

Abstract
Doxorubicin (DOX) is a chemotherapeutic that is used in the treatment of a wide variety of cancers. However, it causes cardiotoxicity partly due to the formation of reactive oxygen species (ROS). CYP2J2 is a human cytochrome P450 that is highly expressed in cardiomyocytes. It converts arachidonic acid (AA) into four different regioisomers of epoxyeicosatrienoic acids (EETs). Using kinetic analyses we show that AA metabolism by CYP2J2 is modulated by DOX. We show that cytochrome P450 reductase (CPR), the redox partner of CYP2J2, metabolizes DOX to 7-deoxydoxorubicin aglycone (7-de-aDOX). This metabolite then binds to CYP2J2, and inhibits and alters the preferred site of metabolism of AA leading to change in the ratio of the EET regioisomers. Furthermore, molecular dynamics (MD) simulations indicate that 7-de-aDOX and AA can concurrently bind to the CYP2J2 active site to produce these changes in the site of AA metabolism. To see if these observations are unique to DOX/7-de-aDOX, we use non-cardiotoxic DOX analogues, zorubicin (ZRN) and 5-iminodaunorubicin (IDN). ZRN and 5-IDN inhibit CYP2J2-mediated AA metabolism, but does not change the ratio of EET regioisomers. Taken together, we demonstrate that DOX and 7-de-aDOX inhibit CYP2J2-mediated AA metabolism and 7-de-aDOX binds close to the active site to alter the ratio of cardioprotective EETs. These mechanistic studies of CYP2J2 can aid in the design of new alternative DOX derivatives.
Keywords: Doxorubicin, arachidonic acid, cardiotoxicity, CYP2J2, epoxyeicosatrienoic acids
Graphical abstract
INTRODUCTION
Anthracyclines are one of the major classes of chemotherapeutics that are used in the treatment of a wide variety of cancers.1, 2 The prototypical anthracyclines are daunorubicin, which was naturally discovered in Streptomyoces peucetius, and the 14-OH derivative, doxorubicin (DOX).3,4 Their anticancer mechanisms involve DNA intercalation and topoisomerase II inhibition.2, 5 DOX produces reactive oxygen species (ROS) leading to nonspecific cytotoxicity. DOX is especially cardiotoxic and causes arrhythmia as a result of QT interval prolongation.6, 7 Though ROS-related injury, including mitochondrial damage and lipid peroxidation, has been suggested to cause cardiotoxicity, there remain unknown mechanisms.8–10 For instance, zorubicin (ZRN) and 5-iminodaunorubicin (5-IDN) are daunorubicin analogues that have reduced cardiotoxicity; however, whereas the former produces ROS, the latter does not,1, 11 indicating there are other potential mechanisms of DOX cardiotoxicity. It is also known that DOX cardiotoxicity is dependent on the cumulative dose, indicating that in addition to DOX, the DOX metabolites are highly likely to be involved in long-term cardiotoxicity.
Cytochromes P450 (CYPs) are involved in first-pass drug metabolism.12 After metabolism by liver, the drug metabolites are delivered to the heart. Therefore, it is important to understand the interaction of drugs and their metabolites with cardiovascular CYPs such as CYP2J2. Similar to other CYPs such CYP3A4,13–17 our hypothesis is that CYP2J2 should be capable of binding several substrates at once, which might explain the potential cardiotoxicity of certain drugs.
There is an absence of any direct metabolism study of DOX with CYPs. In one study it was shown that CYP2B1 was able to reduce DOX to a semiquinone using EPR measurements.18 However, DOX is typically reduced by the CYP redox partner, cytochrome P450 reductase (CPR),19 to a semiquinone, producing ROS and leading to reductive aglycosylation, producing mostly 7-deoxydoxorubicin aglycone (7-de-aDOX) (Figure 1).18, 20, 21 Since CPR reduces DOX directly and CYPs require CPR to do catalysis, delineating the role of CYPs in DOX metabolism has been challenging. Herein, we carefully delineate the role of heart CYP2J2 in the metabolism of DOX, and the modulation of CYP2J2 activity by DOX and its primary metabolite 7-de-aDOX. CYP2J2 is highly expressed in human cardiomyocytes22 and is responsible for the endogenous oxidation of arachidonic acid (AA) into four regioisomers of epoxyeicosatrienoic acids (EETs)22–26 (Figure 1) and two hydroxyeicosatetraenoic acids (HETEs).23, 25, 27, 28 Together, these metabolites are responsible for mediating several cardiac physiologies, including anti-inflammation, vasodilation, and cellular proliferation.29–31 They also protect against post-ischemia reperfusion injury and reduce myocardium infarct size.31–33 Therefore, these CYP2J2-derived metabolites are cardioprotective.34 However, the efficacy of these EET regioisomers are not similar to each other.30 For instance, 5,6-EET and 8,9-EET have been shown to be both vasoconstrictory and vasodilatory depending on the tissue,35, 36 and 8,9-EET did not reduce myocardial infarct size.29 Overexpression of CYP2J2 was found to protect against DOX-induced injury in human-CYP2J2-overexpressed mice with an unknown mechanism.37 In this study, a decrease in mitochondrial damage was observed when 11,12-EET was administered to H9c2 cells. This effectively demonstrates the importance of different regioisomers of EETs (e.g.11,12-EET) in mediating cardioprotection.
Figure 1.
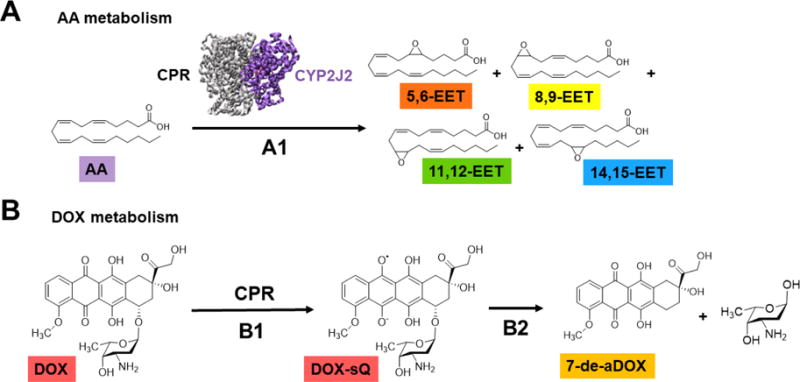
Schematic of DOX and AA metabolism by CYP2J2 and CPR. (A) CYP2J2-CPR metabolizes arachidonic acid (AA) into four regioisomers of epoxyeicosatrienoic acids (EETs) (A1). (B) CPR reduces doxorubicin (DOX) to a semiquinone (DOX-sQ) (B1) that leads to the formation of ROS and the reductive aglycosylation of DOX to form 7-deoxydoxorubicin aglycone (7-de-aDOX) (B2).
CYP2J2 has also been found to metabolize several cardiotoxic drugs, such as terfenadine, astemizole, amiodarone, and dronedarone.38–41 Our hypothesis is if cardiotoxic drugs inhibit CYP2J2-mediated drug metabolism, they should also inhibit CYP2J2-mediated AA metabolism. The inhibition of endogenous AA metabolism by CYP2J2 can have deleterious cardiovascular effects as EETs play an important role in cardiovascular function.
Herein, we use fluorescence polarization (FP) measurements, molecular dynamics (MD) simulations, and substrate kinetics to delineate the mechanism of the DOX/CYP2J2/AA interactions. We demonstrate that DOX inhibits CYP2J2-mediated AA metabolism. Additionally, it alters the site of AA metabolism indirectly through its metabolite 7-de-aDOX. 7-de-aDOX modulates the binding of AA in the active site of CYP2J2 leading to significant change in the regioselectivity of AA epoxidation. We further show that DOX analogues, 5-IDN and ZRN, inhibit CYP2J2 with different potencies, but neither of them alter the regioselectivity of AA metabolism. FP binding measurements and kinetics studies indicate that 7-de-aDOX can bind at a separate site from AA. These studies are supported by MD simulations, which demonstrate that 7-de-aDOX binds into a binding site we had previously determined to be crucial for PUFA binding.25 This positions AA into a conformation to support our observed changes in the site of metabolism.
EXPERIMENTAL PROCEDURES
Materials expression and purification of CYP2J2, CPR, and CYPB5; EBS kinetics and quantification; EET LC-MS/MS quantification; and IC50 calculations
See supplemental information, methods section.
Use of reactive oxygen species (ROS) scavengers in in vitro kinetic experiments
To account for ROS-mediated effects, kinetic experiments were performed with and without ROS scavengers (10 U/mL of catalase and 10 U/mL of superoxide dismutase final, each an order of magnitude larger than required). Scavenger stocks were made in 0.1 M KPi buffer, pH 7.4, and handled and stored according to manufacturer’s recommendations. There were no significant differences in the data with and without the scavengers, and all the data presented is in the presence of scavengers.
Arachidonic Acid (AA) metabolism kinetics
The kinetics of AA metabolism by CYP2J2 with and without DOX were determined as follows. CYP2J2 and CPR (0.6 μM each) were incubated in a 20% POPS lipid reconstituted system (see Supplementary Methods) in 0.5 mL of potassium phosphate buffer (pH 7.4). This mixture was then preincubated with 100 μM AA with varying concentrations of DOX or 7-de-aDOX (0–50 μM from 1 mM and 10 mM stocks in DMSO) for 10 min. Reactions were started with 6 mM NADPH and terminated after 5 min with 100 μL acetic acid. Metabolites were extracted and quantified as previously described25. AA epoxidation by CYP2J2 was determined to be linear up to 8 min under these conditions. IC50 and Ki values were determined as stated in the Supplementary Methods. To investigate the effects of ROS-mediated epoxidation of AA from the reduction of DOX, the same procedure was repeated with CPR and without CYP2J2 and without ROS scavengers.
Fluorescence polarization binding of DOX and 7-de-aDOX
Steady-state fluorescence measurements were conducted with a K2 multi-frequency phase and modulation fluorimeter (ISS, Urbana, IL). Spectra were recorded using Vinci 2 software (Urbana, IL). Measurements were taken with a 1 nm slit width filter. Fluorescence emission spectra between 500 nm and 650 nm of DOX and 7-de-aDOX were determined using an excitation wavelength of 480 nm, with both showing optimal absorbance between 590 and 595 nm. Experiments were conducted at 37°C using a circulating water bath and monomeric protein stocks were solubilized in 0.1% cholate. The presence of CYPs or CYB5 did not alter the fluorescence of DOX or 7-de-aDOX. Polarization measurements were taken using a 594-nm cut-off filter, and the initial G and polarization (r0) values were determined. For all measurements, G ≈ 1.000 and r0 ≈ 0.900 and did not significantly change between experiments. 0–40 μM of CYP2J2 and CYB5 were titrated into 1 μM DOX or 7-de-aDOX in 0.1% cholate and 0.1 M potassium phosphate (KPi) buffer (pH 7.4) at 37°C and incubated for 2 min before each measurement. For concentrations of protein greater than 10 μM, 1 μM of DOX or 7-de-aDOX were titrated into diluted protein stocks. The polarization values were plotted and fitted to either linear (CYB5) or one-binding site (Eq 1) (CYP2J2) equations
| (1) |
where r0 = the intrinsic (initial) polarization value and r1 = the maximum polarization value.
Competitive FP binding
EBS and AA competitive binding experiments to probe where DOX and 7-de-aDOX bind to CYP2J2 were performed as above with the following modifications. CYP2J2 protein stocks were preincubated with 60 μM EBS or 100 μM AA prior to the titration. Initial fluorescence measurements using 1 μM compound were also determined in the presence of 60 μM EBS and 100 μM. The CYP2J2 and substrate mixture was then titrated into the solution and the FP determined as above. Data fit to a competitive binding equation whenever inhibition was present.
Doxorubicin (DOX) Metabolism Kinetics
In vitro DOX metabolism was determined by pre-incubating CPR and CYP2J2 (0.6 μM each) with DOX (10 mM and 1 mM stocks in DMSO) and a 20% POPS lipid reconstituted system for 10 min at 37°C in 0.1 M KPi buffer (pH 7.4). Reactions were initiated upon the addition of 6 mM NADPH based upon our initial NADPH kinetic rates. Reactions were terminated with 100 μL acetic acid and were extracted thrice with equal volumes of ethyl acetate to purify doxorubicin aglycone (aDOX) metabolites. Undigested DOX remained in the aqueous layer with no noticeable accumulation of aDOX metabolites. The organic extracts were then dried under a stream of N2 gas and resuspended in 100 μL acetonitrile for HPLC quantification. Samples were heated with warm water to facilitate solubilization.
HPLC separation and quantification of DOX metabolites
A high-performance liquid chromatography (HPLC) system consisting of an Alliance 2695 analytical separation module (Waters, Milford, MA) and a Waters 996 photodiode diode array detector (Waters) was used for the separation and quantification of DOX metabolites. Metabolites were separated using a Phenomenex Prodigy® 5μm ODS-2, 150 × 4.60 mm column (Phenomenex, PN 00F-3300-E0, Torrance, CA) and a linear-gradient reverse-phase method at a flow rate of 0.5 mL/min. The mobile phase consisted of Mobile Phase A (filtered ultrapure water titrated with formic acid to pH = 3.0) and Mobile Phase B (acetonitrile). The gradient consisted of 95% Mobile Phase A to 100% Mobile Phase B over a 35-min period and held at 100% Solvent B for 5 more min (40 min total) to wash the column. DOX eluted at 15 min and aDOX metabolites (DOX metabolites without the daunosamine sugar) eluted between 17 and 24 min, with a major peak at 21.7 min. Incubations with CPR produced a major 21.7 min peak and a minor peak at 23 min. The major peak was identified as 7-deoxy-doxorubicin aglycone (7-de-aDOX) by comparing elution time and mass spectroscopy fragmentation against an authentic standard. DOX metabolism was quantified at λ = 484 nm using an authentic standard curve.
High-resolution LC-MS/MS identification of Anthracycline metabolites
For high resolution LC/MS, the samples were analyzed by using the Q-Exactive MS system (Thermo. Bremen, Germany) in the Metabolomics Laboratory of Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. Software Xcalibur 3.0.63 was used for data acquisition and analysis. The Dionex Ultimate 3000 series HPLC system (Thermo, Germering, Germany) used includes a degasser, an autosampler, and a binary pump. The LC separation was performed using the same Phenomenex Prodigy® 5μm ODS-2 column as for HPLC. The autosampler was set to 10°C. The injection volume was 10 μL. Mass spectra were acquired under positive electrospray ionization (sheath gas flow rate, 49; aux gas flow rate: 12; sweep gas flow rate, 2; spray voltage, 3.5 kV; capillary temp, 259 °C; Aux gas heater temp, 419 °C). Mass scan range is 70 - 1,000. For full scan, the resolution was set to 70,000, and the AGC target was 1E6 with a maximum injection time of 50 ms. For MS/MS scan, the resolution was set to 17,500 and the AGC target was 5E4 with a maximum injection time of 50 ms. NCE was 25 and 30.
NADPH kinetics
The rate of NADPH consumption in the presence of anthracyclines (0–50 μM from 1 mM and 10 mM stocks in DMSO) was determined using UV-Vis spectroscopy as previously described42. Experiments were performed using 0.6 μM CPR with or without 0.6 μM CYP2J2 and a 20% POPS lipid reconstituted system. 200 μM of NADPH was added to initiate the reaction to keep the absorbance values below 1. Rates were determined based on the initial linear rate of three experiments.
Statistical Analysis
All data were produced from the means of 3 repeats. Statistical significance was based on a two-tailed student’s t-test confidence level of P < 0.05. All data curve fittings were determined using OriginPro 9.1 software (Origin Labs Inc., Northhampton, MA).
Modeling and simulation of membrane-bound CYP2J2 with AA and 7-de-aDOX
Initial structural models of membrane-bound CYP2J2 bound to AA and 7-de-aDOX were generated with molecular docking performed with AutoDock Vina.43 We started by docking 7-de-aDOX to our previously derived models of CYP2J2 bound to AA based upon the homology model of Lafite, P. et al.25, 44 For this docking step, we employed configurations of AA in which sites involved in the regioselectivity shift (i.e., carbons C5,C6 and C8,C9) were close to the heme (with distance < 5 °A), resulting in about 20 initial structures used for docking. A grid box of dimension 22 °A in x, y and z and centered in the active site of CYP2J2 was employed. The resulting docked poses of 7-deaDOX were then clustered based on the RMSD of its heavy atoms, resulting in four clusters. Finally, for each cluster, the configurations of CYP2J2 with both AA and 7-de-aDOX bound with the best docking score were selected as a starting point for MD simulations. Each system was then minimized for 2,000 steps, and equilibrated for 1 ns with the Cα of CYP2J2 and the heavy atoms of AA and 7-de-aDOX harmonically restrained (with force constant k=1 kcal/mol/°A2). Following this preparation step, the four systems were simulated for a short time (20 ns) to assess the stability of 7-de-aDOX binding. After this step, only one system showed stable binding of 7-de-aDOX to CYP2J2, and this system was further extended to 200 ns of simulation without restraints. As a control, an additional simulation in which 7-de-aDOX was removed from this initial model was also performed following the same preparation protocol, with 200 ns of production simulation.
As a control, we also performed docking of DOX both to apo membrane-bound CYP2J2 and to the CYP2J2-AA complex, following the similar protocol as for 7-de-aDOX. Snapshots of apo membrane-bound CYP2J2 were obtained from our simulations previously reported.45 From DOX docking to CYP2J2, two main configurations of DOX in the active site were identified through clustering and employed starting points for MD simulations (see Supplementary Movies). For DOX docking to the CYP2J2-AA complex, only one main configuration of DOX was identified (Figure 7). In this configuration, DOX was located ~17 Å from the heme (center of mass distance) and not within the active site, and thus this configuration was not simulated.
Figure 7.
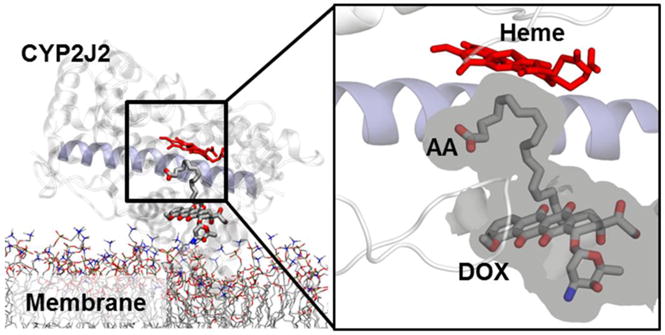
Representative snapshot (i.e., most often observed configuration) of DOX docked pose obtained from docking to membrane-bound CYP2J2-AA structures, with AA located close to the heme (see Methods). The volume available is shown as a grey surface, surrounding the two ligands. The docked poses reveal that DOX cannot be accommodated within the active site when AA is present, and is located away from the heme and at the entrance of the substrate access channel (with center of mass distance ~17 Å).
Docking of EBS to the CYP2J2/7-de-aDOX complex
We generated a model of EBS-bound CYP2J2 by employing the last snapshot of the CYP2J2-7-de-aDOX/AA simulation for molecular docking. For this, we removed AA from the structure, while 7-de-aDOX was preserved. A grid box with same dimensions as previously described and centered in the active site of CYP2J2 was employed for EBS docking. From this step, 10 different docked poses of EBS with the CYP2J2/7-de-aDOX complex were generated, and sorted based on docking score.
Simulation protocol
The simulations were performed using NAMD2.46 The CHARMM27 force field with cMAP47, 48 corrections was used for the protein and the CHARMM3649, 50 force field was used for lipids. Parameters for AA and 7-de-aDOX were obtained by analogy from the CHARMM General Force Field.51 The TIP3P model was used for water.52 Simulations were performed with the NPT ensemble with a time step of 2 fs. A constant pressure of 1 atm was maintained using the Nos´e-Hoover Langevin piston method.53, 54 Temperature was maintained at 310 K using Langevin dynamics with a damping coefficient γ of 0.5 ps-1applied to all atoms. Nonbonded interactions were cut off at 12 °A, with smoothing applied at 10 °A. The particle mesh Ewald (PME) method55 was used for long-range electrostatic calculations with a grid density of > 1 °A-3.
RESULTS
CYP2J2-mediated AA metabolism in the presence of DOX and 7-de-aDOX
We firstly investigated the effect of DOX on CYP2J2-mediated arachidonic acid (AA) metabolism. We used AA at a concentration near the previously determined Km.25 The concentration ranges for DOX covers plasma levels upon immediate exposure (~1–10 μM) to the toxic cumulative dose (~50 μM).56, 57 We see that DOX inhibits the total AA metabolism with an IC50 of 5.48 ± 0.04 μM and a Ki of 3.11 ± 0.02 μM.
DOX is converted to 7-de-aDOX by cytochrome P450 reductase. In order to do the studies, we synthesized 7-de-aDOX from DOX through reduction (Supplementary Methods and Figure S1). We next determined the effect of 7-de-aDOX on AA metabolism. 7-de-aDOX also inhibits AA metabolism, with an IC50 of 10.2 ± 0.1 μM and a Ki of 5.78 ± 0.03 μM. However, the maximum inhibition by 7-de-aDOX appears to be almost half as effective compared to DOX, with a y0 of 42.9 ± 5.6 pmol (min)−1 (nmolCYP2J2) −1 for 7-de-aDOX and a y0 of 19.8 ± 3.8 pmol (min) −1 (nmolCYP2J2) −1 for DOX (Figure 2B). This implies that 51% of the enzyme activity is retained in the presence of saturating amounts of 7-de-aDOX, compared to 23% in the presence of DOX. In theory, competitive inhibition of an enzyme would result in a y0 = 0 due to the inhibitor completely outcompeting the substrate. Since the y0 of the 7-de-aDOX data does not approach 0, this demonstrates that the inhibition is not competitive and suggests a possible second binding site for binding 7-de-aDOX.
Figure 2.
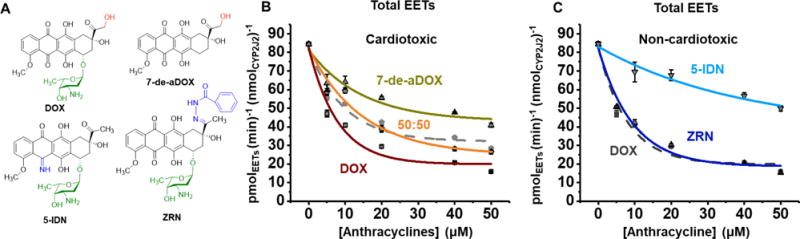
AA Inhibition by DOX, 7-de-aDOX, and the non-toxic analogues ZRN and 5-IDN. (A) Structures of the anthracyclines doxorubicin (DOX), 7-deoxydoxorubicin aglycone (7-de-aDOX), 5-iminodaunorubicin (5-IDN), and zorubicin (ZRN). (B) Rate of epoxidation of 100 μM AA to EETs by CYP2J2-CPr in the presence of increasing concentrations of cardiotoxic DOX, 7-de-aDOX, or a 50:50 mixture of DOX:7-de-aDOX. The theoretical fit based on the linear combination of DOX and 7-de-aDOX is shown as a grey, dashed line to demonstrate the overlap of the theoretical fit to the experimental (see Results for details). (C) Rate of epoxidation in the presence of non-cardiotoxic analogues, zorubicin (ZRN) and 5-iminodaunorubicin (5-IDN) with the DOX data from panel (B) shown in grey for comparison. 100 μM of AA was used in all experiments. Concentrations of anthracyclines represent the total amount of anthracyclines present. All inhibition data fit to eq S1. Error represents the SEM of 3 experiments.
To confirm these observations, we proceeded to measure the AA metabolism with a 50:50 mixture of DOX and 7-de-aDOX, maintaining the total anthracycline concentration (Figure 2B). If the incomplete inhibition of 7-de-aDOX is due to 7-de-aDOX and AA concurrently binding, then the data would be a linear combination of the individual DOX and 7-de-aDOX data. The theoretical values for this mixture based on the DOX and 7-de-aDOX data are as follows: IC50 = 6.68 ± 0.05 μM (Ki = 3.79 ± 0.03 μM) and y0 = 32.1 ± 4.2. Our experimental values were: IC50 = 10.1 ± 0.03 μM (Ki = 5.73 ± 0.02 μM) and y0 = 24.8 ± 5.5. These values agree well to the theoretical values and support that DOX competitively inhibits AA metabolism, and that 7-de-aDOX allows for the concurrent metabolism of AA and an incomplete inhibition.
DOX and 7-de-aDOX changes the regioselectivity of the AA epoxidation
AA metabolism by Formation of different EET regioisomers by CYP2J2-mediated AA metabolism in the presence of DOX or 7-de-aDOX. CYP2J2 leads to the formation of four different EET regioisomers. We further observed that DOX had a concentration-dependent change on the regioselectivity of AA epoxidation (Figure 3B and Figure 3D). With increasing concentrations of DOX, the ratio of 5,6-EET significantly increases (1.41-fold with 50 μM DOX) (Figure 3D) and 14,15-EET significantly decreases (0.78-fold with 50 μM DOX), with less-pronounced albeit significant changes in 8,9-EET and 11,12-EET (Figure 3B and D).
Figure 3.
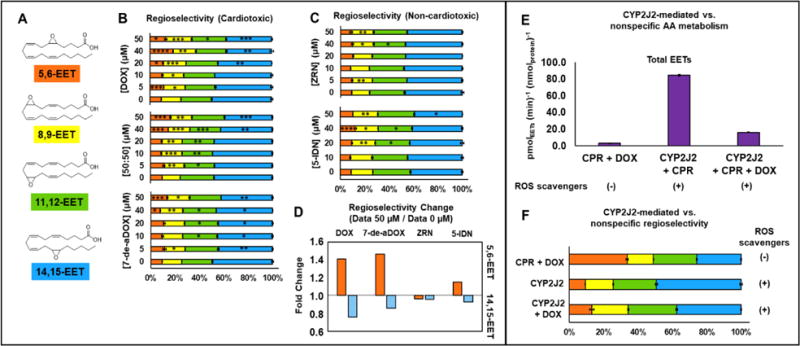
Regioselectivity of the epoxidation of AA by CYP2J2 in the presence of anthracyclines. (A) EET regioisomers. (B) Regioselectivity of EETs in the presence of DOX, 7-de-aDOX, and the 50:50 mixture of DOX:7-de-aDOX. Each EET is shown as a percentage of the total EETs. The colors of the bars represent the colors associated with EETs in panel (A). (C) Regioselectivity of epoxidation in the presence of ZRN and 5-IDN. (D) Fold-change of the regioselectivity for 5,6-EET and 14,15-EET of the data in Panels (B) and (C). Fold change = (Percent of regioisomer in the presence of 50 μM anthracycline) / (Percent of regioisomer in the presence of 0 μM anthracycline). Data presented in (A-D) is obtained from the same dataset as presented in Figure 2. (E) Rate of epoxidation of AA by CYP2J2 and/or CPR in presence and absence of ROS scavengers. Rates are normalized to the amount of CPR (0.3 nmol) or CYP2J2 (0.3 nmol). (F) Regioselectivity of epoxidation by CYP2J2 vs. nonspecific epoxidation. In all these experiments 50 μM DOX and 100 μM of AA was used. Concentrations of anthracyclines represent the total amount of anthracyclines present. Error represents the SEM of 3 experiments. P values: * < 0.05; ** < 0.01; *** < 0.001; **** < 0.0001.
As a control, incubating AA with CPR and 50 μM DOX, without ROS scavengers produces a negligible epoxidation of AA (Figure 3E) with a different distribution of EETs compared to that produced by CYP2J2 (Figure 3F). Furthermore, since all these experiments with CYP2J2 were conducted in the presence of ROS scavengers, this EET regioselective change is CYP2J2-mediated and not ROS-mediated.
The experiments with 7-de-aDOX also resulted in a similar regioselectivity change of the AA epoxidation (1.41-fold for 5,6-EET and 0.86-fold for 14,15-EET with 50 μM 7-de-aDOX) (Figure 3B and D). Similar changes were observed in the 50:50 mixture (Figure 3B). Since DOX displays competitive inhibition and 7-de-aDOX displays incomplete inhibition, 7-de-aDOX appears to be the modulator of the EET regioselectivity. The regioselective change with DOX is likely due to its rapid conversion to 7-de-aDOX. Coincidentally, 11,12-EET and 14,15-EET have been shown to be the most effective at reducing ischemic injury, whereas 5,6-EET and 8,9-EET have not.29
CYP2J2-mediated AA metabolism in the presence of non-cardiotoxic DOX analogues, ZRN and 5-IDN
DOX and 7-de-aDOX inhibited AA metabolism and altered the site of metabolism. In order to determine if these observations are unique to DOX and 7-de-aDOX, we repeated the study using two DOX analogues. The rationale for choosing the analogues is that both ZRN and 5-IDN are chemotherapeutic DOX analogues with lowered cardiotoxicity compared to DOX. ZRN redox cycles and produces ROS as effectively as DOX, while 5-IDN has a lowered rate of redox cycling.1,11, 58 The structural differences are shown in Figure 2A. ZRN contains a large benzohydrazide modification, and the presence of an imino group in place of a keto makes 5-IDN inhibit redox cycling. ZRN inhibits AA metabolism as effectively as DOX, with inhibition parameters of: IC50 = 6.46 ± 0.03 μM (Ki = 3.67 ± 0.02 μM) and y0 = 18.7 ± 3.5 (Figure 2C). 5-IDN displayed a lower inhibition affinity compared to ZRN or DOX, with an IC50 = 27.4 ± 0.03 μM (Ki = 15.6 ± 0.02 μM) (Figure 2C).
However, there is not a significant change in the epoxide regioselectivity for either 5-IDN or ZRN compared to DOX or 7-de-aDOX (Figure 3C and D). The minor changes in the regioselectivity (Figure 3D) are likely due to the conversion of 5-IDN and ZRN to their respective aglycones, which do not modulate AA metabolism as effectively as 7-de-aDOX (Figures S2–S3). For ZRN, the bulky benzohydrazide would significantly change the binding of the ZRN-aglycone. In summary, ZRN and IDN differ in their inhibition potential towards CYP2J2-mediated AA metabolism and do not produce a significant concentration-dependent regioselectivity change (Figure 3 C and D).
Fluorescence polarization (FP) binding of DOX and 7-de-aDOX to CYP2J2
To further investigate the mechanistic details of the AA metabolism modulation by DOX and 7-de-aDOX, we measured their binding to CYP2J2 using FP measurements. Heme Soret titration could not be used to measure the binding, as DOX and its derivatives have a high absorbance between 400–500 nm. Cytochrome b5 (CYB5) was used as a nonspecific binding control, as it does not interact with DOX. None of these hemoproteins altered the fluorescent properties of DOX (Figure S4). CPR, however, has an intrinsic fluorescence in the same region as DOX fluorescence and could not be used in this assay (Figure S4D).
The binding parameters are shown in Table 1. CYB5 showed polarization values, r, that are linear as a function of protein concentration indicating nonspecific binding (Figure S5). CYP2J2 demonstrated one-site binding with a KD of 5.32 ± 0.89 μM (Figure 4A), agreeing well with the IC50/Ki of DOX for the AA inhibition. AA, however, weakly inhibits DOX binding.
Table 1.
Calculated fitting parameters of the fluorescence polarization data of DOX and 7-de-aDOX binding (Figure 4). Ki refers to the inhibition of DOX or 7-de-aDOX by either AA or EBS as indicated. Error represents the SEM of 3 experiments.
| Summary of Binding Parameters
|
||||
|---|---|---|---|---|
| r0 | r1 | KD (μM) | Ki (μM) | |
| DOX | ||||
| DOX | 0.083 ± 0.007 | 0.219 ± 0.010 | 5.32 ± 0.89 | — |
| DOX + AA | 0.094 ± 0.005 | — | — | 176 ± 64 |
| DOX + EBS | 0.101 ± 0.003 | — | — | 13.8 ± 1.6 |
| 7-de-aDOX | ||||
| 7-de-aDOX | 0.055 ± 0.006 | 0.351 ± 0.022 | 13.1 ± 1.1 | — |
| 7-de-aDOX + AA | 0.084 ± 0.002 | — | — | 210 ± 26 |
| 7-de-aDOX + EBS | 0.159 ± 0.006 | 0.383 ± 0.021 (app) | 13.4 ± 1.4 (app) | ∞ |
Figure 4.
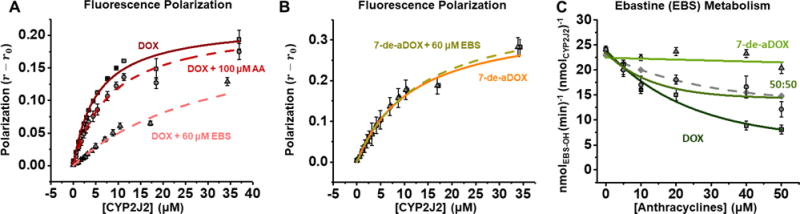
Binding and metabolism studies of DOX and 7-de-aDOX. (A-B) Fluorescence polarization measurements of DOX (A) and 7-de-aDOX (B) binding to CYP2J2. Both DOX and 7-de-aDOX demonstrate one-site binding (eq 1). Binding was repeated in the presence of 60 μM ebastine (EBS) or 100 μM AA. DOX data in the presence of EBS or AA fit to a competitive binding model and 7-de-aDOX is fitted to eq 1 (no inhibition). For all experiments, [DOX] and [7-de-aDOX] = 1 μM. (C) EBS metabolism in the presence of DOX, 7-de-aDOX, and a 50:50 mixture of DOX:7-de-aDOX. DOX data fit to eq S1 and 7-de-aDOX data is linear. The theoretical fit based on the linear combination of DOX and 7-de-aDOX is shown as a grey, dashed line to demonstrate the closeness of the theoretical fit to the experimental fit. Concentrations of anthracyclines represent the total amount of anthracyclines present. Error represents ± SEM of 3 experiments.
To determine if DOX is binding to the active site of CYP2J2, we performed these studies with CYP2J2 and DOX in the presence of 60 μM ebastine (EBS) to saturate the active site (Figure 4A). EBS is a substrate of CYP2J2 with a similar KD as DOX (8.16 μM) and demonstrates one-site binding to the active site.25, 59 EBS competitively inhibited DOX binding to CYP2J2, with a Ki of 13.8 ± 1.6 μM that agrees well with the KD and Km values for EBS.25 These data conclude that DOX and EBS are competing for binding to the active site of CYP2J2 and that AA weakly inhibits DOX binding (Figure 4A).
CYP2J2 had a larger amplitude of change on the 7-de-aDOX polarization compared to DOX and demonstrated one-site binding with a KD of 13.1 ± 1.1 μM (Figure 4B). AA showed little inhibition of 7-de-aDOX binding (Ki > 200 μM). Despite EBS having a tighter KD than 7-de-aDOX (8.16 μM25 vs. 13.1 μM), EBS did not inhibit 7-de-aDOX binding to CYP2J2 (Figure 4B). The inability of EBS to inhibit 7-de-aDOX binding suggests that these two molecules do not compete for binding to the same site. Since EBS binds the active site, this proposes a peripheral binding site for 7-de-aDOX. The presence of peripheral binding sites is common in CYPs.13–17
FP also measures the “rigidity” of binding, where an FP value of r = 0.4 represents the theoretical maximum value and the most restriction to rotational mobility. Therefore, the r maximum (r1) value of the one-site model can be used as a semi-quantitative measurement of the rotational flexibility of the molecule in the active site of the protein60. The binding of DOX to CYP2J2 produced an r1 value of 0.219 ± 0.010 and thus DOX maintains some rotational freedom while bound to the active site. Contrariwise, CYP2J2 binding of 7-de-aDOX showed an r1 value of 0.351 ± 0.022. This indicates that 7-de-aDOX has more rotational restriction when bound to CYP2J2.
Effects of DOX on CYP2J2-mediated EBS metabolism
In order to further probe the inhibitory characteristics of DOX and 7-de-aDOX, we investigated their effects on EBS metabolism by CYP2J2. The binding and kinetics of EBS metabolism are closer to the parameters of DOX and therefore provides clearer insights into the interactions. We determined the Km of EBS metabolism by CYP2J2 to be 19.4 μM.25 Therefore, we looked at the effect that DOX has on the CYP2J2-mediated metabolism with 20 μM of EBS. DOX inhibited EBS metabolism with an IC50 of 17.9 ± 0.0 μM and a Ki of 8.97 ± 0.01 μM (Figure 4C). Incubations with 7-de-aDOX, however, did not result in an inhibition of EBS metabolism (Figure 4C). This is similar to the observations from the FP binding experiments, in which 7-de-aDOX binding to CYP2J2 was not inhibited by EBS. Therefore, EBS and 7-de-aDOX likely bind to different sites in CYP2J2. To confirm, we measured the EBS metabolism in the presence of a 50:50 mixture of DOX and 7-de-aDOX. The theoretical combination of the DOX (y0 = 5.87 ± 2.84) and 7-de-aDOX (b = 22.5 ± 1.2) data results in a y0 = 13.2. The 50:50 DOX:7-de-aDOX data match the theoretical model as a linear combination of the 7-de-aDOX and DOX data quite well, resulting in an IC50 of 9.07 ± 0.08 μM (Ki = 4.53 ± 0.04 μM) and a y0 of 14.2 ± 2.1. This confirms our observations that DOX competes with EBS for binding to the active site of CYP2J2, whereas 7-de-aDOX binds in a site that allows for the binding of a second ligand such as EBS or AA. Furthermore, this demonstrates that the effects of 7-de-aDOX are not universal for all CYP2J2 substrates, since 7-de-aDOX alters the metabolism of AA but not the metabolism of EBS.
Metabolism of DOX, ZRN, and 5-IDN by CPR
We next investigated if CYP2J2 is capable of metabolizing DOX. This is difficult to delineate as CPR, the obligate redox partner of CYPs, can reduce DOX to produce a DOX semiquinone (DOX-sQ) via NADPH oxidation (Figure 1B). DOX-sQ can then produce reactive oxygen species or reductively aglycosylate the daunosamine sugar moiety to produce doxorubicin aglycone (aDOX) metabolites.18, 20, 21 Previously, the steady-state kinetics of these reactions have not been measured. We determined the kinetics of DOX metabolism by CPR. HPLC analysis (Figure S6) of the reaction products showed a major 7-de-aDOX peak (20.5 min) that was confirmed using authentic standards (Figure S7), and a minor peak (22.4 min) (Figure S6). The minor peak had MS/MS fragments that were +14 m/z values of the 7-de-aDOX fragments (Figure S8), which may be due to the oxidation of the 7-OH of doxorubicin aglycone to a ketone. The product was not analyzed further due to poor yields.
A similar metabolism was observed for the reduction of ZRN and 5-IDN by CPR (Figures S2 and S3). ZRN elutes at 17.2 min, and masses corresponding to 7-deoxyzorbucin aglycone were identified at 22.2 and 25.7 min (Figure S2). 5-IDN elutes at 16.4 min and a mass corresponding to the 7-deoxy aglycone analogue was identified at 22.7 min (Figure S3).
Kinetics of DOX, 7-de-aDOX, ZRN, and 5-IDN metabolism by CPR
The rate of NADPH oxidation in the presence of DOX or 7-de-aDOX was negligible in the absence of any enzyme. The rate of NADPH oxidation was linear when CPR is reducing DOX or 7-de-aDOX, indicating that the reduction proceeds without a discernable binding event (Figure 5A–B and Table S1). The rate with 7-de-aDOX (Figure 5B) was approximately tenfold lower than with DOX (Figure 5A) and implies that 7-de-aDOX is not readily reduced by CPR to form ROS. Comparatively, NADPH oxidation in the presence of ZRN is similar to DOX, and with 5-IDN is significantly slower (Figure S9 and Table S1). This confirms the observation that ZRN redox cycles as effectively as DOX and 5-IDN has a much lower rate of redox cycling. The metabolism of DOX to 7-de-aDOX by CPR is also linear, confirming the metabolism of DOX by CPR proceeds without a binding event (Figure 5C and Table S1).
Figure 5. Dox metabolism kinetics.
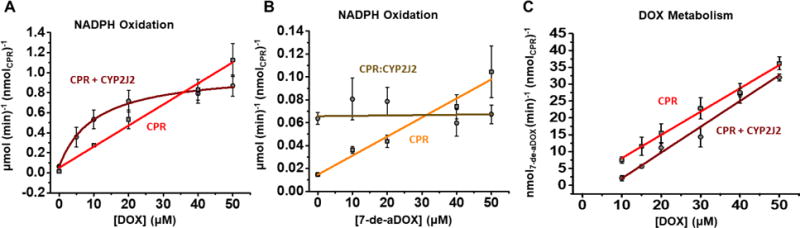
(A) Rate of NADPH oxidation by CPR (red, squares) and by CPR with CYP2J2 (dark red, circles) to reduce DOX. CPR displayed linear kinetics towards the reduction of DOX (equation shown on plot) and the presence of CYP2J2 displayed one-binding-site kinetics. (B) Rate of NADPH oxidation by CPR (orange, squares) and by CPR with CYP2J2 (gold, circles) to reduce 7-de-aDOX. (C) DOX metabolism to 7-de-aDOX by CPR and in the presence of CPR + CYP2J2. Concentrations of anthracyclines represent the total amount of anthracyclines present. Error represents ± SEM of 3 experiments.
NADPH oxidation and Kinetics of DOX metabolism by CYP2J2-CPR
With the addition of CYP2J2, the NADPH oxidation with DOX follows a hyperbolic curve, with an apparent Km of 10.4 ± 1.6 μM (Figure 5A). This implies that CYP2J2 reduces DOX. The NADPH oxidation does not increase when 7-de-aDOX binds CYP2J2 and implies that 7-de-aDOX is not reduced by CYP2J2 (Figure 5B).
The conversion of DOX to 7-de-aDOX remained linear and is inhibited in the presence of CYP2J2 (Figure 5C and Table S1). Therefore, we conclude that CYP2J2 does not contribute towards the reductive aglycosylation of DOX (Figure 5C). Overall, this shows that CYP2J2 does not metabolize DOX or 7-de-aDOX to form new products (none detected by LC-MS/MS analysis), but it reduces DOX (Figure 5A).
Molecular dynamic (MD) simulations to investigate the effect of 7-de-aDOX on AA orientation in the active site of CYP2J2
To gain insight into how 7-de-aDOX may be modulating the AA metabolism by CYP2J2, we employed a combined approach of molecular docking and MD simulations, as described in Experimental Procedures. We have successfully used these techniques to identify key residues that differentially moderate PUFA binding to the CYP2J2 active site.25 To investigate how the presence of 7-de-aDOX modulates AA regioselectivity by CYP2J2, we performed MD simulations starting from structures of membrane-bound CYP2J2 in complex with AA and 7-de-aDOX obtained with molecular docking (see Methods). The initial model included both 7-de-aDOX and AA molecules in the active site of CYP2J2. The initial orientation of AA was selected so that sites involved in the regioselectivity shift (i.e., carbons C5,C6 and C8,C9) were initially close to the heme (with distance < 5 °A) (Figure 6). As a control, a simulation where 7-de-aDOX was removed from the initial model was also performed to assess the stability of AA in its initial orientation.
Figure 6.
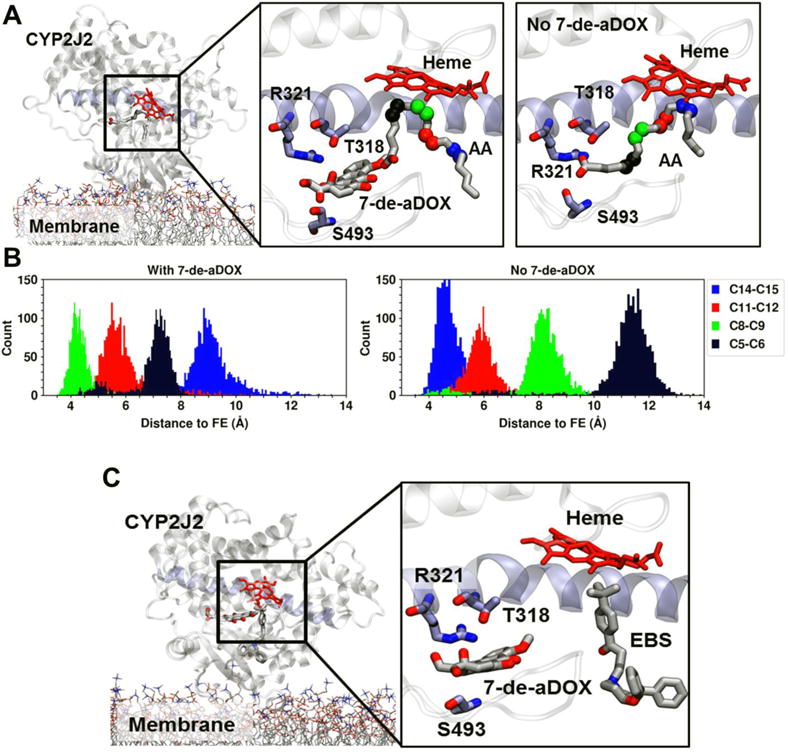
Effect of 7-de-aDOX on AA orientation in CYP2J2 simulations. (A) Representative snapshots (i.e., most often observed configuration) of the active site of membrane-bound CYP2J2 in complex with AA/7-de-aDOX in with AA only. Membrane Lipids are shown in stick representation. CYP2J2 in cartoon representation, with helix I (longest helix in CYP2J2) in purple cartoon. AA and 7-de-aDOX molecules are shown as sticks. Residues involved in AA interactions as previously reported are also shown as sticks. Carbons involved in epoxidation are highlighted as spheres, with colors corresponding to the distributions shown in the next panel. (B) Distribution of carbon-to-heme distances obtained from 200 ns MD simulations for the main epoxidation sites of AA. Colors correspond to the spheres shown in (A). (C) Representative snapshot obtained from molecular docking of EBS to the CYP2J2/7-de-aDOX structure obtained from MD simulations. Docking shows that EBS can be accommodated in a productive orientation (i.e., with its methyl groups close to the heme) with 7-de-aDOX bound to CYP2J2 without hindrance.
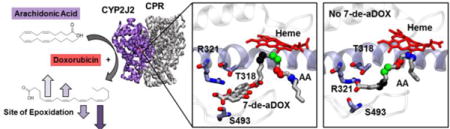
A table of binding interaction energies is shown in Table S2. The simulations suggest that the regioselectivity shift is in part due to how 7-de-aDOX sits in the active site of CYP2J2 (Figure 6A). A specific orientation of 7-de-aDOX located near the active site interacts with key residues that have been shown to be important for AA binding to CYP2J2 (i.e., R321 and S493)25. In this configuration, 7-de-aDOX prevents AA from fully extending to place its main epoxidation sites (i.e., carbons C11,C12 and C14,C15) close to the heme, and instead favors positioning of other sites (carbons C8,C9 and to a lesser extent C5,C6) at potentially productive distances from the heme (< 5 °A for C8,C9) during the majority of the simulation (Figure 6B).
In contrast, by removing 7-de-aDOX from the starting model, AA quickly adopts the orientation that we have previously reported,25 interacting with residues near the active site (including T318, R321 and S493) and placing its main epoxidation sites (carbons C11,C12 and C14,C15) close to the heme (distance < 5 °A) during the simulation (Figure 6B).
Furthermore, docking 7-de-aDOX and EBS together in the active site shows that there is minimal perturbation by 7-de-aDOX on the binding of EBS near the heme, which supports the experimental data (Figure 6C). Simulations of membrane-bound CYP2J2 with DOX revealed that this drug occupies a majority of the active site volume of CYP2J2 (see Supplementary Movies), suggesting that AA and DOX cannot concurrently bind. Indeed, docking of DOX to the CYP2J2-AA structures employed for 7-de-aDOX docking revealed that DOX cannot bind within the active site of CYP2J2 once AA is present (Figure 7), in contrast to the simulated 7-de-aDOX configuration where both AA and 7-de-aDOX are accommodated within the active site volume (Figure 6A). Interestingly, the binding of both AA and 7-de-aDOX does not require a significant conformational change of the active site relative to the apo- (without substrate) or a single-ligand-bound CYP2J2, and relies more on finding the right orientation for both ligands within the active site volume.
DISCUSSION
CYP2J2 metabolizes AA and other PUFAs to produce epoxides, such as EETs, that are cardioprotective. In addition to metabolism of endogenous PUFAs, CYP2J2 is also involved in the metabolism of several drugs that show cardiotoxicity. It is our hypothesis that the cardiotoxicity of these drugs is in part due to modulation of CYP2J2-mediated AA metabolism. Herein, we show that the metabolism of AA by CYP2J2 is inhibited by DOX, a cardiotoxic drug (Figure 2 and Figure 7). This is the first direct measurement of AA inhibition by a cardiotoxic drug for any mammalian CYP epoxygenase. We have determined that DOX potently inhibits AA metabolism by CYP2J2. The mode of inhibition by DOX is competitive, as at saturating concentrations it almost completely inhibits CYP2J2’s AA metabolism. The residual AA metabolism at saturating concentrations is most likely due to the conversion of DOX to 7-de-aDOX during the experiment. Furthermore, FP binding demonstrates that DOX binds CYP2J2 in a one-site model and is competitively inhibited by EBS and weakly by AA (Figure 4, Table 1).
7-de-aDOX, however, shows an incomplete inhibition at saturating concentrations (Figure 2) indicating a second binding site. As determined by FP measurements, AA very weakly inhibits 7-de-aDOX binding to CYP2J2 and EBS does not inhibit 7-de-aDOX binding, despite EBS inhibiting DOX binding (Figure 4, Table 1). These data demonstrate that 7-de-aDOX concurrently binds with EBS and AA. While the inhibition of AA metabolism by DOX and 7-de-aDOX is the most pronounced effect, there is a significant change in the regioselectivity of AA epoxidation by CYP2J2 in the presence of DOX and 7-de-aDOX (Figure 3). The ratio of 5,6-EET and 8,9-EET increases with a concomitant decrease of 11,12-EET and 14,15-EET. The regioselective changes are likely due to 7-de-aDOX. As DOX demonstrates one-site, competitive binding, the regioselective change observed with DOX may be consequence of its conversion to 7-de-aDOX during the CYP2J2-mediated AA metabolism studies.
Although AA and EBS are structurally dissimilar substrates, the effects of DOX and 7-de-aDOX on EBS metabolism parallel those to AA (Figure 4). DOX inhibits EBS metabolism similarly as the inhibition of AA, and demonstrates competitive inhibition. Interestingly, 7-de-aDOX does not inhibit EBS metabolism, and therefore these two molecules are capable of concurrently binding CYP2J2 as supported by kinetics and FP measurements. These studies highlight the importance of determining the effects of cardiotoxic drugs directly on AA metabolism by CYP2J2 instead of using probe substrates such as EBS. The partial inhibition and regioselective changes to AA in the presence of 7-de-aDOX did not manifest with EBS and therefore the effects of 7-de-aDOX on AA metabolism would have been overlooked if EBS was used as the probe substrate.
We further determined that CPR metabolizes DOX to 7-de-aDOX while CYP2J2 does not directly metabolize DOX or 7-de-aDOX. This might be explained by the lack of available sites for metabolism. The NADPH oxidation rate increases on DOX binding to CYP2J2 which indicates that CYP2J2 assists in reduction of DOX similar to CYP2B1.18
These experimental observations are further supported by MD simulations. DOX occludes most of the active site volume and does not allow for AA to bind (Figure 7). 7-de-aDOX, however, binds rather rigidly at the PUFA binding pocket we had previously determined (Figure 6).25 This rigidity in binding is confirmed by our FP measurements (Figure 4), which show that 7-de-aDOX has greater restriction to rotation in the active site compared to DOX. When bound at the active site, 7-de-aDOX positions the binding of AA to favor the formation of 5,6-EET and 8,9-EET. However, since EBS does not bind near this PUFA binding pocket, it is not perturbed by the binding of 7-de-aDOX (Figure 6C). Taken together, DOX and 7-de-aDOX modulate CYP2J2-mediated AA metabolism by inhibiting AA metabolism and by altering the binding of AA to make epoxidation towards the carboxyl head of AA more favorable.
The MD data suggests that the A ring, especially carbons C13 and C14 in 7-de-aDOX is instrumental in perturbing the binding of AA, which produces the regioselective change in the AA epoxidation. It is important to note that the DOX analogues that are least cardiotoxic all have major modifications to carbons C13 and C14, whereas modifications to other parts of the molecule retained cardiotoxicity.1
DOX has been shown to reduce EET levels in acute doses in both liver and heart of rats.61–63 Herein, we present a different mechanism (Figure 8) based on our data by which DOX may alter AA metabolism in humans, namely, through direct inhibition of CYP2J2 and the binding of 7-de-aDOX that concurrently changes the EET regioselectivity. DOX generates ROS that initiates an assault on cardiac cells. Additionally, DOX also inhibits CYP2J2-mediated AA metabolism leading to a decrease in cardioprotective EETs. DOX is converted to 7-de-aDOX by CPR and other reductases. 7-de-aDOX continues to inhibit CYP2J2-mediated AA metabolism and also concurrently binds CYP2J2 to alter the EET ratio, promoting 5,6-EET production. Compared to less cardiotoxic analogues of DOX, 5-IDN weakly inhibits CYP2J2 and ZRN inhibits CYP2J2 as strongly as DOX. Both 5-IDN and ZRN do not change the EET regioisomers ratio significantly. 5-IDN is known to produce less ROS than DOX. ZRN produces ROS, although is not cardiotoxic.
Figure 8.
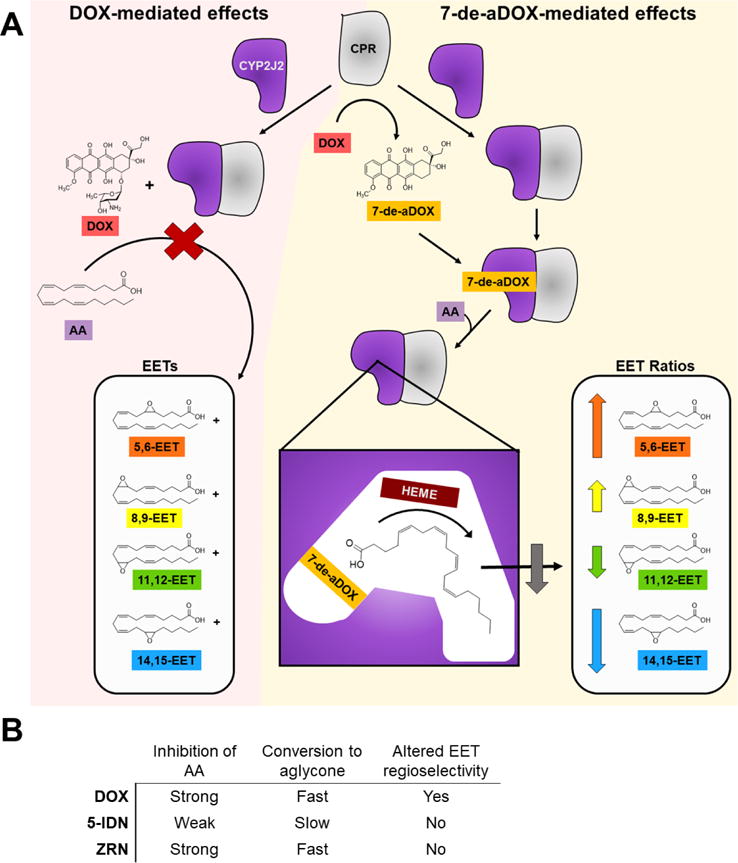
Summary DOX-mediated and 7-de-aDOX-mediated effects on AA metabolism by CYP2J2. (A) DOX-mediated effects. DOX binding from all data displays one-site, competitive inhibition of AA metabolism. When bound to the active site of CYP2J2, DOX occupies a majority of the volume and prevents AA from binding and being converted to EETs. 7-de-aDOX-mediated effects. DOX is reduced to 7-de-aDOX by CPR. 7-de-aDOX then binds into the active site of CYP2J2 into a pocket that directs AA binding. This positions AA with Carbons C5, C6, C8, and C9 closest to the heme. The result is an incomplete inhibition of AA metabolism with a larger percentage of 5,6-EET and a lower percentage of 14,15-EET in the total EETs present. (B) Summary of CYP2J2-mediated AA metabolism by all anthracyclines studied.
Currently, the impact that altering the EET ratios has on the cardiovascular system is not well-understood. The four EET regioisomers have differing affinities for their downstream targets and can also uniquely activate other pathways.30, 64, 65 For instance, 11,12-EET was found to most potently decrease TNF-α levels in human endothelial cells, with 5,6-EET being the weakest; 14,15-EET had no effect.24 5,6-EET and 8,9-EET have been shown to be both vasoconstrictory and vasodilatory depending on tissue, which has been hypothesized to be mediated by their conversion to pro-inflammatory lipid mediators by COX-2.35, 36 8,9-EET and 11,12-EET signal proliferation through the p38 MAPK pathway, whereas 5,6-EET and 14,15-EET signal proliferation through the PI3K pathway.66 Finally, as aforementioned, 8,9-EET was found to not reduce myocardial infarct size.29 It is possible that the modulation of the EET ratio alters the homeostasis in the vasculature. This may then further exacerbate the toxicity of DOX in conjunction to the overall inhibition of EET production. If this is true, then this can also help to explain the reduced cardiotoxicity with ZRN and 5-IDN, insofar as they do not produce a significant EET regioselectivity change.
Therefore, while the overall inhibition of AA metabolism by DOX and 7-de-aDOX would prevent the cells from recovering from DOX-induced cardiotoxicity, the alteration of the EET ratios may be a novel pathway of cardiotoxicity that needs to be tested further. Moreover, the differential effects of the DOX/7-de-aDOX on CYP2J2-mediated metabolism of AA and EBS demonstrates a need for screening of cardiotoxicity using endogenous probe substrates. These data provide valuable insight into drug design and investigating cardiotoxic drug interactions through CYP2J2.
Supplementary Material
Acknowledgments
We thank the National Cancer Institute for providing the 5-IDN and ZRN. We would like to thank Dr. Li Zhong of the UIUC Roy J. Carver Metabolomics Center for the LC-MS/MS analyses. We would like to thank Dr. Ilia Denisov for helpful discussions of the data. We would like to thank Prof. Sligar for providing the CYB5-expressing E. coli.
Funding Sources
American Heart Association Scientist Development grant [15SDG25760064] (AD), NIH R01 GM1155884 (AD) and in part by the National Institutes of Health R01-GM101048, U54- GM087519, and P41-GM104601 (ET). All simulations have been performed using XSEDE resources (grant MCA06N060 to ET).
ABBREVIATIONS
- 5-IDN
5-iminodaunorubicin
- 7-de-aDOX
7-deoxydoxorubicin aglycone
- AA
arachidonic acid
- CYB5
cytochrome b5
- CYP
cytochrome P450
- CPR
cytochrome P450 reductase
- DOX
doxorubicin
- EBS
ebastine
- EET
epoxyeicosatrienoic acid
- EBS-OH
hydroxyebastine
- HETE
hydroxyeicosatetraenoic acid
- LC/MS-MS
liquid chromatography-tandem mass spectrometry
- MD
molecular dynamics
- PUFA
polyunsaturated fatty acid
- ROS
reactive oxygen species
- ZRN.
Zorubicin
Footnotes
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Materials, extra methods (protein expressions, EBS kinetics and quantification), kinetic data, binding data, NADPH kinetic data, MD simulation energies, MD simulation movies. This material is available free of charge via the Internet at http://pubs.acs.org.
CONFLICTS OF INTEREST
Authors declare that they have no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTIONS
WRA wrote the manuscript, designed and performed the experiments, made figures, and analyzed the data. JLB and ET performed MD simulations, and analyzed corresponding data and contributed to making figures and writing the manuscript. AD designed the experiments and wrote the manuscript. All authors analyzed the results and approved the final version of the manuscript.
References
- 1.Singh Y, Ulrich L, Katz D, Bowen P, Krishna G. Structural requirements for anthracycline-induced cardiotoxicity and antitumor effects. Toxicol Appl Pharmacol. 1989;100:9–23. doi: 10.1016/0041-008x(89)90087-2. [DOI] [PubMed] [Google Scholar]
- 2.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 3.Marco DiA, Cassinelli G, Arcamone F. The discovery of daunorubicin. Cancer Treat Rep. 1981;65(Suppl 4):3–8. [PubMed] [Google Scholar]
- 4.Arcamone F, Cassinelli G, Fantini G, Grein A, Orezzi P, Pol C, Spalla C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius, Biotechnol Bioeng. 1969;11:1101–1110. doi: 10.1002/bit.260110607. [DOI] [PubMed] [Google Scholar]
- 5.Rabbani A, Finn RM, Ausio J. The anthracycline antibiotics: antitumor drugs that alter chromatin structure. Bioessays. 2005;27:50–56. doi: 10.1002/bies.20160. [DOI] [PubMed] [Google Scholar]
- 6.Volkova M, Russell R., 3rd Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev. 2011;7:214–220. doi: 10.2174/157340311799960645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nohl H. Identification of the site of adriamycin-activation in the heart cell. Biochem Pharmacol. 1988;37:2633–2637. doi: 10.1016/0006-2952(88)90257-2. [DOI] [PubMed] [Google Scholar]
- 8.Unverferth DV, Leier CV, Balcerzak SP, Hamlin RL. Usefulness of a free radical scavenger in preventing doxorubicin-induced heart failure in dogs. Am J Cardiol. 1985;56:157–161. doi: 10.1016/0002-9149(85)90585-5. [DOI] [PubMed] [Google Scholar]
- 9.Minotti G, Mancuso C, Frustaci A, Mordente A, Santini SA, Calafiore AM, Liberi G, Gentiloni N. Paradoxical inhibition of cardiac lipid peroxidation in cancer patients treated with doxorubicin. Pharmacologic and molecular reappraisal of anthracycline cardiotoxicity. J Clin Invest. 1996;98:650–661. doi: 10.1172/JCI118836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. 2010;115:155–162. doi: 10.1159/000265166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261:3068–3074. [PubMed] [Google Scholar]
- 12.Guengerich FP, Wu ZL, Bartleson CJ. Function of human cytochrome P450s: characterization of the orphans. Biochem Biophys Res Commun. 2005;338:465–469. doi: 10.1016/j.bbrc.2005.08.079. [DOI] [PubMed] [Google Scholar]
- 13.Denisov IG, Baas BJ, Grinkova YV, Sligar SG. Cooperativity in cytochrome P4503A4: linkages in substrate binding, spin state, uncoupling, and product formation. J Biol Chem. 2007;282:7066–7076. doi: 10.1074/jbc.M609589200. [DOI] [PubMed] [Google Scholar]
- 14.Denisov IG, Frank DJ, Sligar SG. Cooperative properties of cytochromes P450. Pharmacol Ther. 2009;124:151–167. doi: 10.1016/j.pharmthera.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacology & Therapeutics. 2013;138:103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 16.Guengerich FP. Cytochrome p450 and chemical toxicology. Chem Res Toxicol. 2008;21:70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 17.Furge LL, Guengerich FP. Cytochrome P450 enzymes in drug metabolism and chemical toxicology: An introduction. Biochem Mol Biol Educ. 2006;34:66–74. doi: 10.1002/bmb.2006.49403402066. [DOI] [PubMed] [Google Scholar]
- 18.Goeptar AR, Te Koppele JM, Lamme EK, Pique JM, Vermeulen NP. Cytochrome P450 2B1-mediated one-electron reduction of adriamycin: a study with rat liver microsomes and purified enzymes. Mol Pharmacol. 1993;44:1267–1277. [PubMed] [Google Scholar]
- 19.Meling DD, McDougle DR, Das A. CYP2J2 epoxygenase membrane anchor plays an important role in facilitating electron transfer from CPR. Journal of Inorganic Biochemistry. 2015;142:47–53. doi: 10.1016/j.jinorgbio.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 20.Bartoszek A. Metabolic activation of adriamycin by NADPH-cytochrome P450 reductase; overview of its biological and biochemical effects. Acta Biochim Pol. 2002;49:323–331. [PubMed] [Google Scholar]
- 21.Deng S, Kruger A, Kleschyov AL, Kalinowski L, Daiber A, Wojnowski L. Gp91 phox-containing NAD(P)H oxidase increases superoxide formation by doxorubicin and NADPH. Free Radic Biol Med. 2007;42:466–473. doi: 10.1016/j.freeradbiomed.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 22.Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J Biol Chem. 1996;271:3460–3468. doi: 10.1074/jbc.271.7.3460. [DOI] [PubMed] [Google Scholar]
- 23.Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, Dechend R, Konkel A, von Schacky C, Luft FC, Muller DN, Rothe M, Schunck WH. Arachidonic Acid-metabolizing Cytochrome P450 Enzymes Are Targets of ω-3 Fatty Acids. J Biol Chem. 2010;285:32720–32733. doi: 10.1074/jbc.M110.118406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold WR, Baylon JL, Tajkhorshid E, Das A. Asymmetric Binding and Metabolism of Polyunsaturated Fatty Acids (PUFAs) by CYP2J2 Epoxygenase. Biochemistry. 2016;55:6969–6980. doi: 10.1021/acs.biochem.6b01037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDougle DR, Kambalyal A, Meling DD, Das A. Endocannabinoids Anandamide and 2-Arachidonoylglycerol Are Substrates for Human CYP2J2 Epoxygenase. J Pharmacol Exp Ther. 2014;351:616–627. doi: 10.1124/jpet.114.216598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation: molecular and functional properties of the arachidonate monooxygenase. Journal of Lipid Research. 2000;41:163–181. [PubMed] [Google Scholar]
- 28.Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. Journal of Lipid Research. 2009;50:S52–S56. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gross GJ, Hsu A, Falck JR, Nithipatikom K. Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J Mol Cell Cardiol. 2007;42:687–691. doi: 10.1016/j.yjmcc.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 31.Xu X, Zhang XA, Wang DW. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv Drug Deliv Rev. 2011;63:597–609. doi: 10.1016/j.addr.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Nithipatikom K, Moore JM, Isbell MA, Falck JR, Gross GJ. Epoxyeicosatrienoic acids in cardioprotection: ischemic versus reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;291:H537–542. doi: 10.1152/ajpheart.00071.2006. [DOI] [PubMed] [Google Scholar]
- 33.Gross GJ, Gauthier KM, Moore J, Falck JR, Hammock BD, Campbell WB, Nithipatikom K. Effects of the selective EET antagonist, 14, 15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am J Physiol Heart Circ Physiol. 2008;294:H2838–2844. doi: 10.1152/ajpheart.00186.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oni-Orisan A, Alsaleh N, Lee CR, Seubert JM. Epoxyeicosatrienoic acids and cardioprotection: the road to translation. J Mol Cell Cardiol. 2014;74:199–208. doi: 10.1016/j.yjmcc.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fulton D, Balazy M, McGiff JC, Quilley J. Possible contribution of platelet cyclooxygenase to the renal vascular action of 5, 6-epoxyeicosatrienoic acid. J Pharmacol Exp Ther. 1996;277:1195–1199. [PubMed] [Google Scholar]
- 36.Takahashi K, Capdevila J, Karara A, Falck JR, Jacobson HR, Badr KF. Cytochrome P-450 arachidonate metabolites in rat kidney: characterization and hemodynamic responses. Am J Physiol. 1990;258:F781–789. doi: 10.1152/ajprenal.1990.258.4.F781. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, El-Sikhry H, Chaudhary KR, Batchu SN, Shayeganpour A, Jukar TO, Bradbury JA, Graves JP, DeGraff LM, Myers P, Rouse DC, Foley J, Nyska A, Zeldin DC, Seubert JM. Overexpression of CYP2J2 provides protection against doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol. 2009;297:H37–46. doi: 10.1152/ajpheart.00983.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evangelista EA, Kaspera R, Mokadam NA, Jones JP, 3rd, Totah RA. Activity, inhibition, and induction of cytochrome P4502J2 in adult human primary cardiomyocytes. Drug Metab Dispos. 2013;41:2087–2094. doi: 10.1124/dmd.113.053389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaspera R, Kirby BJ, Sahele T, Collier AC, Kharasch ED, Unadkat JD, Totah RA. Investigating the contribution of CYP2J2 to ritonavir metabolism in vitro and in vivo. Biochem Pharmacol. 2014;91:109–118. doi: 10.1016/j.bcp.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee CA, Jones JP, 3rd, Katayama J, Kaspera R, Jiang Y, Freiwald S, Smith E, Walker GS, Totah RA. Identifying a selective substrate and inhibitor pair for the evaluation of CYP2J2 activity. Drug Metab Dispos. 2012;40:943–951. doi: 10.1124/dmd.111.043505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karkhanis A, Lam HY, Venkatesan G, Koh SK, Chai CL, Zhou L, Hong Y, Kojodjojo P, Chan EC. Multiple modes of inhibition of human cytochrome P4502J2 by dronedarone, amiodarone and their active metabolites. Biochem Pharmacol. 2016;107:67–80. doi: 10.1016/j.bcp.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Capdevila JH, Wei S, Helvig C, Falck JR, Belosludtsev Y, Truan G, Graham-Lorence SE, Peterson JA. The highly stereoselective oxidation of polyunsaturated fatty acids by cytochrome P450BM-3. J Biol Chem. 1996;271:22663–22671. doi: 10.1074/jbc.271.37.22663. [DOI] [PubMed] [Google Scholar]
- 43.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lafite P, Andre F, Zeldin DC, Dansette PM, Mansuy D. Unusual regioselectivity and active site topology of human cytochrome P4502J2. Biochemistry. 2007;46:10237–10247. doi: 10.1021/bi700876a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDougle DR, Baylon JL, Meling DD, Kambalyal A, Grinkova YV, Hammernik J, Tajkhorshid E, Das A. Incorporation of charged residues in the CYP2J2 F-G loop disrupts CYP2J2-lipid bilayer interactions. Biochim Biophys Acta. 2015;1848:2460–2470. doi: 10.1016/j.bbamem.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 48.Mackerell AD, Jr, Feig M, Brooks CL., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 49.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hart K, Foloppe N, Baker CM, Denning EJ, Nilsson L, Mackerell AD., Jr Optimization of the CHARMM additive force field for DNA: Improved treatment of the BI/BII conformational equilibrium. J Chem Theory Comput. 2012;8:348–362. doi: 10.1021/ct200723y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD., Jr CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 53.Feller SE, Zhang YH, Pastor RW, Brooks BR. Constant-Pressure Molecular-Dynamics Simulation - the Langevin Piston Method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 54.Martyna GJ, Tobias DJ, Klein ML. Constant-Pressure Molecular-Dynamics Algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 55.Darden T, York D, Pedersen L. Particle Mesh Ewald - an NLog(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 56.Rahman AM, Yusuf SW, Ewer MS. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int J Nanomedicine. 2007;2:567–583. [PMC free article] [PubMed] [Google Scholar]
- 57.Barpe DR, Rosa DD, Froehlich PE. Pharmacokinetic evaluation of doxorubicin plasma levels in normal and overweight patients with breast cancer and simulation of dose adjustment by different indexes of body mass. Eur J Pharm Sci. 2010;41:458–463. doi: 10.1016/j.ejps.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 58.Jelic S, Tomasevic Z, Kreacic M, Kovcin V, Radosavljevic D, Vlajic M. A pilot study of high-dose zorubicin in advanced stages of soft tissue sarcoma in adults. Bull Cancer. 1996;83:1002–1007. [PubMed] [Google Scholar]
- 59.McDougle DR, Palaria A, Magnetta E, Meling DD, Das A. Functional studies of N-terminally modified CYP2J2 epoxygenase in model lipid bilayers. Protein Sci. 2013;22:964–979. doi: 10.1002/pro.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Husain N, Agbaria RA, Warner IM. Spectroscopic Analysis of the Binding of Doxorubicin to Human Alpha-1-Acid Glycoprotein. J Phys Chem-Us. 1993;97:10857–10861. [Google Scholar]
- 61.Alsaad AM, Zordoky BN, El-Sherbeni AA, El-Kadi AO. Chronic doxorubicin cardiotoxicity modulates cardiac cytochrome P450-mediated arachidonic acid metabolism in rats. Drug Metab Dispos. 2012;40:2126–2135. doi: 10.1124/dmd.112.046631. [DOI] [PubMed] [Google Scholar]
- 62.Zordoky BN, Anwar-Mohamed A, Aboutabl ME, El-Kadi AO. Acute doxorubicin toxicity differentially alters cytochrome P450 expression and arachidonic acid metabolism in rat kidney and liver. Drug Metab Dispos. 2011;39:1440–1450. doi: 10.1124/dmd.111.039123. [DOI] [PubMed] [Google Scholar]
- 63.Zordoky BN, Anwar-Mohamed A, Aboutabl ME, El-Kadi AO. Acute doxorubicin cardiotoxicity alters cardiac cytochrome P450 expression and arachidonic acid metabolism in rats. Toxicol Appl Pharmacol. 2010;242:38–46. doi: 10.1016/j.taap.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 64.Imig JD. Eicosanoid regulation of the renal vasculature. Am J Physiol Renal Physiol. 2000;279:F965–981. doi: 10.1152/ajprenal.2000.279.6.F965. [DOI] [PubMed] [Google Scholar]
- 65.Thomson SJ, Askari A, Bishop-Bailey D. Anti-inflammatory effects of epoxyeicosatrienoic acids. Int J Vasc Med. 2012;2012:605101. doi: 10.1155/2012/605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pozzi A, Macias-Perez I, Abair T, Wei S, Su Y, Zent R, Falck JR, Capdevila JH. Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids (5,6- and 89-EET) as potent in vivo angiogenic lipids. J Biol Chem. 2005;280:27138–27146. doi: 10.1074/jbc.M501730200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.