief, tumor material was washed, cut into small fragments and digested mechanically using collagenase A (Roche Diagnostics, The Netherlands) based growth medium. Single cell suspensions were obtained by passing the digested material through a 45μm sterile filter after which the samples were frozen under controlled conditions in liquid nitrogen. Before use, the colon cancer samples were thawed, dead cells were removed by a ficoll based density gradient separation and resuspended in DMEM+. The mutational status of KRAS exon 2/3/4, NRAS exon 2/3/4 and BRAF exon 15 was assessed by a high resolution melting (HRM) assay followed by Sanger sequencing of HRM-PCR products with an aberrant melt curve, essentially as described previously.67,68
Flow cytometry and monoclonal antibodies
FITC-labeled anti-TCR Vδ2 (catalog #555738), FITC-labeled anti-IFN-γ (catalog #554700), FITC-labeled anti-CD69 (catalog #347823), PE-labeled anti-CD107a (catalog #555801), PE-labeled anti-CD25 (catalog #55542), PE-labeled pan γδ-TCR (catalog #333141), APC-labeled anti-CD25 (catalog #340907), and 7-AAD (catalog #559925) were obtained from BD Biosciences. PerCP-labeled anti-TCR Vδ2 (catalog #331410), PE-labeled anti-TCR Vγ9 (catalog #331308) and APC-labeled anti-TCR Vγ9 (catalog #331310) were from Biolegend. RPE-labeled goat-anti-mouse F(ab’)2 fragment (catalog #R0480) was obtained from Dako. Anti-Myc tag mAb clone 4A6 (catalog #05-724) was obtained from Merck Millipore and anti-Myc tag mAb clone 9E10 was produced in-house. Alexa488-labeled cetuximab was a kind gift of Rens Braster and Yvette van Kooyk (VUmc, Amsterdam, NL). All stainings for flow cytometry were performed in PBS supplemented with 0.1% BSA and 0.02% sodium-azide. Intracellular IFN-γ production was determined by adding GolgiPlug to the cell culture for the final 4 hrs of the experiment. Cells were fixed and permeabilized with the Fixation/Permeabilization Solution Kit from BD Biosciences(catalog #555028) and stained with anti-IFN-γ mAb.
Stained cells were measured with a FACS Calibur or LSRFortessa (BD Biosciences) and analyzed with CellQuest (BD Biosciences) or Kaluza software (Beckman-Coulter).
Generation of donor-derived Vγ9Vδ2-T and pan γδ-T cell lines
Healthy donor-derived Vγ9Vδ2-T cells were isolated, expanded and cultured as described previously.32 In brief, Vγ9Vδ2-T cells were isolated by magnetic-activated cell sorting from PBMC using FITC-labeled anti-TCR Vδ2 or PE-labeled anti-TCR Vγ9 mAb in combination with anti-mouse IgG MicroBeads (Miltenyi Biotec, catalog #130-048-401). Purified Vγ9Vδ2-T cells were stimulated weekly with irradiated and NBP-pretreated (100 μM Pamidronate for 2–3 hrs, Teva Pharmachemie, catalog #12J08RD) human mature monocyte derived dendritic cells or an irradiated feeder mix (PBMC of 2 healthy human donors and Epstein Barr Virus transformed B cells with addition of 50 ng/ml PHA). Vγ9Vδ2-T cells were used for experiments when Vγ9+Vδ2+-TCR expression was >90% and CD25 expression was <40% as determined by flow cytometry.
Vγ9Vδ2-T cell lines were cultured in Yssels+, i.e. Yssels medium69 supplemented with 1% human AB serum (Cellect, MP Biomedicals, catalog #2931949), 50 U/ml rhIL-2 (Proleukin, Novartis), 0.05 mM β-ME, 100 IU/mL sodium penicillin, 100 μg/mL streptomycin sulfate and 2.0 mM L-glutamine. Vγ9Vδ2-T cell lines and tumor target cell lines were cultured in IMDM+ medium during experiments. The Vγ9Vδ2-T cell lines were maintained at 37 °C with 5% CO2 in a humidified atmosphere and tested mycoplasma negative.
Vγ9+Vδ2+, Vγ9+Vδ2−, Vγ9+Vδ2+ and Vγ9−Vδ2− γδ-T cell lines were obtained from human PBMC as follows. PBMC were stained with PE-labeled pan γδ-TCR antibody and anti-mouse IgG MicroBeads by MACS isolation. This pan γδ-T cell line was expanded with a feeder mix and subsequently stained with FITC-labeled anti-TCR Vδ2 and PE-labeled anti-TCR Vγ9 antibodies to allow flow cytometric cell sorting of 4 separate populations (i.e. Vγ9−Vδ2+,Vγ9+Vδ2−, Vγ9+Vδ2+ and Vγ9−Vδ2− γδ-T cells).
Generation, production and purification of bivalent and bispecific VHHs
To generate bivalent or bispecific VHHs, genes of VHH 6H432, VHH 7D1242 or VHH R244 were PCR-amplified using Phusion High-Fidelity DNA Polymerase (New England Biolabs, catalog #M0530) and appropriate primers encoding the N- or C-terminal end of the VHH gene, a restriction endonuclease site, and a linker sequence (composed of Gly4Ser repeats). PCR products were purified by gel extraction using the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, catalog #740609), digested with restriction endonucleases to allow cloning into appropriate plasmids.
For in vitro experiments, PCR products were cloned to plasmid pMek219, verified by sequencing and produced in TG1 bacteria as described previously32. Produced VHH were derived from the bacterial periplasm by a PBS freeze-thawing step and purified by immobilized metal ion affinity chromatography (IMAC) using Talon resin (Clontech, catalog # 635504). VHH were eluted with 150 mM imidazole and dialyzed twice against PBS.
For in vivo experiments, PCR products were cloned to a modified version of the pFastBac I plasmid (Thermo Fisher Scientific) containing a Honeybee Melittin Signal Sequence (HMSS) and a C-terminal his-tag. Bacmid DNA and virus were essentially prepared according to the Bac-to-Bac manual (Thermo Fisher Scientific). Briefly, the pFastBac constructs were transformed into EMBACY cells70 and bacmid DNA was isolated. Sf9 insect cells were transfected with the bacmid DNA and the virus was subsequently amplified in a sf9 suspension culture. Collected virus was used to infect sf9 cells for protein expression. Medium containing the secreted proteins was harvested 3 days post infection and dialyzed 2 x against 25 mM HEPES pH 7.5 and 200 mM NaCl. Proteins were purified on a 5 ml HiTrap Ni2+ column, eluted with 200 mM imidazol in 25 mM HEPES pH 7.5, 200 mM NaCl. Proteins were further purified by size exclusion chromatography on a S75 16/60 Superdex column (GE Healthcare) equilibrated with PBS buffer. Fractions containing the proteins were pooled, concentrated and passed through a 0.22 µM filter.
The purity of produced protein was checked on a coomassie blue-stained protein gel before use.
Binding analysis of VHH
To determine the binding of VHH to cells, 5*104 Vγ9Vδ2-T cells, A431 cells, Jurkat or JurMa transductants were incubated with VHH at the indicated concentrations for 30 minutes. Bound VHH was detected with anti-Myc-tag antibody clone 4A6 and RPE-labeled goat-anti-mouse F(ab’)2 fragment by flow cytometry.
Functional analysis of monospecific, bivalent and bispecific VHH
To determine the effect of monovalent and bivalent Vγ9Vδ2-TCR specific VHH on Vγ9Vδ2-T cell activation, Vγ9Vδ2-TCR expressing cells were either cultured in the presence of plate bound or soluble VHHs. For plate bound conditions, wells of a 96-well flat-bottom culture plate (Greiner, catalog #655180) were coated with 4 µg/ml mouse-anti-Myc clone 9E10 in PBS overnight at 4 °C. Wells were washed three times with PBS and incubated for 2 hrs with the indicated concentrations of VHH in PBS. After the wells were again washed three times with PBS, 105 Vγ9Vδ2-T cells, 105 Vγ9Vδ2-TCR-G115 transduced JurMa cells or 2.5*105 PBMC were added per well in a final volume of 200 µl IMDM+. For soluble VHH conditions, the indicated concentration of VHH in PBS was added to 105 Vγ9Vδ2-T cells in a 96-well flat-bottom culture plate. For control conditions, Vγ9Vδ2-T cells were co-cultured with untreated (negative control) or NBP-treated (100 μM Pamidronate for 2–3 hrs, positive control) HeLa cells in a 1:1 ratio. Cells were cultured in a final volume of 200 µl IMDM+ for 24 hrs.
To determine the effect of bispecific anti-EGFR-anti-Vγ9Vδ2-TCR VHH on Vγ9Vδ2-T cell activation, degranulation and target cell lysis, 5*104 target cells (A431, JY, HT29, Colo320, SW480 or primary colon cancer cells) were labeled with 40 nM CFSE (Sigma-Aldrich, catalog #21888) or 5 µM PBSE (Thermo Fisher Scientific, catalog #P10163), according to the manufacturer`s protocol and allowed to adhere for 4hrs in a 96-well flat bottom culture well. Vγ9Vδ2-T cells were incubated with the indicated concentrations of VHH for 1 hr at 4 °C, washed three times with PBS and added in a 1:1 ratio to the target cells and cultured for 24 hrs in a final volume of 200 µl IMDM+. In case of keratinocytes, primary keratinocytes were plated 2 days beforehand on collagen IV coated wells to obtain a viable cell pool at the start of the experiment. In case of primary colon cancer cells, these were not adhered to the plate before the addition of Vγ9Vδ2-T cells and cells were co-cultured for 4 hrs instead of 24 hrs. For the NBP-pretreated positive control, target cells were incubated with 100 μM Pamidronate during cell adherence and washed by a 3x PBS rinse before the addition of Vγ9Vδ2-T cells.
To determine degranulation of Vγ9Vδ2-T cells, anti-CD107a mAb and GolgiStop (BD Bioscience, catalog #554724) were added to the co-culture for the final 4 hrs of the experiment. At the end of the experiment, cells were harvested and stained with anti-Vδ2 and/or anti-Vγ9 mAb or CD3 mAb to identify Vγ9+Vδ2+-T cells. CD25 and CD69 expression on Vγ9Vδ2-T cells was determined with an anti-CD25 and anti-CD69 mAb, respectively. Cells were stained with 7-AAD according to the manufacturer`s protocol to distinguish lysed cells. mAbs bound to the cells and 7-AAD staining were analyzed by flow cytometry.
Inhibition of EGF-induced EGFR phosphorylation
Inhibition of EGF-induced EGFR phosphorylation was performed as described before.71 105 Her14 cells were seeded per well in a 12-wells plate in DMEM+ and allowed to adhere. After 8 hrs, the medium was replaced by DMEMmin, i.e. Dulbecco's Modified Eagle's Medium supplemented with 0.1% (v/v) FCS, 100 IU/mL sodium penicillin, 100 μg/mL streptomycin sulfate and 2.0 mM L-glutamine. The following day, cells were washed once with PBS, after which a mixture of the indicated VHHs was added to the cells in combination with 8 nM recombinant human EGF (Peprotech, catalog # AF-100-15) in DMEMmin for 15 minutes at 37 °C. Subsequently the cells were washed three times with ice-cold PBS and resuspended in 2x Laemmli protein sample buffer. Half of the sample was loaded and run on a SDS-PAGE gel and western blotted. Phosphorylated EGFR was detected with an anti-phosphoEGF Receptor (Tyr1068) polyclonal antibody and an anti-rabbit-HRP mAb (both from Cell Signaling Technology, catalog #2234 and #7074, respectively). Blots were stained with an anti-β-actin (Sigma-Aldrich, catalog #21888) and anti-mouse HRP mAb (Cell Signaling Technology, catalog #7076) to demonstrate that equal amounts of cell lysate were loaded on gel.
In vivo studies
Immunodeficient BRGS mice (BALB/c Rag2−/−Il2rg−/−SirpaNOD)72 were housed in isolators under pathogen-free conditions and randomly divided in 4 treatment groups (n = 6/group). At day 0, mice received an intravenous (i.v.) tail vein injection with 0.5*106 SW480Gluc cells. At days 1, 4 and 7 mice were treated with either a) 500 μg cetuximab i.p., b) 1*107 Vγ9Vδ2-T cells i.v., c) 1*107 Vγ9Vδ2-T cells in combination with 1 µg of the bispecific 7D12-5GS-6H4 VHH i.v., or d) an equal volume of sterile PBS i.v.. Mice that received Vγ9Vδ2-T cells were injected at days 1, 4, 7, 10 and 14 with 10,000 U human recombinant IL-2 i.p. to stimulate the proliferation of activated Vγ9Vδ2-T cells. Bioluminescence imaging (BLI) was performed at day 35 in 4 randomly selected mice from each study group. For this procedure, mice were anesthetized with inhalation anesthetics (isofluorane/oxygen) and injected i.v. retro-orbitally with 4 mg/kg coelenterazine (PJK GmbH, native-CTZ). BLI was recorded using an IVIS imaging system (PerkinElmer) and images were analysed using Living Image 4.0 software. BRGS mouse experiments were approved by the animal ethical committee of the Institut Pasteur (Reference # 2007–006), Paris, France, and validated by the French Ministry of Education and Research (Reference # 02162.01).
Statistical analyses
Statistical analyses were performed in GraphPad Prism version 5 (La Jolla, CA, USA). For in vitro analyses, a one-way or two-way ANOVA with a Bonferroni's post-hoc test was used as appropriate. For in vivo data analysis of BLI, an unpaired T-test was used. For the survival analysis, a Mantel-Cox test was used. All findings were considered significant when p-values were <0.05.
Supplementary Material
Acknowledgments
We would like to thank Taco Waaijman (Department of Dermatology, VUmc, Amsterdam, NL) for providing us with primary keratinocyte cultures, the Netherlands Cancer Institute Protein Facility (Amsterdam, NL) for expression and purification of the recombinant VHH proteins for in vivo experiments, Daniëlle Heideman (Department of Pathology, VUmc, Amsterdam, NL) for analysis of the RAS and BRAF mutation status of the primary colon cancers, and Ricardo Vos, Inge de Greeuw and Mariska Verlaan (VUmc, Amsterdam, NL) for technical assistance. This work was supported by grant VU 2010–4728 from the Dutch Cancer Society (KWF) to HJV. ZS and JK were supported by ZonMW 43400003 and VIDI-ZonMW 917.11.337, KWF UU 2010–4669, UU 2013–6426, UU 2014–6790, and UU 2015–7601, and Worldwide Cancer Research grants 10–0736 and 15–0049 to JK. JPD is supported by the Institut Pasteur, INSERM and the Ligue National Contre le Cancer. SLL and JV have been supported by the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7-PEOPLE-2012-ITN under REA grant agreement n° 317013 (NATURIMMUN consortium).
Disclosures
We would like to state that authors Renée de Bruin, Tanja de Gruijl, Henk Verheul and Hans van der Vliet have a patent on Vγ9Vδ2-TCR specific nanobodies. Hans van der Vliet is CSO/CMO of Lava Therapeutics. Author Jürgen Kuball reports grants from Pierre Fabre, Novartis, and Miltenyi Biotech. He is scientific co-founder and CSO of gadeta (www.gadeta.nl) and inventor of multiple patents on γδ TCR receptors and isolation strategies for engineered immune cells. The other authors have no financial conflicts of interest.
Authorship contributions
RB performed research, collected data, analyzed and interpreted data, performed statistical analysis, and wrote the manuscript
JV performed research, analyzed and interpreted data and wrote the manuscript
SL performed research, analyzed and interpreted data and performed statistical analysis and wrote the manuscript.
FS performed research, analyzed and interpreted data and wrote the manuscript
SLL performed research, analyzed and interpreted data and wrote the manuscript
RL performed research, analyzed and interpreted data and wrote the manuscript
AS performed research, analyzed and interpreted data and wrote the manuscript
ZS designed research, performed research and revised the manuscript critically
JK designed research, interpreted data and revised the manuscript critically
CM designed research and revised the manuscript critically
EH designed research and revised the manuscript critically
RR designed research and revised the manuscript critically
JD designed research and revised the manuscript critically
PBH designed research and revised the manuscript critically
HMWV designed research and revised the manuscript critically
TD designed research, analyzed and interpreted data, and wrote the manuscript
HJV designed research, analyzed and interpreted data, and wrote the manuscript
References
- 1.Gründer C, van Dorp S, Hol S, Drent E, Straetemans T, Heijhuurs S, Scholten K, Scheper W, Sebestyen Z, Martens A, et al. . γ9 and δ2CDR3 domains regulate functional avidity of T cells harboring γ9δ2TCRs. Blood. 2012;120:5153-62. doi: 10.1182/blood-2012-05-432427. PMID:23018643 [DOI] [PubMed] [Google Scholar]
- 2.Bouet-Toussaint F, Cabillic F, Toutirais O, Le Gallo M, Thomas de la Pintière C, Daniel P, Genetet N, Meunier B, Dupont-Bierre E, Boudjema K, et al. . Vgamma9Vdelta2 T cell-mediated recognition of human solid tumors. Potential for immunotherapy of hepatocellular and colorectal carcinomas. Cancer Immunol Immunother. 2008;57:531-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viey E, Fromont G, Escudier B, Morel Y, Da Rocha S, Chouaib S, Caignard A. Phosphostim-activated gamma delta T cells kill autologous metastatic renal cell carcinoma. J Immunol. 2005;174:1338-47. doi: 10.4049/jimmunol.174.3.1338. PMID:15661891 [DOI] [PubMed] [Google Scholar]
- 4.Sicard H, Al Saati T, Delsol G, Fournié JJ. Synthetic phosphoantigens enhance human Vgamma9Vdelta2 T lymphocytes killing of non-Hodgkin's B lymphoma. Mol Med. 2001;7:711-22. PMID:11713370 [PMC free article] [PubMed] [Google Scholar]
- 5.Corvaisier M, Moreau-Aubry A, Diez E, Bennouna J, Mosnier J-F, Scotet E, Bonneville M, Jotereau F. V 9 V 2 T Cell Response to Colon Carcinoma Cells. J Immunol. 2005;175:5481-8. doi: 10.4049/jimmunol.175.8.5481. PMID:16210656 [DOI] [PubMed] [Google Scholar]
- 6.Argentati K, Re F, Serresi S, Tucci MG, Bartozzi B, Bernardini G, Provinciali M. Reduced number and impaired function of circulating gamma delta T cells in patients with cutaneous primary melanoma. J Invest Dermatol. 2003;120:829-34. doi: 10.1046/j.1523-1747.2003.12141.x. PMID:12713589 [DOI] [PubMed] [Google Scholar]
- 7.Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, Nieda M. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer. 2011;105:778-86. doi: 10.1038/bjc.2011.293. PMID:21847128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thedrez A, Lavoué V, Dessarthe B, Daniel P, Henno S, Jaffre I, Levêque J, Catros V, Cabillic F. A quantitative deficiency in peripheral blood Vγ9Vδ2 cells is a negative prognostic biomarker in ovarian cancer patients. PLoS One. 2013;8:e63322. doi: 10.1371/journal.pone.0063322. PMID:23717410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cordova A, Toia F, La Mendola C, Orlando V, Meraviglia S, Rinaldi G, Todaro M, Cicero G, Zichichi L, Donni PL, et al. . Characterization of human γδ T lymphocytes infiltrating primary malignant melanomas. PLoS One. 2012;7:e49878. doi: 10.1371/journal.pone.0049878. PMID:23189169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wistuba-Hamprecht K, Martens A, Haehnel K, Geukes Foppen M, Yuan J, Postow MA, Wong P, Romano E, Khammari A, Dreno B, et al. . Proportions of blood-borne Vδ1+ and Vδ2+ T-cells are associated with overall survival of melanoma patients treated with ipilimumab. Eur J Cancer. 2016;64:116-26. doi: 10.1016/j.ejca.2016.06.001. PMID:27400322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chien YH, Jores R, Crowley MP. Recognition by gamma/delta T cells. Annu Rev Immunol. 1996;14:511-32. doi: 10.1146/annurev.immunol.14.1.511. PMID:8717523 [DOI] [PubMed] [Google Scholar]
- 12.Steer HJ, Lake RA, Nowak AK, Robinson BWS. Harnessing the immune response to treat cancer. Oncogene. 2010;29:6301-13. doi: 10.1038/onc.2010.437. PMID:20856204 [DOI] [PubMed] [Google Scholar]
- 13.Constant P, Davodeau F, Peyrat MA, Poquet Y, Puzo G, Bonneville M, Fournié JJ. Stimulation of human gamma delta T cells by nonpeptidic mycobacterial ligands. Science. 1994;264:267-70. doi: 10.1126/science.8146660. PMID:8146660 [DOI] [PubMed] [Google Scholar]
- 14.Tanaka Y, Morita CT, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature. 1995;375:155-8. doi: 10.1038/375155a0. PMID:7753173 [DOI] [PubMed] [Google Scholar]
- 15.Fournié JJ, Bonneville M. Stimulation of gamma delta T cells by phosphoantigens. Res Immunol. 1996;147:338-47. doi: 10.1016/0923-2494(96)89648-9. PMID:8876063 [DOI] [PubMed] [Google Scholar]
- 16.Gober H-J, Kistowska M, Angman L, Jenö P, Mori L, De Libero G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med. 2003;197:163-8. doi: 10.1084/jem.20021500. PMID:12538656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fowler DW, Bodman-Smith MD. Harnessing the power of Vδ2 cells in cancer immunotherapy. Clin Exp Immunol. 2015;180:1-10. doi: 10.1111/cei.12564. PMID:25469879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jomaa H, Feurle J, Lühs K, Kunzmann V, Tony HP, Herderich M, Wilhelm M. Vgamma9/Vdelta2 T cell activation induced by bacterial low molecular mass compounds depends on the 1-deoxy-D-xylulose 5-phosphate pathway of isoprenoid biosynthesis. FEMS Immunol Med Microbiol. 1999;25:371-8. PMID:10497868 [DOI] [PubMed] [Google Scholar]
- 19.Harly C, Guillaume Y, Nedellec S, Peigné C-M, Mönkkönen H, Mönkkönen J, Li J, Kuball J, Adams EJ, Netzer S, et al. . Key implication of CD277/butyrophilin-3 (BTN3 A) in cellular stress sensing by a major human γδ T-cell subset. Blood. 2012;120:2269-79. doi: 10.1182/blood-2012-05-430470. PMID:22767497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palakodeti A, Sandstrom A, Sundaresan L, Harly C, Nedellec S, Olive D, Scotet E, Bonneville M, Adams EJ. The molecular basis for modulation of human Vγ9Vδ2 T cell responses by CD277/butyrophilin-3 (BTN3 A)-specific antibodies. J Biol Chem. 2012;287:32780-90. doi: 10.1074/jbc.M112.384354. PMID:22846996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandstrom A, Peigné C-M, Léger A, Crooks JE, Konczak F, Gesnel M-C, Breathnach R, Bonneville M, Scotet E, Adams EJ. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vγ9Vδ2 T cells. Immunity. 2014;40:490-500. doi: 10.1016/j.immuni.2014.03.003. PMID:24703779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol. 2013;13:88-100. doi: 10.1038/nri3384. PMID:23348415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sebestyen Z, Scheper W, Vyborova A, Gu S, Rychnavska Z, Schiffler M, Cleven A, Chéneau C, van Noorden M, Peigné C-M, et al. . RhoB Mediates Phosphoantigen Recognition by Vγ9Vδ2 T Cell Receptor. Cell Rep. 2016; 15(9):1973-85. doi: 10.1016/j.celrep.2016.04.081. PMID:27210746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wrobel P, Shojaei H, Schittek B, Gieseler F, Wollenberg B, Kalthoff H, Kabelitz D, Wesch D. Lysis of a broad range of epithelial tumour cells by human gamma delta T cells: involvement of NKG2D ligands and T-cell receptor- versus NKG2D-dependent recognition. Scand J Immunol 66:320-8. doi: 10.1111/j.1365-3083.2007.01963.x. PMID:17635809 [DOI] [PubMed] [Google Scholar]
- 25.Caccamo N, Dieli F, Meraviglia S, Guggino G, Salerno A. Gammadelta T cell modulation in anticancer treatment. Curr Cancer Drug Targets. 2010;10:27-36. doi: 10.2174/156800910790980188. PMID:20088797 [DOI] [PubMed] [Google Scholar]
- 26.Spada FM, Grant EP, Peters PJ, Sugita M, Melián A, Leslie DS, Lee HK, van Donselaar E, Hanson DA, Krensky AM, et al. . Self-recognition of CD1 by gamma/delta T cells: implications for innate immunity. J Exp Med. 2000;191:937-48. doi: 10.1084/jem.191.6.937. PMID:10727456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fournié J-J, Sicard H, Poupot M, Bezombes C, Blanc A, Romagné F, Ysebaert L, Laurent G. What lessons can be learned from γδ T cell-based cancer immunotherapy trials? Cell Mol Immunol. 2013;10:35-41. doi: 10.1038/cmi.2012.39. PMID:23241899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Latha TS, Reddy MC, Durbaka PVR, Rachamallu A, Pallu R, Lomada D. γδ T Cell-Mediated Immune Responses in Disease and Therapy. Front Immunol. 2014;5:571. doi: 10.3389/fimmu.2014.00571. PMID:25426120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lameris R, de Bruin RCG, Schneiders FL, van Bergen en Henegouwen PMP, Verheul HMW, de Gruijl TD, van der Vliet HJ. Bispecific antibody platforms for cancer immunotherapy. Crit Rev Oncol Hematol. 2014;92:153-65. doi: 10.1016/j.critrevonc.2014.08.003. PMID:25195094 [DOI] [PubMed] [Google Scholar]
- 30.Wilke CM, Wu K, Zhao E, Wang G, Zou W. Prognostic significance of regulatory T cells in tumor. Int J cancer. 2010;127:748-58. PMID:20473951 [DOI] [PubMed] [Google Scholar]
- 31.Koristka S, Cartellieri M, Theil A, Feldmann A, Arndt C, Stamova S, Michalk I, Töpfer K, Temme A, Kretschmer K, et al. . Retargeting of human regulatory T cells by single-chain bispecific antibodies. J Immunol. 2012;188:1551-8. doi: 10.4049/jimmunol.1101760. PMID:22184723 [DOI] [PubMed] [Google Scholar]
- 32.de Bruin RCG, Lougheed SM, van der Kruk L, Stam AG, Hooijberg E, Roovers RC, van Bergen En Henegouwen PMP, Verheul HMW, de Gruijl TD, van der Vliet HJ. Highly specific and potently activating Vγ9Vδ2-T cell specific nanobodies for diagnostic and therapeutic applications. Clin Immunol. 2016;169:128-138. doi: 10.1016/j.clim.2016.06.012. PMID:27373969 [DOI] [PubMed] [Google Scholar]
- 33.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446-8. doi: 10.1038/363446a0. PMID:8502296 [DOI] [PubMed] [Google Scholar]
- 34.Muyldermans S. Single domain camel antibodies: current status. J Biotechnol. 2001;74:277-302. PMID:11526908 [DOI] [PubMed] [Google Scholar]
- 35.van der Linden RH, Frenken LG, de Geus B, Harmsen MM, Ruuls RC, Stok W, de Ron L, Wilson S, Davis P, Verrips CT. Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim Biophys Acta. 1999;1431:37-46. doi: 10.1016/S0167-4838(99)00030-8. PMID:10209277 [DOI] [PubMed] [Google Scholar]
- 36.Harmsen MM, De Haard HJ. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol. 2007;77:13-22. doi: 10.1007/s00253-007-1142-2. PMID:17704915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Transue TR, De Genst E, Ghahroudi MA, Wyns L, Muyldermans S. Camel single-domain antibody inhibits enzyme by mimicking carbohydrate substrate. Proteins. 1998;32:515-22. doi: 10.1002/(SICI)1097-0134(19980901)32:4%3c515::AID-PROT9%3e3.0.CO;2-E. PMID:9726420 [DOI] [PubMed] [Google Scholar]
- 38.Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier T, de Haard HJ, Leemans CR, van Dongen GAMS. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol Cancer Ther. 2008;7:2288-97. doi: 10.1158/1535-7163.MCT-07-2384. PMID:18723476 [DOI] [PubMed] [Google Scholar]
- 39.Markus Morkel PRHBCS. Similar but different: distinct roles for KRAS and BRAF oncogenes in colorectal cancer development and therapy resistance. Oncotarget. 2015;6:20785. doi: 10.18632/oncotarget.4750. PMID:26299805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Semrad TJ, Kim EJ. Molecular testing to optimize therapeutic decision making in advanced colorectal cancer. J Gastrointest Oncol. 2016;7:S11-20. PMID:27034809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kabelitz D, Kalyan S, Oberg H-H, Wesch D. Human Vδ2 versus non-Vδ2 γδ T cells in antitumor immunity. Oncoimmunology. 2013;2:e23304. doi: 10.4161/onci.23304. PMID:23802074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roovers RC, Vosjan MJWD, Laeremans T, el Khoulati R, de Bruin RCG, Ferguson KM, Verkleij AJ, van Dongen GAMS, van Bergen en Henegouwen PMP. A biparatopic anti-EGFR nanobody efficiently inhibits solid tumour growth. Int J Cancer. 2011;129:2013-24. doi: 10.1002/ijc.26145. PMID:21520037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmitz KR, Bagchi A, Roovers RC, van Bergen en Henegouwen PMP, Ferguson KM. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure. 2013;21:1214-24. doi: 10.1016/j.str.2013.05.008. PMID:23791944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frenken LG, van der Linden RH, Hermans PW, Bos JW, Ruuls RC, de Geus B, Verrips CT. Isolation of antigen specific llama VHH antibody fragments and their high level secretion by Saccharomyces cerevisiae. J Biotechnol. 2000;78:11-21. doi: 10.1016/S0168-1656(99)00228-X. PMID:10702907 [DOI] [PubMed] [Google Scholar]
- 45.Fakih M, Vincent M. Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr Oncol. 2010;17 Suppl 1:S18-30. PMID:20680104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balagula Y, Garbe C, Myskowski PL, Hauschild A, Rapoport BL, Boers-Doets CB, Lacouture ME. Clinical presentation and management of dermatological toxicities of epidermal growth factor receptor inhibitors. Int J Dermatol. 2011;50:129-46. doi: 10.1111/j.1365-4632.2010.04791.x. PMID:21244375 [DOI] [PubMed] [Google Scholar]
- 47.Troiani T, Napolitano S, Vitagliano D, Morgillo F, Capasso A, Sforza V, Nappi A, Ciardiello D, Ciardiello F, Martinelli E. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin Cancer Res. 2014;20:3775-86. doi: 10.1158/1078-0432.CCR-13-2181. PMID:24812410 [DOI] [PubMed] [Google Scholar]
- 48.Murcia O, Juárez M, Hernández-Illán E, Egoavil C, Giner-Calabuig M, Rodríguez-Soler M, Jover R. Serrated colorectal cancer: Molecular classification, prognosis, and response to chemotherapy. World J Gastroenterol. 2016;22:3516-30. doi: 10.3748/wjg.v22.i13.3516. PMID:27053844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chung E, Yamashita H, Au P, Tannous BA, Fukumura D, Jain RK. Secreted Gaussia Luciferase as a Biomarker for Monitoring Tumor Progression and Treatment Response of Systemic Metastases. PLoS One. 2009;4:e8316. doi: 10.1371/journal.pone.0008316. PMID:20016816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Normanno N, De Luca A Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2-16. doi: 10.1016/j.gene.2005.10.018. PMID:16377102 [DOI] [PubMed] [Google Scholar]
- 51.Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637-43. doi: 10.1016/S1357-2725(99)00015-1. PMID:10404636 [DOI] [PubMed] [Google Scholar]
- 52.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160-74. doi: 10.1056/NEJMra0707704. PMID:18337605 [DOI] [PubMed] [Google Scholar]
- 53.Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, Taylor G, Barrett JH, Quirke P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27:5931-7. doi: 10.1200/JCO.2009.22.4295. PMID:19884549 [DOI] [PubMed] [Google Scholar]
- 54.Wangefjord S, Sundström M, Zendehrokh N, Lindquist KE, Nodin B, Jirström K, Eberhard J. Sex differences in the prognostic significance of KRAS codons 12 and 13, and BRAF mutations in colorectal cancer: a cohort study. Biol Sex Differ. 2013;4:17. doi: 10.1186/2042-6410-4-17. PMID:24020794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Krieken JHJ Rouleau E, Ligtenberg MJL, Normanno N, Patterson SD, Jung A. RAS testing in metastatic colorectal cancer: advances in Europe. Virchows Arch. 2016;468:383-96. doi: 10.1007/s00428-015-1876-7. PMID:26573425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lutterbuese R, Raum T, Kischel R, Hoffmann P, Mangold S, Rattel B, Friedrich M, Thomas O, Lorenczewski G, Rau D, et al. . T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc Natl Acad Sci U S A. 2010;107:12605-10. doi: 10.1073/pnas.1000976107. PMID:20616015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oberg H-H, Peipp M, Kellner C, Sebens S, Krause S, Petrick D, Adam-Klages S, Röcken C, Becker T, Vogel I, et al. . Novel bispecific antibodies increase γδ T-cell cytotoxicity against pancreatic cancer cells. Cancer Res. 2014;74:1349-60. doi: 10.1158/0008-5472.CAN-13-0675. PMID:24448235 [DOI] [PubMed] [Google Scholar]
- 58.Demain AL, Vaishnav P. Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv 27:297-306. doi: 10.1016/j.biotechadv.2009.01.008. PMID:19500547 [DOI] [PubMed] [Google Scholar]
- 59.Sanchez-Garcia L, Martín L, Mangues R, Ferrer-Miralles N, Vázquez E, Villaverde A. Recombinant pharmaceuticals from microbial cells: a 2015 update. Microb Cell Fact. 2016;15:33. doi: 10.1186/s12934-016-0437-3. PMID:26861699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barthelemy PA, Raab H, Appleton BA, Bond CJ, Wu P, Wiesmann C, Sidhu SS. Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human VH domains. J Biol Chem. 2008;283:3639-54. doi: 10.1074/jbc.M708536200. PMID:18045863 [DOI] [PubMed] [Google Scholar]
- 61.Kijanka M, Warnders F-J, El Khattabi M, Lub-de Hooge M, van Dam GM, Ntziachristos V, de Vries L, Oliveira S, van Bergen En Henegouwen PMP. Rapid optical imaging of human breast tumour xenografts using anti-HER2 VHHs site-directly conjugated to IRDye 800CW for image-guided surgery. Eur J Nucl Med Mol Imaging. 2013;40:1718-29. doi: 10.1007/s00259-013-2471-2. PMID:23778558 [DOI] [PubMed] [Google Scholar]
- 62.Kijanka M, Dorresteijn B, Oliveira S, van Bergen en Henegouwen PM. Nanobody-based cancer therapy of solid tumors. Nanomedicine. 2015;10:161-74. doi: 10.2217/nnm.14.178. PMID:25597775 [DOI] [PubMed] [Google Scholar]
- 63.Honegger AM, Dull TJ, Felder S, Van Obberghen E Bellot F, Szapary D, Schmidt A, Ullrich A, Schlessinger J. Point mutation at the ATP binding site of EGF receptor abolishes protein-tyrosine kinase activity and alters cellular routing. Cell. 1987;51:199-209. doi: 10.1016/0092-8674(87)90147-4. PMID:3499230 [DOI] [PubMed] [Google Scholar]
- 64.Wurdinger T, Badr C, Pike L, de Kleine R, Weissleder R, Breakefield XO, Tannous BA. A secreted luciferase for ex vivo monitoring of in vivo processes. Nat Methods. 2008;5:171-3. doi: 10.1038/nmeth.1177. PMID:18204457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waaijman T, Breetveld M, Ulrich M, Middelkoop E, Scheper RJ, Gibbs S. Use of a collagen-elastin matrix as transport carrier system to transfer proliferating epidermal cells to human dermis in vitro. Cell Transplant. 2010;19:1339-48. doi: 10.3727/096368910X507196. PMID:20525428 [DOI] [PubMed] [Google Scholar]
- 66.Veluchamy JP, Spanholtz J, Tordoir M, Thijssen VL, Heideman DAM, Verheul HMW, de Gruijl TD, van der Vliet HJ. Combination of NK Cells and Cetuximab to Enhance Anti-Tumor Responses in RAS Mutant Metastatic Colorectal Cancer. PLoS One. 2016;11:e0157830. doi: 10.1371/journal.pone.0157830. PMID:27314237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heideman DAM, Thunnissen FB, Doeleman M, Kramer D, Verheul HM, Smit EF, Postmus PE, Meijer CJLM, Meijer GA, Snijders PJF. A panel of high resolution melting (HRM) technology-based assays with direct sequencing possibility for effective mutation screening of EGFR and K-ras genes. Cell Oncol. 2009;31:329-33. PMID:19759413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heideman DAM, Lurkin I, Doeleman M, Smit EF, Verheul HM, Meijer GA, Snijders PJF, Thunnissen E, Zwarthoff EC. KRAS and BRAF mutation analysis in routine molecular diagnostics: comparison of three testing methods on formalin-fixed, paraffin-embedded tumor-derived DNA. J Mol Diagn. 2012;14:247-55. doi: 10.1016/j.jmoldx.2012.01.011. PMID:22425762 [DOI] [PubMed] [Google Scholar]
- 69.Yssel H, De Vries JE Koken M, Van Blitterswijk W Spits H. Serum-free medium for generation and propagation of functional human cytotoxic and helper T cell clones. J Immunol Methods. 1984;72:219-27. doi: 10.1016/0022-1759(84)90450-2. PMID:6086760 [DOI] [PubMed] [Google Scholar]
- 70.Sari D, Gupta K, Thimiri Govinda Raj DB, Aubert A, Drncová P, Garzoni F, Fitzgerald D, Berger I. The MultiBac Baculovirus/Insect Cell Expression Vector System for Producing Complex Protein Biologics. Adv Exp Med Biol. 2016;896:199-215. doi: 10.1007/978-3-319-27216-0_13. PMID:27165327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roovers RC, Laeremans T, Huang L, De Taeye S, Verkleij AJ, Revets H, de Haard HJ, van Bergen en Henegouwen PMP. Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR Nanobodies. Cancer Immunol Immunother. 2007;56:303-17. doi: 10.1007/s00262-006-0180-4. PMID:16738850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, Strick-Marchand H, de Geus SJ, Pouw SM, Böhne M, Voordouw A, et al. . Functional CD47/signal regulatory protein alpha (SIRPα) interaction is required for optimal human T-and natural killer-(NK) cell homeostasis in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.