

Abstract
Objective
Pulmonary artery smooth muscle cells (PASMCs) from Neprilysin (NEP) null mice exhibit a synthetic phenotype and increased activation of RhoGTPases compared to their wild type (wt) counterparts. Although RhoGTPases are known to promote a contractile SMC phenotype, we hypothesize that their sustained activity decreases SM-protein expression in these cells.
Approach and Results
PASMCs isolated from wt and NEP−/− mice were used to assess levels of SM-proteins (SM-actin, SM-myosin, SM22 and calponin) by Western blotting, and were lower in NEP−/− PASMCs compared to wt. Rac and Rho levels and activity were higher in NEP−/− PASMCs, and ShRNA to Rac and Rho restored SM-protein, and attenuated the enhanced migration and proliferation of NEP−/− PASMCs. SM-gene repressors, p-Elk-1 and Klf4, were higher in NEP−/− PASMCs and decreased by shRNA to Rac and Rho. Co-stimulation of wt PASMCs with PDGF and the NEP substrate, ET-1, increased Rac and Rho activity and decreased SM-protein levels mimicking the NEP KO phenotype. Activation of Rac and Rho and downstream effectors was observed in lung tissue from NEP−/− mice and humans with COPD.
Conclusions
Sustained Rho activation in NEP−/− PASMCs is associated with a decrease in SM-protein levels and increased migration and proliferation. Inactivation of RhoGDI and RhoGAP by phosphorylation may contribute to prolonged activation of Rho in NEP−/− PASMCs. RhoGTPases may thus have a role in integration of signals between vasopeptides and growth factor receptors and could influence pathways that suppress SM-proteins to promote a synthetic phenotype.
Keywords: Rho, Pulmonary artery smooth muscle cells, Signaling, Neprilysin
Subject Code: Smooth muscle cell proliferation and differentiation
Graphical abstract
INTRODUCTION
Neprilysin (NEP), a cell-surface endopeptidase expressed in vascular cells, cleaves and inactivates neuropeptides important for vasopressor responses1, 2. NEP−/− mice show increased pulmonary vascular remodeling in response to hypoxia3. We have shown in NEP−/− pulmonary artery smooth muscle cells (PASMCs), persistent ET-1 signals synergize with PDGF to promote migration and proliferation4.
RhoGTPases maintain vascular tone and a differentiated SMC phenotype by regulating Srf-mediated transcription of SM-genes by contractile agonists5. In pathological states, such as pulmonary hypertension (PHTN) and COPD, elevated levels of contractile agonists such as ET-1, promote vasoconstriction and activation of RhoGTPases6. Prolonged vasoconstriction promotes inward remodeling of resistance arteries7, 8. In animal models of PHTN, inhibition of RhoGTPases decreases neointima formation suggesting a role in vascular remodeling9, 10.
Increased Rho activity inhibits myogenic differentiation of skeletal muscle and maintains bronchial SMCs in a mesenchymal phenotype11, 12,13,14. Whether sustained RhoGTPase activity enhances remodeling of pulmonary arteries by maintaining a mesenchymal/synthetic phenotype of SMCs is not known. In this study, we will begin to test this concept and examine mechanisms by which sustained Rho may suppress SM-protein levels in NEP/- PASMCs thereby promoting a dedifferentiated phenotype.
Our study will identify how sustained levels of a contractile agonist like ET-1 in NEP−/− PASMCs could synergize with PDGF to reduce SM-contractile protein levels. Understanding the role of RhoGTPases in modulating the SMC phenotype may help explain their role in increased vascular remodeling observed in PHTN associated with COPD where co-expression of PDGF and ET-1 occurs15,16.
RESULTS
Decreased levels of SM-proteins in NEP−/− PASMCs
Phenotypic switching of SMC is accompanied by down-regulation of SM-proteins (α-SMA (alpha- smooth muscle actin), SM-myosin(SM-MHCs), SM22, and calponin)17. Compared to wt, NEP−/− PASMCs had significantly lower levels of α-SMA, SM22, and calponin, which were restored by lentiviral expression of NEP (Figure 1A–1C), or recombinant NEP (Supplement Figure IA and IC). SiRNA-induced knockdown of NEP (90% reduced) or treatment with the NEP inhibitor, phosphoramidon, decreased levels of SM-proteins in wt PASMCs (Figure 1D–E, Supplement Figure IB and ID). mRNA for SM-genes (α-SMA, SM-MHCs, and calponin) were lower in NEP−/− PASMCs (Supplement FigureIE-F). Decrease in SM-actin was confirmed by immunohistochemistry (Figure 1F). Although SM-MHCs levels were lower they did not reach significance. The antibody for SM-myosin used recognizes both SM1 and SM2 isoforms. The levels in NEP−/− PASMCs may be the SM1 isoform of SM-myosin as SM2 levels are lost during de-differentiation.
Figure 1. Levels of SM-proteins are lower in NEP−/− PASMCs compared to NEP+/+ PASMCs.
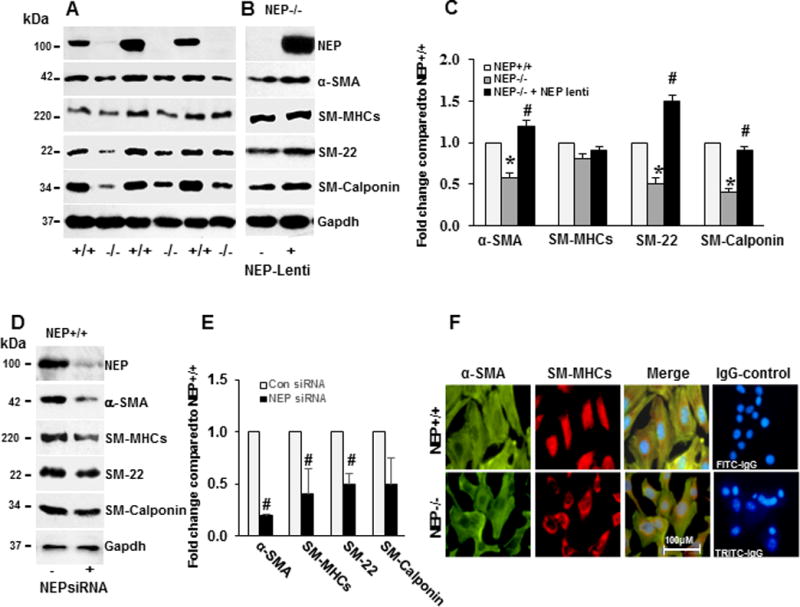
Levels of SM-proteins (α-SMA, SM-myosin, SM-22 and calponin) were measured in lysates from NEP+/+ and −/− PASMCs by Western blotting or immunohistochemistry. Panel A shows levels by Western blotting. Panel B shows effect of expressing NEP in NEP−/− PASMC, on SM-protein levels in NEP−/− PASMCs. Panel C shows average fold change in NEP−/− compared to NEP+/+ PASMCs normalized to Gapdh and effect of NEP expression (n=6). Effect of NEP siRNA (10 nMole/L) on SM-protein levels in NEP+/+ PASMCs is shown in Panel D. Panel E shows average fold change from 3 different transfections normalized to Gapdh. Panel F shows immunostaining for α-SMA and myosin along with IgG controls. (*) represents p≤ 0.05 for comparison between NEP+/+ and NEP −/− PASMC and (#) for comparisons between NEP−/− control and lentivirus-expressing NEP treated cells.
An increase in non-muscle (NM)-myosin and CRBP-1 (markers for synthetic phenotype) was observed in NEP−/− PASMCs as shown in Supplement FigureII. Light microscopy images for NEP+/+, NEP−/− and NEP−/− + lentivirus-NEP are shown in Supplement FigureIIC. Taken together, these data suggest that loss of NEP decreases SM-protein levels in PASMCs.
Increase in baseline activation of RhoGTPases in NEP−/− PASMCs
RhoGTPase (Rac and Rho) signaling plays a critical role in maintaining SM-protein levels through actin remodeling-induced Srf activity5. We measured Rac and Rho activity and downstream effectors (Rock2, p-cofilin, and p-Mlc) in wt and NEP −/− PASMCs at baseline. Rac and RhoA activity were 1.3–1.4 fold higher in NEP−/− PASMCs (Figure 2A–B) compared to wt. Downstream effectors of Rho GTPases, were 1.2–1.5 fold higher at baseline in NEP−/− PASMCs compared to wt (Figure 2C–D). The level of MYPT1Thr696 was 2 fold higher in NEP−/− PASMCs confirming increased Rho activity. In four of the15 isolates of NEP−/− PASMCs tested we observed a decrease in phospho and total cofilin levels compared to wt. Src mediated phosphorylation of cofilin at Tyr86 targets it for degradation and has been suggested as an alternate mechanism for controlling actin dynamics18.
Figure 2. Increased activation of Rho GTPases and downstream effectors in NEP−/− PASMCs.
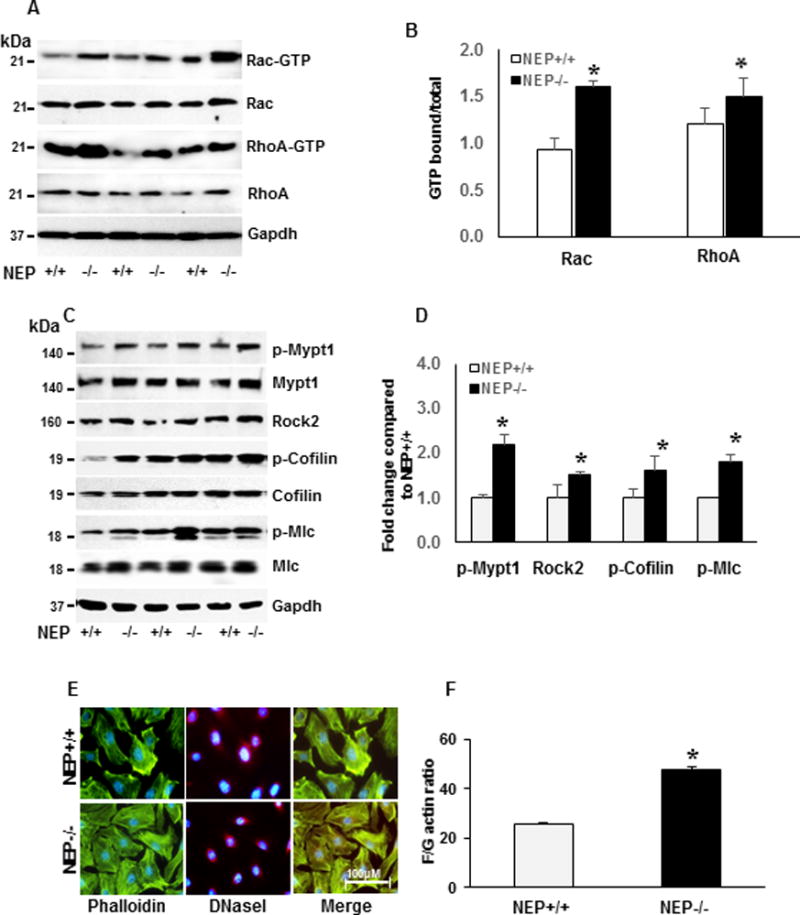
Rac and Rho activity was assessed by measuring levels bound to GTP. Levels from NEP +/+ and −/− PASMCs at baseline are shown in Panel A, and average GTPbound/total from 6 different paired isolates is shown in Panel B. Panel C shows levels of p-Mypt1, Rock2, p-Cofilin, and p-Mlc at baseline. Average levels from 3 different isolates normalized to Gapdh is shown in Panel D. Representative staining for F actin with phalloidin and G-actin with DNaseI in NEP+/+ and −/− PASMCs is shown in Panel E. Levels of F- and G-actin assessed by differential centrifugation is shown in Panel F. *represents p≤ 0.05 for comparison between NEP+/+ and −/− PASMCs.
Decrease in cofilin activity by phosphorylation leads to stabilization of F-actin19. Phalloidin (F-actin) and DNase I (G-actin) stain, were used to visualize F- and G-actin levels. NEP−/− PASMCs showed increased F-actin staining compared to wt (Figure 2E). F/G actin ratio measured by differential centrifugation, showed 2 fold higher levels of F-actin in NEP−/− cells compared to wt (Figure 2F). Our results show that NEP−/− PASMCs exhibit sustained activation of Rac and Rho and downstream effectors.
ShRNA mediated knockdown of Rac and Rho restores SM-protein levels
To determine whether increased Rac and Rho activity suppresses SM-protein, we used shRNA to Rac and Rho, and examined effects on levels, in serum and PDGF-treated NEP−/− PASMCs. There was 70–80% knockdown of Rac and Rho proteins by shRNA treatment (Supplement Figure III A–B). Cofilin phosphorylation was decreased by shRNA to Rho, and Mlc phosphorylation by Rac and Rho (Supplement Figure III A–3D).
In wt PASMCs, shRNA to Rho decreased cofilin phosphorylation. Mlc can be phosphorylated by Rac and Rho. Mlc was weakly phosphorylated at baseline in wt cells and shRNA to either Rac or Rho caused compensatory increase in phosphorylation (Supplement Figure III A–B).
ShRNA to Rac restored SM-proteins levels both at baseline and in PDGF-treated NEP−/− PASMCs. (Figure 3A and Supplement Figure IV). ShRNA to Rho restored α-SMA and SM-myosin levels but suppressed SM-22 and calponin (Figure 3A and Supplement Figure IV). These results suggest that Rho activity is required to maintain baseline expression of SM-22 and calponin in NEP−/− PASMCs.
Figure 3. ShRNA to Rac and RhoA, restores SM-protein levels and attenuates migration and proliferation in NEP−/− PASMCs.
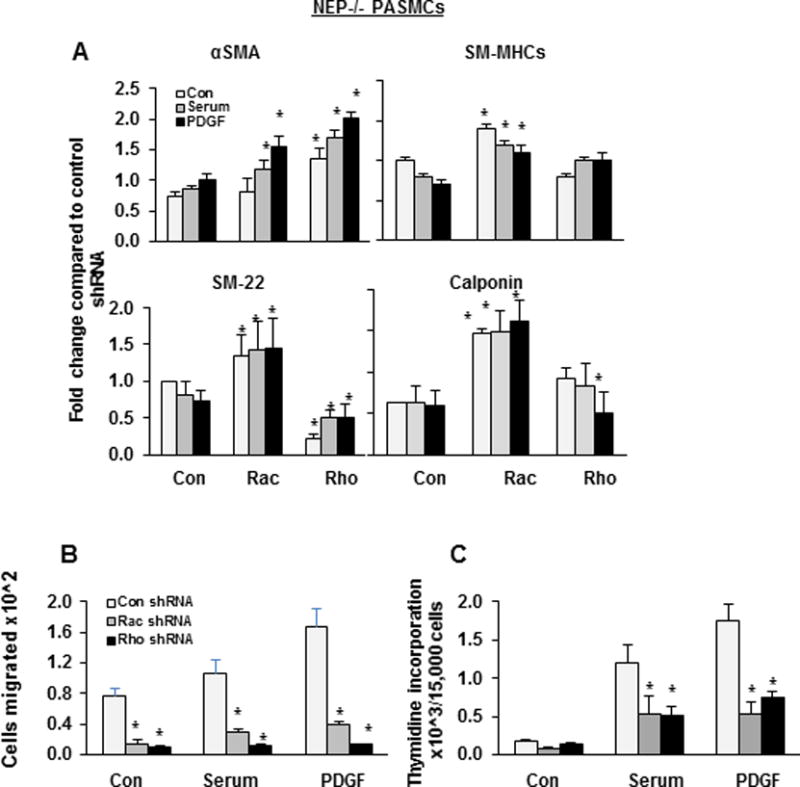
NEP−/− PASMCs were infected with either control shRNA or shRNA to Rac or Rho and selected with puromycin. Cells were treated with serum (0.2%) or PDGF (10ng/ml) for 24h and lysates were analyzed for SM-proteins. Panel A shows effect of shRNA on SM-protein levels from 3 different isolates normalized to Gapdh. Panel B shows the effect of shRNAs on migration and Panel C on proliferation of NEP−/− PASMCs. (*) represents p≤ 0.05 for comparison between control and shRNA treatment.
In wt PASMCs, shRNA to Rac and Rho restored α-SMA levels decreased by PDGF treatment. ShRNA to Rho decreased SM-myosin and calponin levels (Supplement Figure VA–B) suggesting that Rho activity is required for their expression. However, SM-22 levels were increased with Rho shRNA in wt cells suggesting that Rac activity could contribute to expression of SM22. We were unable to knock down both Rac and Rho to confirm role on SM22 levels because the double KO cells did not grow.
RhoGTPases regulate cell migration and proliferation and contribute to mechanisms of vascular remodeling12. ShRNA to Rac and Rho attenuated baseline and serum and PDGF-induced migration and decreased proliferation to wt levels in NEP −/− PASMCs (Figure 3B–C). Similar results were obtained using the Rho kinase inhibitor, Y27362 (Supplement Figure VI).The decreases in proliferation were due to a reduction in cell number. ShRNA to Rac and Rho decreased migration, and PDGF stimulated proliferation was inhibited by Rho shRNA in wt cells (Supplement Figure VIIA–B).
In summary, results from shRNA studies show that sustained activation of Rac and Rho contribute to suppression of SM-protein levels, and increase migration and proliferation of NEP−/− PASMCs. Taken together, the results from shRNA in wt and NEP−/− PASMCs suggest that Rac and Rho have differential effects on SM-proteins under normal physiological conditions as seen in wt cells and in pathological context as seen in NEP−/− cells.
Increased levels of SM-gene repressors, p-Elk-1 and Klf4, in NEP−/− PASMCs
A decrease in mRNA levels for SM-proteins suggested transcriptional repression may contribute to decreased SM-protein levels in NEP−/− PASMCs20. Nuclear levels of SM-gene transcription factors (Srf and myocardin) were 0.5 fold lower, and repressors (Elk1 and Klf4) were 1.5 fold higher in NEP−/− PASMCs compared to wt (Figure 4A–C). Expression of NEP using a lentiviral vector in NEP−/− PASMCs restored levels of Srf and myocardin, while decreasing levels of Klf4 and Elk-1 (Figure 4B–C). SiRNA to NEP significantly decreased Srf and myocardin and increased Klf4 levels in wt PASMCs (Figure 4D–E). Nuclear localization of Srf and p-Elk-1 in wt and NEP−/− PASMCs assessed by immunofluorescence staining is shown in Figure 4F.
Figure 4. NEP−/− PASMCs express higher levels of p-Elk-1 and Klf4 compared to wt.
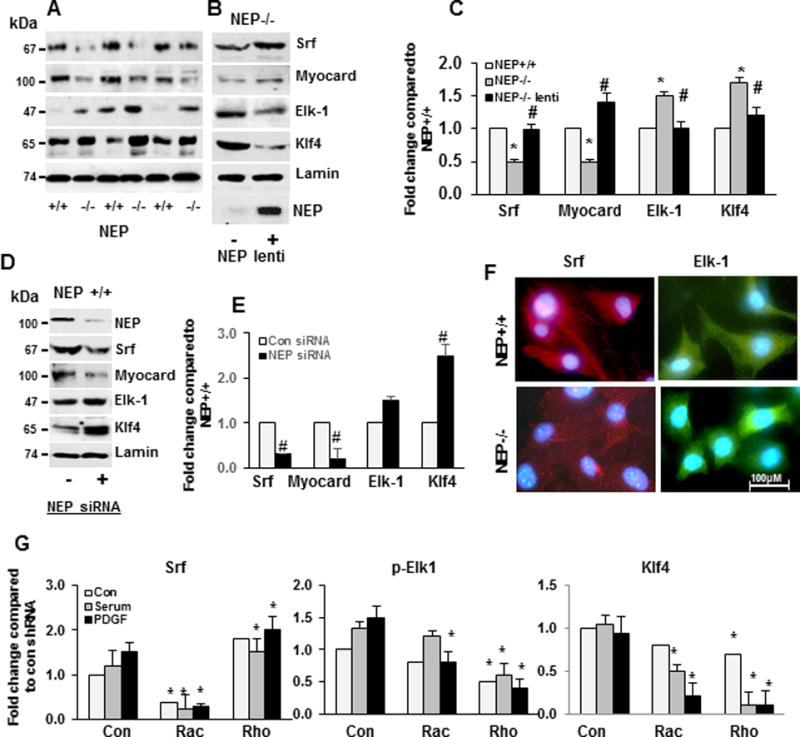
Levels of transcription factors (Srf, myocardin, Elk-1 and Klf4) were measured in nuclear lysates from NEP+/+ and −/− PASMCs and are shown in Panel A. Panel B shows effect of NEP lentivirus on transcription factor levels in NEP −/− PASMCs. Panel C shows average fold change in transcription factor levels in NEP−/− compared to NEP+/+ PASMCs normalized to laminin (n=6). Panel D shows effect of NEP siRNA on transcription factor levels in NEP+/+ PASMCs and Panel E shows average fold change compared to control NEP+/+ PASMCs (n=6). Panel F shows localization of Srf and Elk-1 in NEP+/+ and NEP−/− PASMCs by immunochemistry. Panel G shows effect of shRNAs on Srf, p-Elk-1 and Klf4 from 3 different isolates. Protein levels were normalized to Gapdh. (*) represents p≤ 0.05 for comparison between NEP+/+ and NEP −/− PASMC, and for comparisons between control and shRNA (#) for comparisons between NEP−/− control and lentivirus-expressing NEP treated cells.
Elk-1 and Klf4 inhibit SM-gene expression by competing with myocardin for binding with Srf20,21. We examined the levels of Srf associated with Elk-1, Klf4, and myocardin, by co-immunoprecipitation. There was a 2 fold increase in Srf associated with Elk-1 and Klf4 and a 0.5 fold decrease for myocardin in NEP−/− PASMCs compared to wt (data not shown).
ShRNA to Rac increased Srf expression, and shRNA to Rac and Rho significantly decreased levels of the repressors, p-Elk and Klf4, in NEP−/− PASMCs treated with serum or PDGF (Figure 4G). Baseline levels were not significantly changed in these cells (Figure 4G). These results suggest that enhanced Rac and Rho activity increases levels of SM-gene repressors in PDGF treated NEP−/− PASMCs. In NEP+/+ PASMCs, shRNA to Rho decreased Srf and Klf4 levels (Supplement Figure VIIC–D).
NEP substrate, ET-1, synergizes with PDGF to activate Rac and RhoA and decreases SM-protein levels
Neuropeptide substrates of NEP and Src transactivate growth factor receptors and enhance PDGF-induced migration and proliferation of PASMCs4,22. We tested whether simultaneous treatment with PDGF and ET-1 would suppress SM-protein levels in wt PASMCs, mimicking the NEP−/− phenotype. Cells were treated with PDGF in the absence or presence of ET-1, and activation of Rac and RhoA and downstream effectors, and levels of SM-proteins were measured. PDGF treatment increased RhoGTP levels by 2 fold and ET-1 + PDGF caused a 2-fold increase in Rac and RhoGTP levels at 24h (Figure 5A–B).
Figure 5. PDGF treatment in the presence of NEP substrate, ET-1, increases Rac and Rho activity and decreases SM-protein levels in NEP+/+ PASMCs.
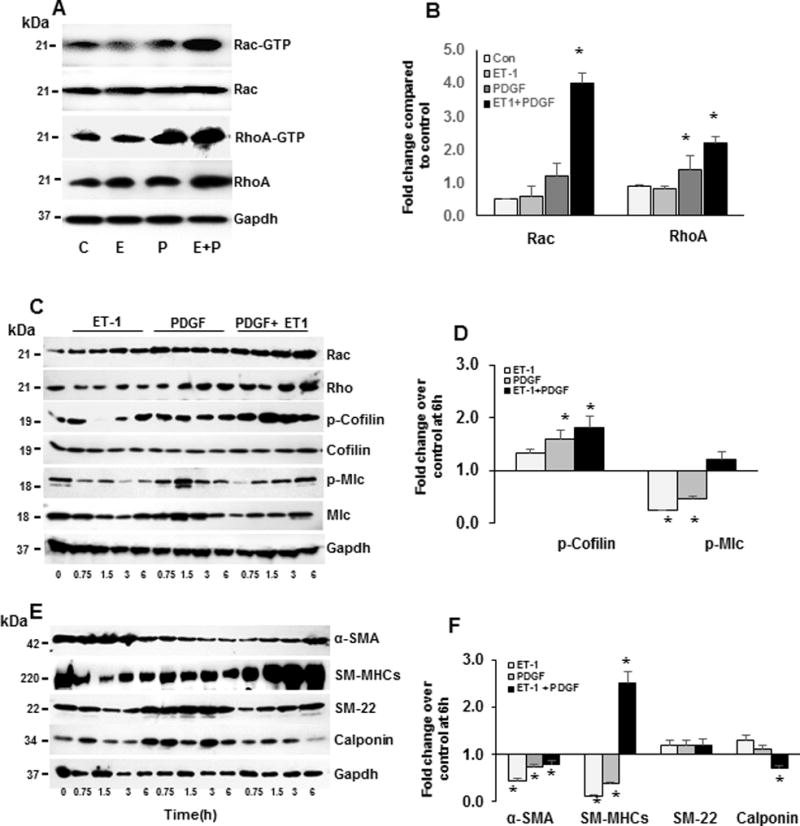
NEP+/+ PASMCs were treated with saline (C) PDGF (P)(10 ng/ml), ET-1(E) (100 nM) and PDGF+ET1 for different time points (0.75h, 1.5h, 3h, and 6h) and lysates probed for downstream effectors of Rho, transcription factors and SM-proteins. Panel A shows levels of Rac and Rho GTP bound form after 24h treatment and graphical representation of average levels of GTPbound/total from 6 isolates is shown in Panel B. Panel C shows effect of agonists on levels of downstream effectors of Rho at different time points and Panel D average fold change at 6h compared to control from 3 different isolates normalized to Gapdh. Panel E shows effect on SM-proteins at different time points and Panel F average fold change at 6h compared to control normalized to Gapdh. *p≤ 0.05 for comparisons between control and treated (n=3).
A time course (0–6h) for activation of downstream effectors of RhoGTPases in response to ET-1, PDGF, and ET-1 + PDGF is shown in Figure 5C and fold change compared to control at 6h in Figure 5D. ET-1 + PDGF increased total levels of Rac, Rho and p–cofilin (Figure 5C–D). ET-1 treatment increased phosphorylation of cofilin and PDGF increased phosphorylation of cofilin and Mlc at times tested (Figure 5C–D). ET-1 and PDGF cause sustained activation of Rac and Rho and downstream effectors. Levels of α-SMA, calponin, and SM22 were also decreased with ET-1+PDGF, as was observed in NEP−/− PASMCs (Figure 5E–F). Surprisingly, ET-1 + PDGF treatment significantly increased SM-myosin which needs further investigation.
Taken together, an NEP substrate, ET-1, synergized with PDGF to increase Rac and Rho activation and cofilin phosphorylation. The increase in Rac and Rho activation coincided with a decrease in SM-proteins as was seen in NEP−/− PASMCs. Similar treatment of NEP−/− PASMCs caused a further increase in p-cofilin and p-Mlc (Supplement FigureVIII). Ambrisentan, an ETRA inhibitor, decreased p-Elk and Klf4 levels in NEP−/− PASMCs, suggesting that ET-1 contributes to the increase in levels of these proteins (Supplement FigureIX).
The presence of NEP in wt PASMCs decreases half-life of ET-1 and is a limitation of this experiment. Whether effects of treatment with ET-1 + PDGF on SM22 in NEP+/+ PASMCs may require prolonged presence of ET-1 is not known.
Altered phosphorylation of Rho regulatory proteins in NEP−/− PASMCs
To further understand the mechanism for sustained activation of Rac and Rho we examined levels and phosphorylation status of their regulators. RhoGTPases are positively regulated by GEFs, and negatively by GAPs and GDIs11,23. GAPs and GDI activities are regulated by phosphorylation by Src, Erk and Rock24 and activity of these kinases is increased in NEP−/− PASMCs.
Tyrosine phosphorylation of RhoGDI by Src inhibits its activity resulting in activation of Rac and Rho. We assessed RhoGDIp-Tyr levels by immunoprecipitation of RhoGDI and probing with a pan-phospho-tyrosine (p-Tyr) antibody due to lack of a commercial site specific one (Figure 6A). Increased p-Tyr levels observed in RhoGDI from NEP−/− PASMCs, and in wt PASMCs treated with ET-1+PDGF suggest that activated Src may inactivate RhoGDI. Serine phosphorylation of RhoGDI inactivates Rho activity. RhoGDISer174 levels were lower in NEP−/− PASMCs and were restored significantly by shRNA to Rac and modestly by shRNA to Rho (Supplemental FigureX).
Figure 6. Decreased tyrosine phosphorylation of RhoGDI and Serine phosphorylation of p190RhoGAP in NEP−/− PASMCs, NEP−/− mouse lungs and human lungs with Copd.
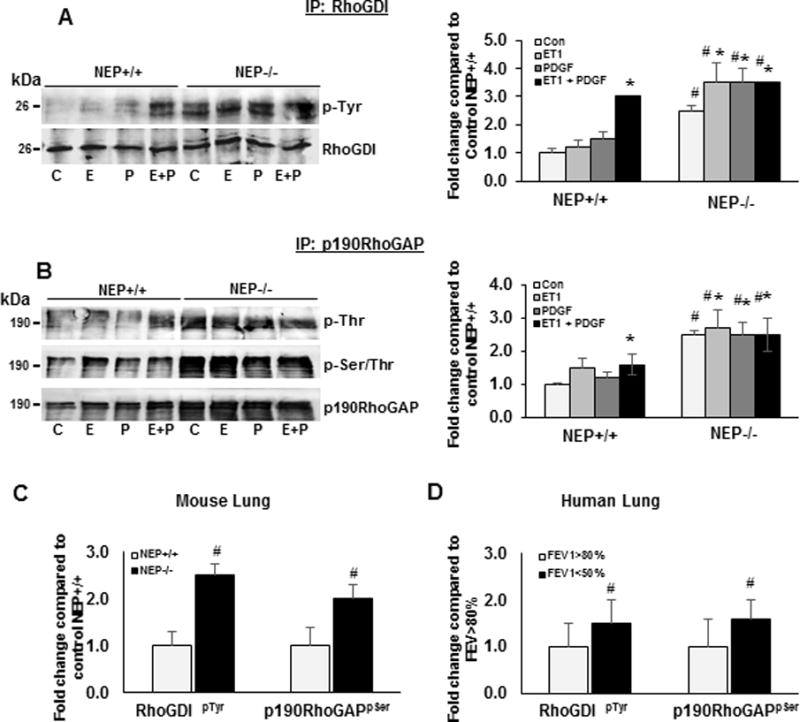
NEP+/+ and −/− PASMCs were treated with saline (C) PDGF (P) (10 ng/ml), ET-1(E) (100 nM), PDGF+ET-1 (E+P) for 24h. Lysates (500 ug) were prepared from lungs of NEP+/+ and −/− mice and Copd patients. Lysates were immunoprecipitated with antibodies to RhoGDI and p190RhoGAP and probed with antibodies to Pan p-Tyr, p-Ser and p-Thr and total RhoGDI and p190RhoGAP. Panel A shows representative Western blot of IP with RhoGDI probed with p-Tyr and fold change from 3 different isolates of PASMCs. Panel B shows representative Western blot of IP with p190RhoGAP probed with p-Thr and p-Ser/Thr blot and fold change from 3 different isolates of PASMCs. Panel C shows levels of p-Tyr in RhoGDI IP and p-Ser in p190RhoGAP IP from lungs of NEP+/+ and −/− mice and Panel D from Copd lungs (n=3). (*) represents p≤ 0.05 for comparison between control and treated and (#) between NEP+/+ and NEP−/− and FEV1>80% to FEV1<50%.
Serine phosphorylation of p190RhoGAP by Rock or Erk inhibits its activity, leading to sustained activation of Rho24. We measured p190RhoGAPpSer/pThr by immune-precipitation with p190RhoGAP antibody and probed with pan-antibodies to p-Thr and p-Ser/Thr. p190RhoGAP was constitutively phosphorylated at Ser/Thr residues in NEP−/− PASMCs suggesting inactivation. As seen in Figure 6B, p-Thr and p-Ser/Thr levels were also increased 2 fold in ET-1 and ET-1+PDGF treated wt PASMCs.
Tyrosine phosphorylation of p190RhoGAP at Y1105 by Src increases its activity causing inhibition of RhoGTPases. Levels of p190RhoGAPY1105 were significantly lower in NEP−/− PASMCs compared to wt. ShRNA to Rac increased p190-RhoGAPY1105 levels 4–6 fold and shRNA to Rho 2–3 fold (Supplement FigureX). We did not detect alterations in the levels of Vav or p115 GEF in these cells.
Rac and Rho and their downstream effectors are increased in lung tissue from NEP−/− mice and humans with COPD
We examined lungs from NEP−/− mice to determine whether RhoGTPases and downstream effectors are activated in vivo. Similar to the observations in SMC, lungs from NEP−/− mice showed increased levels of RhoGTPases and activation of downstream effectors, cofilin and Mlc, which was reversed by treatment with fasudil a Rho kinase inhibitor (Supplement FigureXI). RhoGDIpTyr and RhoGAPpSer levels were higher in lung lysates from NEP−/− mice and is shown in Figure 6C. Levels of RhoGDISer174 and p190RhoGAPY1105 were also decreased in NEP−/− lungs (Supplement FigureXI).
Activity of RhoA/Rho-kinase pathway has been shown to higher in pulmonary arteries from patients with hypoxemic COPD. We examined activation of RhoGTPases and downstream effectors in lungs from COPD patients with FEV>80% and FEV< 50%. Lungs with FEV1<50% showed increased Rac and Rho and downstream effectors p-cofilin and p-Mlc (Supplement FigureXI). RhoGDIpTyr and p190RhoGAPpSer levels were higher and RhoGDISer174 and p190RhoGAPY1105 were lower in FEV1<50 %(Figure 6, Supplement FigureXII and XIII). Lungs of COPD patients with FEV1<50% also showed decrease in NEP (Supplement FigureXI) and increased levels of PDGF and ET-1 compared to lungs from COPD FEV1>80% (data not shown).
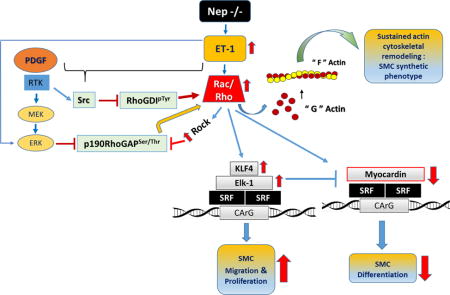
Taken together our results suggest that inactivation of RhoGTPase regulators by increase in phosphorylation of RhoGDIp-Tyr and p190RhoGAPp-Ser/Thr may contribute to sustained activation of RhoGTPases. We speculate that sustained ET-1 and PDGF levels in pathological conditions inactivate RhoGAP and RhoGDI by phosphorylation therefore increasing activities of Rac and Rho, and a dedifferentiated SMC phenotype as shown in the abstract Figure.
DISCUSSION
Differentiated SMCs exist in a quiescent, contractile state and switch to a proliferative one in pathological conditions. The regulation of SMC phenotype is complex and involves transcriptional repressors and signaling pathways, resulting in the downregulation of SM-proteins20, 21. GPCR-coupled contractile agonists including angiotensin II, norepinephrine and ET-1, stimulate RhoA and its downstream effector Rock to maintain vascular tone, and promote a differentiated contractile SMC phenotype. However, studies have shown that prolonged contractile activity leads to musculurization of arteries due to increased SMC proliferation, and contributes to the pathogenesis of systemic and pulmonary hypertension25,26,27.
Our results show NEP−/− PASMCs, and wt PASMCs treated with ET-1 and PDGF have decreased expression of SM-proteins with increased Rac and Rho expression/activity (Figures 1, 2 and 5). Rac and Rho activation is sustained by phosphorylation- dependent inactivation of RhoGDI and RhoGAP in NEP−/− PASMCs and in their wt counterparts treated with ET-1 and PDGF (Figure 6 and graphic abstract). These results suggest that sustained Rac and Rho activity can promote a synthetic SMC phenotype (Figures 1 and 2) and contribute to mechanisms important in vascular remodeling (abstract Figure). Rho and downstream effectors were also activated in lungs from NEP−/− mice.
GWAS studies have reported variants within the FAM13A (family with sequence similarity 13, member A) gene are associated with normal lung function (FEV1/forced vital capacity, FVC) and reduced risk of COPD28. Sequence analysis has identified a RhoGAP domain in the protein suggesting that Rho pathway is involved in lung diseases. Our results show that RhoGDI and RhoGAP were inactivated by phosphorylation in lungs of COPD patients with reduced function (Figure 6D). There was increased activation of downstream effectors cofilin and Mlc (Supplemental FigureXI).
Studies in transgenic mice have suggested a role for both Rac and Rho in vascular remodeling. Prolonged Rho activity is central to increased neointima formation in MLK3−/− mice9. The prevention of medial thickening with Rho inhibitors observed in animal models of hypertension also support a role for Rho in vascular remodeling29. Mice with SM-specific deletion of Rac have enhanced Rho activation and are hypertensive30. Studies with mice overexpressing Vav, a common GEF for Rac and Rho, show that increased Rac activity contributes to vascular remodeling while increased Rho contributes to hypertension31. Kailirin, a GEF for Rac in SMCs, promotes intima formation in atherosclerotic mice10. Our results demonstrate that that sustained Rac in NEP−/− PASMCs is more efficient than Rho in suppressing SM-protein expression (Figure 3).
Rho mediated expression of SM-proteins is mediated by transcription factor, Srf, and repressed by Elk-1 and Klf420. NEP−/− PASMCs express higher levels of SM-gene repressors, Elk-1 and Klf4. Transcriptional repression of SM-genes in response to injury and disease states plays an important role in vascular disease32. Klf4 is a transcription factor that is upregulated in human forms of pulmonary hypertension and in proliferating SMCs33–36. Modulation of Klf4 levels by Rho has not been reported in SMCs. However, sustained Rho activity is required for the repression of protein kinase G (PKG) by Klf4 in SMCs37. Whether increased Klf4 and sustained Rac/Rho activity is required for suppression of SM-proteins expression in NEP−/− PASMCs will need further investigation. Ambrisentan, an ETRA inhibitor, decreased p-Elk and Klf4 levels in NEP−/− PASMCs, suggesting that ET-1 contributes to the increased levels of these proteins (Supplement FigureIX). Muscularization of distal arteries contributes to the pathogenesis of PHTN and other vascular disorders. Understanding SMC phenotypes that contribute to this process maybe important in developing therapeutic strategies.
The Rho/Rho kinase pathway can be activated by several mechanisms involving receptor tyrosine kinases, GPCRs or integrins. It has been suggested that the mechanism by which activity of RhoGTPases is increased, may depend on the stimulus and pathological context31. It is not known if the outcome and coupling to downstream effectors in the activation of Rac/Rho activity may vary depending on whether the mechanism of activation involved GEFs, GAPs or GDIs. Alternatively, matrix proteins like fibronectin activate Rac and Rho and suppress SM-protein expression13, 38. It is possible that alterations in the extracellular matrix of NEP−/− PASMCs couples Rac/Rho to different effectors to promote dedifferentiation compared to that in wt cells. This possibility needs further investigation. Our data show that inactivation of RhoGDI and RhoGAPs by phosphorylation leads to sustained Rac/Rho activity and suppression of SM-contractile protein levels promoting a synthetic SMC phenotype.
Supplementary Material
HIGHLIGHTS.
Under physiological conditions Rho is required for expression of SM-contractile proteins and maintains vascular tone.
Pulmonary artery smooth muscle cells isolated from Neprilysin null mice exhibit a synthetic phenotype with decreased levels of SM-proteins and increased Rac and Rho activity and levels.
ShRNA to Rac and Rho restore SM-protein levels suggesting a role for the GTPases in decreased expression.
The NEP−/− phenotype could be mimicked by treating wild type PASMCs with PDGF in the presence of the NEP substrate, ET-1.
Rho may play an important role in integrating signals from growth factors like PDGF, and GPCRs, like ET-1, to promote a synthetic SM phenotype in these cells.
Acknowledgments
The authors are thankful to Derek Strassheim PhD for critical reading of the manuscript and Andrej Poczobutt MS for administrative assistance. Preliminary results presented in abstract form at 2016 ATS meeting, San Francisco CA (Am J Respir Crit Care Med 193: A7302).
Funding Sources: The work was supported in part by funding from NHLBI - HL078927, HL014985, HL095439, and VA Merit Review to Dempsey EC and DoD-W81XWH-14-1-0451to Fini MA and 1R01HL125642-01A1 to Irwin D.
Abbreviations
- NEP
Neprilysin
- Rho
Ras homology family member
- RhoGDI
Rho guanine dissociation inhibitor
- RhoGAP
Rho GTPase activating protein
- Srf
Serum response factor
- Klf4
Kruppel lung factor 4
Footnotes
Materials and Methods are available in the online-only data supplement
Disclosures: None.
References
- 1.Bayes-Genis A, Barallat J, Richards AM. A test in context: Neprilysin: Function, inhibition, and biomarker. Journal of the American College of Cardiology. 2016;68:639–653. doi: 10.1016/j.jacc.2016.04.060. [DOI] [PubMed] [Google Scholar]
- 2.Turner AJ. Exploring the structure and function of zinc metallopeptidases: Old enzymes and new discoveries. Biochemical Society transactions. 2003;31:723–727. doi: 10.1042/bst0310723. [DOI] [PubMed] [Google Scholar]
- 3.Dempsey EC, Wick MJ, Karoor V, Barr EJ, Tallman DW, Wehling CA, Walchak SJ, Laudi S, Le M, Oka M, Majka S, Cool CD, Fagan KA, Klemm DJ, Hersh LB, Gerard NP, Gerard C, Miller YE. Neprilysin null mice develop exaggerated pulmonary vascular remodeling in response to chronic hypoxia. The American journal of pathology. 2009;174:782–796. doi: 10.2353/ajpath.2009.080345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karoor V, Oka M, Walchak SJ, Hersh LB, Miller YE, Dempsey EC. Neprilysin regulates pulmonary artery smooth muscle cell phenotype through a platelet-derived growth factor receptor-dependent mechanism. Hypertension. 2013;61:921–930. doi: 10.1161/HYPERTENSIONAHA.111.199588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by rhoa-mediated actin polymerization. The Journal of biological chemistry. 2001;276:341–347. doi: 10.1074/jbc.M005505200. [DOI] [PubMed] [Google Scholar]
- 6.Bei Y, Duong-Quy S, Hua-Huy T, Dao P, Le-Dong NN, Dinh-Xuan AT. Activation of rhoa/rho-kinase pathway accounts for pulmonary endothelial dysfunction in patients with chronic obstructive pulmonary disease. Physiological reports. 2013;1:e00105. doi: 10.1002/phy2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bakker EN, van der Meulen ET, van den Berg BM, Everts V, Spaan JA, VanBavel E. Inward remodeling follows chronic vasoconstriction in isolated resistance arteries. Journal of vascular research. 2002;39:12–20. doi: 10.1159/000048989. [DOI] [PubMed] [Google Scholar]
- 8.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circulation research. 2007;100:923–929. doi: 10.1161/01.RES.0000261658.12024.18. [DOI] [PubMed] [Google Scholar]
- 9.Gadang V, Konaniah E, Hui DY, Jaeschke A. Mixed-lineage kinase 3 deficiency promotes neointima formation through increased activation of the rhoa pathway in vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:1429–1436. doi: 10.1161/ATVBAHA.114.303439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu JH, Fanaroff AC, Sharma KC, Smith LS, Brian L, Eipper BA, Mains RE, Freedman NJ, Zhang L. Kalirin promotes neointimal hyperplasia by activating rac in smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:702–708. doi: 10.1161/ATVBAHA.112.300234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loirand G, Pacaud P. Involvement of rho gtpases and their regulators in the pathogenesis of hypertension. Small GTPases. 2014;5 doi: 10.4161/sgtp.28846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu OM, Brown JH. G protein-coupled receptor and rhoa-stimulated transcriptional responses: Links to inflammation, differentiation, and cell proliferation. Molecular pharmacology. 2015;88:171–180. doi: 10.1124/mol.115.097857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beqaj S, Jakkaraju S, Mattingly RR, Pan D, Schuger L. High rhoa activity maintains the undifferentiated mesenchymal cell phenotype, whereas rhoa down-regulation by laminin-2 induces smooth muscle myogenesis. The Journal of cell biology. 2002;156:893–903. doi: 10.1083/jcb.200107049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellani L, Salvati E, Alema S, Falcone G. Fine regulation of rhoa and rock is required for skeletal muscle differentiation. The Journal of biological chemistry. 2006;281:15249–15257. doi: 10.1074/jbc.M601390200. [DOI] [PubMed] [Google Scholar]
- 15.Rautureau Y, Coelho SC, Fraulob-Aquino JC, Huo KG, Rehman A, Offermanns S, Paradis P, Schiffrin EL. Inducible human endothelin-1 overexpression in endothelium raises blood pressure via endothelin type a receptors. Hypertension. 2015;66:347–355. doi: 10.1161/HYPERTENSIONAHA.115.05168. [DOI] [PubMed] [Google Scholar]
- 16.Barst RJ. Pdgf signaling in pulmonary arterial hypertension. The Journal of clinical investigation. 2005;115:2691–2694. doi: 10.1172/JCI26593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owens GK. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Foundation symposium. 2007;283:174–191. doi: 10.1002/9780470319413.ch14. discussion 191–173, 238–141. [DOI] [PubMed] [Google Scholar]
- 18.Yoo Y, Ho HJ, Wang C, Guan JL. Tyrosine phosphorylation of cofilin at y68 by v-src leads to its degradation through ubiquitin-proteasome pathway. Oncogene. 2009;29:263–272. doi: 10.1038/onc.2009.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanellos G, Frame MC. Cellular functions of the adf/cofilin family at a glance. Journal of cell science. 2016;129:3211–3218. doi: 10.1242/jcs.187849. [DOI] [PubMed] [Google Scholar]
- 20.Kawai-Kowase K, Owens GK. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. American journal of physiology. 2007;292:C59–69. doi: 10.1152/ajpcell.00394.2006. [DOI] [PubMed] [Google Scholar]
- 21.Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1495–1505. doi: 10.1161/ATVBAHA.110.221135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pyne NJ, Pyne S. Receptor tyrosine kinase-g-protein-coupled receptor signalling platforms: Out of the shadow? Trends in pharmacological sciences. 2011;32:443–450. doi: 10.1016/j.tips.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, Burridge K. Regulation of rho gtpase crosstalk, degradation and activity by rhogdi1. Nature cell biology. 2010;12:477–483. doi: 10.1038/ncb2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mori K, Amano M, Takefuji M, Kato K, Morita Y, Nishioka T, Matsuura Y, Murohara T, Kaibuchi K. Rho-kinase contributes to sustained rhoa activation through phosphorylation of p190a rhogap. The Journal of biological chemistry. 2009;284:5067–5076. doi: 10.1074/jbc.M806853200. [DOI] [PubMed] [Google Scholar]
- 25.Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. Possible involvement of rho-kinase in the pathogenesis of hypertension in humans. Hypertension. 2001;38:1307–1310. doi: 10.1161/hy1201.096541. [DOI] [PubMed] [Google Scholar]
- 26.Seasholtz TM, Brown JH. Rho signaling in vascular diseases. Molecular interventions. 2004;4:348–357. doi: 10.1124/mi.4.6.8. [DOI] [PubMed] [Google Scholar]
- 27.Connolly MJ, Aaronson PI. Key role of the rhoa/rho kinase system in pulmonary hypertension. Pulmonary pharmacology & therapeutics. 2011;24:1–14. doi: 10.1016/j.pupt.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 28.van der Plaat DA, de Jong K, Lahousse L, Faiz A, Vonk JM, van Diemen CC, Nedeljkovic I, Amin N, Brusselle GG, Hofman A, Brandsma CA, Bosse Y, Sin DD, Nickle DC, van Duijn CM, Postma DS, Boezen HM. Genome-wide association study on the fev1/fvc ratio in never-smokers identifies hhip and fam13a. The Journal of allergy and clinical immunology. 2017;139:533–540. doi: 10.1016/j.jaci.2016.06.062. [DOI] [PubMed] [Google Scholar]
- 29.Abe K, Tawara S, Oi K, Hizume T, Uwatoku T, Fukumoto Y, Kaibuchi K, Shimokawa H. Long-term inhibition of rho-kinase ameliorates hypoxia-induced pulmonary hypertension in mice. Journal of cardiovascular pharmacology. 2006;48:280–285. doi: 10.1097/01.fjc.0000248244.64430.4a. [DOI] [PubMed] [Google Scholar]
- 30.Andre G, Sandoval JE, Retailleau K, Loufrani L, Toumaniantz G, Offermanns S, Rolli-Derkinderen M, Loirand G, Sauzeau V. Smooth muscle specific rac1 deficiency induces hypertension by preventing p116rip3-dependent rhoa inhibition. Journal of the American Heart Association. 2014;3:e000852. doi: 10.1161/JAHA.114.000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fabbiano S, Menacho-Marquez M, Sevilla MA, Albarran-Juarez J, Zheng Y, Offermanns S, Montero MJ, Bustelo XR. Genetic dissection of the vav2-rac1 signaling axis in vascular smooth muscle cells. Molecular and cellular biology. 2014;34:4404–4419. doi: 10.1128/MCB.01066-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miano JM. Vascular smooth muscle cell differentiation-2010. Journal of biomedical research. 2010;24:169–180. doi: 10.1016/S1674-8301(10)60026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Zheng B, Zhang XH, Nie CJ, Li YH, Wen JK. Localization and function of klf4 in cytoplasm of vascular smooth muscle cell. Biochemical and biophysical research communications. 2013;436:162–168. doi: 10.1016/j.bbrc.2013.05.067. [DOI] [PubMed] [Google Scholar]
- 34.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. Klf4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature medicine. 2015;21:628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheikh AQ, Lighthouse JK, Greif DM. Recapitulation of developing artery muscularization in pulmonary hypertension. Cell reports. 2014;6:809–817. doi: 10.1016/j.celrep.2014.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheikh AQ, Misra A, Rosas IO, Adams RH, Greif DM. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Science translational medicine. 2015;7:308ra159. doi: 10.1126/scitranslmed.aaa9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng B, Han M, Wen JK. Role of kruppel-like factor 4 in phenotypic switching and proliferation of vascular smooth muscle cells. IUBMB life. 2010;62:132–139. doi: 10.1002/iub.298. [DOI] [PubMed] [Google Scholar]
- 38.Shi F, Long X, Hendershot A, Miano JM, Sottile J. Fibronectin matrix polymerization regulates smooth muscle cell phenotype through a rac1 dependent mechanism. PloS one. 2014;9:e94988. doi: 10.1371/journal.pone.0094988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.