

Abstract
Almost 20 years after the completion of the C. elegans genome sequence, gene structure annotation is still an ongoing process with new evidence for gene variants still being regularly uncovered by additional in-depth transcriptome studies. While alternative splice forms can allow a single gene to encode several functional isoforms, the question of how much spurious splicing is tolerated is still heavily debated. Here we gathered a compendium of 1682 publicly available C. elegans RNA-seq data sets to increase the dynamic range of detection of RNA isoforms, and obtained robust measurements of the relative abundance of each splicing event. While most of the splicing reads come from reproducibly detected splicing events, a large fraction of purported junctions is only supported by a very low number of reads. We devised an automated curation method that takes into account the expression level of each gene to discriminate robust splicing events from potential biological noise. We found that rarely used splice sites disproportionately come from highly expressed genes and are significantly less conserved in other nematode genomes than splice sites with a higher usage frequency. Our increased detection power confirmed trans-splicing for at least 84% of C. elegans protein coding genes. The genes for which trans-splicing was not observed are overwhelmingly low expression genes, suggesting that the mechanism is pervasive but not fully captured by organism-wide RNA-seq. We generated annotated gene models including quantitative exon usage information for the entire C. elegans genome. This allows users to visualize at a glance the relative expression of each isoform for their gene of interest.
In multicellular organisms, cell differentiation is driven by proteomic diversity. Each cell type and tissue is defined initially by selective expression of gene subsets from the shared genome. Additionally, it is possible for an expressed gene to be subjected to alternative splicing, such that a different subset of exons can be retained or excluded in the final protein-coding mRNAs. Alternative splicing thus allows a single gene to encode several protein variants, called isoforms, with altered stability, localization, specificity, or activity (Kelemen et al. 2013).
The importance of alternative splicing as a mechanism to increase the coding content of genes was emphasized by the accumulation of transcriptomic data over the past decade. In humans ∼95% of genes have detectable alternative splice forms (Pan et al. 2008). The nematode is a powerful model to explore networks involved in alternative splicing regulation and the physiological impact of their perturbation (Barberan-Soler et al. 2009, 2011; Zahler 2012). Recent efforts have been made to systematically identify all possible splice variants of the complete Caenorhabditis elegans genome using transcriptome sequencing (RNA-seq) (Hillier et al. 2009; Ramani et al. 2011; Gerstein et al. 2014; Kuroyanagi et al. 2014; Ragle et al. 2015). Most of these analyses reported previously unannotated splice junctions, indicating that saturation has not yet been reached.
The study of individual alternative splicing events can also be performed using fluorescent reporters in vivo (Kuroyanagi et al. 2006, 2007, 2013; Ohno et al. 2008, 2012; Tomioka et al. 2016). To date, this is the most efficient technique to perform genetic screens in order to identify splicing factors that regulate those events and open the path to study the biochemical details of the regulatory interactions (Amrane et al. 2014; Kuwasako et al. 2014; Mackereth 2014).
While the power of reporter minigene approaches is undeniable, a major drawback of the method is that it relies on accurate genome annotation of alternative splicing events to build functional reporters. Moreover, current genome annotations make it difficult to estimate the functional relevance of predicted isoforms. As a result, the lack of quantitative annotation can lead to research efforts being unnecessarily spent investigating putative isoforms that are too weakly expressed to be detected in vivo.
Here we use the wealth of accumulated RNA-seq data to generate a compendium of quantitative measurements of alternative splicing for each gene in the nematode genome.
Results
Characterization of the spliced RNA-seq compendium
The experiments were selected based on recognition of the keywords “RNA-seq,” “transcriptome,” and “C. elegans” and were downloaded from the NCBI Sequence Read Archive (SRA; https:// www.ncbi.nlm.nih.gov/sra). We collected RNA-seq data from 96 individual studies and retrieved the raw reads from 1682 sequencing runs corresponding to 1185 individual experiments (for full list, see Supplemental Table 1). The cumulative number of sequencing reads at our disposal reached a total of 50,544,023,034 (Fig. 1A). Our goal was to measure the relative exon usage for each alternative splicing event. One potential strategy would have been to use exonic sequence coverage, but this method would miss small variations around exon junctions and is complicated by uneven exon coverage. We thus decided to focus instead on the 6,631,116,146 reads that spanned a junction between two exons in order to also exclude reads potentially corresponding to contamination by genomic DNA.
Figure 1.

(A) Relevant figures regarding the compendium data set used in this study. (B) Distribution of sequencing reads spanning individual exon/exon (top), or splice leader (SL)/exon junctions (bottom). (C) For each splice junction, we measured a usage frequency over the whole compendium (see Methods) and assigned it to a usage category (>99%: constitutive; 50%–99%: major; 5%–50%: minor; 1%–5%: secondary; <1%: rare). We then evaluated for each junction what its usage frequency was in each of the individual 1682 RNA-seq runs. The distribution within each category is represented as pie charts. The breakdown of junctions with a frequency <1% found “nonrare” in individual sets is shown as a stacked graph (see also Supplemental Table 2).
We also identified about 287 million reads corresponding to potential trans-splicing of a splice leader (SL) sequence to an exon (SL-reads) (Conrad et al. 1991, 1995; Spieth et al. 1993). We found that 97.4% of these trans-splicing events were not detected with good reproducibility (less than 100 reads each), whereas 86% of the SL-reads came from the 36,000 most detected junctions (Fig. 1B). These spliced reads were mapped to 667,779 individual splice junctions. We found that ∼79% of these splice junctions were not detected with good reproducibility (less than 100 reads each over 1682 RNA-seq runs). In contrast, 97.6% of the reads came from robustly detected junctions (Fig. 1B).
Absolute read count thresholds are often used in the literature for accepting/rejecting events based on a single RNA-seq experiment. Here, we decided to relate the level of detection for a given isoform to that of other splicing events of the same gene to account for genes with low expression level. Based on the relative usage frequency, we attributed each splicing event to the following categories: constitutive, major, minor, secondary, and rare (in decreasing order of frequency). We checked for the robustness of this classification by comparing it systematically with the usage frequency that would have been obtained using every single RNA-seq run separately (Fig. 1C). This analysis confirmed that the compendium approach provides a more comprehensive evaluation than any single experiment could by improving the dynamic range of detection and averaging out the detection variability between individual sequencing runs. It also shows that the overwhelming majority of the “rare” splicing events are mostly undetected in any given RNA-seq run (see also Supplemental Fig. 1).
Rare isoforms are mostly found in highly expressed genes and are less conserved
While some of the least detected exon junctions come from genes with very low RNA-seq read counts in our compendium—possibly corresponding to genes with restricted spatiotemporal expression, a large fraction come from genes that have been highly covered across a majority of the experiments we analyzed. For each alternative splicing event, we computed a usage frequency defined by the number of reads found for a given junction divided by the number of reads mapping to any junction sharing an acceptor (or donor) site with it. We found that splice junctions detected 100-fold less than the corresponding main product of their gene (i.e., “rare” variants) represent 88% of all detected junctions. To investigate whether gene expression level correlates to the amount of detected splice variants, we generated 20 bins of about 1000 genes grouped according to their expression level in our compendium data set. We then measured the average number of distinct splicing events per gene in each expression bin. The total number of detected junctions per gene increases with the gene expression level (with the highest expression bin having on average around 70 potential introns); however, if we only count the number of junctions with a usage frequency >1%, we see that the average number of introns per gene is almost invariant (between five and seven) regardless of the expression level (Fig. 2A). This seems to indicate that these low frequency junctions do not in fact correspond to functional introns but rather to accidental misfiring of the spliceosome.
Figure 2.

(A) Introns with low inclusion rate are overrepresented in highly expressed genes. All C. elegans genes were ranked according to their expression level (defined by the highest read count for an intronic junction of that gene) and split in 20 bins of about 1000 genes. For each bin (x-axis), the average expression level (black curve; left axis) and the number of observed splice junctions per gene are plotted. Rare junctions (with an inclusion rate <1%) are represented in orange and commonly used junctions in green on the bar graph (axis on the right). (B) Frequently used splice sites are more conserved than rarely used ones. Conservation analysis for pairs of donor and acceptor splice sites grouped by relative inclusion level. For each gene, the genomic sequence was aligned with each available ortholog from seven nematode species (see Methods). The indicated conservation fraction is the number of genes for which both sites were present, divided by the number of orthologous genes identified.
To evaluate the possible functionality of those rare isoforms we analyzed the conservation of all introns boundaries. For each gene with an alternative splice form we retrieved from the WormBase database (http://www.wormbase.org/) the sequence of all available orthologs from seven nematode species (Caenorhabditis briggsae, Caenorhabditis brenneri, Caenorhabditis remanei, Caenorhabditis japonica, Caenorhabditis sinica, Caenorhabditis angaria, and Caenorhabditis tropicalis). After pairwise alignment of the full gene sequences to each of their counterpart, we looked for the presence of paired splice donor and acceptor sites at the expected corresponding positions. As a reference, we measured the conservation level for the constitutive intron boundaries within those genes: ∼28% (9306 junctions) were found in at least two-thirds of the identified orthologs, and only ∼8% (2605) were not found outside C. elegans. For alternative junctions used in >50% of the detected messengers, the conservation level is very similar (24%–25% found in at least two-thirds of orthologs and 12%–14% specific to C. elegans). For junctions with a lower level of inclusion, we observe progressively less conservation and only ∼9% of rare introns are found well conserved, while ∼29% are not found in other species. We also found that the set of 907 predicted junctions (from WormBase) for which no RNA-seq evidence could be found in our compendium shows even less conservation, as could be expected for nonfunctional sequences (Fig. 2B). Overall, we find that “rare” junctions are less evolutionarily conserved than more frequently used junctions.
The number of reads per junction ranges over six orders of magnitude depending of the gene. This means that 1000 reads can be the maximum count for a gene while representing only 0.01% of reads for another; we therefore propose that the usage frequency is a better predictor of the functionality of a splice junction than raw read count.
Trans-splicing is potentially ubiquitous in C. elegans
In C. elegans mRNAs, the 5′ UTR is often cleaved off and replaced by a SL sequence that is provided by an independently transcribed snRNA (Conrad et al. 1991, 1995; Spieth et al. 1993). SL1 is mostly associated with genes linked to a proximal promoter directly upstream, while SL2 is generally associated with genes located downstream in polycistronic operons. Below, we therefore refer to trans-splice sites as indicative of a “gene start” rather than transcription start.
We detected trans-spliced sites for most protein coding genes but found that a vast number of trans-splice sites with very low read counts likely correspond to biological noise. This is supported by the observation that highly expressed genes have a larger number of trans-splice sites, while the average number of robust splice site per gene is independent of the expression level (Fig. 3A).
Figure 3.

(A) All genes for which at least one robust SL site was detected were ranked according to their expression level (as defined by the highest read count for an intronic junction of that gene) then grouped in bins of about 1000 genes. For each bin, the average expression level (left axis) and the average number of SL site per gene found are plotted. Here we counted as high-confidence sites that contained at least 25% of the SL-containing reads of that gene. (B) Genes without detectable trans-splicing have mostly low expression levels. All C. elegans genes were ranked according to their expression level (defined by the highest read count for an intronic junction of that gene) and split in 20 bins of around 1000 genes. For each bin, the average expression level (black curve; left axis) is plotted. For each bin, we represented the fraction of genes for which no SL junction was found (orange) and for which at least one robust SL site was found (blue). For the 1000 genes with the lowest expression level, we inserted a Venn diagram representing the number of genes for which either some predicted cis-junctions were missed and/or no SL site was detected. (C) A scatter-plot of the ratio of SL2/SL1 reads found at each robust splice site against the distance to the closest upstream exon shows a discernible bias for sites with >80% SL2 splicing to be located at ∼100 nt downstream from the nearest exon. Each dot is colored according to the number of sequencing reads supporting the corresponding SL site. A zoomed plot of the number of occurrences of SL sites at each distance shows two sharp peaks indicative of a strong preference for SL2 splicing to occur either at a distance of ∼50 nucleotide (nt) or ∼100 nt downstream from the previous exon.
As for cis-splicing, the wide variation of gene expression levels precludes using an absolute read-count threshold to identify bona fide trans-splicing sites. Therefore, to filter out potential spurious trans-splicing events, we decided to consider “robust” the sites that were corroborated by at least 25% of the SL-containing reads associated to that gene. While this definition is not perfect, it provides a good approximation of the main SL sites used for most genes. We found approximately 20,000 robust trans-splice sites spanning 17,060 genes (84% of protein coding genes). Previous work by Allen et al. (2011) attempted to systematically identify all trans-splicing sites in the C. elegans genome. They detected 70% of genes subjected to trans-splicing and found a tendency for highly expressed genes to be more trans-spliced than the less expressed genes.
In the 2011 study, RNA-seq reads were mapped against a database of potential trans-splice sites generated in silico by joining all SL sequences with all annotated acceptor splice sites. In contrast, our detection method is independent of the accuracy of the genome annotation and therefore allowed us to recover trans-splice sites that had been previously missed. In addition to the difference in mapping strategy, we also benefited from using a 1000-fold larger number of reads. Most of the 15% of genes for which we found no SL site are among the lowest expressed genes in the genome (Fig. 3B). Additionally, for genes in the lowest detection tier (bottom 1000 genes) not only did we not detect a SL junction for 639 genes, we also failed to detect a fraction of the annotated exon junctions for 366 genes (Fig. 3B, inset). This suggests that the RNA-seq coverage for these genes has not been saturated even by our compendium of data sets and that more targeted approaches directed at these low expression genes are needed to determine their trans-splicing status.
For each of the robust SL sites, we determined what proportion of SL1 or SL2 sequence was used at each position (expressed as SL1/SL2 ratio). We then compared this ratio to the distance to the closest upstream exon (Fig. 3C). We found that most positions are strongly favored by one SL sequence rather than the other and that SL2 trans-splicing is strongly preferred when the closest exon is located either ∼50 or ∼100 nucleotides (nt) upstream of the acceptor site, thus confirming and refining the previously reported observations (Allen et al. 2011).
Genome-wide quantitative visualization of differential exon usage
For each C. elegans gene, we generated a visual summary of the splicing data we collected (about 20,000 images collated as Supplemental Fig. S2). For each gene, we represented only the most commonly used exons (i.e., constitutive exons and the most common forms for alternative splicing events) and the quantitative values of all the spliced junctions obtained from our analysis. This allows to see at a glance what is the main product of any given gene and what fraction of the messengers use alternative forms. In addition, we also represented the identified trans-splicing positions and the level at which they were detected. This allows for the first time to directly see how many distinct promoters are used by a given gene and their relative contribution to that gene's expression.
To illustrate the usefulness of our visual representations, we present in Figure 4 three examples together with their current genome browser representation in WormBase, including all annotated isoforms. For the first two examples, alternative isoform expression has been well described and validated by fluorescent reporter constructs:
DAF-2 protein is C. elegans insulin/IGF receptor ortholog well known for its effect on lifespan extension and Dauer formation (Kenyon et al. 1993; Apfeld and Kenyon 1998). Immunodetection of the protein indicates that it is predominantly expressed in a subset of neuronal cells in the animal nerve ring (Kimura et al. 2011). It has recently been shown that an alternative functional isoform generated by inclusion of a cassette exon (exon 11.5) is expressed more specifically in sensory neurons and particularly under starvation conditions (Ohno et al. 2014; Tomioka et al. 2016). We found that this isoform accounts for ∼15% of the detected reads (about 5000 reads for exon 11.5 inclusion vs. about 36,000 for skipping) (Fig. 4A). While we find support for several of the annotated alternative start sites, only one major trans-spliced site stands out as the likely main start site for this gene.
PTB-1 is a hnRNP family protein member whose tissue-specific expression is necessary for the inclusion of exon 11.5 in daf-2 (Tomioka et al. 2016). Tomioka and colleagues demonstrated that ptb-1 was expressed from two alternative promoters active in distinct neuron subsets. Consistent with this observation we found about 15,000 reads for the PTB-1a– or PTB-1b–specific splice junctions and about 30,000 reads for the junctions shared by both isoforms. Our data also support the existence of a third unverified promoter with a lower activity (black arrow in Fig. 4B). We can also observe that a previously unreported skipping of exon 11 accounts for 16% of the detected junctions (5439 reads joining exons 10–12 vs. 28,758 reads joining exons 10–11) (Fig. 4B). This exon skipping causes a frame shift that could generate a truncated version of the protein lacking the last 167 amino acids.
Figure 4.
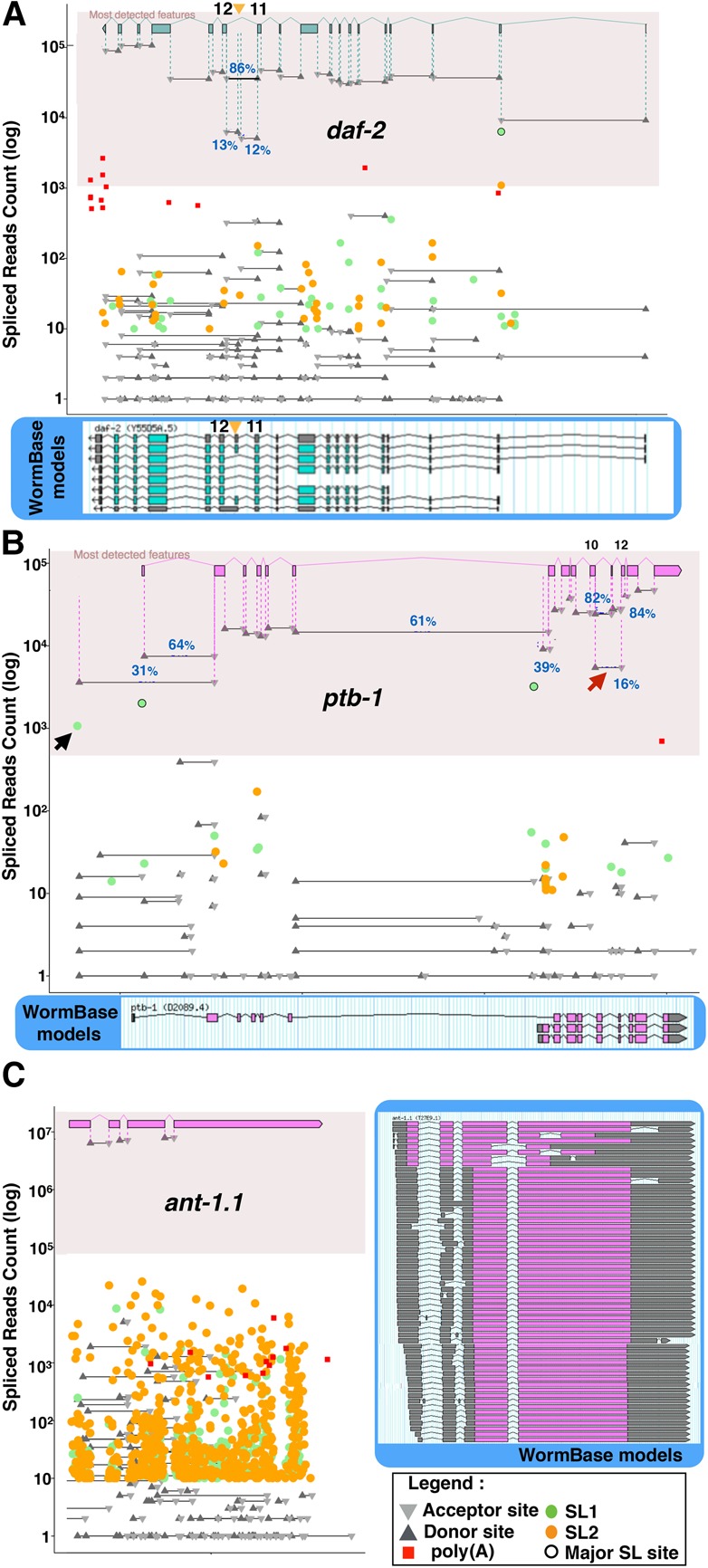
Quantitative visualization of relative splice-sites usage. We present a gene model constituted of the most commonly detected exons, associated with the absolute read count for each cis- and trans-splicing event in the gene, as well as detected poly(A) tail addition, on a logarithmic scale. We highlighted the area containing high-confidence events (detected in at least 1% of the transcripts) and included junction usage frequencies for alternative events. As a reference we include the current WormBase model. (A) daf-2: Exon junctions corresponding to the inclusion of exon 11.5 (orange arrowhead) specific to a small subset of neurons during starvation are detected at ∼15% of the level of other junctions. Evidence of trans-splicing at exon 2 and a 10-fold increase in read numbers between introns 1 and 2 seem to indicate that this is in fact the most used expression start for this gene. (B) The ptb-1 gene has two confirmed alternative promoters active in two distinct neuronal subsets. In our data analysis, this manifests as two major SL1 trans-splicing junctions associated to the detection of the corresponding isoform-specific exon–exon junctions. Note how common junctions are detected with a level corresponding to the sum of both isoform-specific rates downstream from the second promoter. Our data seem to support the existence of a third unverified promoter with a lower activity (black arrow). We also detect an exon junction between exon 10 and exon 12, indicating that ∼16% of the transcripts are skipping exon 11 (red arrow). (C) The ant-1.1 gene had over 50 isoforms predicted in WormBase. Our RNA-seq analysis detected the three constitutive junctions with over 6 million reads each. All other junctions are orders of magnitude below the overall expression level of this gene, indicating that there is only one functional isoform of ANT-1.1.
From these (and several other) examples, we concluded that our visualization tool was indeed suitable to provide a measurement of isoform usage that is consistent with known alternative splicing events even when they are pertaining to a very restricted number of cells.
We next wanted to see if we could easily interpret our data for genes for which no prior information on alternative splicing patterns was available. As an extreme example we show here ant-1.1, which had over 50 predicted isoforms in the C. elegans genome annotation WS251. ANT-1.1 is an essential mitochondrial adenine transporter that is ubiquitously expressed (Farina et al. 2008). Our data clearly discriminate between two classes of exon–exon junctions for ant-1.1: on one hand, three constitutive junctions, for each of which about 6 million reads are found; and on the other hand, 170 junctions supported by a number of reads ranging from one to 200,000. This second class of junctions, amounting to a very small proportion of the detected RNA products (two to six orders of magnitude less than the main product), is likely to result from stochastic misfiring of the splicing machinery as most of the isoforms produced contain frame-shifts and premature stop codons (Fig. 4C). We considered these splice forms as “rare variants” in our curation.
Discussion
In eukaryotes, the accumulation of aberrantly spliced messengers that could encode potentially deleterious truncated proteins is prevented by nonsense-mediated mRNA decay (NMD) (Jaillon et al. 2008; Farlow et al. 2010). Species devoid of NMD tend to almost entirely lack introns (Lynch 2006) indicating that an error proof splicing system has not arisen through natural evolution (or is yet to be discovered). Together, these observations support the widespread existence of biological noise in the splicing process. The sensitivity of modern deep sequencing methods for transcriptome characterization ensures that even rare aberrantly spliced messengers will be detected alongside functional splice forms. Traces of messengers targeted for decay, that are accumulating in NMD mutants, can be detected in wild-type individuals as well (Barberan-Soler et al. 2009; Ramani et al. 2009). Moreover, a recent study unveiled a significant decrease in splicing accuracy correlated with age in C. elegans (Heintz et al. 2016).
We reasoned that by exploring a large collection of RNA-seq data sets, we could compile a nearly comprehensive list of splice junctions in the C. elegans genome. The total amount of data used in our study provides a robust quantitative measure of the frequency of usage of each detected alternative splice junction, and the expanded dynamic range obtained also allows for discrimination between genuine alternative splicing and potential biological noise. Our observation that “rare” junctions come disproportionately from highly expressed genes and are less conserved in other nematode species could indicate that most of these junctions correspond to biological noise causing accidental splicing outside of the preferred functional sites. A similar analysis in human cell lines also found that rare alternative splicing events are more frequently observed in highly expressed genes and tend to be less conserved across species (Pickrell et al. 2010). The investigators of that study also found that the small fraction of reads coming from unannotated rare junctions covered a large number of likely spurious splicing events.
While it is likely that our discrimination between rare and robust splice variants offers a good approximation for the functionality threshold of any given isoform, it is almost certain that some exceptions will apply. It is possible, for example, that a ubiquitously expressed gene also encodes a rare variant with a limited cell specificity, but studying and validating this kind of event will constitute its own challenge. In that context, our classification should be considered as a warning sign: These “rare” events are unlikely to be functional, and studying them will not be trivial as they are not detected in most RNA-seq experiments.
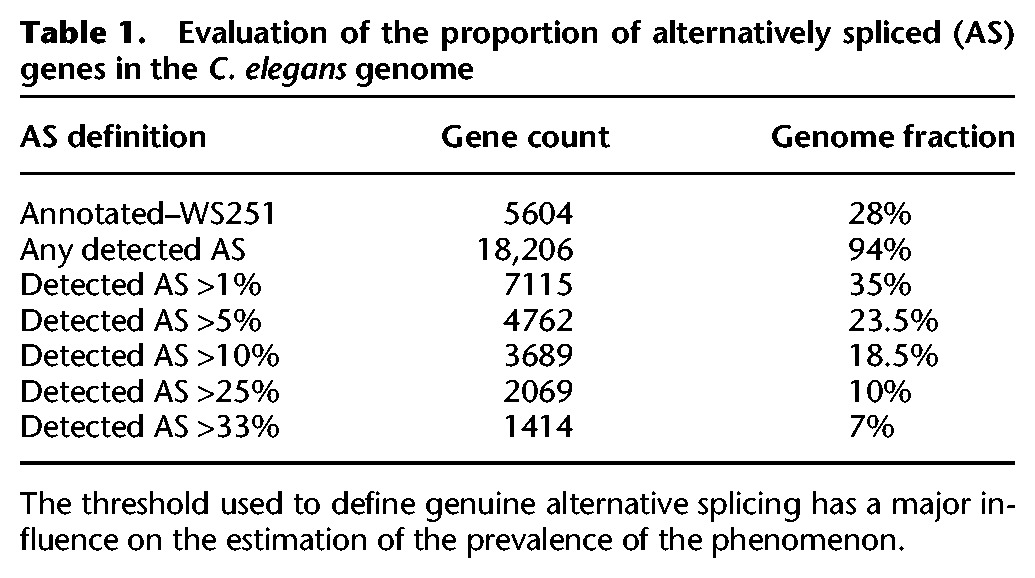
Our compendium-based meta-analysis provides a widely expanded dynamic range of detection with genes having between zero and 107 reads per junction. If we considered every splice junction detected by any RNA-seq experiment in our compendium, we would conclude that ∼94% of C. elegans genes are submitted to alternative splicing (Table 1). If we consider only genes for which there is a second isoform with a frequency of at least 1% of the major isoform, this number drops to 35% (approximately 7000 genes). This could be a valid definition since our analysis suggests that the majority of “rare” splicing events corresponds to biological noise rather than a conserved functional mechanism. However, it is possible that for some genes a rare isoform indeed is critical for a cell-specific function. Conversely, we cannot proclaim that every event that is above the 1% threshold is a genuine alternative splicing event. If we place the bar at 5% of the gene expression level, then only 4700 genes have more than one isoform. There are no objective quantitative criteria that can systematically discriminate between functional and spurious alternative isoforms at this time.
Table 1.
Evaluation of the proportion of alternatively spliced (AS) genes in the C. elegans genome
The visual representation we propose here allows us to immediately see if a gene has a set of genuine introns clearly separated from background splicing noise or if there are intermediate splice variants worthy of investigation. For genes with multiple alternative promoters, our representation also provides a visual ranking of the relative strength of each promoter, which can be indicative of their tissue specify. Such first approximations will be very useful for generating hypotheses and designing experiments to test them. Importantly, these representations are unbiased and objectively derived from a wide range of independent experimental observations.
The combination of increased depth of sequencing data with an unbiased SL site detection strategy led us to find evidence of trans-splicing for 84% of C. elegans protein coding genes (vs. 70% previously). We were not able to detect SL sites mainly for genes that have the lowest expression level. This indicates that despite the large dynamic range of our data compendium, we did not reach sufficient coverage for those genes. Since even the accumulation of 1682 full-animal RNA-seq did not provide the sensitivity needed, more targeted RNA-seq experiments will be necessary to explore the function and specificity of the least expressed genes and their isoforms (Hashimshony et al. 2012; Schwarz et al. 2012; Fox et al. 2005).
Current RNA-seq–based estimations claim that >95% of human multiexon genes express multiple splice isoforms (Pan et al. 2008; Wang et al. 2014), which is very similar to the number we find in C. elegans when considering every detectable isoform. The validity of this evaluation is contested by some other transcriptomic and proteomic studies (Pickrell et al. 2010; Tress et al. 2017). Applying our strategy of aggregating large numbers of RNA-seq data sets to flag potential stochastic splicing could help shed new light on the question of the prevalence of alternative isoforms and proteome complexity in other metazoan organisms.
Methods
Data sets selection
The raw data used in this study are publicly available and downloaded from the NCBI Sequence Read Archive (SRA; https://www. ncbi.nlm.nih.gov/sra; see Supplemental Table 1). Sample experiment and run accession numbers and sequence files were downloaded programmatically using the NCBI “E-utilities” tools “ESearch” and “EFetch” (https://www.ncbi.nlm.nih.gov/books/NBK25501/) and the Unix utility “wget”, and read files in FASTQ format were generated using the “fastq-dump” tool from the NCBI SRA toolkit 2.5.0. Illumina mRNA-seq experiments were selected with a regular expression search of the experiment descriptions, leading to a final compendium of 1682 data sets (1214 single-end and 468 paired-end). Read files were taken as is; no read processing was performed.
Exon–exon junction identification
Junctions containing canonical intron splice sites (GT-AG, GC-AG, and AT-AC) were identified by mapping raw RNA-seq reads to the C. elegans genome sequence (obtained from WormBase release WS251, http://www.wormbase.org/) using TopHat2 2.0.14 (Kim et al. 2013) based on the Bowtie 2 2.2.5 (Langmead and Salzberg 2012) core read aligner. The list of junctions and the number of reads spanning them was retrieved from the “junctions.bed” output file of TopHat2. All data sets were mapped in single-end mode, and the following options were set for TopHat2 (all other parameters as default): “--min-intron-length 10 --max-intron-length 20000 --read-mismatches 3 --read-gap-length 2 --read-edit-dist 3 --max-multihits 2 --b2-sensitive --segment-mismatches 2 --segment-length 15 --min-segment-intron 10 --max-segment-intron 20000 --no-coverage-search.”
RNA-seq reads having only a few matches to one exon side of a junction may be missed by the above mapping strategy. To find additional junctions and additional reads corresponding to the previously detected junctions, unmapped reads were remapped to the C. elegans WS251 genome by setting the following TopHat2 parameters to force the detection of deletions of up to 1 kb in length: “--read-gap-length 1000 --read-edit-dist 1003 --b2-ma 3 --b2-rdg 3,1.” Sequences of 200 bp flanking each side of a given deletion site were joined and then mapped to the C. elegans genome with TopHat2 run with the same parameters as in the original search to recover further reads containing canonical exon–exon junctions.
For reads that were mapped to two distinct junctions, we attributed the reads to single junctions in proportion of the corresponding single mapped reads (if 100 reads map to both junctions A and B while one read maps only to junction A and 99 reads map only to junction B, our final tally will be two reads for junction A and 198 for junction B). If no discriminating reads (mapped to either A or B only) were found, we retained only previously annotated junctions when one existed.
In addition, junctions that were predicted in WormBase WS251 but that were not recovered in our searches (“undetected junctions”) were included.
Junction usage quantification
The relative usage of splice junctions was estimated as follows: First, all junctions for which at least one boundary was within the coordinates of a gene predicted on the same DNA strand in WormBase WS251 were assigned to that gene. Then, for a given gene, a donor ratio was computed for all detected junctions that shared a common donor boundary by dividing the number of RNA-seq reads mapping to a junction by the sum of reads mapping to the set of junctions having the same donor site. An acceptor ratio was similarly calculated for detected junctions sharing a common acceptor site. If both ratios could be computed, then the usage ratio was set to the ratio that was based on the largest number of reads. In the case of junctions that did not share any boundary with any other junction in the gene, the usage ratio was set to one (constitutive junction). We also defined a ratio relative to the junction with the highest number of mapped reads for the gene (“max_junction”), i.e., number of reads for the junction divided by number of reads for max_junction. If max_junction had no reads (in the case of a totally undetected gene), then the usage ratio was set to “NA” (not available). If a junction “Max_Ratio” was <1%, the junction was classified as “rare.” Note that a junction may be assigned to more than one gene (e.g., when genes are overlapping or in close proximity) and will have usage ratios specific to each gene. Supplemental Table 2 contains the genomic positions, the number of supporting reads in our compendium, the number of experiments that detected it, the inclusion ratio, and the curated category for all exon–exon junctions we detected.
Conservation analysis
A comparative analysis was conducted to determine whether exon–exon junctions identified in C. elegans were conserved in other nematodes. The list of genes that were predicted to be orthologous between C. elegans and seven other Caenorhabditis species (C. angaria, C. brenneri, C. briggsae, C. japonica, C. remanei, C. sinica, and C. tropicalis) was retrieved from WormBase WS251, as well as the corresponding gene annotation and genome sequence files. In a given species, there may be several predicted orthologs to the same C. elegans gene. A global and optimal pairwise alignment was computed according to the Needleman and Wunsch algorithm (Needleman and Wunsch 1970) between the full nucleotide sequence of each C. elegans gene and its ortholog(s) using the program “needle” from the EMBOSS 6.6.0 package (Rice et al. 2000). Then alignments were scanned for junctions. For each C. elegans exon–exon junction along the gene, if the four bases at the corresponding alignment positions in the other species matched a canonical splice site (GT-AG, GC-AG, or AT-AC), then the junction was considered as conserved between C. elegans and the other species (the junction need not to be identical between the two species to be considered present). Supplemental Table 3 contains the data pertaining to this conservation analysis.
Trans-splice site identification
Trans-splice sites were identified from the RNA-seq reads that did not map to the C. elegans genome (with or without introns). Our strategy was multistep (see Supplemental Fig. S3). First, cutadapt 1.3 (Martin 2011) was employed to identify all sequencing reads containing a putative SL sequence (or the 3′ end of it) and to extract the sequence downstream from the SL. The search requirements were: a match length of at least 5 nt to the SL with a maximum of 10% mismatches and a downstream sequence of at least 15 nt (options “-e 0.10 -O 5 -m 15 --trimmed-only”).
The following SL1 sequence “CTCAAACTTGGGTAATTAAACCG” and seven SL2 variant sequences (“GGTTTAAAACCCAGTTACCAAGG,” “GGTTTTAACCCAGTTAACCAAGG,” “GGTTTTAACCCAGTTACTCAAGG,” “GGTTTTAACCCAGTTTAACCAAGG,” “GGTTTTAACCCATATAACCAAGG,” “GGTTTATACCCAGTTAACCAAGG,” and “GGTTTTAACCCAGTTAATTGAGG”), as well as their reverse-complement, were used as queries for the search. Then, three mapping algorithms were used in succession to determine the genomic locations of the corresponding trans-splice sites. The read portion downstream from the SL portion was first mapped to the C. elegans WS251 genome with TopHat2 (with the options “--min-intron-length 10 --max-intron-length 20000 --read-mismatches 3 --read-gap-length 2 --read-edit-dist 3 --max- multihits 2 --b2-sensitive --segment-mismatches 2 --segment-length 15 --min-segment-intron 10 --max-segment-intron 20000 --no- coverage-search”), which allows reads to contain introns but requires full alignment. Then, the unmapped SL reads were mapped to C. elegans WS251 RNA transcripts with Bowtie 2, which does not make spliced alignments but allows for partial mapping with soft-clipping. Bowtie 2 was run with the options “--local --sensitive-local” (all other parameters set as default). Finally, the remaining unmapped SL reads were aligned to the C. elegans genome with GSNAP from the GMAP-GSNAP package release 2017-01-14 (Wu and Nacu 2010), which allows for both introns and soft-clipping. The following GSNAP options were set as follows (all other parameters as default): “--nofails --novelsplicing=1 --localsplicedist=20000 --novelend-splicedist=20000 --suboptimal-levels=0 --max- mismatches=3 --indel-penalty=2 --max-middle-insertions=2 --max-middle-deletions=2 --max-end-insertions=2 --max-end- deletions=2 --input-buffer-size=100000 --output-buffer-size= 100000.” In the above mapping strategy, in case of soft-clipping the genomic position of the trans-splice site was shifted according to the length of the clipped region. Otherwise, it was taken as the mapping position of the 5′ end of the SL read (i.e., the 5′ end of the sequence downstream from the SL).
To further improve the quantification of the number of reads spanning a given SL position, full reads that mapped to the C. elegans genome and that extended a few bases over the discovered SL positions were examined. For those reads, the sequence portion extending beyond the SL position was compared with the corresponding genomic sequence at that position and with the 3′ ends of the SL1 and SL2 variants. If the number of mismatches to any of the SL ends was smaller than that to the genomic region, then the read was considered as having a SL piece and was added to the count of reads spanning the given SL position.
Intergenic SL sites were assigned to the nearest downstream gene if it was located within a distance of 2 kb. Supplemental Table 4 contains the data pertaining to the trans-splicing events with a read count greater than 10.
Nongenomic poly(A) site identification
From the set of RNA-seq reads that did not map to the C. elegans genome, cutadapt was used to identify those that harbored a poly(A) stretch at their 3′ end and extract the upstream read region. Reads carrying a stretch of at least 10 A residues, with a maximum of 20% mismatches, and a remaining upstream portion of at least 15 nt were searched for (cutadapt options “-e 0.20 -O 10 -m 15 --trimmed-only”). The upstream portion of these reads was then mapped to the C. elegans genome with TopHat2 (run with the same parameters as for the trans-splice site search), and the poly(A) site location was set to the 3′ end of the read mapping position. Then sites corresponding to genome-encoded poly(A) runs were filtered out. For that, genomic poly(A)s were identified by searching the C. elegans genome sequence by means of the program “fuzznuc” from the EMBOSS package with the query “AAAAAAAAAA” and allowing for two mismatches (options “-pmismatch 2 -pattern AAAAAAAAAA - complement”). Potential poly(A) sites whose coordinates matched the genomic sites were discarded.
Intergenic poly(A) sites were assigned to the nearest upstream gene if it was located within a distance of 2 kb. Supplemental Table 5 contains the genomic location for poly(A) sites with a read count greater than 500.
Graphical representation of quantitative splicing and trans-splicing
For each gene, a graphical representation showing the gene features (exons, splice junctions, SL, and poly(A) sites) along with quantitative usage data was generated using R 3.2.0 (R Core Team 2015). We present a gene model constituted of the most commonly detected exons. We report on a logarithmic scale the absolute read count for each cis- and trans-splicing events, as well as poly(A) additions (the read count for the most detected cis-junction is indicated on the y-axis). Vertical dashed lines connect the nonrare junctions to the gene model. The usage ratio of alternative events is indicated. We highlighted the area containing features detected at a level of at least 1% of the maximum junction read count for the gene (shaded area). All plots are shown in Supplemental Figure 2.
Data access
Graphical summaries of our quantitative spicing analysis for each individual gene are available in Supplemental Figure 2 and at WormBase.
Supplementary Material
Acknowledgments
This work has been funded by Inserm (D.D., N.J.T.) and the French Ministry for Higher Education and Research (J.R.M.M.). We thank Dr. Axel Innis for providing access to his computer cluster at the IECB, and the University of Bordeaux for access to the supercomputer of the Mésocentre de Calcul Intensif Aquitain (MCIA).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.224626.117.
References
- Allen MA, Hillier LW, Waterston RH, Blumenthal T. 2011. A global analysis of C. elegans trans-splicing. Genome Res 21: 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrane S, Rebora K, Zniber I, Dupuy D, Mackereth CD. 2014. Backbone-independent nucleic acid binding by splicing factor SUP-12 reveals key aspects of molecular recognition. Nat Commun 5: 4595. [DOI] [PubMed] [Google Scholar]
- Apfeld J, Kenyon C. 1998. Cell nonautonomy of C. elegans daf-2 function in the regulation of diapause and life span. Cell 95: 199–210. [DOI] [PubMed] [Google Scholar]
- Barberan-Soler S, Lambert NJ, Zahler AM. 2009. Global analysis of alternative splicing uncovers developmental regulation of nonsense-mediated decay in C. elegans. RNA 15: 1652–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan-Soler S, Medina P, Estella J, Williams J, Zahler AM. 2011. Co-regulation of alternative splicing by diverse splicing factors in Caenorhabditis elegans. Nucleic Acids Res 39: 666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad R, Thomas J, Spieth J, Blumenthal T. 1991. Insertion of part of an intron into the 5′ untranslated region of a Caenorhabditis elegans gene converts it into a trans-spliced gene. Mol Cell Biol 11: 1921–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad R, Lea K, Blumenthal T. 1995. SL1 trans-splicing specified by AU-rich synthetic RNA inserted at the 5′ end of Caenorhabditis elegans pre-mRNA. RNA 1: 164–170. [PMC free article] [PubMed] [Google Scholar]
- Farina F, Alberti A, Breuil N, Bolotin-Fukuhara M, Pinto M, Culetto E. 2008. Differential expression pattern of the four mitochondrial adenine nucleotide transporter ant genes and their roles during the development of Caenorhabditis elegans. Dev Dyn 237: 1668–1681. [DOI] [PubMed] [Google Scholar]
- Farlow A, Meduri E, Dolezal M, Hua L, Schlötterer C. 2010. Nonsense-mediated decay enables intron gain in Drosophila. PLoS Genet 6: e1000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RM, Von Stetina SE, Barlow SJ, Shaffer C, Olszewski KL, Moore JH, Dupuy D, Vidal M, Miller DM. 2005. A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics 6: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Rozowsky J, Yan KK, Wang D, Cheng C, Brown JB, Davis CA, Hillier L, Sisu C, Li JJ, et al. 2014. Comparative analysis of the transcriptome across distant species. Nature 512: 445–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Wagner F, Sher N, Yanai I. 2012. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2: 666–673. [DOI] [PubMed] [Google Scholar]
- Heintz C, Doktor TK, Lanjuin A, Escoubas CC, Zhang Y, Weir HJ, Dutta S, Silva-García CG, Bruun GH, et al. 2016. Splicing factor 1 modulates dietary restriction and TORC1 pathway longevity in C. elegans. Nature 541: 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Reinke V, Green P, Hirst M, Marra MA, Waterston RH. 2009. Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res 19: 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Bouhouche K, Gout JF, Aury JM, Noel B, Saudemont B, Nowacki M, Serrano V, Porcel BM, Ségurens B, et al. 2008. Translational control of intron splicing in eukaryotes. Nature 451: 359–362. [DOI] [PubMed] [Google Scholar]
- Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, Stamm S. 2013. Function of alternative splicing. Gene 514: 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. 1993. A C. elegans mutant that lives twice as long as wild type. Nature 366: 461–464. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KD, Riddle DL, Ruvkun G. 2011. The C. elegans DAF-2 insulin-like receptor is abundantly expressed in the nervous system and regulated by nutritional status. Cold Spring Harb Symp Quant Biol 76: 113–120. [DOI] [PubMed] [Google Scholar]
- Kuroyanagi H, Kobayashi T, Mitani S, Hagiwara M. 2006. Transgenic alternative-splicing reporters reveal tissue-specific expression profiles and regulation mechanisms in vivo. Nat Methods 3: 909–915. [DOI] [PubMed] [Google Scholar]
- Kuroyanagi H, Ohno G, Mitani S, Hagiwara M. 2007. The Fox-1 family and SUP-12 coordinately regulate tissue-specific alternative splicing in vivo. Mol Cell Biol 27: 8612–8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi H, Watanabe Y, Hagiwara M. 2013. CELF family RNA-binding protein UNC-75 regulates two sets of mutually exclusive exons of the unc-32 gene in neuron-specific manners in Caenorhabditis elegans. PLoS Genet 9: e1003337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi H, Takei S, Suzuki Y. 2014. Comprehensive analysis of mutually exclusive alternative splicing in C. elegans. Worm 3: e28459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwasako K, Takahashi M, Unzai S, Tsuda K, Yoshikawa S, He F, Kobayashi N, Güntert P, Shirouzu M, Ito T, et al. 2014. RBFOX and SUP-12 sandwich a G base to cooperatively regulate tissue-specific splicing. Nat Struct Mol Biol 21: 778–786. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2006. The origins of eukaryotic gene structure. Mol Biol Evol 23: 450–468. [DOI] [PubMed] [Google Scholar]
- Mackereth CD. 2014. Splicing factor SUP-12 and the molecular complexity of apparent cooperativity. Worm 3: e991240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12. [Google Scholar]
- Needleman SB, Wunsch CD. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443–453. [DOI] [PubMed] [Google Scholar]
- Ohno G, Hagiwara M, Kuroyanagi H. 2008. STAR family RNA-binding protein ASD-2 regulates developmental switching of mutually exclusive alternative splicing in vivo. Genes Dev 22: 360–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno G, Ono K, Togo M, Watanabe Y, Ono S, Hagiwara M, Kuroyanagi H. 2012. Muscle-specific splicing factors ASD-2 and SUP-12 cooperatively switch alternative pre-mRNA processing patterns of the ADF/cofilin gene in Caenorhabditis elegans. PLoS Genet 8: e1002991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Kato S, Naito Y, Kunitomo H, Tomioka M, Iino Y. 2014. Role of synaptic phosphatidylinositol 3-kinase in a behavioral learning response in C. elegans. Science 345: 313–317. [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Pai AA, Gilad Y, Pritchard JK. 2010. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet 6: e1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Ragle JM, Katzman S, Akers TF, Barberan-Soler S, Zahler AM. 2015. Coordinated tissue-specific regulation of adjacent alternative 3′ splice sites in C. elegans. Genome Res 25: 982–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Nelson AC, Kapranov P, Bell I, Gingeras TR, Fraser AG. 2009. High resolution transcriptome maps for wild-type and nonsense-mediated decay-defective Caenorhabditis elegans. Genome Biol 10: R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Calarco JA, Pan Q, Mavandadi S, Wang Y, Nelson AC, Lee LJ, Morris Q, Blencowe BJ, Zhen M, et al. 2011. Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res 21: 342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277. [DOI] [PubMed] [Google Scholar]
- Schwarz EM, Kato M, Sternberg PW. 2012. Functional transcriptomics of a migrating cell in Caenorhabditis elegans. Proc Natl Acad Sci 109: 16246–16251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieth J, Brooke G, Kuersten S, Lea K, Blumenthal T. 1993. Operons in C. elegans: Polycistronic mRNA precursors are processed by trans-splicing of SL2 to downstream coding regions. Cell 73: 521–532. [DOI] [PubMed] [Google Scholar]
- Tomioka M, Naito Y, Kuroyanagi H, Iino Y. 2016. Splicing factors control C. elegans behavioural learning in a single neuron by producing DAF-2c receptor. Nat Commun 7: 11645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress ML, Abascal F, Valencia A. 2017. Alternative splicing may not be the key to proteome complexity. Trends Biochem Sci 42: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zhang P, Shu Y, Yuan F, Zhang Y, Zhou Y, Jiang M, Zhu Y, Hu L, et al. 2014. Alternative splicing at GYNNGY 5′ splice sites: more noise, less regulation. Nucleic Acids Res 42: 13969–13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S. 2010. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler AM. 2012. Pre-mRNA splicing and its regulation in Caenorhabditis elegans. WormBook: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.