

SUMMARY
Epstein-Barr virus (EBV) in tumor cells is predominately in latent phase but the virus can undergo lytic reactivation in response to various stimuli. However, the cellular factors that control latency and lytic replication are poorly defined. In this study, we demonstrated that a cellular factor, PIAS1, restricts EBV lytic replication. PIAS1-depletion significantly facilitated EBV reactivation, while PIAS1-reconstitution had the opposite effect. Remarkably, we found that various lytic triggers promote caspase-dependent cleavage of PIAS1 to antagonize PIAS1-mediated restriction, and that caspase inhibition suppresses EBV replication through blocking PIAS1 cleavage. We further demonstrated that a cleavage-resistant PIAS1 mutant suppresses EBV replication upon B cell receptor activation. Mechanistically, we demonstrated that PIAS1 acts as an inhibitor for transcription factors involved in lytic gene expression. Collectively, these results establish PIAS1 as a key regulator of EBV lytic replication and uncover a mechanism by which EBV exploits apoptotic caspases to antagonize PIAS1-mediated restriction.
In Brief
The lifecycle of Epstein-Barr virus (EBV) is tightly regulated by cellular factors. Zhang et al. identify PIAS1 as a critical host factor that suppresses EBV lytic replication. Upon B cell receptor activation or chemical perturbation, EBV hijacks host caspases to cleave and inactivate PIAS1 for efficient replication.
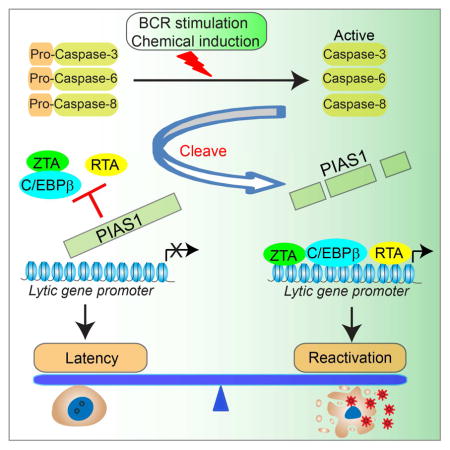
INTRODUCTION
Epstein-Barr virus (EBV) is the causative agent of a number of human cancers, including nasopharyngeal carcinoma, subtypes of gastric carcinoma and several types of lymphomas (Young and Rickinson, 2004; Young et al., 2016). EBV has two distinct phases in its life cycle, latency and lytic replication, which have characteristic viral gene expression profiles. During latency, the EBV genome is maintained as a circular episome and is replicated in synchrony with the host chromosomal DNA once per cell cycle. However, during lytic infection, a cascade of viral genes are expressed with massive induction of viral DNA replication. Two immediate early gene products, ZTA and RTA, are essential for the switch from latency to the lytic cycle (Kenney and Mertz, 2014). Cellular factors that regulate the expression of ZTA and RTA play important roles in EBV lytic cycle activation (Kenney and Mertz, 2014).
Protein inhibitor of activated STAT 1 (PIAS1) was originally identified as a negative regulator of STAT1 signaling (Liao et al., 2000; Liu et al., 1998; Shuai and Liu, 2005). PIAS1 is one of four members of the PIAS family that consists of PIAS1, PIASx/PIAS2, PIAS3 and PIASy/PIAS4. Subsequent studies indicated that all PIAS proteins have Small Ubiquitin-like MOdifier (SUMO) E3 ligase activity with the ability to SUMOylate both viral and cellular proteins to alter their functions (Chang et al., 2004; Kahyo et al., 2001; Kim et al., 2014; Lee et al., 2003). More recent studies suggest that PIAS1 functions as an epigenetic regulator in the self-renewal and differentiation of hematopoietic stem cells, the differentiation of regulatory T cells, and breast tumorigenesis (Liu et al., 2010; Liu et al., 2014a; Liu et al., 2014b). PIAS1 also functions as a chromatin-bound co-regulator for the androgen receptor (Toropainen et al., 2015). EBV RTA has been shown to be SUMOylated by PIAS1, PIAS2 and RanBPM and its SUMOylation correlated with EBV replication in P3HR-1 cells (Chang et al., 2004; Chang et al., 2008; Liu et al., 2006).
PIAS1 has been shown to play a positive role in viral infection for Ebola virus (Chang et al., 2009), Sendai virus (Li et al., 2013) and Murine γ-herpesvirus-68 (MHV-68) (Liu et al., 2004) by inhibiting the host immune response. In contrast, recent studies proposed that PIAS1 is a restriction factor for ICP0-null herpes simplex virus 1 (HSV-1) and possibly adeno-associated virus (Brown et al., 2016; Holscher et al., 2015). However, the underlying mechanisms by which PIAS1 restricts these viruses remain unclear. Moreover, aside from regulating RTA SUMOylation, whether and how PIAS1 regulates the EBV life cycle is not known.
Apoptosis and the EBV life cycle are intrinsically linked together. It was reported that various lytic triggers induce apoptosis together with EBV lytic replication (Feng et al., 2004; Fukuda and Longnecker, 2005; Hui et al., 2012; Inman et al., 2001). Other recent studies suggested that apoptosis triggers the lytic replication of a variety of herpesviruses, possibly through a caspase-activation dependent pathway (Du et al., 2012; Gastaldello et al., 2013; Prasad et al., 2012; Prasad et al., 2013). However, it is a long-standing mystery how these viruses utilize or subvert caspases to facilitate viral replication.
In this study, we present evidence that PIAS1 restricts EBV lytic replication through suppressing the transcriptional activity of key viral and cellular factors. Furthermore, we demonstrate a previously unrecognized regulation of PIAS1 by caspases upon B cell receptor (BCR) activation and chemical induction, which represents a strategy exploited by EBV to promote efficient viral replication.
RESULTS
PIAS1 depletion facilitates EBV lytic replication
PIAS1 plays different and even opposite roles in the infection of different viruses (Brown et al., 2016; Chang et al., 2009; Li et al., 2013; Liu et al., 2004). To test whether PIAS1 regulates EBV lytic replication, we utilized a Burkitt’s lymphoma cell line, Akata (EBV+) cells, as a model system. These cells express surface immunoglobulin receptors of the G(κ) class (IgG) and anti-IgG cross-linking mediated BCR activation is a physiological relevant stimulus for inducing EBV reactivation (Takada, 1984). First, we depleted PIAS1 in Akata (EBV+) cells using a CRISPR/Cas9 genomic editing approach (Figure 1A). Western blot analysis confirmed that two guide RNAs (PIAS1-sg-1 and PIAS1-sg-2) efficiently reduced PIAS1 expression compared with non-targeting control (NC) (Figure 1B). By sequencing the genomic DNA spanning the CRISPR/Cas9 targeting region of the PIAS1-sg1 cell line, we found that 9 out of 12 clones contain PIAS1 gene disruption adjacent to PAM sequence (Figure S1A), further confirming the high depletion efficiency of PIAS1 by CRISPR/Cas9. We then analyzed the expression of Immediate-Early (IE), Early and Late genes by qPCR. Interestingly, the expression of all the lytic genes tested increased in both PIAS1-depleted cells in the absence or presence lytic induction (Figure 1C and Figure S1B), suggesting that PIAS1 depletion facilitates EBV reactivation.
Figure 1. PIAS1 depletion facilitates EBV lytic replication.

(A) Schematic representation of Cas9 target sites for guide RNA sg-1 and sg-2.
(B) Akata (EBV+) cells were used to establish stable cell lines using guide RNA constructs sg-1, sg-2 and a non-targeting control (NC). Western blot analyses showing PIAS1 depletion in the two cell lines as indicated.
(C) PIAS1-depletion leads to enhanced lytic gene expression. PIAS1-depleted (sg-1) and control (NC) cells were either untreated or treated with anti-IgG for 24 hr. RNAs from these cells were extracted and analyzed by RT-qPCR using primers as indicated. The values of NC control without treatment were set as 1.
(D–E) PIAS1 depletion leads to enhanced EBV protein expression and viral DNA replication. The PIAS1-depleted and control cells were treated with anti-IgG induced cross-linking of BCR. Viral protein expression was monitored by WB using antibodies as indicated in (D) and viral DNA replication was measured by qPCR in (E). The value of NC control at 0 hr (lane 3) was set as 1.
(C and E) Representative results from three biological replicates are presented. Error bars indicate the standard deviation. *, p < 0.01; **, p < 0.001
See also Figure S1.
Next, we performed western blot analysis of these cells with or without lytic induction. We detected ZTA expression with longer exposure time without lytic induction in PIAS1-depleted cells, while RTA and BGLF4 proteins were below the detection limit without lytic induction (Figure 1D, lanes 1, 2 vs 3). Moreover, there was a significant increase of viral DNA copies when PIAS1 was knocked down (Figure 1E, lanes 1, 2 vs 3), suggesting that PIAS1-depletion promotes low level viral DNA replication. Interestingly, we also found that PIAS1-depletion significantly facilitates EBV ZTA, RTA and BGLF4 protein expression upon BCR activation triggered by IgG cross-linking (Figure 1D, lanes 4, 5 vs 6 and lanes 7, 8 vs 9). Consistently, the viral DNA copies in two PIAS1-depletion cell lines (sg-1 and sg-2) were significantly higher compared to the non-targeting control cell line (NC) following BCR activation (Figure 1E, lanes 4, 5 vs 6). To further confirm whether PIAS1 depletion facilitates EBV replication, we used Akata-BX1 (EBV+) cells (Molesworth et al., 2000). The recombinant EBV in these cells contains a GFP marker, which is suitable for measuring EBV titer by a Raji cell infection assay (Li et al., 2011; Meng et al., 2010). We depleted PIAS1 in Akata-BX1 (EBV+) cells and, interestingly, we found that PIAS1-depletion dramatically facilitated ZTA, RTA and BGLF4 protein expression (Figure S1C) and mature virus release to the medium (Figure S1D).
To further demonstrate that the observed phenotype was not due to off-target effects, we reconstituted PIAS1 back into the PIAS1-depleted cells. We found that PIAS1 restoration suppressed EBV ZTA, RTA and BGLF4 protein expression compared with PIAS1-depleted cells upon IgG cross-linking (Figure 2A, lanes 2–3 vs 5–6). Moreover, EBV DNA replication was also dramatically reduced upon PIAS1 reconstitution (Figure 2B, lanes 2–3 vs 5–6). Together these results suggest that PIAS1 restricts EBV replication possibly through limiting viral gene expression.
Figure 2. PIAS1 reconstitution suppresses EBV lytic replication.

(A) Akata (EBV+) sg-1 cells were used to establish PIAS1-expressing stable cell lines using pLX-PIAS1 lentiviral construct. Western blot analyses showing PIAS1, ZTA, RTA and BGLF4 expression level in different cell lines upon IgG cross-linking as indicated.
(B)Viral DNA replication was measured by qPCR using primers to BALF5. Representative results from three biological replicates are presented. The value of NC control at 0 hr (lane 7) was set as 1. Error bars indicate the standard deviation. *, p < 0.01.
To demonstrate the effect of PIAS1 in broader settings, we knocked out PIAS1 in four additional cell lines that cover three latency types, Kem-I and P3HR-1 Burkitt’s lymphoma cells (type I), SNU-719 gastric cancer cells (type I/II) and an EBV transformed LCL (type III). We observed universal higher reactivation for EBV in all these cells (Figure S1E–H), reinforcing that PIAS1 is a key EBV restriction factor. In addition to BCR stimulation, we also noticed that chemical inducers also strongly promote EBV lytic protein expression in PIAS1-depleted P3HR-1 and SNU-719 cells (Figure S1G–H).
By analyzing RNA-seq data from over 140 LCLs (Arvey et al., 2012), interestingly, we found that PIAS1 gene level negatively correlates with EBV lytic gene expression (Figure S1I–L and Table S1). We also found that PIAS1, but not PIAS2/3/4 gene level is significantly lower in EBV and KSHV-dually positive cell lines derived from infected huNSG mice, which support higher EBV lytic gene expression, than that in EBV-only cell lines derived from huNSG mice, which have lower EBV lytic gene expression (Figure S1M) (McHugh et al., 2017).
Moreover we found that PIAS1-depletion also facilitates the lytic gene expression of another gamma-herpesvirus, Kaposi’s sarcoma-associated herpesvirus (KSHV), suggesting a conserved role of PIAS1 in suppressing gamma-herpesvirus reactivation (Figure S1N).
PIAS1 is downregulated during EBV lytic replication
During the course of EBV lytic replication, we also noticed that PIAS1 protein level decreased dramatically (Figure 1D, lanes 3, 6 and 9 and Figure 2A lanes 1–3 and 7–9). To test whether the transcription of PIAS1 was affected, we monitored the mRNA level of PIAS1 upon BCR activation and found that PIAS1 mRNA was even slightly upregulated (Figure S2A), suggesting that PIAS1 is regulated at protein level. To further explore whether PIAS1 is regulated by viral replication or BCR activation, we monitored the PIAS1 protein level of both EBV-positive and EBV-negative Akata cells following IgG cross-linking. As shown in Figure S2B, BCR activation induced the decrease of PIAS1 in both Akata (EBV+) and Akata-4E3 (EBV−) cells, suggesting the BCR activation triggers the destabilization of PIAS1. In eukaryotic cells, two major pathways responsible for protein degradation are the ubiquitin-proteasome pathway and lysosomal proteolysis. To test whether PIAS1 degradation observed in our system results from these two pathways, we treated the IgG cross-linked cells together with a proteasome inhibitor (MG132) and a lysosome inhibitor (chloroquine). We found that proteasome and lysosome inhibition could not block PIAS1 degradation (Figure S2C, lanes 3, 4, 7 and 8) in the two Akata cell lines upon BCR activation.
Our recent study suggests that BCR activation triggers apoptosis induction in both EBV-positive and EBV-negative Akata cells (Lv et al., 2017). We reasoned that apoptosis induction may play a role in PIAS1 degradation. To test this possibility, we monitored the degradation kinetics of PIAS1. Interestingly, we found that PIAS1 degradation coincides with the production of cleaved PARP and other cleaved caspase substrates (Figure 3A). Because caspase-3, -6 and -8 were all activated upon BCR activation (Figure 3B), we reasoned that caspase activation may play a role in PIAS1 de-stabilization. To test this possibility, we used a pan caspase inhibitor Z-VAD-FMK to pretreat the Akata (EBV+) and Akata-4E3 (EBV−) cells before IgG cross-linking and we found that PIAS1 degradation was blocked in both cells compared to the non-treatment controls (Figure 3C, lanes 3 and 6). Interestingly, we found that, in addition to the decreased PIAS1 level upon IgG cross-linking, a smaller ~25 kDa fragment was detected using the PIAS1 antibody (Figure 3D, lane 2), and that pretreatment the cells with a caspase-3/7 inhibitor blocked the generation of this fragment (Figure 3D, lane 3). Because the PIAS1 antibody recognizes a C-terminal peptide (a peptide surrounding Ser550), the small fragment observed possibly represents a C-terminal cleaved PIAS1. Together, these results suggest that PIAS1 is cleaved by caspases upon BCR activation.
Figure 3. PIAS1 is downregulated following caspase activation upon BCR activation.

(A) Immunoblot showing PIAS1 degradation, the generation of cleaved-PARP and other cleaved caspase substrates, and the accumulation of EBV ZTA and BGLF4. Akata (EBV+) and Akata-4E3 (EBV−) cells were treated with anti-IgG antibody for 0 to 48 hrs as indicated.
(B) Western blot analyses showing caspase activation upon IgG cross-linking.
(C) Caspase inhibition blocks PIAS1 degradation. The cells were either untreated or pretreated with a pan canspase inhibitor (Z-VAD-FMK, 50 μM) for 1 hr and then anti-IgG antibody was added for 48 hrs. WB was performed using antibodies as indicated.
(D) Caspase inhibition blocks PIAS1 cleavage. Akata (EBV+) cells were either untreated or pretreated with a caspase-3/7 inhibitor (Z-DEVD-FMK, 50 μM) for 1 hr and then anti-IgG antibody was added for 48 hrs. WB was performed using antibodies as indicated. Arrow denotes cleaved PIAS1.
See also Figures S2–S4.
PIAS1 is cleaved by caspase-3, -6 and -8
To further prove that caspases can cleave PIAS1, we performed an in vitro cleavage assay using individual recombinant caspases and PIAS1. To facilitate the detection of cleaved PIAS1 fragments, we generated an N-terminal Halo-V5 tagged PIAS1 construct and purified the PIAS1 protein from transfected cells using a Halo-tag purification method (Li et al., 2015). After TEV protease cleavage, V5-PIAS1 was eluted for the in vitro cleavage assay. Because anti-V5 and anti-PIAS1 antibodies recognize N- and C-terminal of PIAS1 respectively (Figure 4A), both N- and C-terminal cleaved fragments can be detected by using these two antibodies.
Figure 4. Caspase-3, -6 and -8 cleave PIAS1 in vitro.

(A) Schematic representation of the Halo-V5-PIAS1. Red dot denotes the epitope position for anti-PIAS1 antibody.
(B) V5-tagged wild-type PIAS1 was incubated with individual recombinant caspase for 2 hrs at 37°C. WB was performed using either anti-V5 (B) or anti-PIAS1 (C) antibodies. The relative positions of predicted cleaved bands were labeled as indicated.
See also Figure S2.
Interestingly, we found that caspase-1, -3, -6, -7, -8 and-10 all cleave PIAS1 while the major caspases responsible for PIAS1 cleavage are caspase-3, -6 and -8 (Figure 4B and 4C, lanes 3, 6 and 8). By examining the size of these cleaved fragments, we noticed that caspase-3 mediated cleavage generates one N-terminal (Figure 4B, lane 3, 50 kDa) and one C-terminal (Figure 4C, lane 3, 25 kDa) fragments, caspase-6 mediated cleavage generates one N-terminal (Figure 4B, lane 6, 15 kDa) and two C-terminal (Figure 4C, lane 6, 70 kDa and 25 kDa) fragments, and the cleavage by caspase-8 leads to two N-terminal (Figure 4B, lane 6; 15 kDa and 50 kDa) and one C-terminal (Figure 4C, lane 8, 70 kDa) fragments. The 25 kDa fragment generated by caspase-3 and -6 (Figure 4C, lanes 3 and 6) matched with that observed in EBV-replicating cells (Figure 3D, lane 2), suggesting that PIAS1 is also cleaved by caspase-3 or -6 in vivo. Together, these results suggest that caspase-3 cleaves PIAS1 at one major site and caspase-6 and -8 cleave PIAS1 at two major sites with one shared with caspase-3.
Caspases cleave their substrates at specific tetrapeptides (canonical P4-P1 sites) with a highly conserved aspartate (D) at the P1 position, so we used several tools (Cascleave, Cascleave 2.0 and CaspDB) to predict the potential cleavage sites (Kumar et al., 2014; Song et al., 2010; Wang et al., 2014) (Table S2). The top two predicted caspase-3/7 cleavage sites (D100 and D433 obtained by Cascleave 2.0) are presented in Figure 5A. The cleavage on D433 would generate a 23.4 kDa band, which is close to the size of the cleaved fragment observed (Figure 3D, lane 2 and Figure 4C, lanes 3 and 6). The predicted cleavage at D100 would generate a small N-terminal fragment (the calculated molecular weight with V5-tag is 13.0 kDa), which possibly reflects the 15 kDa fragment generated by caspase-6 and -8 mediated cleavage (Figure 4B, lanes 6 and 8). To verify the major cleavage sites of PIAS1, we mutated D100 and D433 together with another potential cleavage site (D148) to alanine individually or in combination. The Halo-V5-tag mutants were purified by a similar Halo-tag purification method (Figure 4A), and the eluted V5-PIAS1 proteins were subjected to an in vitro cleavage assay with caspase-3, -6 and -8. As expected, we found that PIAS1 D100A single mutant, but not D148A, blocked the generation of the 15 kDa fragment (Figure 5B, lanes 2–3 vs 6–7) and the 70 kDa fragment (Figure 5C, lanes 2–3 vs 6–7) by caspase-6 and -8. Similarly, we found that PIAS1 D433A single mutation, blocked the generation of the 50 kDa fragment (Figure 5D, lanes 1–3) and the 25 kDa fragment (Figure 5E, lanes 1–3) by caspase-3, -6 and -8. PIAS1 D100A/D433A double mutation completely blocked cleavage by caspase-3, -6 and -8 (Figure 5F–5G). Together, these results demonstrate that caspase-6 and -8 cleave PIAS1 at D100 and that caspase-3, -6 and -8 all cleave PIAS1 at D433. Interestingly, the cleavage sites of PIAS1 are highly conserved across different species but not among the human PIAS family proteins (PIAS2, PIAS3 and PIAS4) (Figure S2D–F), suggesting that the cleavage of PIAS1 is an evolutionary conserved event during apoptosis.
Figure 5. Caspase-3, -6 and -8 cleave PIAS1 after D100 and D433.

(A) Schematic representation of functional domains and two of the predicted cleavage sites of PIAS1. SAP (SAF-A/B, Acinus, and PIAS): DNA and protein binding domain; PINIT: nuclear localization motif; RING finger: E3 ligase domain for protein SUMOylation; SIM: SUMO interacting motif; S/T Rich: variable Ser/Thr rich region.
(B to G) V5-tagged D100A, D148A, D433A or D100/433A PIAS1 mutants were incubated with individual recombinant caspases for 2 hrs at 37°C. WB was performed using either anti-V5 (B, D and F) or anti-PIAS1 (C, E and G) antibodies. The relative positions of cleaved bands were labeled as indicated.
Caspase activation facilitates EBV lytic replication through promoting PIAS1 degradation
We hypothesize that caspase-3, -6 and -8 activation contributes to PIAS1 degradation and EBV lytic replication upon BCR stimulation. To test this hypothesis, we examined the effects of specific caspase inhibition on caspase activation, PIAS1 stability and EBV protein expression (Figure 6A and Figure S2G). Interestingly, pretreatment the Akata (EBV+) cells with inhibitors that target caspase-3/7, -6 and -8 all led to higher levels of PIAS1 and reduced expression of EBV ZTA and RTA upon BCR activation (Figure 6B, lane 2 vs lanes 3–5). The combination of two inhibitors led to further reduced viral protein expression (Figure 6B, lanes 6–7). The mRNA levels of several EBV lytic genes were dramatically suppressed by caspase inhibition (Figure 6C). Consistently, the caspase-3/7, -6 or -8 inhibition substantially suppressed viral DNA replication and pre-treatment the Akata (EBV+) cells with the combination of two inhibitors led to 20- to 60-fold reductions of viral DNA replication upon BCR activation (Figure 6D). These results suggested that caspase-mediated PIAS1 cleavage might facilitate EBV lytic gene expression. To further prove this possibility, we analyzed EBV ZTA and BGLF4 expression when PIAS1 was depleted and caspase activity was inhibited. Compared to the control cells, caspase inhibition did not fully suppress ZTA and BGLF4 expression when PIAS1 was depleted (Figure 6E, lanes 2–4 vs 6–8). The relative viral DNA copies in caspase inhibitor pretreated PIAS1-depleted cells were still higher than those in the control cells without caspase inhibition (Figure 6E, lanes 3–4 vs 6). These results suggested that caspase-mediated cleavage of PIAS1 contributes to efficient EBV replication.
Figure 6. Caspase activation facilitates EBV replication through PIAS1 cleavage.

(A) Caspase inhibitors and the concentration used in the following experiments.
(B–D) Caspase inhibition blocks EBV lytic replication. Akata (EBV+) cells were either untreated or pretreated with caspase inhibitors individually or in combination for 1 hr and then anti-IgG antibody was added for 48 hrs as indicated. EBV ZTA and RTA expression, and the generation of cleaved PARP were monitored by western blot (B). EBV gene expression was measured by RT-qPCR using primers as indicated (C). Relative EBV DNA copy numbers were measured by qPCR using primers specific for BALF5 and BHLF1 (D). The value of cells without treatment (lane 1) was set as 1.
(E) Caspase activation facilitates lytic replication through PIAS1 cleavage. PIAS1-depleted (sg-1) and control (NC) Akata (EBV+) cells were either untreated or pretreated with caspase inhibitors for 1 hr and then anti-human IgG antibody was added for 48 hrs. Western blot was performed using antibodies as indicated. Relative EBV DNA copy numbers were measured by qPCR using primers specific for BALF5. The value of NC control without treatment (lane 5) was set as 1.
(F–G) Cleavage-resistant PIAS1 further suppresses EBV replication. Akata (EBV+) sg-1 cells were used to establish PIAS1-knockout (PIAS1-KO) single cell clones. PIAS1-KO cells were then reconstituted with wild-type (WT) or cleavage-resistant (D100/433A) PIAS1 using lentiviral constructs. Western blot analyses showing PIAS1, ZTA, RTA and BGLF4 expression level in these cell lines upon IgG cross-linking as indicated (F). Viral DNA replication was measured by qPCR using primers specific to BALF5 and BHLF1 (G). The value of WT PIAS1 at 0 hr (lane 4) was set as 1.
Representative results from three biological replicates are presented. Error bars indicate the standard deviation. *, p < 0.01.
See also Figures S2–S4.
Cleavage-resistant PIAS1 blocks EBV lytic replication
To further prove caspase-mediated PIAS1 cleavage is important for EBV lytic replication, we created a cleavage-resistant PIAS1 construct, pLX-PIAS1 (D100/433A). To provide a clean background for testing the function of this PIAS1 mutant in EBV replication, we utilized a limiting dilution method to establish a PIAS1 knockout single cell clone using the PIAS1-depleted Akata (EBV+) pool cells. We then confirmed the PIAS1 knockout status by western blot and DNA sequencing (Figure S3A–D). As expected, PIAS1 knockout facilitated the expression of EBV ZTA and RTA upon BCR activation (Figure S3A). We then used the PIAS1-knockout cells to reconstitute wild-type and the cleavage-resistant mutant PIAS1 (D100/433A). Interestingly, the protein level of wild-type PIAS1 gradually decreased following BCR activation but the PIAS1 (D100/433A) mutation blocked PIAS1 degradation (Figure 6F, lanes 4–6 vs 7–9), suggesting that D100 and D433 are bona fide caspase cleavage sites in cells. Both wild-type and the cleavage-resistant PIAS1 strongly suppressed EBV ZTA and RTA protein accumulation upon lytic induction (Figure 6F, lanes 1–3 vs 4–9) with a stronger suppression observed for the cleavage-resistant PIAS1. Consistent with the western blot results, EBV DNA replication was dramatically suppressed in PIAS1 (D100/433A)-expressing cells following BCR activation (Figure 6G, lanes 3 vs 6 and 9). Together, these results indicated that caspase-mediated PIAS1 cleavage fosters EBV lytic replication.
BCR activation of lytic infection occurs not only in tumor cell lines but also in freshly isolated B cells from patients (Kosowicz et al., 2017). We reasoned that BCR activation could also trigger the cleavage of PIAS1 in primary human B cells. As expected, we found that anti-IgM induced BCR activation leads to the cleavage of PIAS1 in primary human B cells, which can be blocked by a pan-caspase inhibitor (Figure S3E).
LMP2A has been shown to block strong BCR stimuli (Fukuda and Longnecker, 2005), we reasoned that LMP2A may also suppress PIAS1 degradation, we examined the PIAS1 cleavage and EBV reactivation when LMP2A is stably expressed in Akata cells. Interestingly, we found that LMP2A expression suppressed PIAS1 degradation and thus EBV lytic protein expression upon BCR stimulation (Figure S3F). By using LCLs derived from three different individuals, we noticed that BCR activation by anti-IgM can still partially trigger caspase activation, PARP cleavage and ZTA protein accumulation. This also correlates with a slower but still noticeable degradation of PIAS1 than that seen in the Akata cells (Figure S3G, lanes 1–3, 4–6 and 7–9 vs S3A, lanes 1–3).
In addition to BCR stimulation, we also tested other types of lytic inducer in PIAS1 stability and EBV reactivation. Interestingly, we found that lytic induction by TPA and sodium butyrate triggers PIAS1 degradation in P3HR-1 cells and caspase inhibition suppresses PIAS1 degradation, inhibits EBV lytic protein expression and blocks viral DNA replication (Figure S4A–C). We further showed that lytic induction by gemcitabine also triggers PIAS1 degradation and caspase inhibition suppresses PIAS1 degradation, inhibits EBV lytic protein expression (Figure S4D–E).
PIAS1 suppresses ZTA, RTA and C/EBPβ-mediated EBV lytic gene expression
The above results suggested that PIAS1 suppresses EBV lytic replication through modulating viral gene expression. We hypothesize that PIAS1 directly suppresses lytic gene expression induced by cellular and viral transcription factors. To test this hypothesis, we examined the function of PIAS1 in EBV promoter activity by luciferase reporter assays. We first checked PIAS1 in EBV ZTA-mediated activation of ZTA (Zp) and RTA (Rp) promoters and found that PIAS1 suppresses ZTA-mediated Zp and Rp activation in a dose-dependent manner but has minimal impacts on HSV-TKp activatin (Figure 7A and Figure S5A). To test whether PIAS1 directly interacts with ZTA, we performed co-immunoprecepitation (Co-IP) experiments and found that full-length and truncated PIAS1 did not interact with ZTA (Figure S5B), suggesting that PIAS1 blocks the function of ZTA independent of direct protein-protein interaction.
Figure 7. PIAS1 blocks C/EBPβ, ZTA and RTA-mediated lytic gene activation.

(A) Suppression of Zp- and Rp-Luc reporters by PIAS1. 293T cells were transfected with 250 ng of plasmid DNA encoding Zp or Rp-Luc, and effector plasmid DNA expressing ZTA and increasing amount of PIAS1 as indicated. The total amount of effector plasmid DNA used in each transfection was normalized by adding vector DNA.
(B) PIAS1 suppresses ZTA-C/EBPβ-mediated Zp promoter activation. 293T cells were transfected with 250 ng Zp-Luc and other plasmid DNA as indicated.
(C) PIAS1 suppresses RTA-mediated BGLF2 promoter activation. 293T cells were transfected with 250 ng of BGLF2p-Luc and other plasmid DNA as indicated.
(D) PIAS1 binds to EBV promoters. The relative positions of EBV ZTA, RTA and BHLF1 are depicted in the top panel. ChIP-PCR analysis performed on Akata (EBV+) cells showing PIAS1 binding to ZTA (Zp) and RTA (Rp) promoters but not BHLF1 (BHLF1p) promoter in the PIAS1-expressing control cells (NC). ChIP by a nonspecific IgG was included as a negative control.
Representative results from three biological replicates are presented. Error bars indicate the standard deviation. *, p < 0.01.
See also Figures S5–S7.
Because ZTA synergizes with C/EBPβ to promote EBV lytic replication (Huang et al., 2006; Shirley et al., 2011) and PIAS1 has been shown to suppress the transcriptional activity of C/EBPβ (Liu et al., 2013), we reasoned that PIAS1 may suppress ZTA and C/EBPβ-mediated Zp activation. To test this hypothesis, we co-transfected ZTA, C/EBPβ and/or PIAS1 together with a Zp-luciferase reporter, and we found that PIAS1 strongly suppressed ZTA and C/EBPβ-mediated Zp luciferase activity (Figure 7B, lanes 2 vs 4–5). Because the E3 SUMO ligase activity of PIAS1 has been implicated in suppressing C/EBPβ transactivation, we tested the E3 ligase-dead mutant PIAS1 (C351S) in ZTA-C/EBPβ-mediated Zp activation. Although the expression level of PIAS1 (C351S) was lower than wild-type PIAS1, this mutant still suppressed Zp activation at a higher dosage (Figure 7B, lanes 2 vs 8), suggesting that PIAS1 suppresses ZTA and C/EBPβ-mediated Zp activation in a SUMO ligase activity-independent manner.
PIAS1 has been shown to up-regulate RTA-mediated EBV lytic gene activation through SUMOylation (Chang et al., 2004). We also tested the role of PIAS1 in RTA-mediated activation of a late gene BGLF2 promoter (BGLF2p). In contrast, we found that PIAS1 also suppressed RTA-mediated luciferase activity driven by BGLF2p in a dose-dependent manner (Figure 7C, lanes 3–6). Similarly, the SUMO ligase-deficient PIAS1 also suppressed RTA-mediated BGLF2p activation in a dose-dependent manner (Figure 7C, lanes 7–10). In order to define the domains within PIAS1 responsible for interaction with RTA, we created a series of PIAS1 truncation mutants (Figure S5F). We found that both the N-terminal and central parts PIAS1 interacted with RTA while the C-terminal of PIAS1 did not (Figure S5G, Lanes 1, 2, 4 and 5 vs 3 and 6). To further probe the domains required for PIAS1-mediated repression, we tested the role of both N-terminal and C-terminal PIAS1 in RTA-mediated activation of BGLF2p. Interestingly, we found that the N-terminal PIAS1 failed to suppress the transcriptional activity of RTA, while higher levels of C-terminal domain of PIAS1 still inhibited it (Figure S5H). These results suggested that the N-terminal PIAS1 binds to RTA and the C-terminal PIAS1 blocks RTA activation and both domains are required to maximally suppress RTA. Because caspase-mediated cleavage of PIAS1 separated these domains and the C-terminal PIAS1 was less stable (Figure S3D), suggesting that BCR activation antagonizes PIAS1-mediated repression to facilitate EBV replication. To further confirm our observations, we also repeated the luciferease assays using SNU-719 cells as a physiologically relevant cell line. Similar results were obtained for PIAS1 suppression of ZTA, RTA and C/EBPβ-mediated EBV lytic gene activation (Figure S6A–E).
PIAS1 blocks the recruitment of ZTA, RTA and C/EBPβ to EBV lytic gene promoters
PIAS1 was reported to directly associate with cellular promoters (Liu et al., 2010; Liu et al., 2014b; Toropainen et al., 2015). We reasoned that PIAS1 may also interact with viral promoters in EBV-infected cells. To test this hypothesis, we performed chromatin immunoprecipitation (ChIP) experiments on PIAS1-depleted (PIAS1-sg-1) and control (NC) cells using a PIAS1 antibody and examined the enrichment of PIAS1 on Zp, Rp and Orilyt promoter (BHLF1p) by PCR using specific primers. As expected, we found PIAS1 is enriched in the Zp and Rp in control cells (NC) but not in PIAS1-depleted (sg-1) cells (Figure 7D). However, we did not detect the PIAS1 association with the Orilyt promoter, suggesting that PIAS1 may specifically suppress Zp and Rp activity (Figure 7D).
To further explore the mechanism of PIAS1-mediated restriction, we performed a series of ChIP experiments. Using Akata (EBV+) cells, we demonstrated that BCR crosslinking facilitates the recruitment of ZTA and C/EBPβ to EBV ZTA promoter (Zp) and PIAS1 knockout further enhances their recruitment to Zp (Figure S6F). We also demonstrated C/EBPβ interacts with both ZTA and PIAS1, and RTA interacts with PIAS1 in lytically induced Akata cells in vivo (Figure S6G–H). To further prove whether PIAS1 suppresses the recruitment of ZTA, RTA and C/EBPβ to EBV lytic gene promoters, we performed ChIP assays using EBV-positive SNU-719 cells in conjugation with transfections. We found that PIAS1 expression blocks the recruitment of ZTA, RTA and C/EBPβ to EBV lytic gene promoters, possibly thorough protein-protein interactions (Figure S7). In all cases the further recruitment of PIAS1 to viral promoters was not affected by either IgG crosslinking or overexpression, suggesting that PIAS1 mediated repression was not dependent on further chromatin association (Figures S6F, S7B and S7D, PIAS1 ChIP). These results together suggested that PIAS1 can block the DNA binding activity of ZTA, RTA and C/EBPβ.
DISCUSSION
In this report, we have identified an important mechanism by which PIAS1 restricts EBV lytic replication through modulating viral gene expression. Our investigation into the regulation of PIAS1 upon BCR activation and chemical induction led us to uncover a critical role for caspases in antagonizing PIAS1 function and in facilitating viral replication.
PIAS1 plays a key role in the negative regulation of host immune responses. By inhibiting IRF3, IRF7 and STAT1 mediated host anti-viral responses. PIAS1 facilitates the infections of Ebola virus, Sendai virus, and MHV-68 (Chang et al., 2009; Li et al., 2013; Liu et al., 2004). In contrast, our study revealed that PIAS1 restricts EBV lytic replication upon reactivation (Figures 1, 2 and S1). Mechanistically, PIAS1 suppresses viral gene expression through blocking the DNA binding of cellular factor C/EBPβ and EBV ZTA/RTA, all critical for EBV reactivation (Figures 7 and S6–S7). PIAS1 restriction of herpesviruses has been reported for ICP0-null HSV-1 (Brown et al., 2016). PIAS1 is actively recruited to nuclear sites that contain infecting HSV genomes upon primary infection but it is not further recruited to EBV genomes upon lytic induction or PIAS1 overexpression (Figures S6–S7). Therefore, it appears that PIAS1 affects the EBV replication in a different way compared with that of HSV.
One insight into PIAS1 regulation upon BCR activation and chemical induction comes from the evidence that caspase inhibition blocks PIAS1 degradation (Figures 3 and S4). Although PIAS1 protein stability has been shown to be regulated by the ubiquitin-proteasome pathway and the E3 ubiquitin ligases SIAH1/SIAH2, HECTD2 and adaptor protein necdin are known to promote PIAS1 ubiquitination and subsequent degradation (Coon et al., 2015; Depaux et al., 2007; Gur et al., 2014), our study reveals a surprising regulation of PIAS1 by apoptotic caspases. We further provided evidence that caspase-dependent cleavage of PIAS1 plays a key role in EBV lytic replication (Figure 6).
There is a long-standing mystery regarding the linkage between apoptosis and EBV lytic replication (Feng et al., 2004; Fukuda and Longnecker, 2005; Hui et al., 2012; Inman et al., 2001). Although many viruses, including EBV, have developed mechanisms to suppress apoptosis for their own benefit, evidence has emerged for viral hijacking of host caspases for facilitating viral replication (Connolly and Fearnhead, 2017). Our demonstration of caspase-dependent cleavage of the cellular repressor PIAS1 (Figures 4–6) provides an explanation for the role of apoptotic caspases in the replication of EBV, and possibly other viruses. This is consistent with recent studies showing that apoptotic induction triggers the lytic replication of a variety of herpesviruses (De Leo et al., 2017; Du et al., 2012; Gastaldello et al., 2013; Harold et al., 2016; Kim et al., 2015; Lin et al., 2015; Prasad et al., 2012; Prasad et al., 2013).
The results obtained using specific caspase inhibitors should be interpreted cautiously because the regulation caspase activity is very complex as each of them can possibly regulate the other caspases through cleavage. In addition, specific caspase inhibitors at higher concentration may also inhibit other caspases (Eeva and Pelkonen, 2004; Pereira and Song, 2008). Indeed, in our experiment conditions, we found that caspase-3/7 inhibitor suppresses caspase-8 activation (Figure S2G, lane 3 vs 2), caspase-6 inhibitor suppresses caspase-3 and -8 activation (Figure S2G, lane 4 vs 2), and caspase-8 inhibitor also partially suppresses caspase-3 activation (Figure S2G, lane 5 vs 2).
In addition to PIAS1, caspase inhibition might also regulate the cleavage of other substrates important for EBV reactivation. For examples, a recent publication from the Lieberman group showed that RAD21 cleavage by caspases contributes to KHSV reactivation (De Leo et al., 2017) and the cleavage of RAD21 may also contribute to EBV reactivation. However, when PIAS1 was pre-depleted, caspase inhibition could not fully block viral reactivation (Figure 6E), suggesting that caspase-mediated cleavage of PIAS1 is a key for EBV reactivation and that caspase inhibitor effects on EBV gene expression is at least partially tied to PIAS1.
PIAS1 has been shown to regulate promoter activity through SUMOylation (Toropainen et al., 2015) or through recruiting other factors to alter histone methylation and acetylation (Liu et al., 2010). Although PIAS1 was reported to enhance the RTA transcriptional activity through SUMOylation (Chang et al., 2004), in contrast, our results suggested that PIAS1 functions as an inhibitor for ZTA-C/EBPβ- and RTA-mediated EBV lytic gene expression and that the inhibition function of PIAS1 is largely independent of its SUMO ligase activity (Figure 7, Figures S5H and S6A–E). PIAS1 directly interacts with RTA and inhibits its DNA binding (Figures S6–7), suggesting that PIAS1 blocks RTA-mediated gene activation in a similar way to PIAS1 suppression of STAT1-, NF-κB- and IRF3-mediated gene expression (Li et al., 2013; Liu et al., 1998; Liu et al., 2005). Although PIAS1 depletion did not affect the global SUMOylation level (Figure S5C), it did reduce the level of SUMOylation on EBV Zp promoter (Figure S5D), suggesting that Zp-associated proteins might be regulated by PIAS1-mediated SUMOylation. In contrast to the regulation of histone modifications on cellular Foxp3 promoter (Liu et al., 2010), PIAS1 did not affect the level of H3K4Me3, H3K27Me3, H3K9Me3 and H3K9Ac associated with Zp (Figure S5E), suggesting that PIAS1 regulates viral promoter activity independent of the histone methylation or acetylation.
According to the database from Atlas of Genetics and Cytogenetics in Oncology and Haematology, the human population contains genetic polymorphisms of PIAS1, including the G298A (D100N mutation) and G1297A (D433N mutation) polymorphisms that are predicted to affect PIAS1 cleavage. It will be interesting to examine whether PIAS1 polymorphisms correlate with EBV-associated diseases in the future.
In summary, we have discovered a hitherto unappreciated function for PIAS1 as a key host determinant of EBV lytic replication. A common strategy employed by viruses to counteract host restriction is to manipulate or utilize the existing cellular machinery. Our demonstration that EBV hijacks host caspases to facilitate viral replication through cleaving and inactivating PIAS1 represents a striking example of an emerging category of virus-host interactions, which should offer insights into understanding and controlling viral pathogenesis.
EXPERIMENTAL PROCEDURES
Ethics Statement
CD19-positive primary B cells were purchased from the Precision for Medicine (Cat# 84400 Frederick, MD). As these were derived from anonymous blood donors, no ethical approval is required.
Cell Lines and Cultures
The Akata (EBV+), Akata-4E3 (EBV−) and Akata-BX1 (EBV+) cells were grown in RPMI 1640 media supplemented with 10% FBS in 5% CO2 at 37°C (Li et al., 2015; Li et al., 2012; Li et al., 2011). 293T cells were grown in DMEM media supplemented with 10% FBS in 5% CO2 at 37°C. The P3HR-1 (ATCC), Kem-I (a gift from Jeffery Sample) (Hughes et al., 2012) and SNU-719, a naturally derived EBV-infected gastric carcinoma cell line, were grown in RPMI 1640 media supplemented with 10% FBS. The LCL cells were cultured in RPMI 1640 media supplemented with 15% FBS. The KSHV-positive B-lymphoma cells (BC3 and BCBL1) were maintained in RPMI 1640 media with 10% FBS.
PIAS1 Depletion CRISPR/Cas9 Genome Editing
To knock out PIAS1, two different sgRNAs targeting human PIAS1 were designed and cloned into lentiCRISPR v2 vector (a gift from Feng Zhang) (Sanjana et al., 2014). Packaging 293T cells were transfected with PIAS1 sgRNAs or negative controls (non-targeting sgRNA-NC) and helper vectors (pMD2.G and psPAX2; gifts from Didier Trono) using Lipofectamine 2000 reagent. Medium containing lentiviral particles and 8 μg/mL polybrene was used to infect EBV-positive and KSHV-positive cells. Infected cells were selected in medium containing 2 μg/mL puromycin.
Cloning, Gibson Assembly and Site-directed Mutagenesis
V5-PIAS1 (full length, 1–415, 409–651, 101–433, 434–651) and Halo-V5-PIAS1 plasmids were constructed using Gibson assembly. The V5-PIAS1 mutants (C351S, 1-433 stop) and Halo-V5-PIAS1 mutants (D100A, D148A, D433A and D100/433A) were generated by the QuikChange II site-Directed Mutagenesis Kit according to the manufacturer’s instructions. See the Supplemental Experimental Procedures for additional details.
Cell Treatment
To induce the EBV lytic cycle, Akata (EBV+) cells were treated with anti-IgG antibody (1:200) for 0 to 48 hrs. Akata-4E3 (EBV−) cells were treated similarly as controls. P3HR-1 cells were triggered by addition of TPA (20 ng/ml) and sodium butyrate (3 mM). The EBV lytic replication in SNU-719 cells and LCL cells was induced by addition of gemcitabine (1 μg/mL). To induce the BCR activation, the LCL cells were treated with anti-IgM antibody (20 μg/mL) for 0 to 48 hrs. For caspase inhibition assay, cells were untreated or pretreated with caspase inhibitors for 1 hrs and then treated with anti-IgG (1:200) for additional 48 hrs.
Lentiviral Transduction of PIAS1
To prepare lentiviruses, 293T cells were transfected with lentiviral vector pLX304 containing the gene of wild-type PIAS1 or cleavage-resistant mutant (D100/433A) and the help vectors (pMD2.G and psPAX2) using Lipofectamine 2000 reagent. The supernatants were collected 48 hrs after transfection and used for infection of PIAS-1 knockout cell lines. Infected cells were selected in medium containing 10 μg/mL blasticidin.
Chromatin Immuno-precipitation (ChIP) Assay
ChIP assay was performed using a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Tech) according to the manufacturer’s protocol. See the Supplemental Experimental Procedures for additional details.
In vitro Caspase Cleavage Assay
Purified PIAS protein and caspases (active human caspases group IV; ApexBio, Cat# K2060) were incubated in caspase assay buffer (50 mM HEPES, pH7.2, 50 mM NaCl, 0.1% Chaps, 10 mM EDTA, 5% Glycerol and 10mM DTT) at 37°C for 2 hrs. Reactions were stopped by boiling in 2× SDS sample buffer and samples were analyzed by western blot.
Luciferase Assay
Transfection of 293T or SNU-719 cells was performed with lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. See the Supplemental Experimental Procedures for additional details.
Reverse Transcription and Quantitative PCR (RT-qPCR)
Total RNA was extracted from cells following induction using Isolate II RNA Mini Kit (Bioline). RNA was reverse transcribed to cDNA using the High Capacity cDNA Reverse Transcription Kit. qPCR was performed using Brilliant SYBR Green qPCR master mix with specific primers. The relative expression of mRNA was normalized to β-actin expression using the comparative Ct method. Primers are listed in Table S3.
EBV DNA Detection
To measure cell associated viral DNA, total genomic DNA was extracted using the Genomic DNA Purification Kit (Cat# A1120, Promega). The relative viral genome copy numbers were determined by quantitative PCR using primers to BALF5 and BHLF1 genes with and β-actin as a control. The values were normalized by β-actin. Primers are listed in Table S3.
Quantification and Statistical Analysis
Statistical analyses employed a two-tailed Student’s t test. A p value of ≤ 0.05 was considered statistically significant. Values are given as the mean of replicate experiments. Error bars represent the standard deviation (SD) from triplicate samples.
Supplementary Material
Highlights.
PIAS1 depletion promotes the reactivation of EBV from multiple latency types
Caspases cleave PIAS1 to facilitate viral replication upon lytic induction
Cleavage-resistant PIAS1 blocks EBV replication
PIAS1 is a inhibitor for cellular and viral transactivators
Acknowledgments
We thank S. Diane Hayward (Johns Hopkins), Iain Morgan (Virginia Commonwealth University) and Matthew Weitzman (Children’s Hospital of Philadelphia) for comments and suggestions on the manuscript. We thank Richard Longnecker (Northwestern) for advice on the generation of single B cell clones. We thank Feng Zhang for sharing the lentiCRISPR v2 plasmid. We thank Didier Trono for providing the pMD2.G and psPAX2 plasmids. We thank Karl Munger for providing MSCV-N LMP2A and MSCV-N GFP plasmids and Bob Weinberg for providing pCMV-VSV-G and pUMVC plasmids. We thank Jeffery Sample (Penn State College of Medicine), Lindsey Hutt-Fletcher (Louisiana State University) and S. Diane Hayward (Johns Hopkins) for providing various cell lines. We also thank Heng Zhu (Johns Hopkins) for V5-PIAS1 construct and Mei-Ru Chen (National Taiwan University) for anti-BGLF4 antibody. This work was supported by NIH K99AI104828/R00AI104828 to RL. RL received support from the VCU Philips Institute for Oral Health Research and the VCU NCI Designated Massey Cancer Center (NIH P30 CA016059). The work was also partly supported by Institutional Research Grant IRG-14-192-40 from the American Cancer Society.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures and three tables and can be found with this article online.
AUTHOR CONTRIBUTIONS
Conceptualization, R.L. and K.Z.; Methodology, K.Z. and D.W.L.; Formal Analysis, R.L. and K.Z.; Investigation, K.Z. and D.W.L.; Writing – Original Draft, R.L. and K.Z.; Writing – Review & Editing, R.L. and K.Z.; Visualization, R.L. and K.Z.; Funding Acquisition, R.L.; Resources, K.Z. and D.W.L.; Supervision, R.L.; Project Administration, R.L.; Funding Acquisition: R.L.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe. 2012;12:233–245. doi: 10.1016/j.chom.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JR, Conn KL, Wasson P, Charman M, Tong L, Grant K, McFarlane S, Boutell C. SUMO Ligase Protein Inhibitor of Activated STAT1 (PIAS1) Is a Constituent Promyelocytic Leukemia Nuclear Body Protein That Contributes to the Intrinsic Antiviral Immune Response to Herpes Simplex Virus 1. J Virol. 2016;90:5939–5952. doi: 10.1128/JVI.00426-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LK, Lee YH, Cheng TS, Hong YR, Lu PJ, Wang JJ, Wang WH, Kuo CW, Li SS, Liu ST. Post-translational modification of Rta of Epstein-Barr virus by SUMO-1. J Biol Chem. 2004;279:38803–38812. doi: 10.1074/jbc.M405470200. [DOI] [PubMed] [Google Scholar]
- Chang LK, Liu ST, Kuo CW, Wang WH, Chuang JY, Bianchi E, Hong YR. Enhancement of transactivation activity of Rta of Epstein-Barr virus by RanBPM. Journal of molecular biology. 2008;379:231–242. doi: 10.1016/j.jmb.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Chang TH, Kubota T, Matsuoka M, Jones S, Bradfute SB, Bray M, Ozato K. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009;5:e1000493. doi: 10.1371/journal.ppat.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly PF, Fearnhead HO. Cell Death Differ. 2017. Viral hijacking of host caspases: an emerging category of pathogen-host interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon TA, McKelvey AC, Lear T, Rajbhandari S, Dunn SR, Connelly W, Zhao JY, Han S, Liu Y, Weathington NM, et al. The proinflammatory role of HECTD2 in innate immunity and experimental lung injury. Sci Transl Med. 2015;7:295ra109. doi: 10.1126/scitranslmed.aab3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leo A, Chen HS, Hu CA, Lieberman PM. Deregulation of KSHV latency conformation by ER-stress and caspase-dependent RAD21-cleavage. PLoS Pathog. 2017;13:e1006596. doi: 10.1371/journal.ppat.1006596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaux A, Regnier-Ricard F, Germani A, Varin-Blank N. A crosstalk between hSiah2 and Pias E3-ligases modulates Pias-dependent activation. Oncogene. 2007;26:6665–6676. doi: 10.1038/sj.onc.1210486. [DOI] [PubMed] [Google Scholar]
- Du T, Zhou G, Roizman B. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc Natl Acad Sci U S A. 2012;109:14616–14621. doi: 10.1073/pnas.1212661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeva J, Pelkonen J. Mechanisms of B cell receptor induced apoptosis. Apoptosis. 2004;9:525–531. doi: 10.1023/B:APPT.0000038032.22343.de. [DOI] [PubMed] [Google Scholar]
- Feng WH, Hong G, Delecluse HJ, Kenney SC. Lytic induction therapy for Epstein-Barr virus-positive B-cell lymphomas. J Virol. 2004;78:1893–1902. doi: 10.1128/JVI.78.4.1893-1902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Longnecker R. Epstein-Barr virus (EBV) latent membrane protein 2A regulates B-cell receptor-induced apoptosis and EBV reactivation through tyrosine phosphorylation. J Virol. 2005;79:8655–8660. doi: 10.1128/JVI.79.13.8655-8660.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaldello S, Chen X, Callegari S, Masucci MG. Caspase-1 promotes Epstein-Barr virus replication by targeting the large tegument protein deneddylase to the nucleus of productively infected cells. PLoS Pathog. 2013;9:e1003664. doi: 10.1371/journal.ppat.1003664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur I, Fujiwara K, Hasegawa K, Yoshikawa K. Necdin promotes ubiquitin-dependent degradation of PIAS1 SUMO E3 ligase. PLoS One. 2014;9:e99503. doi: 10.1371/journal.pone.0099503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold C, Cox D, Riley KJ. Epstein-Barr viral microRNAs target caspase 3. Virol J. 2016;13:145. doi: 10.1186/s12985-016-0602-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C, Sonntag F, Henrich K, Chen Q, Beneke J, Matula P, Rohr K, Kaderali L, Beil N, Erfle H, et al. The SUMOylation Pathway Restricts Gene Transduction by Adeno-Associated Viruses. PLoS Pathog. 2015;11:e1005281. doi: 10.1371/journal.ppat.1005281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liao G, Chen H, Wu FY, Hutt-Fletcher L, Hayward GS, Hayward SD. Contribution of C/EBP proteins to Epstein-Barr virus lytic gene expression and replication in epithelial cells. J Virol. 2006;80:1098–1109. doi: 10.1128/JVI.80.3.1098-1109.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DJ, Marendy EM, Dickerson CA, Yetming KD, Sample CE, Sample JT. Contributions of CTCF and DNA methyltransferases DNMT1 and DNMT3B to Epstein-Barr virus restricted latency. J Virol. 2012;86:1034–1045. doi: 10.1128/JVI.05923-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui KF, Ho DN, Tsang CM, Middeldorp JM, Tsao GS, Chiang AK. Activation of lytic cycle of Epstein-Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int J Cancer. 2012;131:1930–1940. doi: 10.1002/ijc.27439. [DOI] [PubMed] [Google Scholar]
- Inman GJ, Binne UK, Parker GA, Farrell PJ, Allday MJ. Activators of the Epstein-Barr virus lytic program concomitantly induce apoptosis, but lytic gene expression protects from cell death. J Virol. 2001;75:2400–2410. doi: 10.1128/JVI.75.5.2400-2410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahyo T, Nishida T, Yasuda H. Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol Cell. 2001;8:713–718. doi: 10.1016/s1097-2765(01)00349-5. [DOI] [PubMed] [Google Scholar]
- Kenney SC, Mertz JE. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin Cancer Biol. 2014;26:60–68. doi: 10.1016/j.semcancer.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ET, Kim YE, Kim YJ, Lee MK, Hayward GS, Ahn JH. Analysis of human cytomegalovirus-encoded SUMO targets and temporal regulation of SUMOylation of the immediate-early proteins IE1 and IE2 during infection. PLoS One. 2014;9:e103308. doi: 10.1371/journal.pone.0103308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Choi H, Lee SK. Epstein-Barr Virus MicroRNA miR-BART20–5p Suppresses Lytic Induction by Inhibiting BAD-Mediated caspase-3-Dependent Apoptosis. J Virol. 2015;90:1359–1368. doi: 10.1128/JVI.02794-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosowicz JG, Lee J, Peiffer B, Guo Z, Chen J, Liao G, Hayward SD, Liu JO, Ambinder RF. Drug Modulators of B Cell Signaling Pathways and Epstein-Barr Virus Lytic Activation. J Virol. 2017;91 doi: 10.1128/JVI.00747-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, van Raam BJ, Salvesen GS, Cieplak P. Caspase cleavage sites in the human proteome: CaspDB, a database of predicted substrates. PLoS One. 2014;9:e110539. doi: 10.1371/journal.pone.0110539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Kang HJ, Lee HR, Choi CY, Jang WJ, Ahn JH. PIAS1 enhances SUMO-1 modification and the transactivation activity of the major immediate-early IE2 protein of human cytomegalovirus. FEBS Lett. 2003;555:322–328. doi: 10.1016/s0014-5793(03)01268-7. [DOI] [PubMed] [Google Scholar]
- Li R, Liao G, Nirujogi RS, Pinto SM, Shaw PG, Huang TC, Wan J, Qian J, Gowda H, Wu X, et al. Phosphoproteomic Profiling Reveals Epstein-Barr Virus Protein Kinase Integration of DNA Damage Response and Mitotic Signaling. PLoS Pathog. 2015;11:e1005346. doi: 10.1371/journal.ppat.1005346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Pan Y, Shi DD, Zhang Y, Zhang J. PIAS1 negatively modulates virus triggered type I IFN signaling by blocking the DNA binding activity of IRF3. Antiviral Res. 2013;100:546–554. doi: 10.1016/j.antiviral.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Li R, Wang L, Liao G, Guzzo CM, Matunis MJ, Zhu H, Hayward SD. SUMO binding by the Epstein-Barr virus protein kinase BGLF4 is crucial for BGLF4 function. J Virol. 2012;86:5412–5421. doi: 10.1128/JVI.00314-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhu J, Xie Z, Liao G, Liu J, Chen MR, Hu S, Woodard C, Lin J, Taverna SD, et al. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe. 2011;10:390–400. doi: 10.1016/j.chom.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Fu Y, Shuai K. Distinct roles of the NH2- and COOH-terminal domains of the protein inhibitor of activated signal transducer and activator of transcription (STAT) 1 (PIAS1) in cytokine-induced PIAS1-Stat1 interaction. Proc Natl Acad Sci U S A. 2000;97:5267–5272. doi: 10.1073/pnas.97.10.5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Tsai MH, Shumilov A, Poirey R, Bannert H, Middeldorp JM, Feederle R, Delecluse HJ. The Epstein-Barr Virus BART miRNA Cluster of the M81 Strain Modulates Multiple Functions in Primary B Cells. PLoS Pathog. 2015;11:e1005344. doi: 10.1371/journal.ppat.1005344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci U S A. 1998;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Mink S, Wong KA, Stein N, Getman C, Dempsey PW, Wu H, Shuai K. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat Immunol. 2004;5:891–898. doi: 10.1038/ni1104. [DOI] [PubMed] [Google Scholar]
- Liu B, Tahk S, Yee KM, Fan G, Shuai K. The ligase PIAS1 restricts natural regulatory T cell differentiation by epigenetic repression. Science. 2010;330:521–525. doi: 10.1126/science.1193787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Tahk S, Yee KM, Yang R, Yang Y, Mackie R, Hsu C, Chernishof V, O’Brien N, Jin Y, et al. PIAS1 regulates breast tumorigenesis through selective epigenetic gene silencing. PLoS One. 2014a;9:e89464. doi: 10.1371/journal.pone.0089464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Yang R, Wong KA, Getman C, Stein N, Teitell MA, Cheng G, Wu H, Shuai K. Negative regulation of NF-kappaB signaling by PIAS1. Mol Cell Biol. 2005;25:1113–1123. doi: 10.1128/MCB.25.3.1113-1123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Yee KM, Tahk S, Mackie R, Hsu C, Shuai K. PIAS1 SUMO ligase regulates the self-renewal and differentiation of hematopoietic stem cells. EMBO J. 2014b;33:101–113. doi: 10.1002/embj.201283326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ST, Wang WH, Hong YR, Chuang JY, Lu PJ, Chang LK. Sumoylation of Rta of Epstein-Barr virus is preferentially enhanced by PIASxbeta. Virus Res. 2006;119:163–170. doi: 10.1016/j.virusres.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhang YD, Guo L, Huang HY, Zhu H, Huang JX, Zhou SR, Dang YJ, Li X, Tang QQ. Protein inhibitor of activated STAT 1 (PIAS1) is identified as the SUMO E3 ligase of CCAAT/enhancer-binding protein beta (C/EBPbeta) during adipogenesis. Mol Cell Biol. 2013;33:4606–4617. doi: 10.1128/MCB.00723-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv DW, Zhong J, Zhang K, Pandey A, Li R. Understanding Epstein-Barr Virus Life Cycle with Proteomics: A Temporal Analysis of Ubiquitination During Virus Reactivation. OMICS: A Journal of Integrative Biology. 2017;21:27–37. doi: 10.1089/omi.2016.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Caduff N, Barros MHM, Ramer PC, Raykova A, Murer A, Landtwing V, Quast I, Styles CT, Spohn M, et al. Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe. 2017;22:61–73. e67. doi: 10.1016/j.chom.2017.06.009. [DOI] [PubMed] [Google Scholar]
- Meng Q, Hagemeier SR, Fingeroth JD, Gershburg E, Pagano JS, Kenney SC. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J Virol. 2010;84:4534–4542. doi: 10.1128/JVI.02487-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J Virol. 2000;74:6324–6332. doi: 10.1128/jvi.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira NA, Song Z. Some commonly used caspase substrates and inhibitors lack the specificity required to monitor individual caspase activity. Biochemical and biophysical research communications. 2008;377:873–877. doi: 10.1016/j.bbrc.2008.10.101. [DOI] [PubMed] [Google Scholar]
- Prasad A, Lu M, Lukac DM, Zeichner SL. An alternative Kaposi’s sarcoma-associated herpesvirus replication program triggered by host cell apoptosis. J Virol. 2012;86:4404–4419. doi: 10.1128/JVI.06617-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Remick J, Zeichner SL. Activation of human herpesvirus replication by apoptosis. J Virol. 2013;87:10641–10650. doi: 10.1128/JVI.01178-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley CM, Chen J, Shamay M, Li H, Zahnow CA, Hayward SD, Ambinder RF. Bortezomib induction of C/EBPbeta mediates Epstein-Barr virus lytic activation in Burkitt lymphoma. Blood. 2011;117:6297–6303. doi: 10.1182/blood-2011-01-332379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- Song J, Tan H, Shen H, Mahmood K, Boyd SE, Webb GI, Akutsu T, Whisstock JC. Cascleave: towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics. 2010;26:752–760. doi: 10.1093/bioinformatics/btq043. [DOI] [PubMed] [Google Scholar]
- Takada K. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int J Cancer. 1984;33:27–32. doi: 10.1002/ijc.2910330106. [DOI] [PubMed] [Google Scholar]
- Toropainen S, Malinen M, Kaikkonen S, Rytinki M, Jaaskelainen T, Sahu B, Janne OA, Palvimo JJ. SUMO ligase PIAS1 functions as a target gene selective androgen receptor coregulator on prostate cancer cell chromatin. Nucleic Acids Res. 2015;43:848–861. doi: 10.1093/nar/gku1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zhao XM, Tan H, Akutsu T, Whisstock JC, Song J. Cascleave 2.0, a new approach for predicting caspase and granzyme cleavage targets. Bioinformatics. 2014;30:71–80. doi: 10.1093/bioinformatics/btt603. [DOI] [PubMed] [Google Scholar]
- Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- Young LS, Yap LF, Murray PG. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer. 2016;16:789–802. doi: 10.1038/nrc.2016.92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.