

Abstract
The Mycobacterium tuberculosis (Mtb) 20S proteasome is vital for the pathogen to survive under nitrosative stress in vitro and to persist in mice. To qualify for drug development, inhibitors targeting Mtb 20S must spare both the human constitutive proteasome (c-20S) and immunoproteasome (i-20S). We recently reported members of a family of noncovalently binding dipeptide proteasome inhibitors that are highly potent and selective for Mtb 20S over human c-20S and i-20S. To understand the structural basis of their potency and selectivity, we have studied the structure–activity relationship of six derivatives and solved their co-crystal structures with Mtb 20S. The dipeptide inhibitors form an antiparallel β-strand with the active site β-strands. Selectivity is conferred by several features of Mtb 20S relative to its mouse counterparts, including a larger S1 pocket, additional hydrogen bonds in the S3 pocket, and hydrophobic interactions in the S4 pocket. Serine-20 and glutamine-22 of Mtb 20S interact with the dipeptides and confer Mtb-specific inhibition over c-20S and i-20S. The Mtb 20S and mammalian i-20S have a serine-27 that interacts strongly with the dipeptides, potentially explaining the higher inhibitory activity of the dipeptides toward i-20S over c-20S. This detailed structural knowledge will aid in optimizing the dipeptides as anti-tuberculosis drugs.
Graphical Abstract
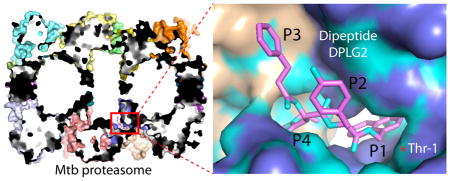
The Mycobacterium tuberculosis (Mtb) 20S proteasome core particle (CP) has emerged as a novel target for the development of anti-tuberculosis agents 1–5. However, humans express a constitutive 20S proteasome (c-20S) as well as an immunoproteasome (i-20S) 6, 7. Therefore, to have potential utility for the treatment of TB, inhibitors have to be species-selective 8. A key to achieving selectivity is to understand the unique features of the active-site pockets of these giant protease complexes. Here we systematically compare their structures in complex with potential inhibitors. The bacterial 20S CP contains 14 copies each of α- and β-subunits that form a cylinder of four stacked rings (α7β7β7α7), with the N-terminal Thr-1 of the β-subunits as the catalytic residue 9, 10. The human c-20S has seven different α- and β-subunits (α1–7β1–7β1–7α1–7), but only β1, β2, and β5 are enzymatically active. In the human i-20S, the three catalytically active β-subunits are replaced by interferon-γ-inducible β1i, β2i, and β5i, respectively. The crystal structures of human c-20S, mouse i-20S, and humanized yeast i-20S have been reported recently 11–13, providing good structural models that we used here as surrogates for the human proteasomes.
We previously reported crystal structures of three covalent Mtb 20S inhibitors. The first inhibitor, a peptide boronate (MLN273), has a low species selectivity and is similar to the anti-myeloma drug Velcade that targets human 20S 9. The second inhibitor, an 1,3,4-oxathiazol-2-one, is highly selective but is unstable in serum 14. The third inhibitor, fellutamide B, is highly potent against Mtb 20S but is not species-selective 15. Recently, noncovalent dipeptide proteasome inhibitors were identified via a high-throughput screen for inhibitors of the ubiquitin-proteasome system in human cells 16. These compounds show nanomolar IC50 values and are selective for the chymotrypsin-like β5 site over the caspase-like β1 and trypsin-like β2 sites of the 20S core particle. The N,C-capped dipeptides bind differently to the β5 sites of the constitutively expressed proteasome and the immunoproteasome 17, 18. Systematic optimization of the N,C-capped dipeptides yielded a compound with a threonine at P3 having an IC50 of 7.4 nM for the human β5 site 18. Because the Mtb 20S CP is chymotryptic 19, the dipeptides provided an attractive scaffold for the development of Mtb 20S–specific inhibitors (Fig. 1A). In previous work, we screened 1600 N,C-capped dipeptides that varied in the P1 (the C-cap), P2, P3, and P4 (the N-cap) residues; variants in the P2, and P3 residues included both natural and unnatural amino acids. We identified several compounds that had 100-fold to over 1000-fold more selectivity for the Mtb 20S “open gate” CP (Mtb20SOG, in which the amino terminal heptapeptide is deleted from the αchains) than for the human c-20S β5 20. One of the compounds, DPLG-2, was shown to penetrate M. tuberculosis to exert bactericidal activity against nonreplicating bacteria under nitrosative stress. Here we report five additional dipeptide compounds—PKS2144, PKS2169, PKS2205, PKS2206, and PKS2208 (Fig. 1B)—that are highly selective for Mtb20SOG over human 20S. The six dipeptides all have a naphthyl ring at the P1 site and inhibit the Mtb20SOG by a time-dependent mechanism rather than the expected rapid equilibrium mechanism. To determine the structural basis for the species selectivity and to assist further improvement of Mtb 20S-selective dipeptides, we solved the crystal structures of the six dipeptides complexed with the Mtb20SOG.
Figure 1. Inhibition of chymotryptic activities of Mtb20SOG and the human c-20S and i-20S by N,C-capped dipeptides.

(A) The four sub-site positions of a 20S CP surrounding a dipeptide inhibitor. (B) Structures of the six dipeptide inhibitors studied in this work. (C) Dose-dependent inhibition curves for Mtb20SOG, human c-20S, and human i-20S. Each curve is a representative of three independent experiments for each compound; each data point is an average of three replicates in those experiments.
MATERIALS AND METHODS
Materials
Proteasome substrates: The fluorogenic substrates Suc-LLVY-AMC, Z-LLE-AMC, Z-VLR-AMC, and Ac-ANW-AMC were from Boston Biochem (Cambridge, MA). Human immunoproteasomes (isolated from peripheral blood mononuclear cells) and human constitutive proteasomes (isolated from red blood cells) were from Boston Biochem Inc. Mtb20SOG was expressed and purified as reported 10.
Synthesis
The synthetic route is described in the supplementary materials.
IC50 determination
IC50 values of compounds against Mtb20SOG, β5i and β5c were determined from tests in a 96-well format as reported 20. In brief, 1 μL of compound in a 3x series dilution in DMSO at concentrations from 100 μM to 0.0017 μM were spotted to the bottom of a black 96-well plate having a solid bottom. Reaction buffer (100 μL; 20 mM HEPES, 0.5 mM EDTA, pH 7.5) containing enzyme and substrate were dispensed into each well, and the plate was then spun in a simple bench top plate centrifuge at 1000x rpm for 1 min, followed by shaking on an orbital shaker for 1 min. The time course of the hydrolysis in each well was followed by recording the fluorescence of the product AMC (λex 360 nm and λem 460 nm) on a SpectraMax M5 plate reader over 1.5–2 h. The initial reaction velocity of each well was fit to a dose-dependent inhibition equation using PRISM to determine the IC50. IC50 values were determined only for β5i and β5c. Inhibitory activities against β1i, β1c, β2i, and β2c were tested at 100 μM, 33.3 μM, and 11.1 μM. All compounds showed < 50% inhibition at 33.3 μM, hence, the IC50 values are presented as > 33.3 μM (Table S1). Suc-LLVY-AMC was used for Mtb20SOG activity at 50 μM and for c-20S β5c at 25 μM. Ac-ANW-AMC was used for i-20S β5i activity at 15 μM. Z-LLE-AMC was used for i-20S β1i and c-20S β1c activity at 50 μM, and Z-VLR-AMC was used for i-20S β1i and c-20S β1c activity at 50 μM. Final concentrations of Mtb20S, β5i and β5c were 2 nM, 0.4 nM and 0.2 nM, respectively. Sodium dodecyl sulfate was used as activator at 0.02% for human β5i, β5c, β1i and β1c. PA28a was used as activator at 4x equivalent of β2i and β2c.
Protein expression and purification
Cloning and expression of Mtb20SOG have been described 10. Briefly, a PACYCDuet-1 vector (Novagen, Madison, WI, USA) consisting of the Mtb prcAOG (encoding truncated-PrcA) and prcB genes was expressed in BL21(DE3) strain of E. coli (Invitrogen, Carlsbad, CA, USA). Cells were grown at 37 °C to OD600 = 0.5–0.6 before being induced with 0.2 mM IPTG. The Mtb proteasome was expressed at 37 °C for 18 h to ensure full processing of β-subunits. Cells were harvested by centrifugation, re-suspended in 10 mM potassium phosphate (pH 8.0), 10 mM imidazole, and 0.25 M NaCl, and lysed by passing through a Microfluidizer cell disruptor. The homogenate was clarified by spinning at 27,000 × g, and the supernatant was applied to a HiTrap-Ni column (GE Healthcare) pre-equilibrated with the lysis buffer. His-tagged proteins were eluted with a 10–300 mM imidazole gradient in 10 mM potassium phosphate (pH 8.0) and 0.25 M NaCl. Eluted fractions containing Mtb proteasomes were applied to a Hi-Trap Q column pre-equilibrated with 10 mM Tris (pH 8.0) and 50 mM NaCl and were eluted with a 50–500 mM NaCl gradient. Mtb20SOG was further purified with a Superdex 200 column (16 × 600 mm, GE Healthcare) in buffer containing 10 mM HEPES (pH 7.5), 1 mM dithiothreitol, and 0.15 M NaCl. For crystallization, the purified Mtb20SOG complex was concentrated to 10 mg/ml.
Crystallization and structure determination
Mtb20SOG was crystallized at 4 °C by the hanging-drop vapor diffusion method using 60 mM sodium citrate (pH 6.2) and 14% PEG-3350 as precipitant. To obtain the inhibitor–proteasome complex, Mtb20SOG crystals were transferred stepwise to the cryo-protectant containing 60 mM sodium citrate (pH 6.2), 14% PEG-3350, 35% dimethylformamide, and 1 mM N,C-capped dipeptide inhibitors. After incubation at 4 °C for 16 h, crystals were flash-frozen in liquid nitrogen. Diffraction data were collected at the X25 and X29A beamlines of National Synchrotron Light Source (NSLS), Brookhaven National Laboratory (BNL). All data were processed with HKL2000. The space groups of inhibitor-soaked Mtb20SOG crystals were all P21. Molecular replacement was carried out to solve the inhibitor-bound Mtb20SOG structures using the program PHASER 21. Mtb20SOG apo-form (PDB ID 3HFA) was chosen as initial search model. After building the corresponding inhibitor models in COOT 22, the refinements were performed using Phenix-refine 23 and the statistics are provided in Table S1.
RESULTS
1. Design rationale, synthesis, and biochemical characterization of PKS2144, PKS2169, PKS2205, PKS2206, and PKS2208
Our previous work suggested that S1-P1 and S3-P3 interactions are important for the potency and selectivity of the N,C-capped dipeptides 20. To further dissect the contribution of S1-P1 and S3-P3, and to understand whether P2 could be used to tune the physicochemical properties of the dipeptides for anti-Mtb drug development, we designed several new compounds based on DPLG-2 (Fig. 1B). We fixed P1 naphthalen-1-ylmethanamine and P4 phenylpropanoyl or isoxazole-3-carboxamid, replaced P2 p-F-phenylalanine with either an alanine or an O-methyl-serine, and replaced P3 N,N-diethyl Asn with either a pyrrolindine Asn or a piperidine Asn. We synthesized the compounds by the typical liquid-phase peptide synthesis approach 18, 20. The synthetic schemes (Schemes S1–S3), experimental details, and spectrometric characterizations (NMR and HR-MS) of these compounds are described in the Supplemental Information.
We found that PKS2144, PKS2169, PKS2205, PKS2206, and PKS2208 all inhibited the Mtb20SOG, the chymotryptic-like β5 active subunits of human constitutive proteasome (c-20S β5c), and the human immunoproteasome (i-20S β5i) in a dose-dependent manner (Fig. 1C), similar to DPLG-2 20. Based on the dose-dependence curves, we calculated the IC50 values of these compounds (Table 1) against Mtb20SOG; all were in the narrow range of 0.010–0.041 μM. However, the apparent selectivity index (SIapp; rightmost two columns) against c-20S and i-20S was from 66- to 4667-fold and 0.7- to 3647-fold higher, respectively, against β5c and β5i. Relative to inhibition by DPLG-2, replacing its P2 p-F phenylalanine with alanine (PKS2169) or O-methyl serine (PKS2205) improved the IC50 value against β5c by 2.4- and 3.3-fold, respectively; the enhancements against β5c were 14.6- and 20.7-fold, respectively. Replacing P3 N,N-diethyl Asn (PKS2205) with piperidine Asn (PKS2206) and pyrrolindine Asn (PKS2208) enhanced the inhibitory activity against β5i by 8.2- and 1.5-fold, respectively, whereas the enhancement against β5c was 60- and 5.2-fold, respectively.
Table 1.
IC50’s of dipeptides against Mtb20SOG and β5c, β5i of the human c-20S and i-20S. The MG-132 values were measured for comparison.
| IC50 (μM) ** | SIapp | SIapp | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Mtb20SOG (LLVY) | c-20S β5c (LLVY) | i-20S β5i (ANW) |
|
|
||
| DPLG-2 * | 0.015 | 70.00 | 54.70 | 4667 | 3647 | |
| PKS2169 | 0.041 ± 0.006 | 29.2 ± 6.6 | 3.75 ± 0.60 | 712 | 91 | |
| PKS2144 | 0.010 ± 0.002 | 0.657 ± 0.087 | 0.007 ± 0.001 | 65.7 | 0.7 | |
| PKS2205 | 0.023 ± 0.002 | 20.95 ± 6.92 | 2.64 ± 0.62 | 911 | 115 | |
| PKS2206 | 0.016 ± 0.001 | 2.57 ± 0.75 | 0.044 ± 0.020 | 161 | 2.8 | |
| PKS2208 | 0.016 ± 0.002 | 14.34 ± 3.81 | 0.512 ± 0.157 | 896 | 32 | |
| MG-132 | 1.714 ± 0.079 | 0.008 ± 0.001 | 0.024 ± 0.001 | 0.005 | 0.014 | |
Data from 20.
Data are given as mean ± SEM.
ANW: Ac-ANW-AMC
LLVY: suc-LLVY-AMC
SIapp: apparent selectivity index
The data appears to indicate that while P3 was the major determinant of the species selectivity, P2 could influence the species selectivity subtly, especially when combined with certain P3 variants, and the influence was greater for i-20S than for c-20S. PKS2144 showed almost identical IC50’s against i-20S and Mtb20SOG (0.007 μM vs 0.010 μM), in agreement with our previous study that the secondary amine of the Asn side chain is essential for species selectivity. PKS2144 potently and selectively inhibited i-20S. A detailed characterization of the compound as a human i-20S selective inhibitor will be described elsewhere. We note that all these compounds are specific inhibitors of the chymotryptic-like β5 of i-20S and c-20S; their IC50 values against tryptic-like β2c and β2i and against caspase-like β1c and β1i were all greater than 33.3 μM.
2. Overall structures of Mtb20SOG in complex with N,C-capped dipeptides
To understand the binding modes of the dipeptide inhibitors, DPLG-2 and the five derivatives were each soaked into crystals of Mtb20SOG. The inhibitor-soaked crystals diffracted x-rays to between 2.8 and 3.1 Å (Table S1). We used the molecular replacement method to solve the crystal structures of inhibitor-bound Mtb20SOG using the Mtb20SOG apo-form (PDB: 3HFA) as the initial search model. After refinement, both the Fo-Fc and 2Fo-Fc density maps were well defined for accurate modeling of the bound inhibitors (Fig. 2). Each of the 14 β-subunits of the inhibitor-proteasome complexes was occupied by one molecule of inhibitor, consistent with the fact that all β-subunits are active in the Mtb proteasome. Unlike the covalent inhibitors GL1 and HT1171, which caused major conformational changes in the substrate binding sites 14, 24, the dipeptides did not significantly alter the active site, which remained similar to that in the apo-Mtb20SOG structure (Fig. 3A).
Figure 2. Electron-density maps of proteasome inhibitors in the active site of the Mtb20SOG core particle.

(A–F) The 2mFo-DFc maps of DPLG-2 (A), PKS2144 (B), PKS2169, (C), PKS2205, (D), PKS2206 (E), and PKS2208 (F) are rendered at 1 σ and shown in grey mesh. The two neighboring β-subunits are green and cyan. The catalytic residue Thr-1 is shown in red.
Figure 3. Overview of dipeptide-20S interactions.

(A) Comparison of DPLG-2 bound active site with apo-Mtb20SOG. No significant conformational change was observed in the S1, S2, S3, or S4 sites. DPLG-2 form is in green; apo-form is in wheat. (B) Overview of inhibitor-occupied binding pockets. All six inhibitors are aligned in accordance with the DPLG-2 bound structure. The two β-subunits forming the inhibitor binding sites are shown in green and cyan; Thr-1 is in magenta. (C) The P1 naphthyl groups in the S1 binding pocket. (D) The P3 groups of six inhibitors in the S3 binding pocket.
3. Common binding features of the six dipeptides
The substrate specificity of a proteasome is predominantly controlled by the S1 and S3 binding pockets. In the Mtb20SOG structure, the S3 and S4 binding sites are formed by elements of two adjacent β-subunits. We previously demonstrated the differential substrate preferences of the Mtb and mammalian proteasomes at the P1 position, with the Mtb proteasome strongly preferring tryptophan 19. The P1 naphthyl residue resembles tryptophan, perhaps explaining why these dipeptides are ideal Mtb proteasome inhibitors. As predicted, the P1 naphthyl residue inserts into the well-defined S1 binding pocket harboring the proteolytic residue Thr-1 (Fig. 3B–C) and establishes multiple Van der Waals interactions with surrounding amino acids. All six inhibitors have a P1 naphthyl residue (Fig. 1B) and adopt the same conformation in the S1 pocket (Fig. 3C). This structural feature, which is not affected by the moieties in the P2, P3, and P4 sites, suggests that the binding of a P1 residue to the S1 pocket is rigid and specific. The P2 residues of all six inhibitors point toward the solvent and display no interactions with the proteasome (Fig. 3B). Therefore, it is unlikely that the shallow S2 site directly contributes to the stabilization of the P2 groups. No significant conformational difference was observed in the S3 pockets of each inhibitor–proteasome complex (Fig. 3D), suggesting that no further conformational change of the S3 pocket is required to accommodate the bulkier P3 N,N-diethylamide moiety in DPLG-2, PKS2169, and PKS2205.
4. Specific interactions of individual dipeptides with Mtb20SOG
DPLG-2, PKS2169, PKS2144, PKS2205, PKS2206, and PKS2208 have IC50 values against Mtb20SOG of 15 nM, 41 nM, 10 nM, 23 nM, 16 nM, and 16 nM, respectively (Table 1). The dipeptide inhibitors mimic the peptide substrate, interacting noncovalently with the Mtb proteasome by forming a short antiparallel β-strand between β-strands B3 and B6 of the Mtb 20S. The inhibitors hydrogen-bond with the backbone atoms of Thr-21, Gly-47, and Ala-49 (Fig. 4A). This basic binding profile has also been observed in the crystal structure of Mtb20SOG complexed with fellutamide B and in the structure of the yeast 20S proteasome complexed with a tripeptide inhibitor 15, 18. In addition to the backbone interactions, several side-chain interactions seem to further enhance these inhibitors’ binding to the Mtb 20S: Ser-20 interacts with the C-terminal carbonyl group of the inhibitors; at the S3 sub-site, Gln-22 and Ser-27 provide extra bonds with the carbonyl group of the P3 moiety; and at the S4 site, Asp-124 of the adjacent β-subunit interacts with the N-terminal amino group of the P4 moiety (Fig. 4A).
Figure 4. The binding mode of the dipeptides in Mtb20SOG.

(A) In addition to the antiparallel bindings between the inhibitor backbone and Thr-21, Gly-47, and Ala-49, also within hydrogen-bonding distance are the surrounding amino acids, including Ser-20, Gln-22, Ser-27, Ala-50, and Asp-124 of neighboring subunits, and a water molecule. Possible hydrogen bonds between the inhibitors and the proteasome are shown as magenta dashed lines; the two neighboring β-subunits are shown in green and cyan; water molecules are shown as red spheres. PKS2144 has extra hydrogen-bonding capacities between tBu-O-NH of the P3-Asn side chain and Asp-124, between tBu-O-NH of the P3-Asn side chain and Gln-22, and between the nitrogen and oxygen atoms of the 5-methylisoxazole of P4 and Ala-125 through a water molecule. (B) The five dipeptides that contain a phenylpropanoyl P4 group can form additional hydrophobic interactions with Leu-98, and with Leu-91, Met-95, Ala-125, and Ala-126 of neighboring subunits. Pictured is DPLG-2; the distances between the P4 3-phenylpropanoyl group and the surrounding amino acids are shown as orange dashed lines. (C) PKS2144 has a 5-methylisoxazole group at the P4 position, which forms hydrophobic interactions with the surrounding amino acids. H-bond interactions are shown in panel A.
In the PKS2144 complex, Gln-22 and Asp-124 provide more interactions with the P3 residue than are seen for the other five inhibitors (Fig. 4A). Also, Asp-124 and Ala-50 interact with a water molecule to further stabilize the bindings with the N-terminus and carbonyl group of the ligand backbone. This water molecule was also observed in the tripeptide–yeast proteasome complex 18. In all six inhibitor–Mtb20SOG structures, there is invariably a water molecule near Ala-125 of the adjacent β-subunit. This water in PKS2144 coordinates three hydrogen bonds between Ala- 125 and the P4 5-methylisoxazole. These extra interactions may contribute to the high affinity of PKS2144 for Mtb 20S.
Hydrophobic interactions also contribute significantly to the tight inhibitor binding at S4. Except for PKS2144, all inhibitors contain an N-cap 3-phenylpropanoyl group. This group is surrounded at the S4 sub-site by Leu-98, plus the Leu-91, Met-95, Ala-125, and Ala-126 of the adjacent β-subunit, at distances between 3.5 Å and 4.1 Å (Fig. 4B). In the case of PKS2144, the P4 5-methylisoxazole is surrounded by Leu-98 plus the Leu-91 and Ala-126 of the adjacent β-subunit. These hydrophobic interactions and the water-mediated H-bond mentioned above stabilize the P4 group in the S4 site (Fig. 4C).
5. Proposed model for DPLG-2 binding in mouse c-20S and i-20S
The dipeptide inhibitors showed differential selectivity among the Mtb proteasome, human c-20S, and human i-20S (Table 1, Fig. 1C). Notably, the affinity of DPLG-2 for Mtb 20S was 4667-fold stronger than for human c-20S and 3647-fold stronger than for human i-20S. In mammalian proteasomes, the β1 and β1i, β2 and β2i, and β5 and β5i subunits have caspase-like, trypsin-like, and chymotrypsin-like activities, respectively; the Mtb β-subunits only have chymotrypsin-like activity. To elucidate the underlying mechanism of the differential selectivity, we computationally modeled our dipeptide binding with the human c-20S, mouse i-20S, and humanized yeast i-20S 11–13. In contrast to the ample space of the S1 pocket in apo-Mtb20SOG (PDB 3HFA), the S1 binding pockets of ligand-free human constitutive proteasome β5/β6 (PDB 4R3O), mouse immunoproteasome β5i/β6 (PDB 3UNH), and humanized yeast immunoproteasome β5i/β6 (PDB 5L5B) are too small to accommodate the P1 naphthyl group of the dipeptide inhibitors. (Fig. 5A). However, in the crystal structures of carfilzomib-bound human β5/β6 (PDB 4R67), PR-957-bound mouse β5i/β6 (PDB 3UNF), and carfilzomib-bound humanized yeast β5i/β6 (PDB 5L5E), the side chain of Met-45 swung outward, resulting in expanded S1 pockets 11. Although the S1 pockets are smaller than that of dipeptide-bound Mtb20SOG, we find that the P1 naphthyl group of the dipeptides is able to fit in the expanded S1 sites (Fig. 5B). Therefore, depending on whether the dipeptides are able to push away the Met-45 side chain in human c-20S and i-20S, the P1 group may or may not contribute to the high selectivity of these dipeptides.
Figure 5. Proposed interactions between dipeptide inhibitors and the chymotrypsin-like active site of c-20S and i-20S.

.(A) The apo-Mtb20SOG (PDB ID 3HFA) structure is aligned with ligand-free human c-20S β5/β6 (PDB ID 4R3O), mouse i-20S β5i/β6 (PDB ID 3UNH), and humanized yeast i-20S (PDB ID 5L5B). The S1 binding pockets of c-20S and i-20S cannot accommodate the P1 naphthyl group without a conformational change. Human β5/β6 is in orange; mouse β5i/β6 is in purple; apo-Mtb20SOG is in green; humanized yeast β5i/β6 is in wheat. (B) DPLG-2-bound Mtb20SOG is modeled into carfilzomib-bound human β5/β6 (PDB ID 4R67), PR-957-bound mouse β5i/β6 (PDB ID 3UNF) and carfilzomib-bound humanized yeast β5i/β6 (PDB ID 5L5E). After conformational changes, the S1 pockets of β5/β6 and β5i/β6 are enlarged to accommodate the naphthyl group. (C) The P3 groups of the six dipeptides are modeled into the S3 pocket of the human c-20S. Both ligand-free and ligand-bound S3 pockets have enough room to accommodate various P3 groups. The apo-β5/β6 is in green and ligand-bound β5/β6 is in wheat. (D) The P3 groups of the six inhibitors are modeled into the S3 pockets of mouse i-20S. No further conformational change is required to fit the P3 groups after ligand binding. The apo- β5i/β6 is in light blue and ligand-bound β5i/β6 is in wheat. (E) The P3 groups of the six inhibitors are modeled into the S3 pockets of the humanized yeast β5i/β6. Tyr107 of β6 rotates to widen the entrance of the S3 pocket, likely facilitating inhibitor binding. The humanized yeast apo-β5i/β6 is in light blue and ligand-bound β5i/β6 is in wheat. (F) Proposed hydrogen bonding interactions between DPLG-2 and the human c-20S β5/β6. The antiparallel bindings between inhibitor backbone and Thr-21, Gly-47, and Ala-49 are conserved in Mtb 20S and in human c-20S and mouse i-20S. The β5 subunit is in green and β6 is in cyan. The grey dashed lines are the potential hydrogen bonds within 3.6 Å distance. (G) Modeled interactions of DPLG-2 with mouse i-20S β5i/β6. Besides the backbone interactions, Ser-27 contributes additional H-bond to the O35 of the P3 group. (H) Upper panel: Sequence alignment of Mtb 20S β subunit, human c-20S β5 subunit, and mouse i-20S β5i subunit. Lower panel: Sequence alignment of Mtb 20S β subunit and human 20S β6 subunit. The orange stars denote the residues involved in backbone antiparallel bindings that are conserved in all three proteasomes. The orange circles indicate that the conserved residues participate in inhibitor binding in the Mtb proteasome and possibly in c-20S and i-20S. Green circles are residues specific for Mtb 20S involved in dipeptide inhibitor binding. Ser-27 (blue circle), which is not conserved in c-20S β5, is essential for P3 binding.
We note that the side chain of Met-31 in the mouse β5i appears to clash with the P1 naphthyl group. However, Met-31 is replaced by a valine in human c-20S and in human i-20S, suggesting that this site does not interfere with P1 insertion in human proteasome. In modeled humanized yeast β5i/β6, the S1 site is enlarged to accept the P1 naphthyl group (Fig. 5B). The S3 pockets of both the human c-20S and mouse i-20S are not significantly changed after ligand binding and appear to be spacious enough to accommodate the variable but largely hydrophobic P3 moieties of the dipeptide inhibitors (Fig. 5C–D). Yet, in the carfilzomib-bound humanized yeast β5i/β6 structure, the side chain of Tyr107 of β6 rotates to allow opening the entrance of S3 site (Fig. 5E). The IC50 values show that the PKS2169 and PKS2205, which have a N,N-diethyl amide of the P3 side chain, are 33- and 41-fold more selective against human i-20S than is PKS2206 (Table 1), which has a piperidin-1-yl P3 side chain, suggesting that small differences in the interaction with the S3 site contribute to species selectivity.
Our modeling suggests that the dipeptide inhibitors form the expected antiparallel β-strand interaction with Thr-21, Gly-47, and Ala-49 of the human c-20S and mouse i-20S (Fig. 5F–G). However, many interactions found at the S3 and S4 sites of the Mtb β-subunit are absent in the mammalian β5/β6 and β5i/β6. For example, Ser-20 and Gln-22, which contribute extra hydrogen bonds in the Mtb β-subunit, are not conserved in β5 and β5i (Fig. 5G). Ser-27 of the immunoproteasome β5i, which is conserved in the Mtb β-subunit but not in constitutive β5, provides an additional hydrogen bond for β5i (Fig. 5F–G). This may account for the higher affinity of the inhibitors for i-20S versus c-20S. In the S4 sub-site, the Mtb β-subunit Ala-125 is replaced by Pro-126 in β6, indicating that the water-mediated hydrogen bonding seen in PKS2144-Mtb is likely absent in β5/β6 and β5i/β6. The S4 sites of human c-20S and i-20S, with only a Val-127 of β6 (corresponding to Ala-126 in Mtb β-subunit) near the P4 group, are much less hydrophobic than that of Mtb20S, suggesting a reduced number of hydrophobic interactions between P4 and S4 in the mammalian 20S. Therefore, P1, P3, and P4 all contributed to the potency and selectivity of these inhibitors.
DISCUSSION
The Mtb Pup-proteasome system is being actively investigated, and several new factors that parallel with the eukaryotic ubiquitin proteasome system have been identified which may provide novel targets for anti-TB drug development 1, 4, 5, 25–32. Selective inhibition of the proteasomes of pathogens over host proteasomes has been demonstrated in vitro and in vivo to be a viable approach for anti-infectives 33. Because of the mechanism-based toxicity of proteasome inhibitors in human cells, inhibitors for pathogen proteasomes must be highly selective. Additionally, reversible inhibition of the proteasome is potentially a better means to avoid the toxicity of accumulated host proteasome inhibition from the chronic use of irreversible inhibitors.
We have developed the first generation of noncovalent bacterial-proteasome-selective inhibitors 20. Here we have shown that the N,C-capped dipeptide inhibitors adopt an antiparallel β-strand configuration at the active site of Mtb 20S; they have far lower affinity for human c-20S and i-20S. Noncovalent binding of these dipeptides without significantly altering the enzyme active site is conducive to reversible inhibition. By analyzing the crystal structures of dipeptide–Mtb proteasome complexes, we found that Ser-20, Gln-22, and Ser-27 at the S3 sub-site and the hydrophobic residues at the S4 sub-site are the major contributors to the differential binding of these dipeptides to the Mtb versus mammalian 20S CPs. Furthermore, Ser-27 confers the relative selectivity of the inhibitors for i-20S over c-20S 34. These observations will guide our effort to develop better Mtb-specific proteasome inhibitors.
The wide variation in species selectivity of the dipeptides suggests that interactions beyond backbone H-bonds are involved in their binding with c-20S and i-20S. The covalently bound tripeptide PR-957 is known to alter the S1 pocket in both c-20S and i-20S 35. The binding poses of the dipeptide inhibitors we studied in human i20S and c-20S are currently unknown, but they may be quite different from that of PR957, as changing the P3 groups had a dramatic impact on IC50’s of the dipeptides. Because the modeled interactions are similar among the six inhibitors, the various P3 groups may induce subtle conformational changes to accommodate the noncovalent inhibitors. PKS2144 differs from the other dipeptides chiefly in the side chain moiety of the P3-Asn: it has a primary amine, tert-Bu-NH-O-NH-Asn, whereas the other five compounds contain a secondary amine. This may explain why PKS2144 is the least selective, as our previous study found that the secondary amine of the Asn is a major determining factor for species selectivity 20. Moreover, Ala-50 of β5 (β5i) and Asp-125 of β6 are conserved among the Mtb β-subunits, β5/β6, and β5i/β6. These residues in the mammalian 20S may undergo binding-induced conformational changes to form H-bonds with the inhibitors similar to those of Mtb20S, thereby increasing their binding affinity (Fig. 5E–F, grey dashed lines). Likewise, in the S4 sub-site, Leu- 95 of β5 (β5i) (corresponding to Leu-98 in Mtb β-subunit) and Leu-92 and Leu-96 of β6 (corresponding to Leu-91 and Met-95 in Mtb β-subunit) may undergo conformational changes to form hydrophobic interactions with the P4 groups of the dipeptide inhibitors.
DPLG-2 is 4-fold more potent against Mtb20SOG than ML9 (which has a 2,4-difluorobenzyl P1 group) and 51-fold more potent than DPLG-4, which has a 4-isopropyl phenyl P1 group 20. This suggests that a bulky group at position-4 of the P1 phenyl ring is not tolerated well by the S1 pocket of Mtb 20S; indeed, our structures show that the S1 pocket has no room for an extra group at that position. However, Mtb 20S has more room at the sub-site of the S1 facing position-3 of the P1 phenyl ring (Fig. 3C, 5A,B). We therefore suggest that adding a moiety at position-3 of P1 phenyl ring may increase the potency and selectivity against Mtb 20S.
Previous studies have largely focused on optimizing the P1 and P3, which interact with the amino acid residues in the S1 and S3 pockets, respectively. In this study, we have shown that the replacement of the P2 hydrophobic 4-F-phenyl ring (DPLG-2) with methyl (PKS2169) or methoxymethyl (PKS2205) moieties retained potency but reduced the selectivity against c-20S by 6- to 5-fold and against i-20S by 40- to 31-fold, respectively. The results indicate that the P2/S2 interaction plays a secondary but important role in species selectivity by enhancing inhibitory activities against the human chymotryptic β5 active subunits, especially the β5i. More SAR studies are needed to address how to maintain selectivity while reducing hydrophobicity at the P2 site. The P3 side chain differences among PKS2205, PKS2206, and PKS2208 produced little effect on potency against Mtb20S yet strongly affected selectivity. This, together with the loss of selectivity of PKS2144, strongly suggests that P3 may hold the key for species selectivity. Finally, we show that hydrophobic interactions in the S4 site and water-assisted hydrogen bonding may contribute significantly to affinity and selectivity. All this information will guide our effort to develop a new generation of Mtb20S-specific inhibitors.
Supplementary Material
1. Table S1: X-ray diffraction data collection and structure refinement statistics.
2. Chemical synthesis route and characterization of the dipeptides and their intermediates, including three synthesis schemes.
3. NMR spectrometric characterization of the dipeptide inhibitors
Acknowledgments
FUNDING
The work was supported by NIH grants R01 AI070285 to H.L., Tri-Institutional TB Research Unit grant U19 AI111143 to C.N., and R21 AI101393 to G.L. and the Milstein Program in Chemical Biology and Translational Medicine, directed by C.N.
We acknowledge access to beamlines X25 and X29A at the National Synchrotron Light Source (NSLS) in the Brookhaven National Laboratory, and we thank the staff at those beamlines. NSLS was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-98CH10886. We thank J. David Warren at The Abby and Howard P. Milstein Synthetic Chemistry Core Facility at Weill Cornell Medicine for assistance. We thank Dr. Kenjiro Sato at Tri-Institution Therapeutics Discovery Institute and David Nadziejka at Van Andel Research Institute for critical reading of the manuscript. We thank Dr. Christopher Tsu at Takeda for Ac-PAL-AMC. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation.
ABBREVIATIONS
- Mtb
Mycobacterium tuberculosis
- 20S CP
20S core particle
- 20SOG
open gate mutant of 20S CP
Footnotes
ACCESSION CODES
The coordinates and associated diffraction data have been deposited in RCSB PDB with accession codes 5TRG, 5TRS, 5TRR, 5THO, 5TRY, and 5TS0.
References
- 1.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C, Gold B, Lin G, Stegman M, de Carvalho LP, Vandal O, Venugopal A, Bryk R. A philosophy of anti-infectives as a guide in the search for new drugs for tuberculosis. Tuberculosis (Edinb) 2008;88(Suppl 1):S25–33. doi: 10.1016/S1472-9792(08)70034-9. [DOI] [PubMed] [Google Scholar]
- 3.Nathan C. Fresh approaches to anti-infective therapies. Sci Transl Med. 2012;4:140sr142. doi: 10.1126/scitranslmed.3003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bai L, Hu K, Wang T, Jastrab JB, Darwin KH, Li H. Structural analysis of the dodecameric proteasome activator PafE in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2016;113:E1983–1992. doi: 10.1073/pnas.1512094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jastrab JB, Wang T, Murphy JP, Bai L, Hu K, Merkx R, Huang J, Chatterjee C, Ovaa H, Gygi SP, Li H, Darwin KH. An adenosine triphosphate-independent proteasome activator contributes to the virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2015;112:E1763–1772. doi: 10.1073/pnas.1423319112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta. 2014;1843:13–25. doi: 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groll M, Huber R, Moroder L. The persisting challenge of selective and specific proteasome inhibition. J Pept Sci. 2009;15:58–66. doi: 10.1002/psc.1107. [DOI] [PubMed] [Google Scholar]
- 9.Hu G, Lin G, Wang M, Dick L, Xu RM, Nathan C, Li H. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol Microbiol. 2006;59:1417–1428. doi: 10.1111/j.1365-2958.2005.05036.x. [DOI] [PubMed] [Google Scholar]
- 10.Lin G, Hu G, Tsu C, Kunes YZ, Li H, Dick L, Parsons T, Li P, Chen Z, Zwickl P, Weich N, Nathan C. Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol Microbiol. 2006;59:1405–1416. doi: 10.1111/j.1365-2958.2005.05035.x. [DOI] [PubMed] [Google Scholar]
- 11.Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, Groll M. Immuno-and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell. 2012;148:727–738. doi: 10.1016/j.cell.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 12.Huber EM, Heinemeyer W, de Bruin G, Overkleeft HS, Groll M. A humanized yeast proteasome identifies unique binding modes of inhibitors for the immunosubunit beta5i. EMBO J. 2016;35:2602–2613. doi: 10.15252/embj.201695222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harshbarger W, Miller C, Diedrich C, Sacchettini J. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure. 2015;23:418–424. doi: 10.1016/j.str.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 14.Lin G, Li D, de Carvalho LP, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. Inhibitors selective for mycobacterial versus human proteasomes. Nature. 2009;461:621–626. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin G, Li D, Chidawanyika T, Nathan C, Li H. Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome. Arch Biochem Biophys. 2010;501:214–220. doi: 10.1016/j.abb.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ, Barrett C, Liu JX, Soucy TA, Sappal DS, Bump N, Olhava EJ, Fleming P, Dick LR, Tsu C, Sintchak MD, Blank JL. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta5-subunit. Biochem J. 2010;430:461–476. doi: 10.1042/BJ20100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groll M, Gallastegui N, Marechal X, Le Ravalec V, Basse N, Richy N, Genin E, Huber R, Moroder L, Vidal J, Reboud-Ravaux M. 20S proteasome inhibition: designing noncovalent linear peptide mimics of the natural product TMC-95A. ChemMedChem. 2010;5:1701–1705. doi: 10.1002/cmdc.201000293. [DOI] [PubMed] [Google Scholar]
- 18.Blackburn C, Barrett C, Blank JL, Bruzzese FJ, Bump N, Dick LR, Fleming P, Garcia K, Hales P, Hu Z, Jones M, Liu JX, Sappal DS, Sintchak MD, Tsu C, Gigstad KM. Optimization of a series of dipeptides with a P3 threonine residue as non-covalent inhibitors of the chymotrypsin-like activity of the human 20S proteasome. Bioorg Med Chem Lett. 2010;20:6581–6586. doi: 10.1016/j.bmcl.2010.09.032. [DOI] [PubMed] [Google Scholar]
- 19.Lin G, Tsu C, Dick L, Zhou XK, Nathan C. Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-acetyl tripeptide substrates. J Biol Chem. 2008;283:34423–34431. doi: 10.1074/jbc.M805324200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin G, Chidawanyika T, Tsu C, Warrier T, Vaubourgeix J, Blackburn C, Gigstad K, Sintchak M, Dick L, Nathan C. N,C-Capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: role of S3 and S1 binding pockets. J Am Chem Soc. 2013;135:9968–9971. doi: 10.1021/ja400021x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 23.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li D, Li H, Wang T, Pan H, Lin G, Li H. Structural basis for the assembly and gate closure mechanisms of the Mycobacterium tuberculosis 20S proteasome. EMBO J. 2010;29:2037–2047. doi: 10.1038/emboj.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolten M, Delley CL, Leibundgut M, Boehringer D, Ban N, Weber-Ban E. Structural Analysis of the Bacterial Proteasome Activator Bpa in Complex with the 20S Proteasome. Structure. 2016 doi: 10.1016/j.str.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 26.Delley CL, Laederach J, Ziemski M, Bolten M, Boehringer D, Weber-Ban E. Bacterial proteasome activator bpa (rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLoS One. 2014;9:e114348. doi: 10.1371/journal.pone.0114348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samanovic MI, Li H, Darwin KH. The pup-proteasome system of Mycobacterium tuberculosis. Subcell Biochem. 2013;66:267–295. doi: 10.1007/978-94-007-5940-4_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Striebel F, Hunkeler M, Summer H, Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup’s N-terminus. EMBO J. 2010;29:1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burns KE, Pearce MJ, Darwin KH. Prokaryotic ubiquitin-like protein provides a two-part degron to Mycobacterium proteasome substrates. J Bacteriol. 2010;192:2933–2935. doi: 10.1128/JB.01639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerda-Maira F, Darwin KH. The Mycobacterium tuberculosis proteasome: more than just a barrel-shaped protease. Microbes Infect. 2009;11:1150–1155. doi: 10.1016/j.micinf.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearce MJ, Arora P, Festa RA, Butler-Wu SM, Gokhale RS, Darwin KH. Identification of substrates of the Mycobacterium tuberculosis proteasome. EMBO J. 2006;25:5423–5432. doi: 10.1038/sj.emboj.7601405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Festa RA, Pearce MJ, Darwin KH. Characterization of the proteasome accessory factor (paf) operon in Mycobacterium tuberculosis. J Bacteriol. 2007;189:3044–3050. doi: 10.1128/JB.01597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khare S, Nagle AS, Biggart A, Lai YH, Liang F, Davis LC, Barnes SW, Mathison CJ, Myburgh E, Gao MY, Gillespie JR, Liu X, Tan JL, Stinson M, Rivera IC, Ballard J, Yeh V, Groessl T, Federe G, Koh HX, Venable JD, Bursulaya B, Shapiro M, Mishra PK, Spraggon G, Brock A, Mottram JC, Buckner FS, Rao SP, Wen BG, Walker JR, Tuntland T, Molteni V, Glynne RJ, Supek F. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature. 2016;537:229–233. doi: 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh PK, Fan H, Jiang X, Shi L, Nathan CF, Lin G. Immunoproteasome beta5i-Selective Dipeptidomimetic Inhibitors. ChemMedChem. 2016;11:2127–2131. doi: 10.1002/cmdc.201600384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huber EM, de Bruin G, Heinemeyer W, Paniagua Soriano G, Overkleeft HS, Groll M. Systematic Analyses of Substrate Preferences of 20S Proteasomes Using Peptidic Epoxyketone Inhibitors. J Am Chem Soc. 2015;137:7835–7842. doi: 10.1021/jacs.5b03688. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1. Table S1: X-ray diffraction data collection and structure refinement statistics.
2. Chemical synthesis route and characterization of the dipeptides and their intermediates, including three synthesis schemes.
3. NMR spectrometric characterization of the dipeptide inhibitors