

Abstract
Objective
The noncoding single nucleotide polymorphism rs12740374 has been hypothesized to be the causal variant responsible for liver-specific modulation of SORT1 expression (i.e., expression quantitative trait locus, or eQTL) and, by extension, the association of the SORT1 locus on human chromosome 1p13 with low-density lipoprotein cholesterol levels and coronary heart disease. The goals of this study were to compare three different hepatocyte models in demonstrating that the rs12740374 minor allele sequence is responsible for transcriptional activation of SORT1 expression.
Approach and Results
We found that whereas primary human hepatocytes of varied rs12740374 genotypes strongly replicated the SORT1 eQTL previously observed in whole-liver samples, a population cohort of induced pluripotent stem cell (iPSC)-derived hepatocyte-like cells (HLCs) poorly replicated the eQTL. In primary human hepatocytes from multiple individuals heterozygous at rs12740374, we used CRISPR-Cas9 to specifically target the rs12740374 minor allele sequence ex vivo, resulting in a reproducible reduction in SORT1 expression. We generated a locus-humanized transgenic mouse with a bacterial artificial chromosome bearing the human SORT1 locus with the rs12740374 minor allele. In this mouse model, we used CRISPR-Cas9 to target the rs12740374 minor allele sequence in the liver in vivo, resulting in a substantial reduction of hepatic SORT1 expression.
Conclusions
The rs12740374 minor allele sequence enhances SORT1 expression in hepatocytes. CRISPR-Cas9 can be used in primary human hepatocytes ex vivo and locus-humanized mice in vivo to interrogate the function of noncoding regulatory regions. iPSC-derived HLCs suffer from limitations that prevent faithful modelling of some hepatocyte eQTLs.
Keywords: Gene expression, gene regulation, genetically altered mice, transgenic model, liver
Subject Codes: Functional Genomics, Gene Expression and Regulation, Genetically Altered and Transgenic Models
INTRODUCTION
Low-density lipoprotein cholesterol (LDL-C) is one of the best-established causal risk factors for coronary heart disease (CHD). Genome-wide association studies have reproducibly identified a locus on human chromosome 1p13 as being strongly associated with both LDL-C and CHD.1,2 Within the locus, sortilin 1 (SORT1) has been established to be the causal gene responsible for modulation of blood LDL-C levels and, by extension, CHD risk.3,4 SORT1 regulates both the hepatic secretion of very-low-density lipoprotein particles into the bloodstream and the clearance of LDL particles from the bloodstream.4,5
The best candidate for the causal DNA variant responsible for the association of the SORT1 locus on chromosome 1p13 with LDL-C and CHD is rs12740374, a noncoding single nucleotide polymorphism (SNP) that lies ~120 kb away from the promoter of the SORT1 gene.3 A series of experiments have suggested that the minor allele of rs12740374 creates a binding site for CCAAT-enhancer-binding protein (C/EBP) transcription factors, resulting in liver-specific transcriptional activation of SORT1, i.e., a liver-specific expression quantitative trait locus (eQTL). However, these experiments did not demonstrate a direct causal link between rs12740374 and SORT1 by perturbing the site of the SNP in the genome and evoking an alteration of SORT1 expression. A significant challenge to achieving this is that the strong association between rs12740374 genotype and SORT1 expression has previously been observed only in liver and presumably is specific to hepatocytes.3,6 Indeed, querying of all 44 tissue types available in the Gene-Tissue Expression (GTEx) project’s database (https://www.gtexportal.org/) as of the time of this writing shows evidence for a substantial eQTL only in liver. All commonly used cultured human hepatoma cell lines (e.g., HepG2, HuH-7, Hep3B) are homozygous for the major allele of rs12740374 and thus should have no enhancement of SORT1 expression;3 moreover, these cell lines do not represent authentic, fully functional hepatocytes. Adding to the challenge, the sequence surrounding rs12740374 in human chromosome 1p13 is poorly conserved in the orthologous region in the mouse genome, preventing the use of wild-type mice to study the rs12740374-SORT1 relationship in vivo.
Here we employ three hepatocyte model systems to interrogate the role of the hypothesized rs12740374 minor allele enhancer vis-à-vis human SORT1 expression. The first is a collection of primary human hepatocytes with varied rs12740374 genotypes. The second is a population cohort of induced pluripotent stem cell (iPSC)-derived hepatocyte-like cells (HLCs). The third is a mouse model in which the human SORT1 locus with the rs12740374 minor allele has been added to the genome via bacterial artificial chromosome (BAC) transgenesis. With two of the model systems, we use CRISPR-Cas9 ex vivo or in vivo to specifically target the rs12740374 minor allele sequence and assess for an effect on hepatic SORT1 expression, thus establishing complementary approaches to study human-specific regulatory regions in hepatocytes.
MATERIALS AND METHODS
Materials and Methods are available in the online-only Data Supplement.
RESULTS
Of the genes in the SORT1 locus (Figure 1A), SORT1 and PSRC1 have previously been reported to display a strong correlation of expression levels with rs12740374 genotype in liver samples (with the minor allele associated with increased expression), with CELSR2 showing weaker correlation and SARS, MYBPHL, and PSMA5 showing no correlation.3 Of note, we found that CELSR2 and MYBPHL expression levels were very low and could not be reliably measured by qRT-PCR in primary human hepatocytes and BAC transgenic mice (i.e., very high Ct values), and so they were not analyzed for this study.
Figure 1. Expression of SORT1 and PSRC1 in primary human hepatocytes and HLCs of varied rs12740374 genotypes.
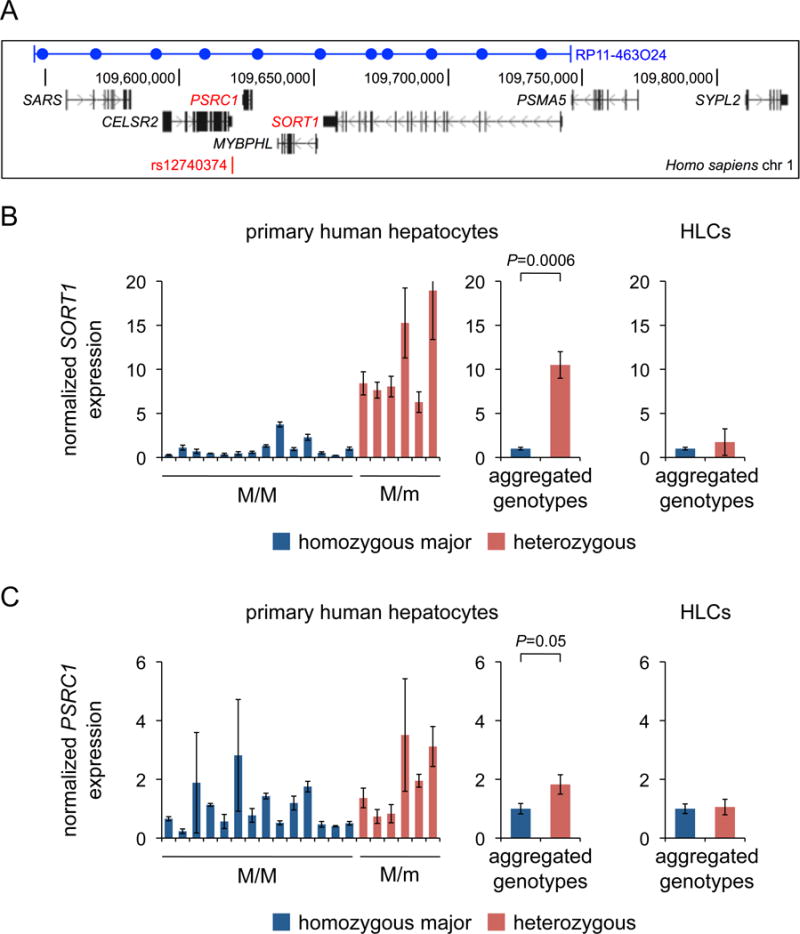
A shows the genes in or near the SORT1 locus on human chromosome 1p13. The site of the rs12740374 SNP and the boundaries of the insert of BAC RP11-463O24 (which has the minor allele of rs12740374) are indicated, with sites of PCR within the BAC shown as blue circles. B and C show the expression levels of SORT1 and PSRC1, respectively, in either primary human hepatocytes (left and middle panels) or HLCs (right panel), normalized to the mean of the expression levels of all of the homozygous major samples of each cell type. The data are displayed as means and standard errors of the mean (s.e.m.) for each of the 20 lots (N = 3 samples per lot) in the left panel and as means and s.e.m. for all of the samples within each genotype group in the middle and right panel. P values were calculated using the Mann-Whitney U test to compare the two genotype groups for each gene.
We initially screened primary human hepatocytes from 20 individuals for rs12740374 genotype, plated in triplicate format. Six of the lots were heterozygous and 14 were homozygous for the major allele; none were homozygous for the minor allele. Three days after plating, we found that the heterozygous cells, with one copy of the putative transcriptional enhancer, had 10.5-fold higher SORT1 expression on average compared to homozygous major cells (P = 0.0006) as assessed by quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR), signifying strong replication of the previously observed liver-specific eQTL (Figure 1B). We also observed 1.8-fold higher PSRC1 expression (P = 0.05) (Figure 1C). Of note, these findings do not establish that rs12740374 is the causal variant responsible for the increased SORT1 and PSRC1 expression in the heterozygous cells; rather, rs12740374 is a tag SNP for the haplotype of the SORT1 locus, with any of the SNPs in the minor haplotype potentially being the causal variant.
In a recent report, HLCs derived from 17 individuals homozygous for the rs12740374 minor allele and 17 individuals homozygous for the rs12740374 major allele were compared with respect to SORT1 expression.7 By qRT-PCR, there was only a 1.3-fold increase in SORT1 expression in the homozygous minor HLCs, with nominal statistical significance (P = 0.05). Given the enormous discrepancy between this finding and our finding in the primary human hepatocytes, we assessed the rs12740374-SORT1 eQTL in a much larger population cohort of iPSC-derived HLC samples (N = 86 individuals).8 Using RNA-seq data from the HLC cohort, we observed only a weak relationship between rs12740374 genotype and SORT1 expression, with nominal statistical significance (P = 0.05) (Supplemental Figure I). By way of direct comparison, we used RNA-seq data from the whole-liver cohort of the GTEx project (N = 96) and observed a strong rs12740374-SORT1 eQTL (P = 7 × 10−18) (Supplemental Figure I). We also used qRT-PCR to assess SORT1 and PSRC1 expression in the HLC cohort and confirmed that the differences between heterozygous HLCs (N = 27) and major allele homozygous HLCs (N = 55) were small (for SORT1, 1.7-fold increase, P = 0.15; for PSRC1, no increase, P = 0.94) in direct comparison to the differences observed in primary human hepatocytes (Figure 1B–C).
Given the poor replication of the rs12740374-SORT1 eQTL in two different HLC cohorts, we chose to instead use the primary human hepatocytes to attempt functional validation of the rs12740374 minor allele sequence as a transcriptional enhancer of SORT1 expression, employing CRISPR-Cas9 to disrupt the sequence. In order to achieve genome editing of the rs12740374 minor allele sequence, we used a validated guide RNA designed to cleave 1 bp upstream of rs12740374, with a protospacer overlapping the position of rs12740374 and complementary to the minor allele of the SNP (Figure 2A). In principle, this guide RNA should target Cas9 specifically to a chromosome with the minor allele of rs12740374 and spare a chromosome with the major allele. We recognized that genome editing of primary human hepatocytes would be challenging, due to (1) the difficulty of delivering CRISPR-Cas9 into primary cells and (2) the hepatocytes being only rated to survive in culture for three days following plating. To assess whether we could achieve a high infection rate of primary human hepatocytes in culture, we treated samples of cells with GFP-encoding adenovirus one day after plating and observed GFP expression in virtually all of the cells on the third day (Supplemental Figure II). We treated replicates of the heterozygous human hepatocytes with either the CRISPR-SNP adenovirus or the CRISPR-control adenovirus one day after plating and harvested the cells on the third day, allowing for only ~48 hours of expression of the CRISPR-Cas9 system in the cells. We performed deep sequencing of the rs12740374 site in a representative sample, which demonstrated (1) essentially exclusive targeting of minor allele-bearing chromosomes in preference to major allele-bearing chromosomes (>99% proportion) and (2) an estimated 6% indel rate in minor allele-bearing chromosomes (Online Dataset).
Figure 2. Effects of CRISPR-Cas9 genome editing of the rs12740374 minor allele sequence in primary human hepatocytes.
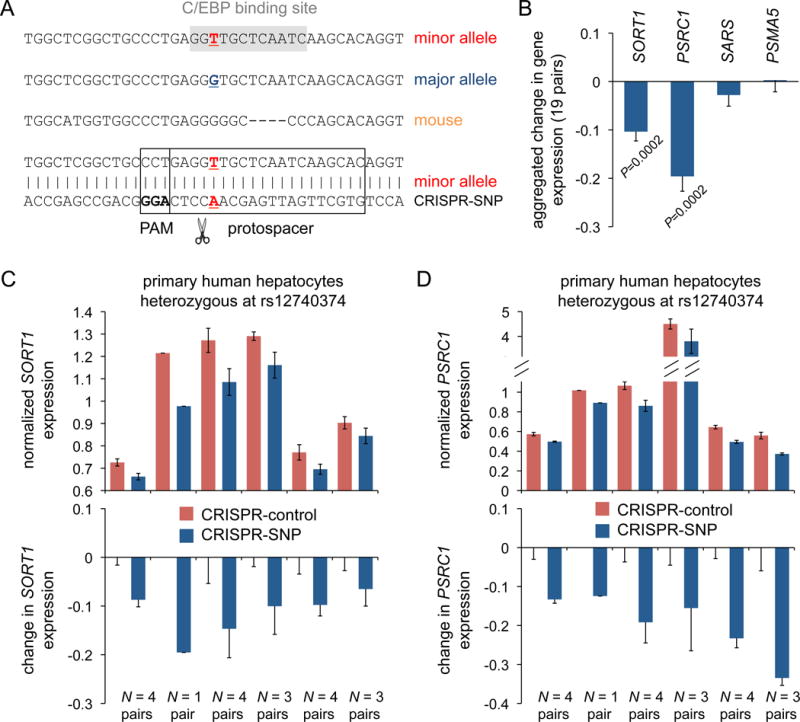
A shows the human rs12740374 minor and major allele sequences and the orthologous sequence in mouse. The rs12740374 SNP position is bold and underlined. The C/EBP consensus binding site, intact with the minor allele and disrupted with the major allele, is shown with grey shading. The bottom minor allele sequence is shown as double-strand DNA, with boxes indicating the chosen CRISPR-Cas9 protospacer sequence and protospacer-adjacent motif (PAM) used for the CRISPR-SNP adenovirus. The predicted CRISPR-Cas9 cleavage site is indicated with scissors. B shows the aggregated differences in gene expression between the CRISPR-control-treated and CRISPR-SNP-treated samples within each pair of samples of heterozygous primary human hepatocytes (N = 19 pairs total); the aggregated differences are expressed as the means and s.e.m. of the proportions of change. P values were calculated using the Wilcoxon signed-rank test for the 19 pairs for each gene. C and D show the expression levels of SORT1 and PSRC1, normalized to the mean of the expression levels of all of the CRISPR-control-treated samples. The data are displayed as means and s.e.m. for the pairs of samples within each of the 6 heterozygous lots on the top. The same data is shown on the bottom, with the means for the CRISPR-control-treated samples within each of the 6 lots set to zero and the means for the CRISPR-SNP-treated samples of the 6 lots shown as proportional changes from the CRISPR-control-treated samples. The numbers of sample pairs for each lot are shown at the bottom.
Despite the low level of CRISPR-Cas9-mediated mutagenesis, we observed highly reproducible decreases in SORT1 and PSRC1 expression in the CRISPR-SNP-treated cells compared to the CRISPR-control-treated cells (Figure 2B–D). Each of the six lots of heterozygous human hepatocytes, when replicates were averaged, displayed lower expression of both genes with CRISPR-SNP treatment (Figure 2C–D).
When the data from all of the replicates of all of the lots were aggregated (N = 19 pairs across six lots), SORT1 expression was reduced by 11% (P = 0.0002), and PSRC1 expression was reduced by 20% (P = 0.0002) (Figure 2B). We also assessed two control genes in the SORT1 locus whose expression levels in liver are not correlated with rs12740374 genotype, SARS and PSMA5. For these genes, there were negligible, non-significant differences between the treatment groups (Figure 2B).
As a complementary approach, we sought to study the interaction of the rs12740374 minor allele sequence and hepatic SORT1 expression in vivo (Figure 3A). Given the lack of conservation of the rs12740374 site in the mouse genome (Figure 2A), we generated BAC transgenic mice with the human SORT1 locus integrated into the mouse genome. BAC clone RP11-463O24 (boundaries indicated in Figure 1A), which has the minor allele of rs12740374, was injected into single-cell embryos of the C57BL/6J background, yielding 10 founder mice. Upon breeding of the mice, we identified the stable transgenic line with the lowest copy number of the complete BAC transgene (Figure 3B) and confirmed the presence of the minor allele of rs12740374. We used mice of the F2 generation of this line for somatic in vivo genome editing experiments.
Figure 3. Effects of CRISPR-Cas9 genome editing of the rs12740374 minor allele sequence in locus-humanized mice.
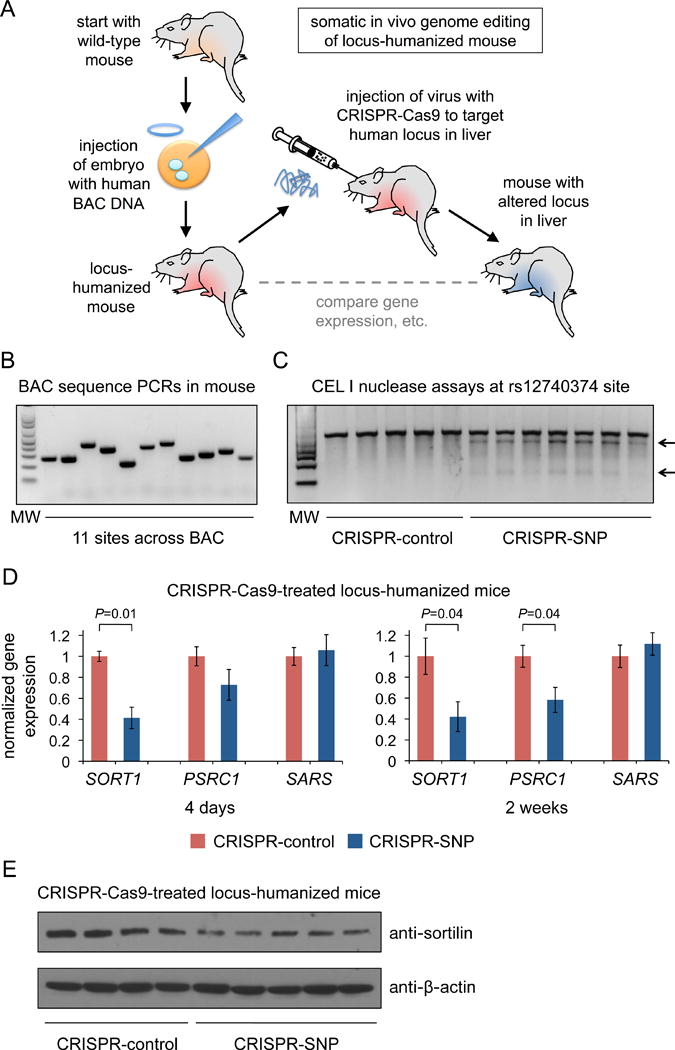
A shows a schematic for the generation and testing of locus-humanized mice. B shows PCR assays confirming the presence of the entire BAC RP11-463O24 (rs12740374 minor allele) insert in the genome in F2 mice. The 11 PCR amplicons shown correspond to the sites of the 11 blue circles in Figure 1A. C shows CEL-I nuclease assays performed with PCR amplicons around the rs12740374 SNP site from liver genomic DNA from locus-humanized mice administered CRISPR-control or CRISPR-SNP adenovirus. Arrows show the cleavage products resulting from the CEL-I nuclease assays; the intensity of the cleavage product bands relative to the uncleaved product band corresponds to the mutagenesis rate. D shows the hepatic expression levels of SORT1, PSRC1, and SARS from the human BAC locus, normalized to the mean of the expression levels of all of the CRISPR-control mice. The left panel shows hepatic gene expression four days after virus treatment (N = 5 CRISPR-control mice and 7 CRISPR-SNP mice). The right panel shows hepatic gene expression two weeks after virus treatment (N = 4 CRISPR-control mice and 5 CRISPR-SNP mice). The data are displayed as means and s.e.m. for the mice within each treatment group. P values were calculated using the Mann-Whitney U test to compare the two treatment groups for each gene. E shows Western blot analysis of sortilin and β-actin in liver samples from the mice represented in the right panel of D.
The CRISPR-SNP adenovirus or the CRISPR-control adenovirus was administered to mice at two to three months of age. After either four days or two weeks, we sacrificed the mice in order to harvest liver tissue. Whereas there was no evidence of mutagenesis in the control mice, the CRISPR-SNP-treated mice displayed consistent and substantial levels of mutagenesis by CEL-I nuclease assay (Figure 3C). Deep sequencing of the rs12740374 site in a representative CRISPR-SNP-treated mouse documented a 33% indel rate (Online Dataset), a degree of mutagenesis concordant with previous studies of in vivo hepatic genome editing with virally delivered CRISPR-Cas9.9–11 We observed 60% reduction (4 days) and 58% reduction (2 weeks) in human SORT1 expression in CRISPR-SNP-treated mice compared to CRISPR-control-treated mice (Figure 3D), with concordant reduction of hepatic sortilin protein levels observed by Western blot analysis (Figure 3E). We also observed 30% reduction (4 days, non-significant P value) and 42% reduction (2 weeks) in human PSRC1 expression, whereas there was minimal change in expression of the control human SARS gene at either time point (PSMA5 is not contained within the BAC clone RP11-463O24 and so could not be used as an additional control gene). No differences were observed in blood cholesterol levels, as expected from the small effect of rs12740374 in humans (5.65 mg/dL change in cholesterol per allele1). In a parallel experiment, we used an adenovirus with a guide RNA similar to the CRISPR-SNP adenovirus but matched to the rs12740374 major allele rather than the minor allele (CRISPR-major adenovirus). Compared to CRISPR-control, CRISPR-major produced no discernible mutagenesis and did not decrease SORT1 or PSRC1 expression after 2 weeks (Supplemental Figure III).
DISCUSSION
Our motivation to compare three different hepatocyte model systems came from recognition of the shortcomings of available model systems. Cultured human hepatoma cell lines have profound karyotypic abnormalities and do not faithfully replicate many aspects of hepatocyte biology.12 Even if they did represent authentic hepatocytes, none of the commonly used hepatoma cell lines are homozygous for the minor allele or even heterozygous at rs12740374. This is a challenge common to any SNP for which the minor allele and not the major allele has biological activity. One could use genome editing to knock in the minor allele at rs12740374 in one of the cell lines, but besides being very inefficient due to the limitations of homology-directed repair, this would not address the other problems of hepatoma cells.
In principle, iPSCs have substantial advantages over human hepatoma cell lines: they have normal karyotypes that can be maintained over many passages, even through expansion from a single cell to millions of cells; thousands of iPSC lines have been made, and so it is feasible to identify existing cell lines that are homozygous minor for a common SNP of interest; they are quite amenable to genome editing; and they can be readily differentiated into cells resembling hepatocytes. However, such differentiated cells tend to be heterogeneous, immature, and lacking in important aspects of hepatocyte biology.13 These shortcomings are evident in the inability of HLCs to faithfully model the strong rs12740374-SORT1 eQTL observed in both whole-liver samples and primary human hepatocytes. This has now been observed in two separate studies (ref. 7 and the current study) despite the investigators generating HLCs with two distinct differentiation protocols, with the earlier study using a protocol in which the HLCs were sorted by expression of ASGR1 (hepatocyte-specific marker) in an attempt to obtain more mature cells.7 We speculate that HLCs lack a factor or factors present in primary human hepatocytes and liver needed to fully reproduce the SORT1 eQTL. It should be noted that HLCs have been observed to faithfully model eQTLs with other lipid-associated genes, e.g., the hepatocyte-specific gene ANGPTL3.8 This suggests that HLCs can be useful for establishing causal SNP-gene relationships for some loci but not for others, and their experimental application to the study of genetic variation should be carefully considered on a case-by-case basis.
Primary human hepatocytes are generally regarded as the gold standard, but they are difficult to procure and for that reason are expensive, which makes performing studies with large numbers of samples prohibitive. Furthermore, plated hepatocytes are difficult to maintain in culture, typically surviving for only a few days,12 which militates against prolonged expression of CRISPR-Cas9 to achieve efficient genome editing. Furthermore, gene delivery into primary human hepatocytes is challenging; this prompted us to deliver CRISPR-Cas9 with adenovirus, which is uniquely efficient and can infect 100% of human hepatocytes in culture.14,15 Adenovirus infection can mildly alter some of the properties of cultured hepatocytes,16 although in our experiments we accounted for this with the use of a control adenovirus. However, we found that even with adenovirus we achieved a low degree of genome editing in the hepatocytes, which raises the question of their general suitability for the study of SNP variants with modest effects on gene expression and other phenotypes.
Although our experiments with primary human hepatocytes provided clear evidence that the rs12740374 minor allele sequence enhances SORT1 expression, the limitations of the approach argued for a complementary strategy to assess the function of noncoding regulatory regions in hepatocytes. The downsides of studying hepatocytes or HLCs in culture make studying authentic hepatocytes in vivo in an animal model attractive. In the case of rs12740374 (as is the case with most of the noncoding variants identified in genome-wide association studies), the lack of conservation in the mouse genome required us to introduce the human SORT1 locus via BAC transgenesis. One disadvantage of this approach is that the BAC incorporates in an uncontrolled manner into the genome, i.e., a random insertion site. Although the large size of the BAC guards against position effects, i.e., the influence of surrounding DNA elements and chromatin structure on gene expression from within the transgene,17 it does not eliminate the possibility.18 Ideally, one would wish to cleanly replace the orthologous mouse locus with the human locus, so that the human locus lies in the appropriate genomic context, but this remains a considerable technical challenge.19
Another issue with studying a human locus in the mouse genome is the assumption that the transcriptional regulatory machinery is conserved between human and mouse (i.e., transcription factors have the same sequence specificity, have the same binding partners, etc.). While this may be a reasonable assumption a priori, there is little existing information that bears on the validity of this assumption. Indeed, our finding that the rs12740374 minor allele sequence transcriptionally enhances hepatic SORT1 expression in the locus-humanized mouse provides some of the first information suggesting conservation between human and mouse in this respect.
Another strategy to study human loci in hepatocytes would be to generate liver-humanized mice in which endogenous mouse hepatocytes have been replaced with transplanted primary human hepatocytes.20 Somatic in vivo genome editing of the human hepatocytes in these mice has been demonstrated.11 The major disadvantage of this strategy is that it requires a large supply of primary human hepatocytes of a defined genotype, which might be difficult to procure. In the case of rs12740374-SORT1, homozygous minor hepatocytes would be optimal for use in liver-humanized mice; CRISPR-SNP adenovirus could be used to disrupt the endogenous minor allele sequences with the expectation of reducing SORT1 expression in the human hepatocytes in vivo. Because we were unable to identify any lot of hepatocytes from a homozygous minor individual, we did not pursue this strategy.
Finally, it is worth noting that while clean knock-in of the major allele to replace the minor allele of rs12740374 in either primary human hepatocytes or the livers of locus-humanized mice would have been preferable to disruption of the rs12740374 minor allele sequence, the efficiency of such knock-in (which relies on homology-directed repair) is far lower than the efficiency of disruption with indels (which relies on non-homologous end-joining). As a practical matter, only disruption is feasible in the context of ex vivo targeting of primary cells or somatic in vivo targeting, given the current state of genome-editing technology and in the absence of a method to purify or select for the correct knock-in cells.
With large collaborative efforts such as genome-wide association studies, the GTEx project, and the Encyclopedia Of DNA Elements (ENCODE) Project pinpointing numerous candidate noncoding regulatory regions, novel approaches are needed to interrogate whether these regions are causal for gene expression effects and other phenotypes. Here we used CRISPR-Cas9 genome editing in primary human hepatocytes ex vivo and in locus-humanized mice in vivo to address whether a putative regulatory region in the human SORT1 locus is truly causal for liver-specific modulation of SORT1 expression. Experiments in both models confirmed that the rs12740374 minor allele sequence enhances SORT1 expression in hepatocytes. When employed judiciously, such models could be broadly useful to assess loci associated with a wide variety of cardiovascular and metabolic traits and diseases.
Supplementary Material
HIGHLIGHTS.
Primary human hepatocytes exhibited a strong expression quantitative trait locus (eQTL) for rs12740374 and SORT1
CRISPR-Cas9 targeting of the rs12740374 minor allele sequence in primary human hepatocytes reduced SORT1 expression
In locus-humanized mice with the human SORT1 locus incorporated into the mouse genome, CRISPR-Cas9 targeting of the rs12740374 minor allele sequence reduced hepatic SORT1 expression
Induced pluripotent stem cell-derived hepatocyte-like cells failed to faithfully model the SORT1 eQTL, highlighting a potential limitation of these cells for human genetic studies
Acknowledgments
None.
SOURCES OF FUNDING
This work was supported by an American Heart Association Postdoctoral Fellowship (X.W.); the Howard Hughes Medical Institute Medical Research Fellows Program (A.R.); grant U01-HG006398 from the United States National Institutes of Health (NIH) (D.J.R.); grants R01-HL118744, R01-GM104464, R01-DK099571, and R01-HL126875 from the NIH (K.M.); and funds from Harvard University and from the University of Pennsylvania.
ABBREVIATIONS
- BAC
bacterial artificial chromosome
- CHD
coronary heart disease
- CRISPR-Cas9
clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated 9
- eQTL
expression quantitative trait locus
- GTEx
Gene-Tissue Expression
- HLC
hepatocyte-like cell
- iPSC
induced pluripotent stem cell
- LDL-C
low-density lipoprotein cholesterol
- SNP
single nucleotide polymorphism
- qRT-PCR
quantitative reverse-transcriptase polymerase chain reaction
Footnotes
DISCLOSURES
E.E.P. is now an employee of Pfizer.
References
- 1.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Musunuru K, Strong A, Frank-Kamenetsky M, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kjolby M, Andersen OM, Breiderhoff T, Fjorback AW, Pedersen KM, Madsen P, Jansen P, Heeren J, Willnow TE, Nykjaer A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010;12:213–223. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Strong A, Ding Q, Edmondson AC, et al. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J Clin Invest. 2012;122:2807–2816. doi: 10.1172/JCI63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schadt EE, Molony C, Chudin E, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warren CR, O’Sullivan JF, Friesen M, et al. Induced pluripotent stem cell differentiation enables functional validation of GWAS variants in metabolic disease. Cell Stem Cell. 2017;20:547–557. doi: 10.1016/j.stem.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 8.Pashos EE, Park Y, Wang X, et al. Large, diverse population cohorts of hiPSCs and derived hepatocyte-like cells reveal functional genetic variation at blood lipid-associated loci. Cell Stem Cell. 2017;20:558–570. doi: 10.1016/j.stem.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y, Wang L, Bell P, McMenamin D, He Z, White J, Yu H, Xu C, Morizono H, Musunuru K, Batshaw ML, Wilson JM. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, Cowan CA, Rader DJ, Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Raghavan A, Chen T, Qiao L, Zhang Y, Ding Q, Musunuru K. CRISPR-Cas9 targeting of PCSK9 in human hepatocytes in vivo. Arterioscler Thromb Vasc Biol. 2016;36:783–786. doi: 10.1161/ATVBAHA.116.307227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guguen-Guillouzo C, Guillouzo A. General review on in vitro hepatocyte models and their applications. Methods Mol Biol. 2010;640:1–40. doi: 10.1007/978-1-60761-688-7_1. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz RE, Fleming HE, Khetani SR, Bhatia SN. Pluripotent stem cell-derived hepatocyte-like cells. Biotechnol Adv. 2014;32:504–513. doi: 10.1016/j.biotechadv.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozarsky K, Grossman M, Wilson JM. Adenovirus-mediated correction of the genetic defect in hepatocytes from patients with familial hypercholesterolemia. Somat Cell Mol Genet. 1993;19:449–458. doi: 10.1007/BF01233250. [DOI] [PubMed] [Google Scholar]
- 15.Morsy MA, Alford EL, Bett A, Graham FL, Caskey CT. Efficient adenoviral-mediated ornithine transcarbamylase expression in deficient mouse and human hepatocytes. J Clin Invest. 1993;92:1580–1586. doi: 10.1172/JCI116739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castell JV, Hernández D, Gómez-Foix AM, Guillén I, Donato T, Gómez-Lechón MJ. Adenovirus-mediated gene transfer into human hepatocytes: analysis of the biochemical functionality of transduced cells. Gene Ther. 1997;4:455–464. doi: 10.1038/sj.gt.3300416. [DOI] [PubMed] [Google Scholar]
- 17.Giraldo P, Montoliu L. Size matters: use of YACs, BACs and PACs in transgenic animals. Transgenic Res. 2001;10:83–103. doi: 10.1023/a:1008918913249. [DOI] [PubMed] [Google Scholar]
- 18.Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 19.Lee EC, Liang Q, Ali H, et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat Biotechnol. 2014;32:356–363. doi: 10.1038/nbt.2825. [DOI] [PubMed] [Google Scholar]
- 20.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, Strom S, Kay MA, Finegold M, Grompe M. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat Biotechnol. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.