

ABSTRACT
Purpose: We conducted a phase I vaccine trial to determine safety, toxicity, and immunogenicity of autologous Langerhans-type dendritic cells (LCs), electroporated with murine tyrosinase-related peptide-2 (mTRP2) mRNA in patients with resected AJCC stage IIB, IIC, III, or IV (MIa) melanoma. Experimental Design: Nine patients received a priming immunization plus four boosters at three week intervals. Vaccines comprised 10 × 106 mRNA-electroporated LCs, based on absolute number of CD83+CD86brightHLA-DRbrightCD14neg LCs by flow cytometry. Initial vaccines used freshly generated LCs, whereas booster vaccines used viably thawed cells from the cryopreserved initial product. Post-vaccination assessments included evaluation of delayed-type hypersensitivity (DTH) reactions after booster vaccines and immune response assays at one and three months after the final vaccine. Results: All patients developed mild DTH reactions at injection sites after booster vaccines, but there were no toxicities exceeding grade 1 (CTCAE, v4.0). At one and three months post-vaccination, antigen-specific CD4 and CD8 T cells increased secretion of proinflammatory cytokines (IFN-γ, IL-2, and TNF-α), above pre-vaccine levels, and also upregulated the cytotoxicity marker CD107a. Next-generation deep sequencing of the TCR-V-β CDR3 documented fold-increases in clonality of 2.11 (range 0.85–3.22) for CD4 and 2.94 (range 0.98–9.57) for CD8 T cells at one month post-vaccines. Subset analyses showed overall lower fold-increases in clonality in three patients who relapsed (CD4: 1.83, CD8: 1.54) versus non-relapsed patients (CD4: 2.31, CD8: 3.99). Conclusions: TRP2 mRNA-electroporated LC vaccines are safe and immunogenic. Responses are antigen-specific in terms of cytokine secretion, cytolytic degranulation, and increased TCR clonality, which correlates with clinical outcomes.
KEYWORDS: Langerhans-type dendritic cell, clinical immunology, cancer vaccines, phase I trial, melanoma
Introduction
The advent of immune checkpoint inhibitors targeting anti-cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell-death protein 1 (PD-1) has revolutionized the treatment landscape for advanced melanoma, with a subset of patients achieving long-term survival.1,2 A significant number of patients do not benefit, however, due to the low frequency or absence of tumor antigen-specific cytotoxic T lymphocytes (CTLs) at baseline.3-6 Priming patients with a vaccine to induce or augment the pool of CTLs against a tumor is one strategy to complement or synergize with checkpoint blockade and/or other therapeutic modalities to improve clinical responses.
Dendritic cells (DCs) are key regulators of immunity, and translational studies have demonstrated the safety and feasibility of DC-based immunization in patients with cancer.7 These trials have achieved mixed success with regard to immune and objective clinical responses,7 which may well depend on the type of DC used. Most DC cancer vaccines have used monocyte-derived DCs (moDCs), but Langerhans-type DCs (LCs) derived from CD34+ hematopoietic progenitor cells (HPCs) are superior at stimulating antigen-specific CTLs against tumor antigens in vitro.8-11 LCs, but not moDCs, can also stimulate CTLs de novo against a self-differentiation tumor antigen like Wilms tumor 1 (WT1) in vitro by an IL15Rα/IL15-dependent mechanism.10 Clinical trial data have also shown greater efficacy of LC-containing vaccines,12 as well as greater tetramer reactivity stimulated by LCs when compared with moDCs.13
Electroporation of DCs with mRNA encoding tumor-associated antigens effectively induces T cell responses in vitro and in vivo.14,15 Antigen loading by electroporation is more efficient than peptide pulsing14 and avoids the risk of genome integration seen with retroviral transgenes.16,17 Electroporation with full-length mRNA also facilitates DC presentation of a more diverse array of multiple peptides for presentation, obviating the need to predetermine class I and II MHC restrictions. This in turn supports a more robust immune response than achieved by single class I MHC-peptide pulsing.
Tumor-infiltrating lymphocytes from patients with melanoma recognize tyrosinase-related peptide 2 (TRP2), a melanosomal differentiation antigen.18 In addition, vaccination with highly homologous xenogeneic antigen can augment immune responses against differentiation antigens, like TRP219,20.
We performed a phase I vaccine study to determine the safety and toxicity of autologous LCs electroporated with murine TRP2 mRNA in patients with resected American Joint Committee on Cancer (AJCC) stage IIB, IIC, III, or IV (MIa) melanoma. Post-vaccination studies included evaluation of delayed type hypersensitivity (DTH) reactions and immune response assessments. Clinical outcomes were also monitored after completion of vaccines.
Results
LCs electroporated with TPR2 mRNA are safe and well-tolerated
Figure 1 outlines the treatment schedule. Tables 1a and 1b summarize the characteristics of the ten patients enrolled in this study, which comprised an equal number of male and female participants, with a median age of 56 (range, 34 to 69). None of the patients received adjuvant therapy, with a median interval of 3.3 months (range 1.9 to 11.9) from surgical resection to start of vaccination. Nine patients completed all five scheduled vaccines. One patient received three vaccines before being withdrawn from the study due to disease recurrence. All patients developed some degree of erythema, induration, and mild pruritus at the injection sites after booster vaccines (Figure S1). No patient developed toxicities greater than grade 1, possibly or probably related to these vaccines.
Figure 1.
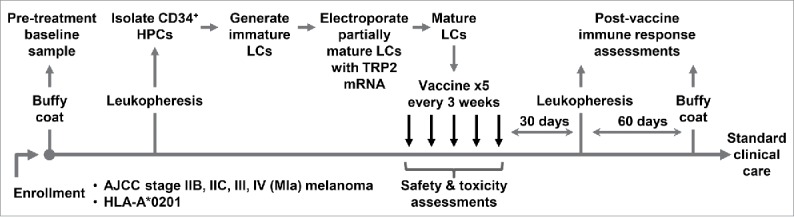
Study schema.
Table 1a.
Patient demographics and disease characteristics.
| Patient | Age | Gender | Stage | KPS | Disease status at study entry | LDH at study entry | Disease status at EOS (3 months after last vaccine) | Interval from surgical resection to first vaccine* (months) | RFS from surgical resection* (months) | OS from surgical resection* (months) | F/u from last vaccine* (months) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 54 | M | IIIA | 100 | NED | 177 | NED | 2.9 | 67.3 | 67.3 | 61.6 |
| 2 | 65 | F | IIB | 100 | NED | 186 | NED | 3.2 | 55.7 | 55.7 | 49.8 |
| 3 | 62 | M | IV (MIc) | 100 | NED | 161 | RelapsedA | 5.1 | 8.3 | 55.8 | 47.9 |
| 4 | 69 | M | IIIB | 100 | NED | 137 | RelapsedB | 3.3 | 7.4 | 51.6 | 45.4 |
| 5 | 58 | F | IIB | 100 | NED | 212 | NED | 3.1 | 50.5 | 50.5 | 44.6 |
| 6 | 47 | M | IIIC | 100 | NED | 158 | NED | 11.9 | 57.7 | 57.7 | 43.7 |
| 7 | 41 | M | IIIB | 100 | NED | 152 | NED | 4.9 | 44.6 | 44.6 | 36.9 |
| 8 | 46 | F | IIC | 100 | NED | 172 | RelapsedC | 1.9 | 5.2 | 23.4 | 18.7 |
| 9 | 59 | F | IIIA | 100 | NED | 187 | NED | 3.6 | 37.6 | 37.6 | 31.3 |
| 10 | 34 | F | IIIB | 100 | NED | 103 | RelapsedD | 2.7 | 4 | 45.6 | 41.6 |
| Median: | 56 | M 50% | — | 100 | — | 167 | NED: 6 | 3.3 | 41.1 | 51.1 | 44.2 |
| Range: | (34–69) | F 50% | — | — | — | (103–212) | Relapsed: 4 | (1.9–11.9) | (4–67.3) | (23.4–67.3) | (18.7–61.6) |
Table 1b.
Relapsed patients (A-D from Table 1a).
| Patient | Time of relapse | Site(s) of relapse | Treatment | Treatment duration | Response | RFS from start of treatment* (months) | RFS from end of treatment* (months) |
|---|---|---|---|---|---|---|---|
| A | 1 mo after last vaccine | Lung, spleen | Ipilimumab + nivolumab | 14 months | CR | 44 | 30 |
| 8.3 mo from resection | |||||||
| B | 1 mo after last vaccine | Liver, spleen | Ipilimumab + nivolumab | 4 months | CR | 43 | 39.3 |
| 7.4 mo from resection | |||||||
| C | 1 mo after last vaccine | Bone | Multiple including ipilimumab & pembrolizumab | 16 months total | POD | NA | NA |
| 5.2 mo from resection | |||||||
| D | On study after 3rd vaccine | Skin, CNS | Lirilumab + nivolumab | One dose | CR | 40.2 | 40.2 |
| 4 mo from resection |
CR = complete response; EOS = end of study; F/u = follow-up; NA = not applicable; NED = no evidence of disease; POD = progression of disease; RFS = recurrence-free survival
cutoff date 07/01/2017, approximately 31 months from the end of the study
Post-vaccination T-cell subset phenotype analysis
Unmanipulated PBMCs were analyzed by flow cytometry to assess for changes in lymphocyte composition at one and three months after completion of vaccines. There were no significant fold-changes from pre-vaccine baseline in the levels of naïve (CCR7+CD45ROneg), central memory (CCR7+CD45RO+), effector (CCR7negCD45ROneg), or effector memory (CCR7negCD45RO+) CD8 T cells (Figure 2A). CD8 T cells maintained relatively static inhibitory receptor expression at one and three months after vaccination (Figure 2B). The post-vaccination trends of CD4 T-cell subsets and inhibitory receptor expression were similar to those of CD8 T cells (data not shown). Inducible co-stimulator (ICOS) expression by CD4 and CD8 T cells increased 2.51- and 1.99-fold, respectively, at one month, indicating T-cell activation (Figure 2C). Vaccines did not significantly alter regulatory T cell (CD3+CD4+CD25brightCD127neg) to CD3+CD8+CD25+ effector T cell ratios (Figure 2D).
Figure 2.

CD8 T cell subset analysis and Treg content after LC vaccines. PBMCs were analyzed by flow cytometry to assess fold-changes in lymphocyte composition at one and three months post-vaccination, compared with pre-vaccine baseline. (A) CD8 T-cell subsets: naïve (TN; CCR7+CD45ROneg), central memory (TCM; CCR7+CD45RO+), effector (TEF; CCR7negCD45ROneg), and effector memory (TEF; CCR7negCD45RO+). (B) Expression of inhibitory receptors (CTLA-4, LAG-3, PD-1, and TIM-3) by CD8 T cells. (C) ICOS expression by CD4 and CD8 T cells. (D) Regulatory T cell (Treg; CD3+CD4+CD25bright CD127neg) to CD3+CD8+CD25+ effector T cell ratios. For all panels (A-D), pooled data (mean ± SD; P = NS) from seven patients are shown.
LC vaccines induce CD4 and CD8 T-cell cytokine secretion and expression of activation epitopes
To amplify antigen-specific reactivity from the pool of responder T cells, PBMCs obtained at one and three months post-vaccination underwent two rounds of restimulation in vitro with freshly generated LCs electroporated with TRP2 mRNA. Compared with pre-vaccine levels, CD4 T-cell secretion of IFN-γ, IL-2, and TNF-α increased 6.15-, 2.27-, and 2.25-fold, respectively, at one-month post-vaccination, and remained 3-, 2.92-, and 2.3-fold elevated, respectively, at three-months post-vaccination (Figure 3A). CD4 T cells also upregulated CD107a 1.84- and 1.7-fold, as a marker of degranulation, and Mip-1-β 3.47- and 2.67-fold, as a marker of activation, at the one and three-month time points, respectively (Figure 3A). Similar to CD4 T cell responses, antigen-specific CD8 T-cell secretion of IFN-γ, IL-2, and TNF-α increased 1.71-, 2.05-, and 1.37-fold, respectively, at one-month post-vaccination, and further increased 2.28-, 3-, and 1.61-fold, respectively, at three-months post-vaccination (Figure 3B). CD8 T cells upregulated CD107a 1.55- and 1.6-fold, and Mip-1-β 1.72- and 1.24-fold, at the one and three-month time points, respectively (Figure 3B). On an individual basis, cytokine secretion and activation epitope markers in both the CD4 and CD8 T-cell compartments increased in all patients, including all five patients (#5–9) with sufficient cells for replicate analyses showing positive T-cell responses, defined as greater than 2 standard deviation increases over pre-vaccine levels (Figure 3C-E). A limited assessment of tetramer-specific reactivity against two different TRP2 residues showed no change (data not shown). Available cell yields precluded evaluation of antigen-specific CTL activity in killing assays.
Figure 3.
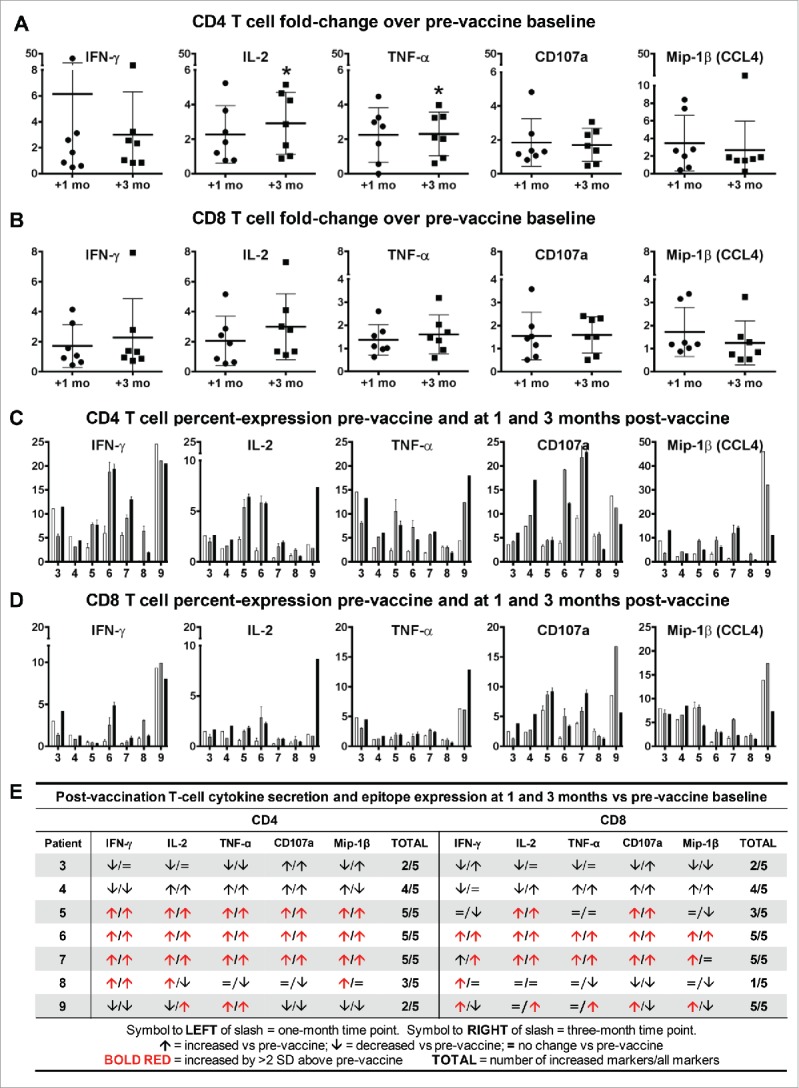
LC vaccines stimulate CD4 and CD8 T-cell cytokine secretion and activation epitope expression in patients with melanoma. Responses against TRP2 mRNA-electroporated LCs were measured at approximately one and three months after completion of vaccines, and compared with pre-vaccine levels. (A) CD4 and (B) CD8 T-cell pro-inflammatory cytokine (IFN-γ, IL-2, and TNF-α) secretion and activation epitope (CD107a and Mip-1-β) expression, fold-change. For panels A and B, pooled data (triplicate means ± SEM; *P < .05) from seven patients are shown. (C) CD4 and (D) CD8 T-cell pro-inflammatory cytokine secretion and activation epitope expression, percent-expression for individual patients pre-vaccine (white bar) and at one (gray bar) and three (black bar) months after completion of vaccines. For panels C and D, Y-axis indicates percent-expression for each marker, and X-axis indicates patient number. For patients #3 and 4, single data points are shown. For patients #5–9, pooled data (triplicate means ± SD) are shown. (E) Composite cytokine secretion and activation epitope expression profile of individual patients.
CD4 and CD8 T-cell clonality increases after vaccination with TRP2 mRNA-electroporated LCs
Clonality, as defined in Methods, is a composite measure of the abundance (frequency) and diversity (uniqueness) of the TCR-V-β repertoire.21 CD4 and CD8 T cells isolated from PBMCs collected at one month after vaccination underwent next-generation deep sequencing of the TCR-V-β CDR3. Pre- to post-vaccination clonality increased in all cases, except one CD4 and one CD8 pair (different subjects); and clonality was significantly higher in the CD8 (P = 0.0097) but not in the CD4 compartment (P = 0.1084) (Figure 4A). Clonality increased 2.11-fold (range 0.85–3.22) for CD4 T cells and 2.94-fold (range 0.98–9.57) for CD8 T cells (Figure 4B). Subset analysis showed a trend in overall lower fold-increases in clonality in patients who relapsed (CD4: 1.83, CD8: 1.54; n = 3) versus non-relapsed patients (CD4: 2.31, CD8: 3.99; n = 4) (Figure 4C). CD8 T cells showed higher clonal frequencies than CD4 T cells one month after LC vaccines (Figure 4D). The mean frequencies in the top clones of post-vaccine samples were higher in both CD4 and CD8 T cells but did not reach statistical significance due to the small sample size (Figure 4D).
Figure 4.
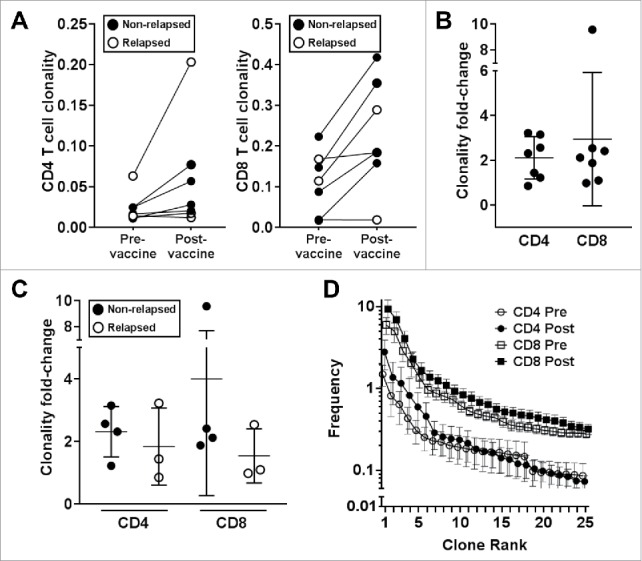
T-cell clonality increases after vaccination with TRP2 mRNA-electroporated LCs. Next-generation deep sequencing of the TCR-V-β CDR3 was performed on CD4 and CD8 T cells isolated from PBMCs obtained from seven patients at one month post-vaccination and compared for changes from pre-vaccine levels. (A) CD4 (left panel; P = 0.1084) and CD8 (right panel; P = 0.0097) T-cell clonal frequency for individual patients. (B) CD4 and CD8 T-cell clonality fold-change. (C) CD4 and CD8 T cell clonality fold-change comparing patients with no evidence of disease (non-relapsed; n = 4) with patients who relapsed (n = 3) after completing vaccines. (D) Mean frequency of each of the top 25 CD4 and CD8 T-cell clones before and after vaccination. Pooled data (mean ± SEM; P = NS).
Clinical outcomes
Patient outcomes are summarized in Tables 1a/1b and Figure 5. At a median follow-up of 18.7 to 61.6 months, six of the nine patients who completed the study remain free of disease recurrence, with a median of 51.1 months (range, 34.6 to 67.3) from initial surgical resection. The other three patients who completed the study (patients 3, 4, and 8) relapsed at approximately one month post-vaccines, which was a median of 7.4 months (range, 5.2 to 8.3) from initial surgical resection. An additional participant (patient 10) received three vaccines before withdrawing from the study due to cutaneous and CNS metastases. All relapsed patients (patients 3, 4, 8, and 10) subsequently received checkpoint inhibitor-based therapy. Patient 8 died of progressive disease, while the others (patients 3, 4, and 10) are in complete remission, with a median recurrence-free survival of 39.3 months (range, 30 to 40.2) from the completion of last therapy.
Figure 5.
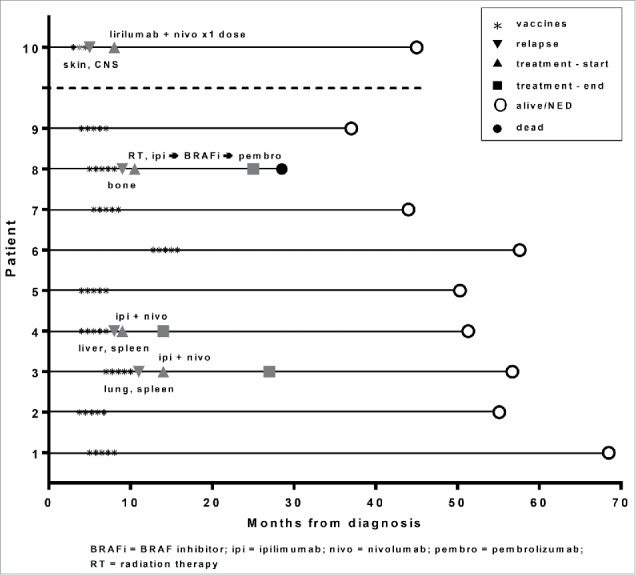
Clinical outcomes. The clinical status timeline of the nine patients who completed treatment on study, from diagnosis to vaccinations, relapse and treatment (if applicable), and follow-up. Also shown is a tenth patient who received 3/5 planned vaccines but relapsed and was removed from the study. Site of relapse/metastasis is indicated under the down-pointing triangle. Treatment is indicated over the up-pointing triangle.
Discussion
This phase I clinical trial demonstrates the safety and immunogenicity of mRNA-electroporated LC vaccines. Aside from mild DTH reactions at injection sites after booster vaccines, there were no toxicities. Post-vaccination immune responses detected enhanced CD4 and CD8 T-cell activation and proinflammatory cytokine secretion, as well as increased T-cell clonality that correlated with clinical outcomes.
The vaccine formulation in this study combined the superior potency of LCs8-11 with electroporation of full-length tumor antigen mRNA14,15,22 to stimulate a more diverse T-cell response. In addition to promoting CD4 and CD8 T-cell activation by traditional phenotypic measures, mRNA-electroporated LCs induced greater CD4 and CD8 T-cell clonality in patients who remained free of disease. Greater T-cell diversity after vaccination correlates with clinical benefit,23,24 indicating an advantage of oligoclonal expansion of several T-cell clones over a more restricted clonal profile. In addition, low baseline levels of TCR diversity associate with inferior outcomes after treatment with ipilimumab,25 and maintenance of high-frequency TCR clones after CTLA-4 blockade correlates with superior outcomes.26 In this study, LC vaccines also induced CD4 and CD8 T-cell expression of ICOS, which predicts response to ipilimumab therapy.27,28 These findings, as well as emerging preclinical29-31 and clinical32-34 data, support the sequential administration of vaccines followed by immune checkpoint blockade to augment T-cell responses and optimize antitumor immunity.
T-cell tetramer reactivity against two well-defined TRP2 epitopes restricted by HLA-A*0201 did not increase after vaccination. This was not unexpected in the setting of antigen presentation by LCs after electroporation with mRNA encoding full-length antigen, where expansion of antigen-specific CTLs recognizing other immunologically relevant TRP2 epitopes is supported by the TCR clonality results. This highlights another challenge in monitoring immune therapy, however, which is the need for more robust assessment tools that increase sensitivity for detecting subtle but significant changes after an intervention.
Four patients in this trial relapsed, three after completing vaccines and one while on study. All received checkpoint inhibitor-based therapy, and three are alive with no evidence of disease, including one patient who had CNS metastases. In a previous study comparing melanoma peptide-pulsed moDCs and LCs in patients with AJCC stage III/IV melanoma,13 clinical responses, although not the primary endpoint of the trial, were observed solely in the LC recipients. For the current study, pre/post-treatment samples from relapsed patients were not available for analyses. Thus, whether the LC vaccines altered local or systemic T-cell subset composition in patients treated with checkpoint inhibitors for relapsed disease is an important unknown that merits future investigation.
Investigators recently discovered that a neoantigen-based DC vaccine increases the breadth and diversity of antigen-specific T-cell responses in melanoma.35 Another study has found induction of DC-mediated antitumor immunity after tumor mRNA delivery to DCs in vivo with nanoparticles.36 These studies and our results with LCs underscore the potential of DC-based vaccines to stimulate immune responses against cancer. For LCs, we acknowledge that resource limitations at most centers to collect and process CD34+ HPCs, as well as the labor, time, and cost of manufacturing LCs in vitro, restrict their exportability for broad clinical application. Strategies aimed at activating LCs directly in vivo to circumvent the production process ex vivo or other approaches to modify more simply derived DC subsets to mimic the superior antigen presentation and accessory functions of LCs for CTL stimulation are underway.
This pilot study highlights the feasibility of immunizing patients with mRNA-electroporated LCs to induce measurable cellular immune responses. Future progress in vaccine development for melanoma and other malignancies will require the identification of rational combinations and sequencing of immune-directed agents to improve clinical outcomes.
Materials and methods
Patients and clinical trial design
Patients expressing HLA-A*0201 with resected AJCC stage IIB, IIC, III, or IV (MIa) melanoma were enrolled in a phase I clinical trial (NCT01456104) to test safety and toxicity and to assess immune responses stimulated by TRP-2 mRNA-electroporated LCs. The use of influenza matrix peptide as a positive control was the only reason necessitating HLA-A*0201-expressing patients. The trial was conducted at Memorial Sloan Kettering Cancer Center (MSKCC), approved by the Institutional Review and Privacy Board of Memorial Hospital/MSKCC and the Food and Drug Administration, and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Patients received a total of five vaccines, comprising a priming immunization followed by four boosters at 3-week intervals (Figure 1). Target enrollment was nine patients, with the trial designed to identify a promising agent for further study from a large pool of candidates, while minimizing the total number of patients studied.37 Of note, a total of ten patients accrued to the trial, including an extra patient enrolled to replace a patient who relapsed before completing vaccines. For this trial, the vaccine was considered promising if more than four out of the nine patients had an immunologic response. For cytokine secretion and activation epitope expression, a positive T-cell response was defined as greater than 2 standard deviations over pre-vaccine replicates. The decision rule aimed to identify an agent that produced an immunologic response rate greater than 50%. The error rates of missing a promising agent or selecting a non-promising agent were set to be at most 10%. The decision rule and error rates were devised using a historical database of vaccine trials performed at MSKCC.37 For clonality, a positive T-cell response required an increased above baseline without respect to degree of increase, as there are no validated metrics to provide cutoffs for determining significance.
Vaccine production and administration
Generation of human LCs
G-CSF-elicited CD34+ HPCs collected from patients by leukopheresis were used to generate LCs with media, media supplements, and cytokines, exactly as published.8 In brief, CD34+ HPCs were cultured in serum-free X-VIVO 15, supplemented with GM-CSF, TGF-β, and TNF-α, to which c-kit-ligand and FLT-3-ligand were added for the initial 5–6 days of a 10–12-day culture. LCs underwent terminal maturation with TNF-α, IL-1β, IL-6, and prostaglandin E2. All cultures used autologous plasma or serum, avoiding the introduction of any allogeneic or xenogeneic plasma or serum (e.g., fetal calf serum).
Several epitopes distinguish LCs from other conventional DC subtypes. LCs are consistently negative for CD11b, express e-cadherin before exposure to terminal maturation stimuli, and express CD207/Langerin primarily by less mature forms and with apparent asynchrony during differentiation in culture.8 LCs also express the chemokine receptor, CCR3, independent of maturation status,38 and upregulate the chemokine receptor, CCR7, with maturation (Figure S2).
Production of in vitro–transcribed mRNA
A pING plasmid expression vector containing the gene for murine TRP2 under the control of a T7 promoter was used to synthesize TRP2 mRNA. The murine TRP2 cDNA was cloned in the laboratory of Dr. Alan Houghton (MSKCC). The plasmid was propagated in Max Efficiency DH5-α competent cells (Invitrogen) and purified using a Plasmid Maxi Kit (QIAGEN). For TRP2 mRNA transcription, the plasmid was linearized with Hind III (New England Biolabs) before mRNA transcription in vitro with T7 RNA polymerase (mMessage mMachine T7 kit; Ambion). Production of full-length capped mRNA was confirmed by agarose gel electrophoresis, and mRNA concentration was determined by spectrophotometry.
Electroporation of LCs
Partially-matured LCs were electroporated with TRP2 mRNA on day 12–13.22 LCs were harvested, washed twice, and resuspended in OptiMEM (Gibco, Invitrogen) at 15 × 106 cells/ml. 200 μL of cell suspension were then mixed with 40 μg mRNA and electroporated in a 2 mm gap cuvette at 700 V for two pulses, using a BTX ECM 830 square-wave electroporator (BTX Harvard). After electroporation, cells were immediately transferred to culture and terminally matured with inflammatory cytokines for 24–48 hours.8
Control antigen-loading of LCs
Synthetic influenza matrix peptide (fluMP58-66 GILGFVFTL; Multiple Peptide Systems) served as a positive control for HLA-A*0201-restricted responses. Peptide (1 μM) was added to terminally maturing LCs in cultures overnight at 37°C.13
Vaccine administration
Cells were washed and met release criteria (Table S1A) before proceeding with vaccination. Each vaccine consisted of 10 × 106 mRNA-electroporated LCs/vaccine, based on the absolute number of CD83+CD86brightHLA-DRbrightCD14neg LCs by flow cytometry (Table S1B). The initial vaccines used freshly generated LCs, whereas booster vaccines used viably thawed cells from the initial product, which was cryopreserved using controlled-rate freezing followed by storage in liquid nitrogen. Vaccines were administered as ten x 0.1 ml, deep intradermal injections, five of which were given at each of two sites adjacent to axillary or inguinofemoral nodes. Areas of prior lymph node resections were excluded.
Clinical assessments
Patients were monitored with serial vital sign measurements once every 15 minutes for one hour after vaccine administration. Surrounding erythema and induration at vaccine injections sites were measured for DTH reactions 48 hours after each booster vaccine. Toxicities were graded according to Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. For disease status monitoring, patients were followed for melanoma recurrence according to National Comprehensive Cancer Network (NCCN) Guidelines for Melanoma.
Post-vaccination immune response assessments
Response assessments used peripheral blood mononuclear cells (PBMCs) collected at pre-vaccine baseline and then approximately one and three months after the fifth and final vaccine. Two of the nine patients (#1 and #2) who received all five vaccines were inevaluable for immune response assessments because of inadequate cell yields.
Flow cytometric analysis
PBMCs were incubated with fluorochrome-conjugated antibodies and analyzed on an LSRFortessa flow cytometer (Becton Dickinson). FITC-, PE-, PE-Texas Red-, ECD-, APC-, PE-Cy5-, PE-Cy7-, PerCP-Cy5.5-, Pacific Blue-, and AF700-conjugated mouse anti-human mAbs included anti-CD3, anti-CD4, anti-CD8, anti-CD14, anti-CD25, anti-CD45RA, anti-CD45RO, anti-CD80, anti-CD86, anti-CTLA-4, anti-HLA-DR, anti-IL-2, anti-CD83, anti-Mip-1-β (Beckman Coulter), anti-CD127, anti-IFN-γ, anti-LAG-3, anti-PD-1, anti-TNF-α (eBioscience), anti-CCR7, and anti-TIM-3 (R&D Systems). LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Life Technologies) was used to exclude dead cells. Gates were set for collection and analysis of at least 50,000 live events. Data were analyzed with FlowJo software (TreeStar).
T-cell subset analysis and intracellular cytokine secretion
PBMCs were restimulated with TRP2 mRNA-electroporated autologous LCs generated from the same CD34+ HPCs used to make patient vaccines. Responder PBMCs were resuspended in 10% autologous serum in complete RPMI-1640 and plated at a 1:10 ratio of LCs:PBMCs in a 96-round bottomed well plate (Costar). Cultures were supplemented with recombinant human IL-2 (10 IU/ml; Chiron, Emeryville, CA) and IL-15 (10 ng/ml; R&D Systems). After 6–7 days, fresh LCs were added for a second round of stimulation in the same culture conditions, followed by harvesting of cells for staining and analysis by flow cytometry. T-cell subsets and intracellular cytokine levels were compared for fold-changes over pre-vaccine baseline. Two HLA-A*0201-restricted TRP2 tetramers, one PE-labeled for TRP2 residues 180–188 and one APC-labeled for TRP2 residues 360–368 (TC Matrix), were used for tetramer analyses.
T-cell receptor variable beta chain sequencing
PBMCs from the one-month post-vaccine time point were separated into CD4 and CD8 T-cell fractions using MS columns (Miltenyi Biotec), then cryopreserved and shipped to Adaptive Biotechnologies (Seattle, WA) for immunosequencing of the CDR3 of human TCR-V-β chains using the ImmunoSEQ™ Assay. Extracted genomic DNA was amplified in a bias-controlled multiplex PCR, followed by high-throughput sequencing. Sequences were collapsed and filtered to identify and quantitate the absolute abundance of each unique TCR-V-β CDR3 for further analysis.39-41
Statistics
For immune response assessments, replicate means from ≥3 independent experiments were averaged and SEM calculated as the measure of variability. Otherwise, SD was calculated for the replicate mean of a single representative experiment of at least three. Unpaired or paired t tests assessed differences in expression levels. Statistical significance required a P value less than 0.05. All statistical analyses were calculated using Prism 6 software (GraphPad).
Statistical Analyses of TCR-β sequencing results
Clonality was defined as 1- Peilou's eveness21 and was calculated on productive rearrangements by:
where pi is the proportional abundance of rearrangement i and N is the total number of rearrangements. Clonality values range from 0 to 1 and describe the shape of the frequency distribution. Clonality values approaching 0 indicate a very even distribution of frequencies, whereas values approaching 1 indicate an increasingly asymmetric distribution in which a few clones are present at high frequencies. Statistical analyses were performed in R version 3.2.
Supplementary Material
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgments
We thank the patients for participating in the clinical trial. We gratefully acknowledge the various contributions of membersof the Laboratory of Cellular Immunobiology to the development of this work. We also thank the nurses, advanced practice providers, research staff, and physicians of the Melanoma and Immunotherapeutics Service, the Blood Bank Donor Room, and the Cell Therapy Laboratory.
Supported by
This work was supported by R01-CA118974 from the National Cancer Institute, NIH (JWY), R21-CA119528 from the National Cancer Institute, NIH (JWY), and Swim Across America (JWY). This research was also funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
References
- 1.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al.. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol. 2015;33(17):1889-94. PMID:25667295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maio M, Grob JJ, Aamdal S, Bondarenko I, Robert C, Thomas L, Garbe C, Chiarion-Sileni V, Testori A, Chen TT, et al.. Five-Year Survival Rates for Treatment-Naive Patients With Advanced Melanoma Who Received Ipilimumab Plus Dacarbazine in a Phase III Trial. J Clin Oncol. 2015;33(10):1191-6. PMID:25713437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, Zheng L, Jr Diaz LA, Donehower RC, Jaffee EM, et al.. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36(7):382-9. PMID:23924790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibney GT, Kudchadkar RR, DeConti RC, Thebeau MS, Czupryn MP, Tetteh L, Eysmans C, Richards A, Schell MJ, Fisher KJ, et al.. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res. 2015;21(4):712-20. PMID:25524312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, Solt S, Dorman A, Wamwea A, Yager A, et al.. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2(7):616-31. PMID:24942756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan J, Adamow M, Ginsberg BA, Rasalan TS, Ritter E, Gallardo HF, Xu Y, Pogoriler E, Terzulli SL, Kuk D, et al.. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci U S A. 2011;108(40):16723-8. PMID:21933959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265-77. PMID:22437871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ratzinger G, Baggers J, de Cos MA, Yuan J, Dao T, Reagan JL, Münz C, Heller G, Young JW. Mature human Langerhans cells derived from CD34+ hematopoietic progenitors stimulate greater cytolytic T lymphocyte activity in the absence of bioactive IL-12p70, by either single peptide presentation or cross-priming, than do dermal-interstitial or monocyte-derived dendritic cells. J Immunol. 2004;173:2780-91(Erratum in J Immunol. 005; 174:3818). PMID:15294997 [DOI] [PubMed] [Google Scholar]
- 9.Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson-Snipes L, Briere F, Chaussabel D, Zurawski G, Palucka AK, et al.. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29(3):497-510. PMID:18789730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romano E, Cotari JW, Barreira da Silva R, Betts BC, Chung DJ, Avogadri F, Fink MJ, St Angelo ET, Mehrara B, Heller G, et al.. Human Langerhans cells use an IL-15R-alpha/IL-15/pSTAT5-dependent mechanism to break T-cell tolerance against the self-differentiation tumor antigen WT1. Blood. 2012;119(22):5182-90. doi: 10.1182/blood-2011-09-382200. PMID:22510877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banchereau J, Thompson-Snipes L, Zurawski S, Blanck J-P, Cao Y, Clayton S, Gorvel JP, Zurawski G, Klechevsky E. The differential production of cytokines by human Langerhans cells and dermal CD14+ DCs controls CTL priming. Blood. 2012;119(24):5742-9. doi: 10.1182/blood-2011-08-371245. PMID:22535664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, Taquet S, Coquery S, Wittkowski KM, Bhardwaj N, et al.. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res. 2001;61(17):6451-8. PMID:11522640 [PubMed] [Google Scholar]
- 13.Romano E, Rossi M, Ratzinger G, de Cos MA, Chung DJ, Panageas KS, Wolchok JD, Houghton AN, Chapman PB, Heller G, et al.. Peptide-Loaded Langerhans Cells, Despite Increased IL15 Secretion and T-Cell Activation In Vitro, Elicit Antitumor T-Cell Responses Comparable to Peptide-Loaded Monocyte-Derived Dendritic Cells In Vivo. Clin Cancer Res. 2011;17(7):1984-97. doi: 10.1158/1078-0432.CCR-10-3421. PMID:21355077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nair SK, Boczkowski D, Morse M, Cumming RI, Lyerly HK, Gilboa E. Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfected with RNA. Nat Biotech. 1998;16:364-9. doi: 10.1038/nbt0498-364. [DOI] [PubMed] [Google Scholar]
- 15.Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. Induction of tumor immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Res. 2000;60(4):1028-34. PMID:10706120 [PubMed] [Google Scholar]
- 16.Muul LM, Tuschong LM, Soenen SL, Jagadeesh GJ, Ramsey WJ, Long Z, Carter CS, Garabedian EK, Alleyne M, Brown M, et al.. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood. 2003;101(7):2563-9. doi: 10.1182/blood-2002-09-2800. PMID:12456496 [DOI] [PubMed] [Google Scholar]
- 17.Hacein-Bey-Abina S, Von Kalle C Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, et al.. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415-9. doi: 10.1126/science.1088547. PMID:14564000 [DOI] [PubMed] [Google Scholar]
- 18.Wang R-F, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med. 1996;184:2207-16. doi: 10.1084/jem.184.6.2207. PMID:8976176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber LW, Bowne WB, Wolchok JD, Srinivasan R, Qin J, Moroi Y, Clynes R, Song P, Lewis JJ, Houghton AN. Tumor immunity and autoimmunity induced by immunization with homologous DNA. J Clin Invest. 1998;102:1258-64. doi: 10.1172/JCI4004. PMID:9739060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowne WB, Srinivasan R, Wolchok JD, Hawkins WG, Blachere NE, Dyall R, Lewis JJ, Houghton AN. Coupling and uncoupling of tumor immunity and autoimmunity. J Exp Med. 1999;190(11):1717-22. doi: 10.1084/jem.190.11.1717. PMID:10587362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirsch I, Vignali M, Robins H. T-cell receptor profiling in cancer. Molecular oncology. 2015;9(10):2063-70. doi: 10.1016/j.molonc.2015.09.003. PMID:26404496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung DJ, Romano E, Pronschinske KB, Shyer JA, Mennecozzi M, St Angelo ET, Young JW. Langerhans-type and monocyte-derived human dendritic cells have different susceptibilities to mRNA electroporation with distinct effects on maturation and activation: implications for immunogenicity in dendritic cell-based immunotherapy. J Transl Med. 2013;11:166. doi: 10.1186/1479-5876-11-166. PMID:23837662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheikh N, Cham J, Zhang L, DeVries T, Letarte S, Pufnock J, Hamm D, Trager J, Fong L. Clonotypic Diversification of Intratumoral T Cells Following Sipuleucel-T Treatment in Prostate Cancer Subjects. Cancer Res. 2016;76(13):3711-8. doi: 10.1158/0008-5472.CAN-15-3173. PMID:27216195 [DOI] [PubMed] [Google Scholar]
- 24.GuhaThakurta D, Sheikh NA, Fan LQ, Kandadi H, Meagher TC, Hall SJ, Kantoff PW, Higano CS, Small EJ, Gardner TA, et al.. Humoral Immune Response against Nontargeted Tumor Antigens after Treatment with Sipuleucel-T and Its Association with Improved Clinical Outcome. Clin Cancer Res. 2015;21(16):3619-30. doi: 10.1158/1078-0432.CCR-14-2334. PMID:25649018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Postow MA, Manuel M, Wong P, Yuan J, Dong Z, Liu C, Perez S, Tanneau I, Noel M, Courtier A, et al.. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer. 2015;3:23. doi: 10.1186/s40425-015-0070-4. PMID:26085931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, Fong L. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med. 2014;6(238):238ra70. doi: 10.1126/scitranslmed.3008211. PMID:24871131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. 2014;211(4):715-25. doi: 10.1084/jem.20130590. PMID:24687957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng Tang D, Shen Y, Sun J, Wen S, Wolchok JD, Yuan J, Allison JP, Sharma P. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol Res. 2013;1(4):229-34. doi: 10.1158/2326-6066.CIR-13-0020. PMID:24777852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73(12):3591-603. doi: 10.1158/0008-5472.CAN-12-4100. PMID:23633484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW, Behrens MD, Knutson KL. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res. 2014;74(11):2974-85. doi: 10.1158/0008-5472.CAN-13-2564. PMID:24728077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-cell Activity and Tumor Eradication. Cancer Immunol Res. 2016;4(2):95-100. doi: 10.1158/2326-6066.CIR-14-0126. PMID:26669718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madan RA, Mohebtash M, Arlen PM, Vergati M, Rauckhorst M, Steinberg SM, Tsang KY, Poole DJ, Parnes HL, Wright JJ, et al.. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(5):501-8. doi: 10.1016/S1470-2045(12)70006-2. PMID:22326924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilgenhof S, Corthals J, Heirman C, van Baren N, Lucas S, Kvistborg P, Thielemans K, Neyns B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients With Pretreated Advanced Melanoma. J Clin Oncol. 2016;34(12):1330-8. doi: 10.1200/JCO.2015.63.4121. PMID:26926680 [DOI] [PubMed] [Google Scholar]
- 34.Boudewijns S, Koornstra RH, Westdorp H, Schreibelt G, van den Eertwegh AJ, Geukes Foppen MH, Haanen JB, de Vries IJ, Figdor CG, Bol KF, et al.. Ipilimumab administered to metastatic melanoma patients who progressed after dendritic cell vaccination. Oncoimmunology. 2016;5(8):e1201625. doi: 10.1080/2162402X.2016.1201625. PMID:27622070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, et al.. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803-8. doi: 10.1126/science.aaa3828. PMID:25837513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al.. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534(7607):396-401. doi: 10.1038/nature18300. PMID:27281205 [DOI] [PubMed] [Google Scholar]
- 37.Yao T-J, Begg CB, Livingston PO. Optimal sample size for a series of pilot trials of new agents. Biometrics. 1996;52:992-1001. doi: 10.2307/2533060. PMID:8805764 [DOI] [PubMed] [Google Scholar]
- 38.Beaulieu S, Robbiani DF, Du X, Rodrigues E, Ignatius R, Wei Y, Ponath P, Young JW, Pope M, Steinman RM, et al.. Expression of a functional eotaxin (CC chemokine ligand 11) receptor CCR3 by human dendritic cells. J Immunol. 2002;169(6):2925-36. doi: 10.4049/jimmunol.169.6.2925. PMID:12218106 [DOI] [PubMed] [Google Scholar]
- 39.Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, Steen MS, LaMadrid-Herrmannsfeldt MA, Williamson DW, Livingston RJ, et al.. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4:2680. doi: 10.1038/ncomms3680. PMID:24157944 [DOI] [PubMed] [Google Scholar]
- 40.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114(19):4099-107. doi: 10.1182/blood-2009-04-217604. PMID:19706884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robins H, Desmarais C, Matthis J, Livingston R, Andriesen J, Reijonen H, Carlson C, Nepom G, Yee C, Cerosaletti K. Ultra-sensitive detection of rare T cell clones. J Immunol Methods. 2012;375(1–2):14-9. doi: 10.1016/j.jim.2011.09.001. PMID:21945395 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.