

ABSTRACT
Introduction: Hematopoietic neoplasms are often driven by gain-of-function mutations of the JAK-STAT pathway together with mutations in chromatin remodeling and DNA damage control pathways. The interconnection between the JAK-STAT pathway, epigenetic regulation or DNA damage control is still poorly understood in cancer cell biology.
Areas covered: Here, we focus on a broader description of mutational insights into myeloproliferative neoplasms and peripheral T-cell leukemia and lymphomas, since sequencing efforts have identified similar combinations of driver mutations in these diseases covering different lineages. We summarize how these pathways might be interconnected in normal or cancer cells, which have lost differentiation capacity and drive oncogene transcription.
Expert opinion: Due to similarities in driver mutations including epigenetic enzymes, JAK-STAT pathway activation and mutated checkpoint control through TP53, we hypothesize that similar therapeutic approaches could be of benefit in these diseases. We give an overview of how driver mutations in these malignancies contribute to hematopoietic cancer initiation or progression, and how these pathways can be targeted with currently available tools.
KEYWORDS: Epigenetic target, hematopoietic cancer, MPN, mutational landscape, PTCL, therapeutic target
1. Introduction
Hematopoietic malignancies arise when somatic hematopoietic stem cells (HSCs) or lymphoid or myeloid progenitor cells acquire driver mutations that change cellular differentiation fates and overcome senescence. During leukemic development, healthy hematopoietic cells switch from normal to enhanced transcription by increasing the number of super- or stretched-enhancers at promoter or enhancer elements. Subsequently, new chromatin loop structures form that change topologically associated domains, which trigger reprogramming of cancer cells [1,2]. We are starting to gain insights into organized chromatin regulatory circuits that not only contain proteins and DNA, but also structural or regulatory RNA, to promote oncogenic gene transcription [3].
During the evolution of hematopoietic diseases, physiologic polyclonal hematopoiesis switches to abnormal monoclonal or oligoclonal hematopoiesis, which involves an increased response to cytokine signaling that is often associated with mutated tyrosine kinases (TKs) and GTPases. Despite advances in understanding the pathophysiology of hematopoietic diseases, developing new therapeutics for patients remains challenging. Not all mutated key genes are ‘easy targets’ and patients still frequently relapse. However, we understand that increased oncogene transcription and silenced tumor suppressor genes facilitate neoplastic growth, survival, and clonal expansion. Furthermore, changes in the epigenome result in abnormal transcription factor/cofactor/corepressor networks at promoter–enhancer interactions. Here, we provide an overview on novel insights and novel targeting approaches against drivers of myeloproliferative neoplasms (MPNs), secondary acute myeloid leukemia (sAML), and peripheral T-cell leukemia and lymphomas (PTCLs). We also discuss the feasibility of targeting new players as the focus of new therapeutic developments.
2. Identification of key driver mutations in hematopoietic cancer
The last decade of cancer research has been significantly shaped by major advances in next-generation sequencing technologies that have led to the analysis of more than 100,000 whole cancer genomes [4]. Approximately 4 million coding mutations have been identified, defining 500 genes that act as functional cancer drivers [5,6], many of which participate in core cancer pathways [7]. The ability to define hyperactivated signaling pathways that are commonly deregulated allows for development of new therapies. One strategy focuses on the development of drugs designed to directly target driver oncoproteins that cancer cells are addicted to. In contrast, specific targeting of a tumor suppressor to restore normal function has been reported but remains challenging. However, loss of tumor suppressors, such as SOCS2 or PRC2, leads to hyperactivation of the JAK-STAT pathway which could be targeted instead [8,9].
We follow the current paradigm of somatic mutation theory in cancer development, which is based on clonal expansion upon the occurrence of mutations rather than mutations being a consequence of cancer itself. Current cancer drug development is primarily focused on the finding and targeting of driver mutations. However, evidence of both mutation-free tumors and the presence of driver mutations in healthy patients (discussed in more detail in the TP53 section), may support alternative theories. For example, epigenetic gene regulation is tightly linked with metabolism, steered by complex cytokine, adipokine, growth factor, and/or hormone signaling, which can also influence cancer cells. It is known that cancer cells may harbor a variety of somatic alterations in various biological pathways, but only part of these mutations are related to cancer initiation and development and only a few are driving the cancer progression. Accordingly, upon treatment with a single targeted drug, it is often difficult to predict the outcome because of the compensatory pathways and feedback loops that are still poorly understood in most cancers [10–12].
The landmark demonstration of targeting a driver oncogene product was the development of Imatinib to inhibit BCR-ABL1 in chronic myeloid leukemia (CML). This led to a dramatic improvement in CML patient survival. It is therefore not surprising that multiple small molecule inhibitors were developed to target a range of TKs with remarkable clinical success. However, despite the exceptional activity of Imatinib in CML, drug resistance develops rather frequently in these patients. This clinically challenging condition is often associated with further genetic aberrations in the driver itself (BCR-ABL1) or with mutations in other critical target genes. Moreover, epigenetic and other mechanisms may promote upregulation of the STAT3/5 pathway, allowing cancer cells to escape drug action [8]. Combining different therapies against multiple ‘oncogene addictions’ could be a possibility to overcome primary or acquired resistance. Targeted approaches in solid cancers inhibit common downstream mediators of known oncogenes, such as MEK-ERK or the PI3K-AKT-mTOR kinase pathways, and similar pathways may also play a role in oncogenesis in hematopoietic malignancies [13]. Last, newer therapeutic strategies aim to exploit the specific dependency of cancer cells on basic cellular processes such as cell division, chromatin regulation, and metabolism.
We will first describe key genetic drivers in MPN (Figure 1) and subsequently discuss driver events in PTCL (Figure 2).
Figure 1.
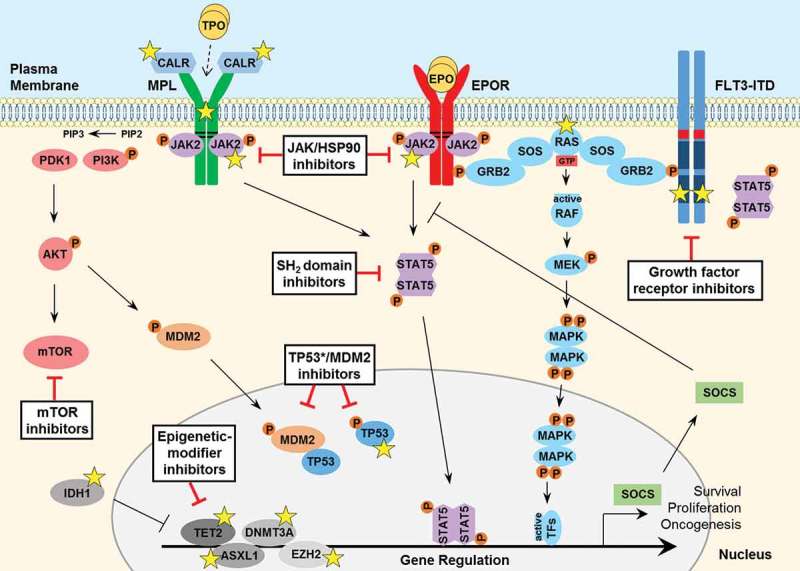
Signaling pathways involved in the pathogenesis of MPNs and secondary AML. JAK2 binds to the cytosolic juxta-membrane region of dimeric cytokine receptors such as MPL (TPOR) and EPOR, via the BOX1 and BOX2 receptor motifs (black lines). JAK2 activation (via receptor-ligand binding or gain-of-function mutation such as JAK2 V617F) promotes various downstream signaling pathways, via STAT5, including RAS-MAPK and PI3K-AKT. These pathways facilitate oncogenic gene transcription and promote cancer cell survival, proliferation or migration. The expression of negative regulators such as the SOCS proteins are induced by the JAK-STAT pathway, however they are not sufficient to block hyperactive JAK-STAT signaling and cannot bind JAK2 V617F. The FLT3-ITD mutant growth factor receptor commonly found in AML patients signals independently of ligand-binding, as a result of the internal tandem duplication (ITD) found within the juxta-membrane domain (red box) and point mutations that occur within the kinase domain (most frequently at D835; dark blue box) of the FLT3 receptor. FLT3-ITD hyperactivation promotes RAS-MAPK, PI3K-AKT as well as STAT5 signaling. A number of important somatic mutations have been reported in various oncogenes and tumor suppressor proteins within these pathways (yellow stars), where such mutations are known to contribute to disease initiation and progression. For further details on these mutations, see Table 1. Mutated calreticulin (CALR), frequently found in MPN patients, interacts with the extracellular portion of the MPL receptor at the Endoplasmic Reticulum-Golgi apparatus and also at the cell surface, promoting direct dimerization, activation of JAK2 and downstream signaling, independently of TPO binding (which is required for normal MPL signaling, indicated by a dashed arrow). Loss-of-function mutations in the critical tumor suppressor protein TP53 are also reported generally in MPN patients that progress to secondary AML. Furthermore, various epigenetic-modifier proteins are found to be mutated in MPN patients, including isocitrate dehydrogenase 1 (IDH1), methylcytosine dioxygenase TET2, DNA methyltransferase 3A (DNMT3A), Polycomb group protein ASXL1 and the histone methyltransferase protein of polycomb repressive complex 2 (PRC2) EZH2. Promising therapeutic agents to target these key proteins/pathways in MPN/AML have been developed and are summarized here (black boxes). TPO, thrombopoietin; EPO, erythropoietin; GTP, guanosine triphosphate; TF, transcription factor.
Figure 2.
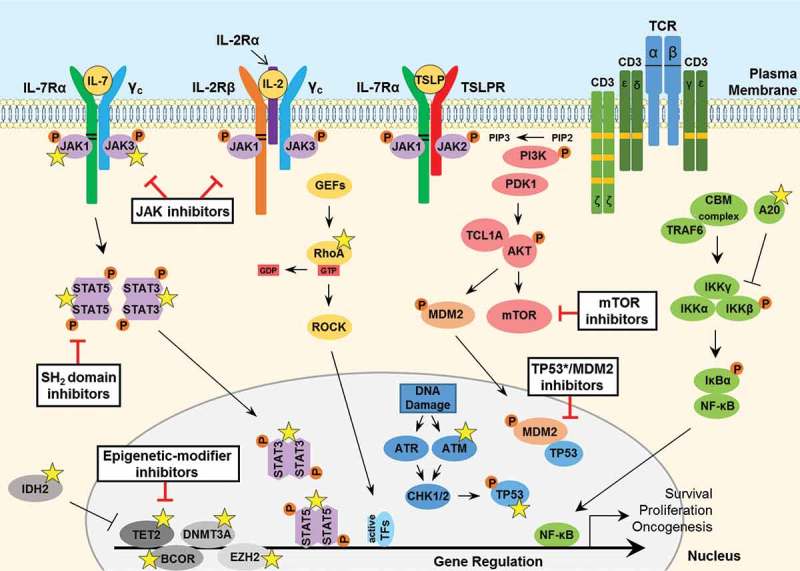
Signaling pathways involved in the pathogenesis of PTCL. JAK tyrosine kinases bind to the cytosolic juxta-membrane region of dimeric T-cell cytokine receptors such as IL-7Rα, IL-2Rβ, TSLPR and the common gamma chain (γc). Conserved juxta-membrane BOX1 and BOX2 cytokine receptor motifs known to bind JAKs are indicated with black lines. Cytokine receptor-ligand binding promotes STAT3/5 tyrosine phosphorylation to facilitate gene transcription to promote cancer cell survival, proliferation or migration. A number of important somatic mutations have been reported in various oncogenes and tumor suppressor proteins within these pathways (yellow stars), where such mutations are known to contribute to disease initiation and progression. For further details on these mutations, see Table 2. GTPase signaling through RAS-RAF (not shown) or mutated RhoA-ROCK pathways are frequently activated in PTCL. T-cell receptor (TCR) activation, involving phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs; orange boxes), triggers various downstream pathways including PI3K-AKT and NF-κB signaling. Furthermore, overexpression of the AKT-activating protein TCL1A, resulting from rearrangements between a TCL1 family gene and TCR loci rendering it under the control of TCR expression-regulating elements, can contribute to aberrant survival signaling and enhanced TCR activation. Loss-of-function mutations in the critical tumor suppressor proteins TP53 and ATM are reported in PTCL. Moreover, various epigenetic-modifier proteins are found to be mutated in PTCL patients, including isocitrate dehydrogenase 2 (IDH2), methylcytosine dioxygenase TET2, DNA methyltransferase 3A (DNMT3A), BCL-6 corepressor (BCOR) and the histone methyltransferase protein of polycomb repressive complex 2 (PRC2) EZH2. Promising therapeutic agents to target these key proteins/pathways in PTCL have been developed and are summarized here (black boxes). IL, interleukin; TSLP, thymic stromal lymphopoietin; CBM, CARMA3-BCL10-MALT1; GEF, guanine nucleotide exchange factor; GTP, guanosine triphosphate; GDP, guanosine diphosphate; TF, transcription factor.
3. Targets in MPN and driver mutations
MPNs are characterized by enhanced proliferation and reduced differentiation and cell death in one myeloid lineage, leading to the outgrowth of a dominant myeloid cell type in addition to extramedullary hematopoiesis. The World Health Organization (WHO) classification recognizes three main variants of BCR-ABL1-negative MPN: essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis (MF) [14]. They are defined by excessive production of leukocytes, platelets, and/or erythrocytes in the bone marrow as well as by extramedullary myelopoiesis [14]. In MF patients, bone marrow fibrosis is also seen [14]. In the initial (chronic) phase of MPN, cellular differentiation and maturation is largely preserved and the expanded cell populations are functionally intact [14]. Apart from severe thromboembolic events, transformation to sAML is the most devastating complication experienced by MPN patients. AML evolution is seen in ~20% of patients with MF, ~5% of PV, and ~1% of ET patients [15–18]. During the early stage of MPN, clonal cells are usually responsive to hydroxyurea (HU), interferon-alpha (IFN-A), and/or Ruxolitinib, but this is not the case in most patients with advanced MPN or sAML. Several new clinical trials in advanced MPN focus on combination trials using various targeted drugs, including JAK2 TK inhibitors (TKI).
The main mutations that are currently used as clinically relevant diagnostic markers are driver mutations in JAK2, CALR, and MPL [14]. We describe below recurrent hotspot mutations in MPN, in key genes that constitute core cancer pathways.
3.1. JAK2 point mutations or exon 12 mutations
The JAK2 V617F mutation is present in 95% of PV, ~60% of ET, and ~45% of MF patients [15,19]. Surprisingly, the JAK2 V617F mutation has no clear association with survival or sAML transformation. The second most frequent mutation in JAK2 occurs in exon 12 with a small deletion causing similar functional consequences as JAK2 V617F. This deletion occurs in a small percentage of JAK2 V617F-negative PV patients, but not in ET or MF. JAK2 activates STAT3/5A/5B transcription factors, which can directly induce target genes to accelerate cell cycle progression, survival, and cancer cell metabolism. It was shown through genetic experiments that particularly the activation of the two STAT5 transcription factors is crucial for PV [20]. Hyperactive JAK2 promotes prominent activation of the PI3K-AKT-mTOR and the RAS-RAF/MAPK-ERK pathways, among other less prominent signaling pathways, and evades negative-regulation by SOCS proteins [13].
JAK2 may be involved in directly or indirectly reprogramming epigenetic gene regulation; however, this is still controversial [21]. JAK2 is known to phosphorylate histone H3, thereby disrupting the binding of heterochromatin protein 1 alpha (HP1α) to chromatin [21,22]. Furthermore, JAK2 phosphorylates the arginine methyltransferase PRMT5, impairing its ability to methylate histone substrates, ultimately driving myeloproliferation [22].
3.2. CALR exon 9 mutations
The CALRETICULIN (CALR) driver mutation was identified in approximately 73% of JAK2/MPL mutation-negative ET and MF patients [23]. Mutations occur in exon 9 of CALR in the majority of JAK2 wild-type MPN cases. CALR constitutes a key component of the quality-control machinery that ensures proper glycoprotein folding and Ca2+ homeostasis. In MPN, mutant CALR interacts with the thrombopoietin receptor (MPL/TPOR) promoting direct dimerization and activation of JAK2 at the endoplasmic reticulum (ER)–Golgi apparatus. The capacity of CALR to bind Ca2+ and regulate its homeostasis is lost due to a frame shift mutation in the carboxy-terminal Ca2+-binding domain [24]. Surprisingly, a functional cytokine-TK-STAT signaling hub at the cell membrane seems to be dispensable in CALR-mutated cells. Interestingly, and very reminiscent, STAT5 activation at the ER–Golgi was also described in Flt3-ITD+ or KIT D816V+ AML cases. Analysis of patient data suggests that CALR mutation-positive patients have a more favorable clinical outcome than patients with JAK2 or MPL mutation-positive MPNs due to a lower risk of thrombosis [23].
3.3. MPL/TPOR point mutations
Somatic mutations affecting MPL are seen in up to 15% of JAK2 V617F-negative ET and MF patients. The most common gain-of-function mutation W515L leads to hyperphosphorylation of JAK2, STAT3, STAT5, ERK, and AKT proteins [25–27].
3.4. Chromatin remodeler mutations in MPN
Emerging evidence suggests that MPNs are likely the result of combined genetic deregulation of several mutated genes encoding for epigenetic regulators. Mutations in epigenetic remodelers have been described for TET2 (12%), ASXL1 (5%), DNMT3A (5%), EZH2 (~3%), and IDH1 (~1.5%) [28]. All of these epigenetic modifiers act either on DNA or histone/transcription factor methylation. Interestingly, they appear to be the most frequent somatic mutations after JAK2 and CALR in MPN [29]. However, these mutations are not restricted to MPN and are also found in a wide spectrum of other neoplasms, including AML. It is thought that the development of clonal evolution in MPN is slow and often includes a clinically ‘silent’ phase. As a result, most mutations are already present at diagnosis. Interestingly, the order in which mutations are acquired may play an important role in the development of the disease phenotype. The reversible nature of epigenetic changes may make them good potential therapeutic targets. An overview of the described mutations as well as other relevant mutations not mentioned here is shown in Table 1.
Table 1.
Mutational landscape of myelofibrosis (MF), essential thrombocytopenia (ET), and polycythemia vera (PV).
| Ref | Gene | Function | Frequency % |
||
|---|---|---|---|---|---|
| MF | ET | PV | |||
| [30] | JAK2 | Tyrosine kinase | 55–60 | 50–60 | 95–97 |
| [31] | CALR | Endoplasmic chaperone | 25–30 | 20–25 | <1 |
| [32] | MPL | Growth factor receptor | 5–10 | 3–5 | <1 |
| [30] | SF3B1 | Splicing regulator | 5–10 | ~1 | ~1 |
| [33] | TP53 | DNA damage response | 2–4 | <1 | <1 |
| [34] | CBL | E3 ubiquitin ligase | 5–10 | 0–2 | Rare |
| [35] | DNMT3A | DNA methyltransferase | 5–12 | 1–5 | 5–10 |
| [35] | TET2 | Methylcytosine dioxygenase | 10–20 | 5 | 10–20 |
| [36] | EZH2 | Chromatin regulator | 5–10 | ~2 | ~2 |
| [37] | IDH1/2 | Isocitrate dehydrogenase | 3–5 | <1 | ~2 |
| [37] | ASXL1 | Chromatin regulator | 15–35 | 5–10 | 2–7 |
3.5. TP53 point mutations in MPN and secondary AML
TP53 senses DNA damage and mitotic checkpoint control, and mutations in the TP53 gene (TP53*) are most frequent in patients with sAML. The TP53 mutations are represented by bi-allelic or homozygous mutations [38]. Interestingly, TP53* heterozygosity is detected in MPNs, but homozygous or compound mutations are only detected in sAML [28]. Notably, loss-of-function mutations in TP53 appear to emerge during disease progression. It is currently under discussion whether cytoreduction upon HU therapy selects for TP53* mutated cells. A recent study analyzed the impact of TP53* in MPN patients and, although it is common that at least one somatic TP53* allele is transcribed in patient cells, the authors did not find a direct association between TP53 inactivation and HU resistance or blast transformation [33]. TP53 can also interact with STAT3 and STAT5 [39,40] and it induces mRNA expression of STAT5A, but not of STAT5B [41]. Overall, current sequencing data suggest that the age of patients is the strongest factor affecting low-burden TP53* incidence in MPN, which may persist for years without an immediate risk of progression.
3.6. GTPase gain-of-function mutations
GTPases are among the most frequently mutated genes in cancer. The amino acid sequences of the entire family are highly conserved making their targeting very difficult. Although infrequent in MPN, more than 10% of AML cases harbor activating RAS mutations. RAS-RAF signaling can be triggered by normal cytokine, growth factor, or hyperactive JAK action and it constitutes a core cancer pathway [7]. RAS-RAF is upstream of MAPK-ERK signaling and it can further activate RHO and RAC GTPase proteins, which were reported to be essential for nuclear shuttling of STAT5A [42]. Furthermore, oncogenic RAS requires mitochondrial functions of STAT3, illustrating that the RAS-RAF pathway is interconnected with aberrant JAK-STAT, cytokine, or growth factor signaling. Similarities in the GTPase superfamily and their pleiotropic biological functions have made targeting attempts in clonal myeloid diseases unsuccessful to date [43].
4. Targets in PTCL and driver mutations
PTCLs represent a heterogeneous mature T-cell disease group of 15% of all non-Hodgkin lymphomas, which are often accompanied by aggressive organ infiltrations. PTCL can be variable in terms of immunophenotypic, morphological, and molecular features [44–46]. The 2016 WHO classification of lymphoid neoplasms distinguishes more than 20 mature T- and natural killer-cell neoplasms [47]. Within this rather rare disease group, the most common subtype is PTCL-not otherwise specified (NOS), which summarizes cases not attributable to other entities, followed by angioimmunoblastic T-cell lymphoma (AITL), ALK+ anaplastic large cell lymphoma (ALCL), and ALK− ALCL [46,48,49]. Further subtypes include adult T-cell lymphoma/leukemia (ATLL), T-cell prolymphocytic leukemia (T-PLL), and cutaneous T-cell lymphoma (CTCL) including mycosis fungoides and Sézary syndrome (SS) [50,51]. The only frequent recurring chromosomal translocation identified in the PTCLs is the t(2;5)(p23;q35) NPM-ALK fusion characteristic of ALK+ ALCL. So far, no other genetic alterations (e.g. ITK-SYK translocation [52], IRF4 rearrangements [53], abnormalities, or deletions in chromosome 6q, 7q, or 9q [54–56]) have been linked to diagnosis, complicating clinical decisions in the treatment of PTCL patients. Moreover, combined chemotherapy (CHOP; Cyclophosphamide, Doxorubicin, Vincristine, Prednisolone; or CHOEP with Etoposide) often results initially in favorable response rates, however relapses and refractory disease are frequently observed [45,57]. Stem cell transplantations can only be offered to certain patients, and it can therefore be speculated that beneficial outcomes after transplantation may be attributed to patient selection [58]. So far, there is no optimal therapeutic approach to treating PTCL patients and the identification of molecular targets is of great importance.
High-throughput methodologies were used to identify the cell of origin and to characterize commonly altered pathways in PTCL [51,59–67]. These efforts were successful in defining characteristic gene expression profiles for AITL, ALK+ ALCL, ATLL, and PTCL-NOS, identifying recurrent mutations and recognizing specific subtypes, which can now help to support correct diagnosis and classification of patients to distinct disease subgroups with different prognoses [46,59,60,62,63].
PTCL patient numbers in sequencing studies are limited, but certain classes of genes and pathways are commonly affected in a majority of cases. The emerging understanding of mutations involves signaling pathways and epigenetic reprogramming, which highlight new targeting concepts. This is an intensive research area and many new reagents have been developed to define novel combinatorial treatments [68]. In the following we focus on PTCL, but we exclude detailed descriptions for ALK+ and ALK− ALCL, since this has been extensively reviewed elsewhere [69].
4.1. TCR activating mutations
Mutations constitutively activating TCR signaling are most frequent in T-PLL, where the TCL1A inversions/translocations upstream of mTOR signaling rearrange with the TCR locus [70]. Enhanced TCL1A expression in T-PLL also amplifies TCR signaling. Moreover, TCR activation was linked to PTCL-NOS with the ITK-SYK fusion gene being present in approximately 10% of cases [52,71], or MYC overexpression due to IRF4 activating fusions [72] mimicking survival signals normally emanating from antigen receptor signaling. Furthermore, missense mutations in TNFAIP3, encoding the negative regulator of NF-κB activation A20 in T-cells after TCR stimulation [73], mutations in WNT/β-Catenin negative regulators APC and CHD8, and other genes with known suppressive roles in TCR activation were disease associated [74]. GATA3 and TBX21 expression are both important in T-cell development, and mutations in these genes may be associated with the PTCL-NOS subgroups, representing potential diagnostic predictors and possibly also therapeutic targets [59].
4.2. Gain-of-function JAK/STAT pathway mutations
Reports on patient mutation sequencing analyses of various T-cell lymphoma subtypes frequently include the JAK/STAT pathway, where JAK1, JAK3, STAT3, and STAT5B are predominantly mutated to cause hyperactivation [64,70,75–78]. Importantly, ALK− ALCL is associated with STAT3 activation [64] and recurrent, somatic activating mutations in the closely related STAT5B gene were reported. The STAT5B N642H mutation occurs with the highest frequency in PTCL, whereby most mutations cluster in the SH2 and C-terminal transactivation domain [70,75,78–82]. A recent study found that ~70% of T-PLL patients carry JAK-STAT hyperactivating mutations [70]. Deep sequencing of known recurrent somatic mutations in T-PLL revealed a mutational burden of 4% in IL2RG, the JAK3 binding receptor chain that is shared by interleukins that use the common gamma chain. JAK1 is less frequently mutated (10%) compared with JAK3 (30%) and STAT5B (36%) [70]. Indeed, studies have linked JAK3 and STAT5B mutations with poorer patient survival [83,84]. STAT5 activation was also linked to an autocrine PDGF signaling loop in PTCL-NOS [85]. Enhanced STAT5 signaling was also linked to overexpression of oncogenic miR-155 in CTCL [86], associated with downregulation of tumor-suppressive miR-22 [87], or enhanced disease progression caused by Lymphotoxin-α-dependent lymphangiogenesis [88]. STAT5-dependent CD80 expression was also linked to resistance to Vorinostat and risk of disease progression in PTCL [84,89,90].
4.3. Chromatin remodeler mutations in PTCL
Like in other hematopoietic neoplasms [91], TET2, DNMT3A, and IDH2 mutations occur frequently across PTCL subtypes, although certain mutations seem to be confined to T-cell lymphoma cases [92] (see Table 2 for overview).
Table 2.
Mutational landscape of angioimmunoblastic T-cell lymphoma (AITL) and peripheral T-cell lymphoma not otherwise specified (PTCL-NOS).
TET2 frameshift and nonsense mutations were frequently identified in AITL (~70%), PTCL-NOS (~60% in TFH cell marker expressing subtype), and CTCL (~10%) [75,96–98]. In AITL, IDH2 and TET2 mutations were detected in the same patients, which is not the case in myeloid malignancies [77]. In AITL and PTCL-NOS, TET2 mutations were associated with a worse prognosis [96]. Tet2-deficient mouse models elicit altered T-cell differentiation and can develop T-cell lymphoma with TFH-like features [99,100]. In Tet2-knockdown mice, the outgrowth of TFH-like tumor cells was connected to methylation changes of BCL-6 [100], the locus repressor of STAT5A/B.
Cancer genome sequencing efforts identified DNMT3A as one of the most frequently mutated genes in hematological malignancies, which raises the question of how these lesions promote malignant cell growth. DNMT3A functions as a de novo DNA methylation enzyme, but it also interacts with histone modifiers promoting gene repression [101,102] in cooperation with STAT5 [103] and EZH2 [104]. In AITL, PTCL-NOS and CTCL subtypes, DNMT3A mutations cluster in the methyltransferase domain. Interestingly, only about 20% of these mutations are at position R882 [75,77,94,95,97], the variant commonly found in myeloid diseases acting as a negatively regulating hypomorphic protein [105]. Dnm3a-deficient mice develop a PTCL-like disease at a frequency of 12% and heterozygous animals at a rate of 10%, associated with hypomethylation and decreased TP53 activity [106]. TET2 and DNMT3A mutations likely occur early during evolution of hematopoietic neoplasms and are even detectable in apparently healthy individuals [99,107]. These mutations can also co-occur which emphasizes the importance of disrupted DNA and histone methylation in PTCL [77].
IDH2, normally catalyzing the conversion of isocitrate to alpha-ketoglutarate in the Krebs cycle, is frequently mutated in hematopoietic neoplasms resulting in novel enzymatic activity producing 2-hydroxyglutarate (2-HG). This oncometabolite represses H3K and DNA 5-mC demethylation by inhibiting TET2, thus leading to abnormal regulation of gene transcription which potentially promotes lymphomagenesis. This is underpinned by the finding that about 30% of AITL cases possess IDH2 mutations [65,108]. Furthermore, TCR signaling and T-cell differentiation promoting genes are hypermethylated [65]. Interestingly, no IDH1 mutations have been mapped in AITL as yet, and only the IDH2 R172 mutant but not IDH2 R140 (a frequent mutation in myeloid neoplasms) has been documented. An explanation may be given by murine knock-in models of the common IDH2 variants, which identified IDH2 mutated at R172 to produce the highest 2-HG levels in T-cells, thereby impairing lymphopoiesis [109]. Interestingly, AITL cases with IDH2 R172 mutations show a distinct gene expression signature with downregulated TH2 differentiation genes (e.g. IFNG and STAT1) and upregulated IL-12 target genes [109]. In addition, mutations in epigenetic regulators catalyzing methylation and acetylation changes such as EZH2, TET2, and BCOR were found in a number of T-PLL patients [76].
4.4. TP53 and diminished DNA damage response pathways
TP53 mutations were found in 14% of cases categorized as T-PLL [83]. However, overexpression and accumulation of wild-type TP53 is common in T-PLL [110]. In ALK+ ALCL, which is driven by NPM-ALK, the fusion kinase can efficiently block wild-type TP53 function [111]. Still, in other PTCL diseases TP53 mutations are not as frequent as in MPN. In addition, patient age could bias the analysis of TP53 mutations.
The ataxia telangiectasia mutated (ATM) tumor suppressor frequently displays loss-of-function mutations or is deleted in PTCL [70]. ATM is centrally involved, in conjunction with ATR and CHK2, in activation of the DNA damage checkpoint control, and it maintains the balance between thymocyte survival and apoptosis, especially during V(D)J recombination [112]. DNA double-strand breaks result in rapid activation of ATM/ATR, in turn activating substrates that regulate cell-cycle progression, DNA repair, and cell death. Interestingly, ATM is also known to interact with TCL1 which was described to result in enhanced NF-κB activity and cell proliferation in association with TCR signaling in PTCL. In addition to immune defects, ataxia-telangiectasia (AT) patients and Atm–/– mice share a predisposition to T-cell malignancies, pointing toward a common etiology for these two consequences of ATM inactivation. The risk of developing a lymphoid neoplasm is increased approximately 200-fold in AT patients compared with the normal population. The frequency of T-cell tumors in AT patients exceeds that of B-cell malignancies by fourfold, and myeloid cancers have yet to be reported. The vast majority of lymphoid tumors that develop in children with AT are T-cell ALL/lymphoma, while young adults are mostly predisposed to T-PLL [112].
In this context, it is of mechanistic interest to link JAK and STAT3/5 activation to regulation of reactive oxygen species (ROS) generation, which is known to cause DNA damage, promoting mutations, oxidization of lipids, or silencing of phosphatases by catalytic cysteine oxidization [113–116]. Surprisingly, wild-type JAK2 decreases detoxifying glutathione S-transferases in epithelial cells, enhancing oxidative damage. In contrast, expression of anti-oxidative scavengers are under the control of STAT5, illustrating the interplay between JAK2 and STAT5 in balancing ROS action [117,118]. RAD51 members are conserved down to the E. coli RecA proteins and they are essential for DNA repair, which is downstream of cytokine- or TK-STAT signaling in mammalian cells [119,120]. However, the link between a hyperactive JAK-STAT pathway, TP53*, and mutated ATM/CHK2 is poorly understood. These core cancer pathways need further characterization in order to understand drug actions, to overcome resistance mechanisms, and to finally eradicate cancer (stem) cells.
4.5. GTPase gain-of-function mutations
RHOA mutations are the only frequent GTPase mutations described in PTCL, occurring predominantly in up to 70% of AITL patients as well as 20% of PTCL-NOS and 15% of ATLL cases [93–95,121]. RHOA is a member of the Rho family of small GTPases that links cell-surface receptors to different intracellular signaling proteins. In its active GTP-bound state, RHOA functions in controlling the actin cytoskeleton and stress fibers [122]. The most prominent mutation is RHOA G17V which acts as a dominant-negative molecule, underpinning its tumor-suppressive function in T-cells [93–95]. Other mutations found frequently in ATLL were mostly located within the GTP-binding pocket, with the gain-of-function variant C16R as the most recurrent. However, loss-of-function mutations have also been detected [121]. Of note, in AITL and related lymphomas, RHOA mutations were accompanied by TET2 mutations, suggesting that TET2 and subsequent RHOA mutations may pave the way for T-cell transformation [95]. The RHOA G17V mouse model has reduced T-cell numbers, but these cells display increased activation upon stimulation and skew toward TFH cell differentiation.
5. Current therapies and novel approaches
5.1. STAT3/5 mutations and inhibition
The STAT protein family is composed of seven members. They share five structural domains: amino-terminal, coiled-coil, DNA-binding, SH2, and carboxy‑terminal transactivation/stability domain. The C-terminal domain of STAT3/5 proteins contains two or three amino acid residues that are phosphorylated and crucial for activity, translocation, and gene regulation. Phosphorylation of an essential tyrosine residue promotes parallel dimerization, whereas phosphorylation of serine residues enhances transcriptional elongation and translocation to mitochondria (in the case of STAT3) or the nucleus (in the case of STAT5A).
Normal STAT action is rapid and transient upon response to cytokines/growth factors. Recycling occurs through tyrosine phosphatases and inhibition by degradation is more associated with JAK and receptor proteins. STATs display a tight regulation of the expression of genes whose protein products regulate critical processes such as proliferation, survival, differentiation, senescence, metabolism, angiogenesis, and invasion [123]. Constitutive activation of STAT3/5 is commonly found in MPN and PTCL [20,124]. Consistent with the prediction that oncogenic transcription factors are triggered downstream of many activated drivers, STATs are activated much more commonly than any single genetic driver mutation [125].
Importantly, recurrent somatic STAT3/5 gain-of-function mutations were found in the SH2 domain or their extreme C-terminus [126], acting as driver genes predominantly in PTCL. Understanding how mutations within the JAK-STAT pathway alter chromatin via epigenetic changes is key to gaining insight into reprogrammed gene regulation in cancer to tailor patient-specific therapies.
The final consideration in developing an anticancer therapy concerns the therapeutic index. While a drug may inhibit a pathway critical for cancer cell proliferation or survival, it is equally important that it is not toxic to normal cells. Evidence from experimental systems to human genetic analyses has provided strong support for reasoning that the activity of specific STAT family members can be lost from normal cells without severe consequence, likely due to redundancies in transcriptional regulation under physiologic conditions [127,128]. Taken together, these findings suggest that STATs are valuable targets for cancer therapy.
It has often been argued that STAT transcription factors are not optimal targets for pharmacological inhibition, because their function is not dependent on small surfaces or pockets to which drug-like molecules can bind. However, STATs clearly have discrete domains necessary for their function, including the SH2, DNA binding and N-terminal oligomerization domains [129]. These sites can certainly be blocked using a number of strategies that hold promise for therapeutic development.
The first inhibitor of a STAT protein was a peptide molecule [130] and efforts to target STAT signaling for therapeutic purposes are ongoing. To date, inhibition of STAT function has been attempted through several approaches, including N-terminal domain binders [131], oligonucleotides targeting the DNA binding domain [132], and most effectively through use of small molecule compounds that bind the SH2 domain to block STAT phosphorylation, dimerization, nuclear transport, and target gene expression [133–135].
The vast majority of medicinal chemistry efforts to target STAT proteins have been conducted to develop specific inhibitors against STAT3 [130–132,136–148], with fewer reports of inhibitory modulators of STAT5A/B. The FDA-approved neuroleptic agent Pimozide was identified in a high-throughput screen as an inhibitor of STAT5 phosphorylation and an inducer of apoptosis in CML cell lines [149]. The underlying mechanism of action is unknown but was suggested to be upstream of STAT5. Furthermore, a non-peptidic chromone-based nicotinyl hydrazine, discovered through a screen of chemical libraries, was shown to weakly inhibit STAT5 activity [150]. This agent selectively inhibited the phosphorylation of STAT5 in lymphoma cell lines by unknown mechanisms. Inhibition of STAT5 activity was also reported for Indirubin derivatives, including E804, which blocked STAT5 phosphorylation and STAT5 DNA-binding activity in CML cells [151], associated with downregulation of MCL-1 and BCL2L1 expression. Based on the structure of the compound, the mechanism of inhibition of STAT5 here is most likely suppression of TK activities.
More recently, a number of promising covalent STAT3/5 SH2 domain-binding inhibitors have been described [8,133,134,152]. These compounds exhibit potent and selective binding activity for STAT3/5 by effectively disrupting phosphopeptide interactions. The lead agent 13a suppresses STAT3/5 tyrosine phosphorylation and inhibits STAT3/5-mediated gene expression, including downregulation of MYC, Cyclin D1, Cyclin D2, and MCL-1 oncoproteins. Importantly, the dual inhibitory function of STAT3/5 inhibitors is of high clinical relevance since Imatinib-resistant CML cells upregulate and activate STAT3, which represents a major signaling node conferring TKI resistance [8]. Moreover, a feedback upregulation of STAT3 as a common cause of resistance to receptor TK/MEK-targeted therapy was described [153]. Overall, high levels of both STAT3/5 activity are found in most cancer types or stroma cells surrounding MPN or PTCL cells. Taken together, new data and mutational landscape studies provide a rationale for targeting both STAT3 and STAT5 [154]. Furthermore, combining potential STAT3/5 inhibitors with approved TKIs might be beneficial in treating cancer. STAT dimerization and signaling can also be blocked by inhibiting upstream JAK kinases, which we discuss next.
5.2. JAK kinase inhibitors
Ruxolitinib partially inhibits the activity of JAK1/2 and is the first drug approved by the FDA for MPN patients. It is prescribed as a targeted therapy for treatment of patients with primary MF, PV, and ET [155]. During clinical trials, it was shown to reduce spleen size, abdominal discomfort, bone pain, night sweats, and itching, as well as diminish the level of inflammatory cytokines in MPN patients.
A number of other drugs that inhibit JAK kinases are currently in clinical trials, including Pacritinib, Momelotinib and NS-018. Pacritinib, a dual JAK2 and FLT3 TKI, is being compared with best available therapy in Phase III trials in patients with MF [156]. Momelotinib performed better than Ruxolitinib with respect to anemia-related end points, but formal statistical testing was not undertaken. How Momelotinib might improve anemia despite inhibiting JAK1/2 is not well understood, but one putative mechanism could be the inhibition of ALK2-mediated hepcidin expression in the liver, which in turn results in increased release of storage iron and promotion of erythropoiesis. NS-018 is a JAK2-selective inhibitor with an IC50 of <1 nM and it has 30- to 50-fold greater selectivity for JAK2 than for other JAK family kinases (JAK1, JAK3, TYK2), and can also inhibit SRC-family kinases. NS-018 potently decreases viability of cell lines expressing constitutively activated JAK2, suppresses endogenous erythroid colony formation by primary cells from PV patients, reduces leukocytosis and splenomegaly, improves BM fibrosis, and prolongs survival in a mouse model of JAK2 V617F-driven MF without causing peripheral anemia or thrombocytopenia [157]. Still, Ruxolitinib remains superior in clinical use and will be challenging to improve upon.
5.3. mTOR inhibitors
Everolimus (also known as RAD001) is a broadly used inhibitor of the mTOR/AKT pathway, which is commonly upregulated in MF hematopoietic cells and appears to contribute to abnormal cell growth. Everolimus was well tolerated in phase I and II clinical trials and was able to reduce both spleen size and systemic symptoms. However, no major sustained responses were seen in these patients [158].
5.4. Epigenetic drugs
Epigenetic drugs change the way genes are organized to make them more or less accessible for use by the cell. Studies have found that Givinostat (HDAC inhibitor) and two hypomethylating drugs, Azacitidine and Decitabine, were minimally effective in treating MF, in contrast to their effectiveness in treating PV. Another HDAC inhibitor, Panobinostat, is currently under investigation. HDAC inhibitors are pleiotropic agents that have multiple potential mechanisms of action in MPN cells, prominent among them being downregulation of JAK2 via inhibition of the chaperone protein function of HSP90. Givinostat and Vorinostat are clearly active in patients with PV and ET, producing both spleen and hematologic responses in a substantial proportion of patients, apparently without regard to the mutational status of JAK2.
Vorinostat, a class I and II HDAC inhibitor, was approved more than 10 years ago for the treatment of CTCL [159]. Romidepsin, a pan-HDAC inhibitor, is also approved for use in CTCL patients as well as for relapsed and refractory PTCL. A third inhibitor, the pan-HDAC inhibitor Belinostat, was also more recently approved for relapsed and refractory PTCL cases [160,161]. The overall responses were 25% for Romidepsin and 26% for Belinostat [162,163]. Additional indications for Romidepsin are currently being evaluated as a combinatorial treatment, for instance, with Bortezomib, Carfilzomib (both proteasome inhibitors), 5-Azacytidine, or CHOP. Chidamide, as well as acting as an HDAC inhibitor, is so far only approved for PTCL treatment in China and used as a monotherapy or in combination with chemotherapy [164,165].
Because JAK2 interacts with the chaperone HSP90, pharmacologic inhibition of HSP90 was proposed to cause misfolding and degradation of JAK2. This was shown in MPN cell lines, primary MPN patient samples, and mouse models of PV and ET treated with the HSP90 inhibitor PU-H71, without degradation of JAK2 in normal tissues or substantial toxicity. Degradation of JAK2 via HSP90 inhibition has also been shown to be a way of circumventing persistent signaling with JAK2 inhibition. Synergism between the HSP90 inhibitor AUY922 and the JAK2 inhibitor TG101209 was demonstrated in human CD34+ MPN cells, which exhibited significantly greater apoptosis than did normal hematopoietic progenitor cells. Combination therapy with PU-H71 and Ruxolitinib was shown to be more potent in inhibiting JAK2 downstream signaling than Ruxolitinib alone [166]. This translated to improvements in blood counts, spleen weights, and BM fibrosis in transgenic mice. The combination of Ruxolitinib and Decitabine appears promising in patients with accelerated or blast phase MPN (post-MPN acute myeloid leukemia) in small studies [167].
5.5. TP53 reactivation and TP53* targeting
Rescuing unstable TP53 protein pools upon hotspot mutation demonstrates feasibility to target transcription factor function. This is independent of whether TP53 was mutated or aberrantly activated due to upstream mutations in negative-regulators. PRIMA-1 and its derivative PRIMA-1MET (also called APR-246) can restore wild-type protein conformation to TP53*. This restores transcriptional activity of normal TP53 that senses DNA damage, leading to expression of PUMA, NOXA, and BAX in TP53-mutated cancer cells [168,169]. PRIMA-1 compounds are converted intracellularly to the Michael acceptor methylene quinuclidinone, subsequently binding covalently to cysteines of TP53*. It will be important in a clinical setting to tailor the strategy to specific MPN or PTCL subtypes dependent on the TP53 mutational status. Furthermore, MDM2 and MDMX expression levels are of relevance, which we discuss next.
MDM2 is an important negative regulator of TP53, and small-molecule inhibitors of MDM2 can trigger apoptosis in cells with intact TP53 function through TP53-activation. Because type I interferons (IFN) target JAK2 V617F+ progenitors in PV through activation of MAPK and STAT1, thereby increasing TP53 transcription, the combination of IFN with MDM2 inhibitors, which prevent the degradation of TP53, provides an opportunity to induce TP53-dependent apoptosis [170]. Indeed, combination treatment with IFN and the MDM2 antagonist Nutlin-3 triggered apoptosis in PV CD34+ cells and inhibited proliferation of these cells to a greater extent than normal CD34+ cells [170]. The combination also reduced the proportion of JAK2 V617F progenitors in PV patients. Combination treatment of PV and primary MF CD34+ cells, followed by transplantation into immunodeficient mice, decreased the extent of donor-derived chimerism as well as the JAK2 V617F allele burden, suggesting that such combinatorial approaches may deplete MPN hematopoietic stem cells [170]. The clinical candidate MDM2 antagonist Idasanutlin is currently in a phase I trial in patients with PV or ET, with a provision for adding pegylated IFN in subjects without or with partial remission after three cycles of therapy.
An overview of the drugs currently undergoing clinical trials for MPN and PTCL is displayed in Table 3.
Table 3.
List of drugs and their targets currently undergoing clinical trials for MPN and/or PTCL disease as mono- and/or combination therapies as of October 2017 (https://clinicaltrials.gov/). Drugs that are involved in clinical trials for both MPN and PTCL are highlighted in italic. Only targeted therapy drugs are listed (no chemotherapy or immunotherapy drugs included).
| MPN |
PTCL |
||
|---|---|---|---|
| Drug | Target | Drug | Target |
| Ruxolitinib | JAK1/2 | Ruxolitinib | JAK1/2 |
| Momelotinib | Cerdulatinib | SYK/JAK | |
| Itacitinib | JAK1 | ASN002 | |
| NS-018 | JAK2 | Everolimus | mTOR/AKT pathway |
| Pacritinib | Temsirolimus | ||
| LY2784544 | BMS-906024 | Notch | |
| Umbralisib (TGR-1202) | PI3Kδ | LY3039478 | |
| Idelalisib | Tipifarnib | Ras (posttranslational modification) | |
| INCB050465 | Umbralisib (TGR-1202) | PI3Kδ | |
| Rigosertib | PI3K and PLK pathways | CPI-618 | α-ketoglutarate dehydrogenase |
| Glasdegib (PF-04449913) | Sonic hedgehog pathway | Alisertib | Aurora A kinase |
| Sonidegib (LDE225) | DS-3201b | EZH2 | |
| LCL161 | cIAP1 and cIAP2 | Decitabine | Hypomethylation |
| Idasanutlin (RG7388) | TP53–MDM2 | Panobinostat | Pan-HDAC |
| PRIMA-1MET (APR-246) | TP53 | Romidepsin | |
| IMG-7289 | LSD1 | Belinostat | |
| Givinostat (ITF2357) | Class I and class II HDACs | AR-42 | |
| Panobinostat | Pan-HDAC | Chidamide | |
| Azacitidine | Hypomethylation | Bortezomib | Proteasome |
| Decitabine | Carfilzomib | ||
| PU-H71 | HSP90 | Ixazomib (MLN 9708) | |
| AUY922 | |||
6. Expert opinion
Small molecule inhibitors targeting key drivers in MPN or PTCL hold the greatest promise to reach the clinic. Their size, polarity, solubility, pharmacokinetics and pharmacodynamics, and toxic side-effects can be improved through medicinal chemistry approaches, and structural modeling based on lead compounds could improve targeting efficacy. We need to better understand and map how mutated disease drivers such as epigenetic remodelers, JAK-STAT gain-of-function mutations, TP53*, and hyperactive GTPases interact and cooperate in specific cell types. In summary, structural modeling and protein interaction studies can reveal a detailed, atomic-level understanding of vulnerable nodes. During therapy with targeted drugs, new subclones with other driver mutations may escape and lead to relapses, which points to the need to develop new drugs with broader multi-target activities. Furthermore, in vivo targeting of suitable animal models, accurate biological read out systems with the right combination of driver mutations, and related early phase clinical trials focused on specific patient subgroups could increase the repertoire of therapeutic approaches to target MPN and PTCL. We stand at a crossroads in understanding key drivers in cancer biology, but we still need to understand how they cooperate to manifest into neoplasia. How interactions between the DNA damage and checkpoint control machinery, or epigenetic gene regulation influences or connects to JAK-STAT driver mutations is still under investigation. Hematologic cancer research defines MPN and PTCL as different disease entities; however, insights into epigenetics and mutational landscapes with expression profiling point to more similarity between the two diseases, suggesting the potential for common targeting strategies.
Funding Statement
B. Wingelhofer, H. A. Neubauer, B. Maurer, and R. Moriggl are supported by the Austrian Science Fund (FWF) [SFB-F4707-B20, SFB-F06105, SFB-F06107]. P. Valent is supported by FWF SFB-F4704-B20. A. Orlova is supported by the Austrian Research Promotion Agency [FFG, 854452]. G. M. Keserü is supported by the Hungarian Scientific Research Fund [OTKA, K116904]. A. Berger-Becvar and P. T. Gunning are sponsored by CIHR, Canada Foundation for Innovation [497203, 495468], a Tier II Canada Research Chair, a MITACS fellowship and the Leukemia Lymphoma Society [IT05960].
Article highlights
MPN and PTCL are both aggressive hematopoietic malignancies, requiring targeted treatment.
High occurrence of resistance mechanisms in MPN patients limits effective use of Ruxolitinib in clinics.
Treatment options for PTCL patients are limited to chemotherapy due to a lack of potential molecular targets for this disease.
High mutational rates in epigenetic regulators, as well as other common signaling pathways, have been reported in both MPN and PTCL.
Potential strategies to improve treatment of both MPN and PTCL patients include drugs targeting these commonly mutated pathways.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1. Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hnisz D, Weintraub AS, Day DS, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351(6280):1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suzuki HI, Young RA, Sharp PA.. Super-enhancer-mediated RNA processing revealed by integrative microRNA network analysis. Cell. 2017;168(6): 1000–1014. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forbes SA, Beare D, Boutselakis H, et al. COSMIC: high-resolution cancer genetics using the catalogue of somatic mutations in cancer. Curr Protoc Hum Genet. 2016;91 . [DOI] [PubMed] [Google Scholar]
- 7. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eiring AM, Page BDG, Kraft IL, et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia. 2015;29(3):586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Danis E, Yamauchi T, Echanique K, et al. Ezh2 controls an early hematopoietic program and growth and survival signaling in early T cell precursor acute lymphoblastic leukemia. Cell Rep. 2016;14(8):1953–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baker SG. A cancer theory kerfuffle can lead to new lines of research. J Natl Cancer Inst. 2015;107(2):1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenfeld S. Biomolecular self-defense and futility of high-specificity therapeutic targeting. Gene Regul Syst Bio. 2011;5:89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jing T, Tero A. Network pharmacology strategies toward multi-target anticancer therapies: from computational models to experimental design principles. Curr Pharm Des. 2014;20(1):23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fiskus W, Verstovsek S, Manshouri T, et al. Dual PI3K/AKT/mTOR inhibitor BEZ235 synergistically enhances the activity of JAK2 inhibitor against cultured and primary human myeloproliferative neoplasm cells. Mol Cancer Ther. 2013;12:577–588. [DOI] [PubMed] [Google Scholar]
- 14. Michiels JJ, Tevet M, Trifa A, et al. 2016 WHO clinical molecular and pathological criteria for classification and staging of myeloproliferative neoplasms (MPN) caused by MPN driver mutations in the JAK2, MPL and CALR genes in the context of new 2016 WHO classification: prognostic and therapeutic implications. Maedica (Buchar). 2016;11(1):5–25. [PMC free article] [PubMed] [Google Scholar]; • A comprehensive review on WHO 2016 classification of MPN according to both genetic drivers and pathological picture.
- 15. Them NC, Kralovics R. Genetic basis of MPN: beyond JAK2-V617F. Curr Hematol Malig Rep. 2013;8(4):299–306. [DOI] [PubMed] [Google Scholar]
- 16. Barbui T, Thiele J, Passamonti F, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. 2011;29(23):3179–3184. [DOI] [PubMed] [Google Scholar]
- 17. Crisa E, Venturino E, Passera R, et al. A retrospective study on 226 polycythemia vera patients: impact of median hematocrit value on clinical outcomes and survival improvement with anti-thrombotic prophylaxis and non-alkylating drugs. Ann Hematol. 2010;89(7):691–699. [DOI] [PubMed] [Google Scholar]
- 18. Mesa RA, Li C-Y, Ketterling RP, et al. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3):973–977. [DOI] [PubMed] [Google Scholar]
- 19. Plo I, Vainchenker W. Molecular and genetic bases of myeloproliferative disorders: questions and perspectives. Clin Lymphoma Myeloma. 2009;9(Suppl 3):S329–S339. [DOI] [PubMed] [Google Scholar]
- 20. Walz C, Ahmed W, Lazarides K, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood. 2012;119(15):3550–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dawson MA, Bannister AJ, Göttgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461(7265):819–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu F, Zhao X, Perna F, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19(2):283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rumi E, Cazzola M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood. 2017;129(6):680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stanley RF, Steidl U. Molecular mechanism of mutant CALR-mediated transformation. Cancer Discov. 2016;6(4):344–346. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This review describes in detail the molecular consequences of CALR mutation.
- 25. Milosevic JD, Kralovics R. Genetic and epigenetic alterations of myeloproliferative disorders. Int J Hematol. 2013;97(2):183–197. [DOI] [PubMed] [Google Scholar]
- 26. Pecquet C, Staerk J, Chaligné R, et al. Induction of myeloproliferative disorder and myelofibrosis by thrombopoietin receptor W515 mutants is mediated by cytosolic tyrosine 112 of the receptor. Blood. 2010;115(5):1037–1048. [DOI] [PubMed] [Google Scholar]
- 27. Staerk J, Lacout C, Sato T, et al. An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Blood. 2006;107(5):1864–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lundberg P, Karow A, Nienhold R, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123(14):2220–2228. [DOI] [PubMed] [Google Scholar]
- 29. McPherson S, McMullin MF, Mills K. Epigenetics in myeloproliferative neoplasms. J Cell Mol Med. 2017;21(9):1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. [DOI] [PubMed] [Google Scholar]
- 31. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kubesova B, Pavlova S, Malcikova J, et al. Low-burden TP53 mutations in chronic phase of myeloproliferative neoplasms: association with age, hydroxyurea administration, disease type and JAK2 mutational status. Leukemia. 2017;1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This research work investigates the consequences of TP53 mutations in MPN progression.
- 34. Niemeyer CM, Kang MW, Shin DH, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42(9):794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study investigated TET2 mutations upon aging in the hematopoietic compartment.
- 36. Deininger MWN, Tyner JW, Solary E. Turning the tide in myelodysplastic/myeloproliferative neoplasms. Nat Rev Cancer. 2017;17:425–440. [DOI] [PubMed] [Google Scholar]
- 37. Patel U, Luthra R, Medeiros LJ, et al. Diagnostic, prognostic, and predictive utility of recurrent somatic mutations in myeloid neoplasms. Clin Lymphoma Myeloma Leuk. 2017;17:S62–S74. [DOI] [PubMed] [Google Scholar]
- 38. Harutyunyan A, Klampfl T, Cazzola M, et al. p53 lesions in leukemic transformation. N Engl J Med. 2011;364(5):488–490. [DOI] [PubMed] [Google Scholar]
- 39. Girardot M, Pecquet C, Chachoua I, et al. Persistent STAT5 activation in myeloid neoplasms recruits p53 into gene regulation. Oncogene. 2015;34(10):1323–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Walerych D, Lisek K, Sommaggio R, et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol. 2016;18(8):897–909. [DOI] [PubMed] [Google Scholar]
- 41. Mukhopadhyay UK, Cass J, Raptis L, et al. STAT5A is regulated by DNA damage via the tumor suppressor p53. Cytokine. 2016;82:70–79. [DOI] [PubMed] [Google Scholar]
- 42. Berger A, Hoelbl-Kovacic A, Bourgeais J, et al. PAK-dependent STAT5 serine phosphorylation is required for BCR-ABL-induced leukemogenesis. Leukemia. 2014;28(3):629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sellar R, Losman JA. Targeting aberrant signaling in myeloid malignancies: promise versus reality. Hematol Oncol Clin North Am. 2017;31(4):565–576. [DOI] [PubMed] [Google Scholar]
- 44. Foss FM, Zinzani PL, Vose JM, et al. Peripheral T-cell lymphoma. Blood. 2011;117(25):6756–6767. [DOI] [PubMed] [Google Scholar]
- 45. Armitage J, Tefferi A. The aggressive peripheral T-cell lymphomas: 2013. Am J Hematol. 2013;88(10):910–918. [DOI] [PubMed] [Google Scholar]
- 46. Armitage JO. The aggressive peripheral T-cell lymphomas: 2015. Am J Hematol. 2015;90(7):665–673. [DOI] [PubMed] [Google Scholar]
- 47. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Horwitz SM. Current and novel treatment options for peripheral T-cell lymphoma. Clin Adv Hematol Oncol. 2015;13(7):463–466. [PubMed] [Google Scholar]
- 49. Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–4130. [DOI] [PubMed] [Google Scholar]
- 50. Gaulard P, de Leval L. Pathology of peripheral T-cell lymphomas: where do we stand? Semin Hematol. 2014;51(1):5–16. [DOI] [PubMed] [Google Scholar]
- 51. Piccaluga P, Tabanelli V, Pileri S. Molecular genetics of peripheral T-cell lymphomas. Int J Hematol. 2014;99(3):219–226. [DOI] [PubMed] [Google Scholar]
- 52. Streubel B, Vinatzer U, Willheim M, et al. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia. 2006;20(2):313–318. [DOI] [PubMed] [Google Scholar]
- 53. Feldman AL, Law M, Remstein ED, et al. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia. 2009;23(3):574–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zettl A, Ott G, Makulik A, et al. Chromosomal gains at 9q characterize enteropathy-type T-cell lymphoma. Am J Pathol. 2002;161(5):1635–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Macon WR, Levy NB, Kurtin PJ, et al. Hepatosplenic alphabeta T-cell lymphomas: a report of 14 cases and comparison with hepatosplenic gammadelta T-cell lymphomas. Am J Surg Pathol. 2001;25(3):285–296. [DOI] [PubMed] [Google Scholar]
- 56. Siu LL, Wong KF, Chan JK, et al. Comparative genomic hybridization analysis of natural killer cell lymphoma/leukemia. Recognition of consistent patterns of genetic alterations. Am J Pathol. 1999;155(5):1419–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reddy NM, Evens AM. Chemotherapeutic advancements in peripheral T-cell lymphoma. Semin Hematol. 2014;51(1):17–24. [DOI] [PubMed] [Google Scholar]
- 58. d’Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093–3099. [DOI] [PubMed] [Google Scholar]
- 59. Iqbal J, Wright G, Wang C, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123(19):2915–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study provides an outstanding comparison of gene expression profiles between subtypes of PTCL.
- 60. Iqbal J, Weisenburger DD, Greiner TC, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood. 2010;115(5):1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laimer D, Dolznig H, Kollmann K, et al. PDGFR blockade is a rational and effective therapy for NPM-ALK-driven lymphomas. Nat Med. 2012;18(11):1699–1704. [DOI] [PubMed] [Google Scholar]
- 62. Wang T, Feldman AL, Wada DA, et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood. 2014;123(19):3007–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Laginestra MA, Piccaluga PP, Fuligni F, et al. Pathogenetic and diagnostic significance of microRNA deregulation in peripheral T-cell lymphoma not otherwise specified. Blood Cancer J. 2014;4(11):259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27(4):516–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang C, McKeithan TW, Gong Q, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood. 2015;126(15):1741–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wilcox RA. A three signal model of T-cell lymphoma pathogenesis. Am J Hematol. 2016;91(1):113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47(11):1304–1315. [DOI] [PubMed] [Google Scholar]
- 68. Casulo C, O'Connor O, Shustov A, et al. T-cell lymphoma: recent advances in characterization and new opportunities for treatment. J Natl Cancer Inst. 2017;109(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Merkel O, Hamacher F, Sifft E, et al. Novel therapeutic options in anaplastic large cell lymphoma: molecular targets and immunological tools. Mol Cancer Ther. 2011;10(7):1127–1136. [DOI] [PubMed] [Google Scholar]
- 70. Kiel MJ, Velusamy T, Rolland D, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124(9):1460–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dierks C, Adrian F, Fisch P, et al. The ITK-SYK fusion oncogene induces a T-cell lymphoproliferative disease in mice mimicking human disease. Cancer Res. 2010;70(15):6193–6204. [DOI] [PubMed] [Google Scholar]
- 72. Boddicker RL, Kip NS, Xing X, et al. The oncogenic transcription factor IRF4 is regulated by a novel CD30/NF-kappaB positive feedback loop in peripheral T-cell lymphoma. Blood. 2015;125(20):3118–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schatz JH, Horwitz SM, Teruya-Feldstein J, et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia. 2015;29(1):237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kiel MJ, Sahasrabuddhe AA, Rolland DCM, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sezary syndrome. Nat Commun. 2015;6:8470. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This manuscript integrated different sequencing approaches to identify common mutations in Sézary syndrome patients.
- 76. Lopez C, Bergmann AK, Paul U, et al. Genes encoding members of the JAK-STAT pathway or epigenetic regulators are recurrently mutated in T-cell prolymphocytic leukaemia. Br J Haematol. 2016;173(2):265–273. [DOI] [PubMed] [Google Scholar]
- 77. Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123(9):1293–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nicolae A, Xi L, Pittaluga S, et al. Frequent STAT5B mutations in [gamma][delta] hepatosplenic T-cell lymphomas. Leukemia. 2014;28(11):2244–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rajala HLM, Eldfors S, Kuusanmäki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bandapalli OR, Schuessele S, Kunz JB, et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica. 2014;99(10): e188–e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kontro M, Kuusanmäki H, Eldfors S, et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia. 2014;28(8):1738–1742. [DOI] [PubMed] [Google Scholar]
- 82. Küçük C, Jiang B, Hu X, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat Commun. 2015;6(6025). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper provides sequencing results that identify mutations in STAT5B and STAT3 in NK/T-cell and γδ-T-cell lymphomas.
- 83. Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016;55(1):82–94. [DOI] [PubMed] [Google Scholar]
- 84. Fantin VR, Loboda A, Paweletz CP, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008;68(10):3785–3794. [DOI] [PubMed] [Google Scholar]
- 85. Piccaluga PP, Rossi M, Agostinelli C, et al. Platelet-derived growth factor alpha mediates the proliferation of peripheral T-cell lymphoma cells via an autocrine regulatory pathway. Leukemia. 2014;28(8):1687–1697. [DOI] [PubMed] [Google Scholar]
- 86. Kopp KL, Ralfkiaer U, Gjerdrum LMR, et al. STAT5-mediated expression of oncogenic miR-155 in cutaneous T-cell lymphoma. Cell Cycle. 2013;12(12):1939–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sibbesen NA, Kopp KL, Litvinov IV, et al. Jak3, STAT3, and STAT5 inhibit expression of miR-22, a novel tumor suppressor microRNA, in cutaneous T-cell lymphoma. Oncotarget. 2015;6(24):20555–20569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lauenborg B, Christensen L, Ralfkiaer U, et al. Malignant T cells express lymphotoxin α and drive endothelial activation in cutaneous T cell lymphoma. Oncotarget. 2015;6(17):15235–15249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang Q, Wang HY, Wei F, et al. Cutaneous T cell lymphoma expresses immunosuppressive CD80 (B7-1) cell surface protein in a STAT5-dependent manner. J Immunol. 2014;192(6):2913–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Litvinov IV, Netchiporouk E, Cordeiro B, et al. The use of transcriptional profiling to improve personalized diagnosis and management of cutaneous T-cell lymphoma (CTCL). Clin Cancer Res. 2015;21(12):2820–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Goyama S, Kitamura T. Epigenetics in normal and malignant hematopoiesis: an overview and update 2017. Cancer Sci. 2017;108(4):553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ondrejka SL, Hsi ED. T-cell lymphomas: updates in biology and diagnosis. Surg Pathol Clin. 2016;9(1):131–141. [DOI] [PubMed] [Google Scholar]
- 93. Yoo HY, Sung MK, Lee SH, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46(4):371–375. [DOI] [PubMed] [Google Scholar]
- 94. Palomero T, Couronné L, Khiabanian H, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46(2):166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study performed targeted deep sequencing to identify new genetic and epigenetic alterations in PTCL.
- 95. Sakata-Yanagimoto M, Enami T, Yoshida K, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46(2):171–175. [DOI] [PubMed] [Google Scholar]
- 96. Lemonnier F, Couronné L, Parrens M, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120(7):1466–1469. [DOI] [PubMed] [Google Scholar]
- 97. Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med. 2012;366(1):95–96. [DOI] [PubMed] [Google Scholar]
- 98. Hildyard CAT, Shiekh S, Browning JAB, et al. Toward a biology-driven treatment strategy for peripheral T-cell lymphoma. Clin Med Insights. 2017;10(10.1177_1179545X17705863.xml):1179545X17705863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Quivoron C, Couronné L, Della Valle V, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1):25–38. [DOI] [PubMed] [Google Scholar]
- 100. Muto H, Sakata-Yanagimoto M, Nagae G, et al. Reduced TET2 function leads to T-cell lymphoma with follicular helper T-cell-like features in mice. Blood Cancer J. 2014;4:e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Karimi MM, Goyal P, Maksakova IA, et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8(6):676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chang Y, Sun L, Kokura K, et al. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat Commun. 2011;2:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Takeuchi A, Nishioka C, Ikezoe T, et al. STAT5A regulates DNMT3A in CD34(+)/CD38(-) AML cells. Leuk Res. 2015;39(8):897–905. [DOI] [PubMed] [Google Scholar]
- 104. Rush M, Appanah R, Lee S, et al. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of Dnmt3a but not de novo DNA methylation. Epigenetics. 2009;4(6):404–414. [DOI] [PubMed] [Google Scholar]
- 105. Holz-Schietinger C, Matje DM, Reich NO. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem. 2012;287(37):30941–30951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Haney SL, Upchurch GM, Opavska J, et al. Dnmt3a is a haploinsufficient tumor suppressor in CD8+ peripheral T cell lymphoma. PLoS Genet. 2016;12(9):e1006334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Cairns RA, Iqbal J, Lemonnier F, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119(8):1901–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lemonnier F, Cairns RA, Inoue S, et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci USA. 2016;113(52):15084–15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Brito-Babapulle V, Hamoudi R, Matutes E, et al. p53 allele deletion and protein accumulation occurs in the absence of p53 gene mutation in T-prolymphocytic leukaemia and Sezary syndrome. Br J Haematol. 2000;110(1):180–187. [DOI] [PubMed] [Google Scholar]
- 111. Cui Y-X, Kerby A, McDuff FKE, et al. NPM-ALK inhibits the p53 tumor suppressor pathway in an MDM2 and JNK-dependent manner. Blood. 2009;113(21):5217–5227. [DOI] [PubMed] [Google Scholar]
- 112. Matei IR, Guidos CJ, Danska JS. ATM-dependent DNA damage surveillance in T-cell development and leukemogenesis: the DSB connection. Immunol Rev. 2006;209:142–158. [DOI] [PubMed] [Google Scholar]
- 113. Bourgeais J, Ishac N, Medrzycki M, et al. Oncogenic STAT5 signaling promotes oxidative stress in chronic myeloid leukemia cells by repressing antioxidant defenses. Oncotarget. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cholez E, Debuysscher V, Bourgeais J, et al. Evidence for a protective role of the STAT5 transcription factor against oxidative stress in human leukemic pre-B cells. Leukemia. 2012;26(11):2390–2397. [DOI] [PubMed] [Google Scholar]
- 115. Jayavelu AK, Moloney JN, Böhmer F-D, et al. NOX-driven ROS formation in cell transformation of FLT3-ITD positive AML. Exp Hematol. 2016;44(12):1113–1122. [DOI] [PubMed] [Google Scholar]
- 116. Zhang Q, Raje V, Yakovlev VA, et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem. 2013;288(43):31280–31288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Themanns M, Mueller KM, Kessler SM, et al. Hepatic deletion of Janus Kinase 2 counteracts oxidative stress in mice. Sci Rep. 2016;6:34719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Friedbichler K, Themanns M, Mueller KM, et al. Growth-hormone-induced signal transducer and activator of transcription 5 signaling causes gigantism, inflammation, and premature death but protects mice from aggressive liver cancer. Hepatology. 2012;55(3):941–952. [DOI] [PubMed] [Google Scholar]
- 119. Slupianek A, Dasgupta Y, Ren S-Y, et al. Targeting RAD51 phosphotyrosine-315 to prevent unfaithful recombination repair in BCR-ABL1 leukemia. Blood. 2011;118(4):1062–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Slupianek A, Schmutte C, Tombline G, et al. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol Cell. 2001;8(4):795–806. [DOI] [PubMed] [Google Scholar]
- 121. Nagata Y, Kontani K, Enami T, et al. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood. 2016;127(5):596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hanna S, El-Sibai M. Signaling networks of Rho GTPases in cell motility. Cell Signal. 2013;25(10):1955–1961. [DOI] [PubMed] [Google Scholar]
- 123. Darnell JE., Jr. STATs and gene regulation. Science. 1997;277(5332):1630–1635. [DOI] [PubMed] [Google Scholar]
- 124. Kucuk C, Jiang B, Hu X, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun. 2015;6:6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Levy DE, Inghirami G. STAT3: a multifaceted oncogene. Proc Natl Acad Sci USA. 2006;103(27):10151–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Andersson E, Kuusanmäki H, Bortoluzzi S, et al. Activating somatic mutations outside the SH2-domain of STAT3 in LGL leukemia. Leukemia. 2016;30(5):1204–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Garcia R, Yu CL, Hudnall A, et al. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997;8(12):1267–1276. [PubMed] [Google Scholar]
- 128. Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1608–1619. [DOI] [PubMed] [Google Scholar]
- 129. Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13(2):211–217. [DOI] [PubMed] [Google Scholar]
- 130. Turkson J, Ryan D, Kim JS, et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276(48):45443–45455. [DOI] [PubMed] [Google Scholar]
- 131. Timofeeva OA, Gaponenko V, Lockett SJ, et al. Rationally designed inhibitors identify STAT3 N-domain as a promising anticancer drug target. ACS Chem Biol. 2007;2(12):799–809. [DOI] [PubMed] [Google Scholar]
- 132. Huang W, Dong Z, Wang F, et al. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem Biol. 2014;9(5):1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Page BDG, Khoury H, Laister RC, et al. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J Med Chem. 2012;55(3):1047–1055. [DOI] [PubMed] [Google Scholar]
- 134. Fletcher S, Drewry JA, Shahani VM, et al. Molecular disruption of oncogenic signal transducer and activator of transcription 3 (STAT3) protein. Biochem Cell Biol. 2009;87(6):825–833. [DOI] [PubMed] [Google Scholar]
- 135. Haftchenary S, Avadisian M, Gunning PT. Inhibiting aberrant Stat3 function with molecular therapeutics: a progress report. Anticancer Drugs. 2011;22(2):115–127. [DOI] [PubMed] [Google Scholar]
- 136. Auzenne EJ, Klostergaard J, Mandal PK, et al. A phosphopeptide mimetic prodrug targeting the SH2 domain of Stat3 inhibits tumor growth and angiogenesis. J Exp Ther Oncol. 2012;10(2):155–162. [PMC free article] [PubMed] [Google Scholar]
- 137. Chen J, Bai L, Bernard D, et al. structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors. ACS Med Chem Lett. 2010;1(2):85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Coleman DRT, Ren Z, Mandal PK, et al. Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J Med Chem. 2005;48(21):6661–6670. [DOI] [PubMed] [Google Scholar]
- 139. Fossey SL, Bear MD, Lin J, et al. The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer. 2011;11:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Gunning PT, Katt WP, Glenn M, et al. Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: structural recognition of STAT SH2 domains. Bioorg Med Chem Lett. 2007;17(7):1875–1878. [DOI] [PubMed] [Google Scholar]
- 141. Hao W, Hu Y, Niu C, et al. Discovery of the catechol structural moiety as a Stat3 SH2 domain inhibitor by virtual screening. Bioorg Med Chem Lett. 2008;18(18):4988–4992. [DOI] [PubMed] [Google Scholar]
- 142. Lin L, Hutzen B, Zuo M, et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 2010;70(6):2445–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Onimoe G-I, Liu A, Lin L, et al. Small molecules, LLL12 and FLLL32, inhibit STAT3 and exhibit potent growth suppressive activity in osteosarcoma cells and tumor growth in mice. Invest New Drugs. 2012;30(3):916–926. [DOI] [PubMed] [Google Scholar]
- 144. Schust J, Sperl B, Hollis A, et al. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13(11):1235–1242. [DOI] [PubMed] [Google Scholar]
- 145. Siddiquee KAZ, Gunning PT, Glenn M, et al. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2(12):787–798. [DOI] [PubMed] [Google Scholar]
- 146. Song H, Wang R, Wang S, et al. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci USA. 2005;102(13):4700–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Zhang X, Yue P, Page BDG, et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci USA. 2012;109(24):9623–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Turkson J, Kim JS, Zhang S, et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3(3):261–269. [PubMed] [Google Scholar]
- 149. Nelson EA, Walker SR, Weisberg E, et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117(12):3421–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Müller J, Sperl B, Reindl W, et al. Discovery of chromone-based inhibitors of the transcription factor STAT5. Chembiochem. 2008;9(5):723–727. [DOI] [PubMed] [Google Scholar]
- 151. Nam S, Scuto A, Yang F, et al. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of Stat5 signaling. Mol Oncol. 2012;6(3):276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Cumaraswamy AA, Lewis AM, Geletu M, et al. Nanomolar-potency small molecule inhibitor of STAT5 protein. ACS Med Chem Lett. 2014;5(11):1202–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Lee H-J, Zhuang G, Cao Y, et al. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26(2):207–221. [DOI] [PubMed] [Google Scholar]
- 154. Gleixner KV, Schneeweiss M, Eisenwort G, et al. Combined targeting of STAT3 and STAT5: a novel approach to overcome drug resistance in chronic myeloid leukemia. Haematologica. 2017;102(9):1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Vannucchi AM, Harrison CN. Emerging treatments for classical myeloproliferative neoplasms. Blood. 2017;129(6):693–703. [DOI] [PubMed] [Google Scholar]; •• A comprehensive review about treatment options in MPN patients.
- 156. Verstovsek S, Komrokji RS. A comprehensive review of pacritinib in myelofibrosis. Future Oncol. 2015;11(20):2819–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Verstovsek S, Talpaz M, Ritchie E, et al. A phase I, open-label, dose-escalation, multicenter study of the JAK2 inhibitor NS-018 in patients with myelofibrosis. Leukemia. 2017;31(2):393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Guglielmelli P, Barosi G, Rambaldi A, et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood. 2011;118(8):2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. O’Connor OA, Heaney ML, Schwartz L, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24(1):166–173. [DOI] [PubMed] [Google Scholar]
- 160. Barbarotta L, Hurley K. Romidepsin for the treatment of peripheral T-cell lymphoma. J Adv Pract Oncol. 2015;6(1):22–36. [PMC free article] [PubMed] [Google Scholar]
- 161. Sawas A, Radeski D, O’Connor OA. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: a perspective review. Ther Adv Hematol. 2015;6(4):202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. O’Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631–636. [DOI] [PubMed] [Google Scholar]
- 164. Lu X, Ning Z, Li Z, et al. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis Res. 2016;5(3):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Shi Y, Jia B, Xu W, et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: a multicenter real-world study in China. J Hematol Oncol. 2017;10(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bhagwat N, Koppikar P, Keller M, et al. Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood. 2014;123:2075–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Daver N, Verstovsek S. Ruxolitinib and DNA methyltransferase-inhibitors: a foray into combination regimens in myelofibrosis. Leuk Lymphoma. 2015;56(2):279–280. [DOI] [PubMed] [Google Scholar]
- 168. Shen J, Vakifahmetoglu H, Stridh H, et al. PRIMA-1MET induces mitochondrial apoptosis through activation of caspase-2. Oncogene. 2008;27(51):6571–6580. [DOI] [PubMed] [Google Scholar]
- 169. Wang T, Lee K, Rehman A, et al. PRIMA-1 induces apoptosis by inhibiting JNK signaling but promoting the activation of Bax. Biochem Biophys Res Commun. 2007;352(1):203–212. [DOI] [PubMed] [Google Scholar]
- 170. Lu M, Wang X, Li Y, et al. Combination treatment in vitro with Nutlin, a small-molecule antagonist of MDM2, and pegylated interferon-alpha 2a specifically targets JAK2V617F-positive polycythemia vera cells. Blood. 2012;120(15):3098–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]