

Abstract
Sequencing complete genomes of all major phylogenetic groups of organisms opens unprecedented opportunities to study evolution and genetics. We report draft genomes of Calephelis nemesis and Calephelis virginiensis, representatives of the family Riodinidae. They complete the genomic coverage of butterflies at the family level. At 809 and 855 Mbp, respectively, they become the largest available Lepidoptera genomes. Comparison of butterfly genomes shows that the divergence between Riodinidae and Lycaenidae dates to the time when other families started to diverge into subfamilies. Thus, Riodinidae may be considered a subfamily of Lycaenidae. Calephelis species exhibit unique gene expansions in actin-disassembling factor, cofilin, and chitinase. The functional implications of these gene expansions are not clear, but they may aid molting of caterpillars covered in extensive setae. The two Calephelis species diverged about 5 million years ago and they differ in proteins involved in metabolism, circadian clock, regulation of development, and immune responses.
Keywords: phylogeny, molecular dating, next-generation sequencing, Lepidoptera, Calephelis
INTRODUCTION
Unprecedented advances in sequencing recast the ways biological questions are posed. Determination of complete genomes and comparative analyses will guide our understanding of genotypic determinants of phenotypic traits. Butterflies are well-suited for such work because they are diverse in colors, patterns and shapes, their genomes are of moderate size and they are closely related to the model organism: Drosophila. Genomes have been reported for five (out of six) butterfly families: the swallowtails (Papilionidae) (Cong et al., 2015a; Li et al., 2015; Mallet, 2015), the Whites and Sulphurs (Pieridae) (Cong et al., 2016a), the Blues (Lycaenidae) (Cong et al., 2016b), the Brushfoots (Nymphalidae) (Ahola et al., 2014; Heliconius Genome, 2012; Zhan et al., 2011), and the Skippers (Hesperiidae) (Cong et al., 2015b). The Brushfoots include Heliconius and the Monarch (Danaus plexippus), which are the most thoroughly studied (Nadeau et al., 2014; Zhan et al., 2014). For meaningful genomic comparisons, it is essential to obtain reference genomes of all butterfly families.
The only remaining butterfly family without a reference genome, the Metalmarks (Riodinidae) includes about 1500 species, mostly in the Neotropics (Espeland et al., 2015; Zhang, 2013). Traditionally placed phylogenetically closest to the Blues (Lycaenidae) (De Jong et al., 1996), Metalmarks are rather small butterflies known for exceptional diversity of wing shapes, colors, and patterns (DeVries, 1997; Espeland et al., 2015). While most possess typical roundish wings, some have wings that are irregular, angular and even with long tails as in Papilionidae. Many Metalmarks are patterned with metallic spots, hence the name. Taxonomically, Metalmarks are sometimes treated as a subfamily of Blues (Lycaenidae) (Ackery et al., 1999; Ehrlich, 1958; Scott, 1986), although most recent studies, including those based on DNA work, assigned family ranks to Metalmarks (Espeland et al., 2015; Robbins, 1988; Wahlberg et al., 2005; Zhang, 2013). Due to all these reasons, Metalmarks are interesting targets for comparative genomics. Genomic studies on Metalmarks may shed light on wing shape and pattern determination by the genotype and may resolve the question about their best taxonomic rank.
For genomic studies, we have selected a genus Calephelis. Called “Scintillants”, these small reddish-brown above and mostly yellow below butterflies are marked with several rows of metallic spots (Glassberg, 2007; Hall and Harveyb, 2002; McAlpine, 1971). Genus Calephelis is distributed throughout the Americas and is mostly Neotropical, but several species reach into the US, with three being eastern USA endemics (C. virginiensis, C. borealis and C. muticum) (McAlpine, 1971; Scott, 1986). At least one of them, C. muticum, is considered as a conservation concern (Bess, 2005). Overall, the genus is rich in species and is poorly understood. Most of about 50 known Calephelis species are cryptic and can be distinguished only upon inspection of genitalia (McAlpine, 1971), and the validity of some of these species is in question (Hall and Harveyb, 2002). We hope that the complete genome reference will catalyze studies of this interesting genus and further its understanding at all levels.
To launch Riodinidae genomics, we sequenced and annotated the complete genomes of Fatal Metalmark (C. nemesis) and Little Metalmark (C. virginiensis). At 809 and 855 Mbp, respectively, they are the largest among available Lepidoptera genomes. Genomic data confirm that Riodinidae and Lycaenidae are more closely related to each other than any other pairs of families and timing of their split suggests that they may better be treated as subfamilies. Calephelis (Riodinidae) and Calycopis (Lycaenidae) genomes encode more copies of Cytochrome P450 and Glutathione S-transferases, which might be related to higher tolerance of their caterpillars to rotting food. The two Calephelis species we sequenced are rather distantly related, and they differ in proteins involved in metabolism, circadian rhythm, and regulation of development.
RESULTS AND DISCUSSION
Genome assembly, annotation, and comparison to other Lepidoptera genomes
We assembled 809 and 855 Mbp reference genomes of Calephelis nemesis (Cne) and Calephelis virginiensis (Cvi), respectively (Fig. 1), the largest among currently sequenced Lepidoptera genomes (Ahola et al., 2014; Cong et al., 2015a; Cong et al., 2016b; Duan et al., 2010; Heliconius Genome, 2012; International Silkworm Genome, 2008; Tang et al., 2014; You et al., 2013; Zhan et al., 2011; Zhan and Reppert, 2013). Representing the family Riodinidae, these genomes complete the genomic coverage of butterflies at the family level. The scaffold N50 of Cne and Cvi genome assemblies are 206 kb and 175 kb, respectively, similar to many other published Lepidoptera genomes. The genome assembly is comparable to other published Lepidoptera genomes in terms of completeness measured by the presence of Core Eukaryotic Genes Mapping Approach (CEGMA) genes (supplemental Table S1B) (Parra et al., 2007), cytoplasmic ribosomal proteins and independently assembled transcripts (Table 1). The genome sequences have been deposited at DDBJ/EMBL/GenBank under the accessions NJDD00000000 and NJDC00000000. The versions described in this paper are versions NJDD01000000 and NJDC01000000. In addition, the main results from genome assembly, annotation and analysis can be downloaded at http://prodata.swmed.edu/LepDB/.
Figure 1. Specimens of Calephelis.

a-f C. nemesis, USA: Texas, Hidalgo Co., Penitas, GPS 26.22615, -98.43653: a, b male, left wings of voucher NVG-3574, 13-Jun-2015; others are reared from eggs: c, d male, eclosed 9-Feb-2005; e, f female, eclosed 19-Feb-2005. g-l C. virginiensis, USA: Texas, Hardin Co., along FM770 4.4 mi southwest of Kountze, GPS 30.33832, -94.37046: g, h male, left wings of voucher NVG-3639, 7-Jun-2015; i, j male 7-Jun-2015; k, l female, reared from caterpillar, eclosed on 24-Jun-2015. Dorsal (a, c, e, g, i, k) and ventral (b, d, f, h, j, l) views of each specimen are shown.
Table 1.
Quality and composition of Lepidoptera genomes.
| Feature | Cne | Cvi | Cce | Pra | Pse | Aly | Lac | Pgl | Ppo | Pxu | Dpl | Hme | Mci | Bmo | Mse | Pxy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genome size (Mb) | 809 | 855 | 729 | 246 | 406 | 567 | 298 | 375 | 227 | 244 | 249 | 274 | 390 | 481 | 419 | 394 |
| Genome size w/o gap (Mb) | 783 | 824 | 689 | 243 | 347 | 536 | 290 | 361 | 218 | 238 | 242 | 270 | 361 | 432 | 400 | 387 |
| Heterozygosity (%) | 0.5 | 1.3 | 1.2 | 1.5 | 1.2 | 1.5 | 1.5 | 2.3 | n.a. | n.a. | 0.55 | n.a. | n.a. | n.a. | n.a. | ~2 |
| Scaffold N50 (kb) | 206 | 175 | 233 | 617 | 257 | 558 | 525 | 231 | 3672 | 6199 | 716 | 194 | 119 | 3999 | 664 | 734 |
| CEGMA completeness1 (%) | 99.6 | 99.6 | 99.3 | 99.6 | 99.3 | 99.6 | 99.6 | 99.6 | 99.3 | 99.6 | 99.6 | 98.9 | 98.9 | 99.6 | 99.8 | 98.7 |
| CEGMA continuity2 (%) | 85.8 | 84.8 | 84.6 | 88.7 | 87.4 | 87.1 | 86.6 | 86.9 | 85.8 | 88.8 | 87.4 | 86.5 | 79.2 | 86.8 | 86.4 | 84.1 |
| Ribosomal Proteins (%) | 98.9 | 98.9 | 98.9 | 98.9 | 98.9 | 98.9 | 98.9 | 98.9 | 98.9 | 97.8 | 98.9 | 94.6 | 94.6 | 98.9 | 98.9 | 93.5 |
| De novo transcripts (%) | 99 | 99 | 97 | 99 | 97 | 98 | 98 | 98 | n.a. | n.a. | 96 | n.a. | 97 | 98 | n.a. | 83 |
| GC content (%) | 34.9 | 35.0 | 37.1 | 32.7 | 39.0 | 35.3 | 34.4 | 35.4 | 34.0 | 33.8 | 31.6 | 32.8 | 32.6 | 37.7 | 35.3 | 38.3 |
| Repeat (%) | 34.8 | 38.8 | 34.1 | 22.7 | 17.2 | 25.0 | 15.5 | 22.0 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| Exon (%) | 2.25 | 2.17 | 2.59 | 7.91 | 6.20 | 3.57 | 6.96 | 5.07 | 5.11 | 8.59 | 8.40 | 6.38 | 6.36 | 4.03 | 5.34 | 6.35 |
| Intron (%) | 19.6 | 20.5 | 20.1 | 33.3 | 25.5 | 28.4 | 31.6 | 25.6 | 24.8 | 45.5 | 28.1 | 25.4 | 30.7 | 15.9 | 38.3 | 30.7 |
| Number of proteins (k) | 15.4 | 15.6 | 14.9 | 13.2 | 16.5 | 15.9 | 17.4 | 15.7 | 15.7 | 13.1 | 15.1 | 12.8 | 16.7 | 14.3 | 15.6 | 18.1 |
n.a. data not available; Cce: Calycopis cecrops; Aly: Achalarus lyciades; Pra: Pieris rapae; Lac: Lerema accius; Pgl: Pterourus glaucus; Dpl: Danaus plexippus; Hme: Heliconius melpomene; Mci: Melitaea cinxia; Bmo: Bombyx mori; Pxy: Plutella xylostella; Mse: Manduca sexta; Ppo: Princeps polytes; Pse: Phoebis sennae; Pxu: Papilio xuthus.
Heterozygosity: Calculated as the percent of heterozygous positions detected by the Genome Analysis Toolkit (GATK)(McKenna et al., 2010) for Pgl, Lac, Cce, Pra and Pse; or taken from information in the literature for Dpl(Zhan et al., 2011); or estimated based on the histogram of K-mer frequencies for Pxy(Kajitani et al., 2014; You et al., 2013).
We also assembled the transcriptomes of Cne and Cvi. Based on the transcriptomes, homologs from other Lepidoptera and Drosophila melanogaster, de novo gene predictions, and repeat identification (supplemental Table S2A,B), we predicted 15430 and 15587 protein-coding genes and annotated the putative functions of 12006 and 11710 proteins encoded in Cne and Cvi genomes, respectively (supplemental Table S2C,D). Although the genome size of Cne is larger than that for other Lepidoptera genomes, the number of proteins encoded by the genome is comparable to others. The large difference in genome size is likely related to the different amount of transposon-like repetitive DNA in the genomes (Neafsey and Palumbi, 2003). The difference between (46 Gbp) the genome sizes of Cne and Cvi can be explained by the different total length (50 Gbp) of the repetitive regions in them. The fraction of repetitive regions in the butterfly genomes that we sequenced and annotated is listed in Table 1, and this faction is positively correlated (Pearson correlation coefficient: 0.92) with the genome size. This fraction only includes repetitive regions that are no more than 30% divergent from the consensus sequences, and many more ancient transposon-derived repetitive regions may exist in the genome.
Phylogeny of Lepidoptera
We identified orthologous proteins encoded by 16 Lepidoptera genomes (Plutella xylostella, Bombyx mori, Manduca sexta, Lerema accius, Acharlarus lyciades, Pterourus glaucus, Princeps polytes, Papilio xuthus, Phoebis sennae, Pieris rapae, Melitaea cinxia, Heliconius melpomene, Danaus plexippus, Calycopis cecrops, Calephelis nemesis, and Calephelis virginiensis) and detected 1,624 universal orthologous groups consisting of a single-copy gene in each species. A phylogenetic tree built from the concatenated alignment of the single-copy orthologs using RAxML places Calephelis as the sister to Calycopis (Fig. 2 left), the representative of the Lycaenidae family. The two families, Riodinidae and Lycaenidae, are known to be close to each other from morphological and DNA evidence (Alexander et al., 2017; Robbins, 1988; Wahlberg et al., 2005). Terminal branches leading to Calephelis and Calycopis are long compared to others, as well as the branch leading to the ancestor of Riodinidae and Lycaenidae. The relative length of these branches suggests an elevated evolutionary rate starting from the common ancestor of both families.
Figure 2. Phylogenetic trees of the Lepidoptera species with complete genome sequences.
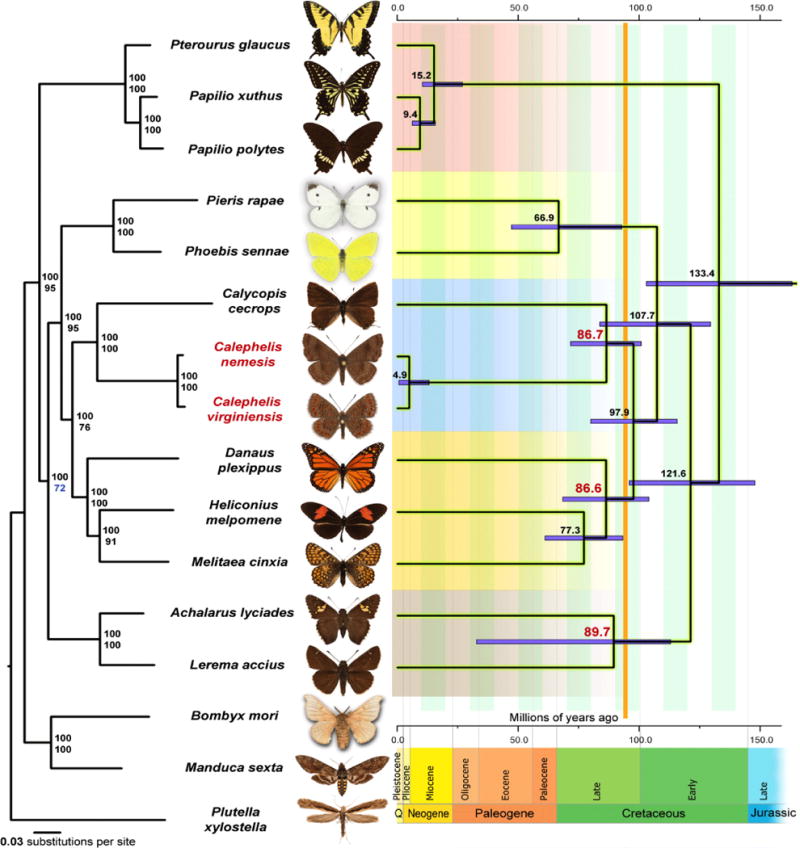
Majority-rule consensus tree of the maximal likelihood trees constructed by RAxML on the concatenated alignment of universal single-copy orthologous proteins is shown on the left. Numbers by the nodes refer to bootstrap percentages. The numbers above are obtained from complete alignments, the number below are obtained on 1% of the dataset. Dated phylogeny of butterflies is shown on the right and the estimated ages (in Myr) are shown by the nodes. Ages of nodes corresponding to subfamily diversification (among taxa with available genomes) of Nymphalidae and Hesperiidae and the split between Riodinidae and Lycaenidae are shown in red. Error bars are shown in violet. Geological time scale is placed at the bottom. Periods and Epochs are shown below and above, respectively. “Q” stands for Quaternary period. Vertical orange line at about 90 million years ago corresponds to the time when butterfly families have diverged, but before diversification of families into subfamilies, suggesting that Riodinidae may be considered a subfamily of Lycaenidae.
The Riodinidae and Lycaenidae families are closer to each other than any other pair of butterfly families. Molecular dating (Fig. 2 right) suggests that Riodinidae and Lycaenidae families diverged from each other around 87 million years ago (95% confidence interval: 72 Myr ~ 101 Myr), which is comparable to the time when the subfamilies within other families started to diverge: 87 Myr (95% confidence interval: 69 Myr ~ 104 Myr) for Nymphalidae and 90 Myr (95% confidence interval: 33 Myr ~ 113 Myr) for Hesperiidae. Taking into account the similarity in morphology, treating metalmarks (Riodinidae) as a subfamily within Lycaenidae might be appropriate, a view expressed in several publications (Scott, 1986; Zhao et al., 2013). The two sequenced species, Calephelis nemesis and Calephelis virginiensis, diverged from each other about 5 million years ago. A comprehensive analysis of Polyommatus Blues (Lycaenidae) suggests that different genera separated from each other 4-5 million years ago (Talavera et al., 2012). Therefore, the two Calephelis species are rather distantly related and may even be classified into different genera.
Blues and Metalmarks: comparison with Calycopis
Compared to other Lepidoptera, Calycopis (Lycaenidae) and Calephelis (Riodinidae) genomes encode almost twice as many copies of (CP450) and Glutathione S-transferases (GST). Other Lepidoptera genomes typically encode around 100 (76 to 117, supplemental Table S3A) copies of CP450, while Calephelis and Calycopis have 150 to 200 copies of them: 156 for Cvi, 192 for Cne and 200 for Calycopis cecrops. Similarly, the number of GSTs in the Calycopis or Calephelis genome is also higher than any other genomes (1.5-2 times of the average, supplemental Table S3A). GSTs are mostly known as detoxifying enzymes (Sheehan et al., 2001). CP450s perform many essential functions, from the synthesis and degradation of hormones to metabolizing foreign chemicals (Meunier et al., 2004), and thus additional CP450s possibly also contribute to the degradation of toxins from food and insecticides. It is possible that the additional GSTs and CP450s in Calycopis and Calephelis are related to their ability to feed on more toxic foods not palatable to other species: Calycopis species are known to be detritivores. Although Calephelis do not feed on detritus, they survive well on old and even partly rotten leaves (Kendall, 1959) and the sister genus of Calephelis, Detritivora, is a detritus feeder (Espeland et al., 2015; Hall and Harvey, 2002).
Several gene expansions seem to be unique to either Calycopis or Calephelis. For instance, only Calycopis shows expansion in salivary secreted peptides and galactosyltransferases (Fig. 3a,b and supplemental table S3B,C). The functional relevance of these expansions remains to be studied, and we hypothesize that the additional salivary secreted peptidases may protect the caterpillars against the bacteria, fungi and their toxins taken in with the food (Francischetti et al., 2007). In contrast, expansion in genes encoding chitinase and the actin disassembling factor, cofilin (Fig. 3c,d and supplemental table S3D,E) are only observed in Calephelis and not in Calycopis. “Hairy” caterpillars are characteristic of many metalmarks, especially Calephelis (McAlpine, 1971). In Drosophila, the hairs on the body are extensions of cells, shaped by actin filament bundles (Guild et al., 2005; Tilney et al., 2000), and covered by chitin on the surface (Nagaraj and Adler, 2012). It is likely that the setae on the surface of Calephelis caterpillar have similar structure. We speculate that the additional chitinases and cofilins in Calephelis may support efficient disassembly of actin filaments and digestion of chitin and are possible requirements for proper molting of the larva that is covered by extensive setae.
Figure 3. Protein families that underwent gene expansion in Calycopis or Calephelis.
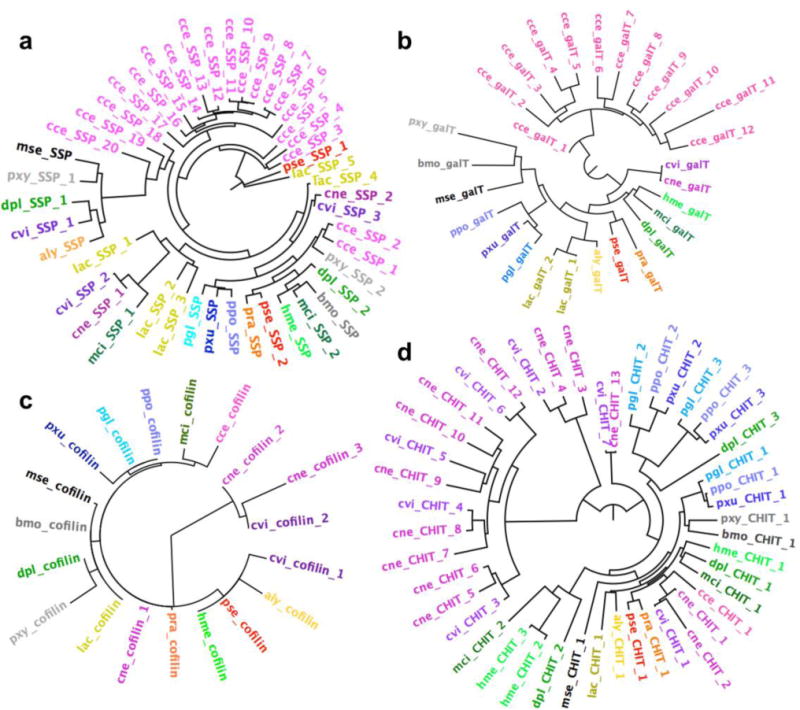
Phylogenetic trees depict (a) putative salivary secreted peptides, (b) galactosyltransferases, (c) actin disassembling factor, cofilin, and (d) chitinases encoded by Lepidoptera genomes. Abbreviation of the species and protein names are used as labels (colored by the species) in the phylogenetic trees.
Molecular mechanisms behind the divergence between Calephelis nemesis and C. virginiensis
While most Calephelis species are of southern and southwestern US origin, three species are exclusively eastern US in distribution (Scott, 1986). Calephelis nemesis and C. virginiensis are the representatives of the southern species and eastern species, respectively. In addition to those used for reference genomes (NVG-3574 and NVG-3639), we sequenced 2 specimens of C. nemesis (NVG-3585 and NVG-3847) and 1 of C. virginiensis (NVG-3505) at about 10-fold coverage (supplemental Table S4A), and mapped the reads of all specimens to both Cne and Cvi reference genomes. Over 90% (supplemental Table S4A) of the genomes can be obtained by mapping the reads of a specimen to the reference from the same species. The southern species, C. nemesis shows much lower heterozygosity (0.5% – 0.6%) than the eastern species, C. virginiensis (heterozygosity: 1.2% – 1.3%). This finding contradicts the trend we observed before in several pairs of sister species, where the southern species living in warmer climates and having larger population size, shows higher heterozygosity (Cong et al., 2016b; Cong et al., 2016c). The heterozygosity and divergence between specimens in different localities for C. nemesis is low compared to other wild-caught species (Table 1), and the reason behind this low genetic diversity remains to be studied, but may be related to some population bottlenecks. It may also be connected to inbreeding in C. nemesis populations caused by the sedentary habits of adults: they do not fly far from the place of their emergence.
It is frequently feasible to obtain a rather complete genome of a specimen using the reference genome from the same genus (Cong et al., 2016a; Cong et al., 2016b). However, due to the high divergence between the two Calephelis species, only about 30%–50% of the genomic regions allows for confident mapping the reads of one species to the genome of another. In contrast, the coding regions in these genomes are less divergent, and therefore we can obtain the alignment between C. virginiensis and C. nemesis specimens for 90% (89.5% for NVG-3505 and 94.2% for NVG-3639 with higher coverage) of the coding positions by mapping the reads of Cvi specimens to the Cne reference genome (Supplemental Table S4A). Mapping the reads of Cne to the Cvi genome behaves similarly: while it is difficult to obtain complete genomes of Cne specimens by mapping to the Cvi reference genome, the coding sequences can be mostly (over 90%) derived.
Requiring at least 90 aligned positions from both species, we obtained the alignments of 14885 protein-coding genes using the Cne reference genome, which were used in the following analyses. Similar analyses were performed using the Cvi reference genome, and the results were consistent regardless of which reference genome was used. The overall divergence in coding gene between the two species is about 3.7%–4.5%, resulting in 3.3%–3.6% different positions in the protein sequences. The divergent positions between the two species are distributed unevenly among proteins, with 1862 proteins significantly enriched in (P < 0.01) in such positions. We further selected proteins that are relatively conserved within species (the ratio between intraspecific and interspecific divergence is lower than 0.1). A total of 475 proteins passed the two criteria, and we term them “interspecific divergence hotspots”. They possibly include genes that are important for the speciation, divergence and adaptation of the two species. (supplemental Table 4B).
The biological processes that are performed by these interspecific divergence hotspots in are shown in Fig. 4 (P < 0.01. supplemental Table S4C lists the GO terms in all categories). These GO terms suggest that the two species show differences in lipid metabolism, circadian clock system, muscle development, assembly of cell projection and androgen receptor signaling pathway. Divergence in circadian clock protein has been repeatedly found in several pairs of sister species that are distributed in different latitudes (Cong et al., 2015a; Cong et al., 2016b). Adaptation to different latitudes and climates may lead to divergence in circadian clock proteins, which possibly contribute to the hybrid incompatibility. Androgen receptor signaling pathway regulates the development and maintenance of the male sexual phenotype, and thus differences in this pathway possibly underplay the obvious difference in male sex organs of the two species. Interestingly, the transcriptional regulator for male sex organ development, spalt-related protein (SALL), is one of the most rapidly evolving group (top 20) among universal single-copy orthologs in Lepidoptera. SALL is also the only protein that is related to male genitalia development among the universal single-copy orthologs. We speculate that the rapid divergence of SALL is one possible reason for the observed high divergence in the shape of male sex organ among Lepidoptera species.
Figure 4. Enriched (P < 0.01) GO terms associated with interspecific divergence hotspots for the two Calephelis species.
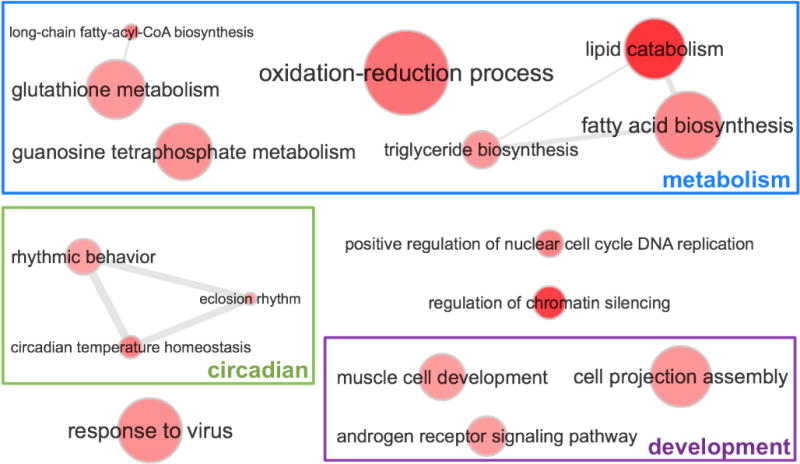
The darkness of the color indicates the significance level (P-value) of the enrichment, with darker color corresponding to lower P-value. The size of the dots correlates to the number of proteins that are associated with this GO term encoded in the Drosophila melanogaster genome. The GO terms that are frequently associated with the same proteins are linked by the grey lines, and we also manually grouped the GO terms with similar biological meanings.
METHODS
Library preparation and sequencing
We removed and preserved the wings and genitalia of freshly caught Calephelis specimens, and the rest of the bodies were stored in RNAlater solution. Wings and genitalia of these specimens will be deposited in the National Museum of Natural History, Smithsonian Institution, Washington, DC, USA (USNM). The information about these specimens is listed in supplemental Table S4A. We mainly used specimens NVG-3574 and NVG-3639 to assemble the reference genomes for C. nemesis and C. virginiensis, respectively. The paired-end libraries used to construct the contigs of the assemblies were made exclusively from these specimens, while mate pair libraries were prepared using a mixture of genomic DNA from multiple specimens. Specimens NVG-3574 and NVG-4212 were used for RNAseq libraries for C. nemesis and C. virginiensis, respectively.
We extracted genomic DNA from them with the ChargeSwitch gDNA mini tissue kit. 250 and 500 bp paired-end libraries were prepared using NEBNext Modules and following the Illumina TruSeq DNA sample preparation guide. 2 kb, 6 kb and 15 kb mate pair libraries were prepared using a protocol similar to previously published Cre-Lox-based method (Van Nieuwerburgh et al., 2012). We extracted RNA from specimens using QIAGEN RNeasy Mini Kit, isolated mRNA using NEBNext Poly(A) mRNA Magnetic Isolation Module, and prepared RNA-seq libraries with NEBNext Ultra Directional RNA Library Prep Kit for Illumina following manufactory’s protocol.
Genome and transcriptome assembly
Mate pair libraries were processed by the Delox script (Van Nieuwerburgh et al., 2012) to remove the loxP sequences and to separate true mate pair from paired-end reads. All reads were processed by mirabait (Chevreux et al., 1999) to remove contamination from the TruSeq adapters, an in-house script to remove low quality portions (quality score < 20) at the ends of both reads, JELLYFISH (Marcais and Kingsford, 2011) to obtain k-mer frequencies in all the libraries, and QUAKE (Kelley et al., 2010) to correct sequencing errors. The data processing resulted in seven libraries that were supplied to Platanus (Kajitani et al., 2014) for genome assembly: 250 bp and 500 bp paired-end libraries, 2 kbp, 6kbp, 15k bp true mate pair libraries, a library containing all the paired-end reads from the mate pair libraries, and a single-end library containing all reads whose pairs were removed in the process (supplemental Table S1A).
We mapped these reads to the initial assembly with Bowtie2 (Langmead and Salzberg, 2012) and calculated the coverage of each scaffold with the help of SAMtools (Janzen et al., 2009). Many short scaffolds in the assembly showed coverage that was about half of the expected value; they likely came from highly heterozygous regions that were not merged to the equivalent segments in the homologous chromosomes. We removed them if they could be fully aligned to another significantly less covered region (coverage > 90% and uncovered region < 500 bp) in a longer scaffold with high sequence identity (>95%) in the longer scaffolds. Similar problems occurred in the Heliconius melpomene, Pterourus glaucus and Lerema accius genome projects, and similar strategies were used to improve the assemblies (Cong et al., 2015a; Cong et al., 2015b; Heliconius Genome, 2012). In case that the genomes contain contaminating fragments from bacteria, fungi and plants, we identified scaffolds that are more similar to sequences from bacteria, fungi and plants than those from other insects by BLAST search against all sequences in the nt database of NCBI. We visualized the results with the help of blobtools (Kumar et al., 2013), and manually curated the results to select the ones that are more likely to be contaminants. In addition to the BLAST results, we considered coverage of the scaffolds and whether the scaffolds were covered by sequences from multiple specimens to make the judgments.
The RNA-seq reads were processed using a similar procedure as the genomic DNA reads. We applied three methods to assemble the transcriptomes: (1) de novo assembly by Trinity (Haas et al., 2013), (2) reference-based assembly by TopHat (Kim et al., 2013) (v2.0.10) and Cufflinks (Roberts et al., 2011) (v2.2.1), and (3) reference-guided assembly by Trinity. The results from all three methods were then integrated by Program to Assemble Spliced Alignment (PASA) (Haas et al., 2008).
Identification of repeats and gene annotation
Two approaches were used to identify repeats in the genome: the RepeatModeler (Smit and Hubley, 2008–2010) pipeline and in-house scripts that extracted regions with coverage 3 times higher than expected. These repeats were submitted to the CENSOR (Jurka et al., 1996) server to assign them to the repeat classification hierarchy. The species-specific repeat library and all repeats classified in RepBase (Jurka et al., 2005) (V18.12) were used to mask repeats in the genome by RepeatMasker (Smit et al., 1996–2010).
We obtained two sets of transcript-based annotations from two pipelines: TopHat followed by Cufflinks and Trinity followed by PASA. In addition, we obtained eight sets of homology-based annotations by aligning protein sets from Drosophila melanogaster (Misra et al., 2002) and seven published Lepidoptera genomes (Bombyx mori, Lerema accius, Princeps polytes, Papilio glaucus, Papilio xuthus, Heliconius melpomene, and Danaus plexippus) to the Calephelis genomes with exonerate (Slater and Birney, 2005). To annotate the proteins in Cvi, the annotated proteins from Cne are also used for homology-based annotation by exonerate. Proteins from insects in the entire UniRef90 (Suzek et al., 2007) database were used to generate another set of gene predictions by genblastG (She et al., 2011). We manually curated and selected 1030 and 1122 confident gene models for Cne and Cvi, respectively, by integrating the evidence from transcripts and homologs to train de novo gene predictors: AUGUSTUS (Stanke et al., 2006), SNAP (Korf, 2004) and GlimmerHMM (Majoros et al., 2004). These trained predictors, the self-trained Genemark (Besemer and Borodovsky, 2005) and a consensus-based pipeline Maker (Cantarel et al., 2008), were used to generate another five sets of gene models. Transcript-based and homology-based annotations were supplied to AUGUSTUS, SNAP and Maker to boost their performance. We generated 15 and 16 sets of gene predictions in total for the Cne and Cvi genomes, respectively, and integrated them with EvidenceModeller (Haas et al., 2008) to generate the final gene models.
We predicted the function of proteins by transferring annotations and GO-terms from the closest BLAST (Altschul et al., 1990) hits (E-value < 10−5) in both the Swissprot (UniProt, 2014) database and Flybase (St Pierre et al., 2014). Finally, we performed InterproScan (Jones et al., 2014) to identify conserved protein domains and functional motifs, to predict coiled coils, transmembrane helices and signal peptides, to detect 3D structure templates, to assign proteins to protein families and to map them to metabolic pathways.
Identification of orthologous proteins and gene expansion
We identified the orthologous groups from 16 Lepidoptera genomes using OrthoMCL (Li et al., 2003). If two OrthoMCL-defined orthologous groups overlapped in the Drosophila proteins that they mapped to, we merged them into one family. The total number and total length of proteins in a family were used to identify expanded gene families in Calephelis and Calycopis. If the total number and length of proteins from Calephelis or Calycopis in a family were more than 1.5 times of the average number and length across other Lepidoptera species, we considered this protein family to have undergone expansion in Calephelis or Calycopis. The most interesting gene expansions discussed in the paper were further investigated to include all relevant proteins using reciprocal BLAST results and function annotations. Proteins encoded by the genome but missed in the protein sets were predicted with the help of genblastG. Protein sequences from each family were aligned with MAFFT (Katoh and Standley, 2013). Evolutionary trees were built with RAxML (Stamatakis, 2014) (-m PROTGAMMAAUTO) and visualized in FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
For some large and diverse protein families, such as Cytochrome P450 and Glutathione S-transferases, in order to efficiently identify all the members in all the Lepidoptera genomes, we used HMMER (Eddy, 1998) to scan the pfam database (Finn et al., 2016) using every protein in each genome as a query. Proteins that identify Cytochrome P450 (PF00067) or Glutathione S-transferases (PF00043, PF02798, PF13409, PF13410, PF13417, PF14497, PF14834, PF16865, PF17171, or PF17172) as the first confident hits were considered belonging to that family. The number of proteins belonging to these families were counted and compared among species.
Phylogeny of Lepidoptera and molecular dating
1624 orthologous groups consisting of single-copy genes from every species were used for phylogenetic analysis. An alignment was built for each universal single-copy orthologous group using both global sequence aligner MAFFT and local sequence aligner BLASTP. Positions that were consistently aligned by both aligners were extracted from each individual alignment and concatenated to obtain an alignment containing 252,520 positions. The concatenated alignment was used to obtain a phylogenetic tree using RAxML (parameter: -m PROTGAMMAAUTO, which allowed the program to select the most suitable model based on the data, and JTTDCMUT was selected). Bootstrap resampling of the aligned positions was performed to assign the confidence level of each node. All the nodes received 100% bootstrap support if all the data were used. In addition, we evaluated the evolutionary rate of each orthologous group based on the average pair-wise protein sequence identity. We partitioned the 1624 orthologous protein sets into three groups with different evolutionary rate, and obtained evolutionary trees for each group, respectively. The trees showed different absolute branch length, but maintained similar relative branch length and the same topology. Finally, in order to detect the weakest nodes in the tree, we reduced the amount of data by randomly splitting the concatenated alignment into 50 alignments and applied RAxML to each alignment. We obtained a 50% majority rule consensus tree and assigned confidence level to each node based on the percent of individual trees supporting it.
61 alignments of single-copy orthologous groups consisting of more than 500 aligned positions each were used to date the evolutionary history of butterflies. We selected the best substitution models and partitioning schemes using the software Partitionfinder (Lanfear et al., 2012). We applied BEAST v1.8.1 (Drummond et al., 2012) to infer a dated phylogeny using this partition and the substitution models selected for each partition by Partitionfinder. We chose an uncorrelated relaxed clock model and set the tree prior to be birth–death with incomplete sampling. We used default values for all other parameters. We used two calibration points: one is the fossil of butterfly dated to the late Paleocene Fur Formation (56 million years ago) (Grimaldi and Engel, 2005), and thus the most recent common ancestor (MRCA) of butterflies should have originated more than 56 million years ago (Mya); another is the MRCA of Lycaenidae and Riodinidae, which was previously dated to the around 88 Mya, with a 95% confidence interval of 73.2–102.5 Mya (Espeland et al., 2015).
Comparison of the two Calephelis species
We mapped the sequencing reads of all 5 Calephelis specimens to both Cne and Cvi reference genomes using BWA (Li and Durbin, 2010) and detected SNPs using the Genome Analysis Toolkit (GATK) (DePristo et al., 2011). We deduced the genomic sequences for each specimen based on the result of GATK. We used two sequences to represent the paternal and maternal DNA in each specimen. For heterozygous positions, each possible nucleotide was randomly assigned to either paternal or maternal DNA. Based on the mapping results and gene annotation of either reference genome, we further deduced the alignments of gene and protein sequences from all the specimens.
Alternatively, we attempted to map the reads to the reference genome of each species, respectively, derive the protein-coding sequences for each species and align them using MAFFT. However, manual curation of highly diverged proteins between species that were identified using this approach revealed cases of wrong alignments caused by inconsistency in the gene models of the two reference genomes. In contrast, alignments generated by mapping the sequence reads from two different species to the same reference bypassed such problems caused by differences in gene models of the two references, producing higher interspecific sequence identity. Therefore, the results described in this manuscript were based on the alignments derived by mapping the sequencing reads of both C. nemesis and C. virginiensis specimens to the Cne reference genome, but the conclusions remained the same if Cvi reference genome was used instead.
We used two criteria to identify the diverged proteins between C. nemesis and C. virginiensis. First, we estimated the fixation index for both species using the following formula: where πbetween is the average divergence between species, and πwithin is the average divergence within species. We required the divergence hotspots to have fixation index above 0.9. Second, we detected all the positions that are conserved (sharing a common amino acid in over 80% of sequences, which in our case allows no more than one haplotype to show a different amino acid) within but different between species, and required the interspecific divergence hotspots to be significantly enriched (p < 0.01) in such positions. The enrichment is quantified using a binomial test (p = averge rate of divergent positions in all proteins, m = the number of divergent positions in a protein, n = the total number of aligned positions in a protein).
We identified the enriched GO terms associated with these “interspecific divergence hotspots” using binomial tests (m = the number of “interspecific divergence hotspots” that were associated with this GO term, N = number of “interspecific divergence hotspots”, p = the probability for this GO term to be associated with any gene). GO terms with P-values lower than 0.01 were considered enriched. Significantly enriched GO terms (p < 0.01) were visualized in REVIGO (Supek et al., 2011).
Supplementary Material
Highlights.
The first sequenced genome for butterflies in the family Riodinidae
Phylogenetic analysis suggests Riodinidae may be treated as a subfamily of Lycaenidae
Gene expansions related to the detritus feeding behavior of Riodinidae and Lycaenidae
Completes the genomic coverage for butterflies at the family level
Acknowledgments
We acknowledge Texas Parks and Wildlife Department (Natural Resources Program Director David H. Riskind) for the permit #08-02Rev that makes research based on material collected in Texas State Parks possible. We thank Lisa N. Kinch, R. Dustin Schaeffer and Raquel Bromberg for critical suggestions and proofreading of the manuscript; Qian Cong is a Howard Hughes Medical Institute International Student Research fellow. This work was supported in part by the National Institutes of Health (GM094575 to NVG) and the Welch Foundation (I-1505 to NVG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Q.C. and J.S. designed and carried out the experiments, performed the computational analyses and drafted the manuscript; W.L. helped to detect and remove contaminations in the genome; D.B. and Z.O. designed and supervised experimental studies; N.V.G. directed the project and drafted the manuscript. All authors wrote the manuscript.
References
- Ackery PR, De Jong R, Vane-Wright RI. The Butterflies: Hedyloidea, Hesperioidea, Papilionoidea. In: Kristensen NP, editor. Lepidoptera, Moths and Butterflies Volume 1: Evolution, Systematics, and Biogeography. Berlin and New York: De Gruyter; 1999. [Google Scholar]
- Ahola V, Lehtonen R, Somervuo P, Salmela L, Koskinen P, Rastas P, Valimaki N, Paulin L, Kvist J, Wahlberg N, et al. The Glanville fritillary genome retains an ancient karyotype and reveals selective chromosomal fusions in Lepidoptera. Nat Commun. 2014;5:4737. doi: 10.1038/ncomms5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander AM, Su YC, Oliveros CH, Olson KV, Travers SL, Brown RM. Genomic data reveals potential for hybridization, introgression, and incomplete lineage sorting to confound phylogenetic relationships in an adaptive radiation of narrow-mouth frogs. Evolution. 2017;71:475–488. doi: 10.1111/evo.13133. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic acids research. 2005;33:W451–454. doi: 10.1093/nar/gki487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bess J. In: Conservation Assessment for The Swamp Metalmark (Calephelis mutica McAlpine) E.R. USDA Forest Service, editor. 2005. [Google Scholar]
- Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sanchez Alvarado A, Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Wetter T, Suhai S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. Computer Science and Biology: Proceedings of the German Conference on Bioinformatics. 1999;99:45–56. [Google Scholar]
- Cong Q, Borek D, Otwinowski Z, Grishin NV. Skipper genome sheds light on unique phenotypic traits and phylogeny. BMC Genomics. 2015a;1(6):639. doi: 10.1186/s12864-015-1846-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Q, Pei J, Grishin NV. Predictive and comparative analysis of Ebolavirus proteins. Cell Cycle. 2015b;14:2785–2797. doi: 10.1080/15384101.2015.1068472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Q, Shen J, Borek D, K RR, Otwinowski Z, Grishin NV. Speciation in Cloudless Sulphurs gleaned from complete genomes. Genome Biology and Evolution. 2016a;8 doi: 10.1093/gbe/evw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Q, Shen J, Borek D, Robbins RK, Otwinowski Z, Grishin NV. Complete genomes of Hairstreak butterflies, their speciation, and nucleo-mitochondrial incongruence. Sci Rep. 2016b;6:24863. doi: 10.1038/srep24863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Q, Shen J, Warren AD, Borek D, Otwinowski Z, Grishin NV. Speciation in Cloudless Sulphurs Gleaned from Complete Genomes. Genome Biol Evol. 2016c;8:915–931. doi: 10.1093/gbe/evw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong R, Vane-Wright RI, R AP. The higher classification of butterflies (Lepidoptera): problems and prospects. Insect Systematics & Evolution. 1996;27:65–101. [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries PJ. The Butterflies of Costa Rica and Their Natural History, Vol II: Riodinidae. Princeton University Press; 1997. [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Li R, Cheng D, Fan W, Zha X, Cheng T, Wu Y, Wang J, Mita K, Xiang Z, et al. SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic acids research. 2010;38:D453–456. doi: 10.1093/nar/gkp801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Ehrlich PR. The comparative morphology, phylogeny and higher classification of the butterflies (Lepidoptera: Papilionoidea) University of Kansas Science Bulletin. 1958;39:305–370. [Google Scholar]
- Espeland M, Hall JP, DeVries PJ, Lees DC, Cornwall M, Hsu YF, Wu LW, Campbell DL, Talavera G, Vila R, et al. Ancient Neotropical origin and recent recolonisation: Phylogeny, biogeography and diversification of the Riodinidae (Lepidoptera: Papilionoidea) Mol Phylogenet Evol. 2015;93:296–306. doi: 10.1016/j.ympev.2015.08.006. [DOI] [PubMed] [Google Scholar]
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. The Pfam protein families database: towards a more sustainable future. Nucleic acids research. 2016;44:D279–285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francischetti IM, Lopes AH, Dias FA, Pham VM, Ribeiro JM. An insight into the sialotranscriptome of the seed-feeding bug, Oncopeltus fasciatus. Insect biochemistry and molecular biology. 2007;37:903–910. doi: 10.1016/j.ibmb.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassberg J. A Swift Guide to the Butterflies of Mexico and Central America (Swift Guide) Sunstreak Books; 2007. [Google Scholar]
- Grimaldi D, Engel MS. Evolution of the Insects. Cambridge University Press; 2005. [Google Scholar]
- Guild GM, Connelly PS, Ruggiero L, Vranich KA, Tilney LG. Actin filament bundles in Drosophila wing hairs: hairs and bristles use different strategies for assembly. Mol Biol Cell. 2005;16:3620–3631. doi: 10.1091/mbc.E05-03-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JPW, Harvey DJ. A Phylogenetic Review of Charis and Calephelis (Lepidoptera: Riodinidae) Entomological Society of America. 2002;95:407–421. [Google Scholar]
- Hall JPW, Harveyb DJ. A Phylogenetic Review of Charis and Calephelis (Lepidoptera: Riodinidae) Annals of the Entomological Society of America. 2002;95:407–421. [Google Scholar]
- Heliconius Genome, C. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–98. doi: 10.1038/nature11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Silkworm Genome, C. The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect biochemistry and molecular biology. 2008;38:1036–1045. doi: 10.1016/j.ibmb.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Janzen DH, Hallwachs W, Blandin P, Burns JM, Cadiou JM, Chacon I, Dapkey T, Deans AR, Epstein ME, Espinoza B, et al. Integration of DNA barcoding into an ongoing inventory of complex tropical biodiversity. Mol Ecol Resour. 2009;9(Suppl s1):1–26. doi: 10.1111/j.1755-0998.2009.02628.x. [DOI] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Jurka J, Klonowski P, Dagman V, Pelton P. CENSOR–a program for identification and elimination of repetitive elements from DNA sequences. Computers & chemistry. 1996;20:119–121. doi: 10.1016/s0097-8485(96)80013-1. [DOI] [PubMed] [Google Scholar]
- Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, Yabana M, Harada M, Nagayasu E, Maruyama H, et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014;24:1384–1395. doi: 10.1101/gr.170720.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome biology. 2010;11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall RO. More larval food plants from Texas. Journal of the Lepidopterists’ Society. 1959;13:221–228. [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. Gene finding in novel genomes. BMC bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front Genet. 2013;4:237. doi: 10.3389/fgene.2013.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SY, Guindon S. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29:1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Fan D, Zhang W, Liu G, Zhang L, Zhao L, Fang X, Chen L, Dong Y, Chen Y, et al. Outbred genome sequencing and CRISPR/Cas9 gene editing in butterflies. Nat Commun. 2015;6:8212. doi: 10.1038/ncomms9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- Mallet J. New genomes clarify mimicry evolution. Nat Genet. 2015;47:306–307. doi: 10.1038/ng.3260. [DOI] [PubMed] [Google Scholar]
- Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine WS. A revision of the butterfly genus Calephelis Riodinidae. Journal of Research on the Lepidoptera. 1971;10:3–125. [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier B, de Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, et al. Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome biology. 2002;3 doi: 10.1186/gb-2002-3-12-research0083. RESEARCH0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau NJ, Ruiz M, Salazar P, Counterman B, Medina JA, Ortiz-Zuazaga H, Morrison A, McMillan WO, Jiggins CD, Papa R. Population genomics of parallel hybrid zones in the mimetic butterflies, H. melpomene and H. erato. Genome Res. 2014;24:1316–1333. doi: 10.1101/gr.169292.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj R, Adler PN. Dusky-like functions as a Rab11 effector for the deposition of cuticle during Drosophila bristle development. Development. 2012;139:906–916. doi: 10.1242/dev.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Palumbi SR. Genome size evolution in pufferfish: a comparative analysis of diodontid and tetraodontid pufferfish genomes. Genome Res. 2003;13:821–830. doi: 10.1101/gr.841703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- Robbins RK. Comparative morphology of the butterfly foreleg coxa and trochanter (Lepidoptera) and its systematic implications. Proceedings of the Washington entomological Society. 1988;90:133–154. [Google Scholar]
- Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- Scott JA. The Butterflies of North America: A Natural History and Field Guide. Stanford, Calif: Stanford University Press; 1986. [Google Scholar]
- She R, Chu JS, Uyar B, Wang J, Wang K, Chen N. genBlastG: using BLAST searches to build homologous gene models. Bioinformatics. 2011;27:2141–2143. doi: 10.1093/bioinformatics/btr342. [DOI] [PubMed] [Google Scholar]
- Sheehan D, Meade G, Foley VM, Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R. 2008–2010 ( http://www.repeatmasker.org/) RepeatModeler Open-1.0.
- Smit AFA, Hubley R, Green P. 1996–2010 ( http://www.repeatmasker.org/) RepeatMasker Open-3.0.
- St Pierre SE, Ponting L, Stefancsik R, McQuilton P, FlyBase C. FlyBase 102–advanced approaches to interrogating FlyBase. Nucleic acids research. 2014;42:D780–788. doi: 10.1093/nar/gkt1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Schoffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC bioinformatics. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS one. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- Talavera G, Lukhtanov VA, Pierce NE, Vila R. Establishing criteria for higher-level classification using molecular data: the systematics of Polyommatus blue butterflies (Lepidoptera, Lycaenidae) Cladistics. 2012;29:166–192. doi: 10.1111/j.1096-0031.2012.00421.x. [DOI] [PubMed] [Google Scholar]
- Tang W, Yu L, He W, Yang G, Ke F, Baxter SW, You S, Douglas CJ, You M. DBM-DB: the diamondback moth genome database. Database: the journal of biological databases and curation. 2014;2014:bat087. doi: 10.1093/database/bat087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Connelly PS, Vranich KA, Shaw MK, Guild GM. Regulation of actin filament cross-linking and bundle shape in Drosophila bristles. J Cell Biol. 2000;148:87–100. doi: 10.1083/jcb.148.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt, C. Activities at the Universal Protein Resource (UniProt) Nucleic acids research. 2014;42:D191–198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nieuwerburgh F, Thompson RC, Ledesma J, Deforce D, Gaasterland T, Ordoukhanian P, Head SR. Illumina mate-paired DNA sequencing-library preparation using Cre-Lox recombination. Nucleic acids research. 2012;40:e24. doi: 10.1093/nar/gkr1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlberg N, Braby MF, Brower AV, de Jong R, Lee MM, Nylin S, Pierce NE, Sperling FA, Vila R, Warren AD, et al. Synergistic effects of combining morphological and molecular data in resolving the phylogeny of butterflies and skippers. Proc Biol Sci. 2005;272:1577–1586. doi: 10.1098/rspb.2005.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Yue Z, He W, Yang X, Yang G, Xie M, Zhan D, Baxter SW, Vasseur L, Gurr GM, et al. A heterozygous moth genome provides insights into herbivory and detoxification. Nat Genet. 2013;45:220–225. doi: 10.1038/ng.2524. [DOI] [PubMed] [Google Scholar]
- Zhan S, Merlin C, Boore JL, Reppert SM. The monarch butterfly genome yields insights into long-distance migration. Cell. 2011;147:1171–1185. doi: 10.1016/j.cell.2011.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan S, Reppert SM. MonarchBase: the monarch butterfly genome database. Nucleic acids research. 2013;41:D758–763. doi: 10.1093/nar/gks1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan S, Zhang W, Niitepold K, Hsu J, Haeger JF, Zalucki MP, Altizer S, de Roode JC, Reppert SM, Kronforst MR. The genetics of monarch butterfly migration and warning colouration. Nature. 2014;514:317–321. doi: 10.1038/nature13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZQ. Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness (Addenda 2013) Zootaxa. 2013;3703:1–82. doi: 10.11646/zootaxa.3703.1.1. [DOI] [PubMed] [Google Scholar]
- Zhao F, Huang DY, Sun XY, Shi QH, Hao JS, Zhang LL, Yang Q. The first mitochondrial genome for the butterfly family Riodinidae (Abisara fylloides) and its systematic implications. Dongwuxue Yanjiu. 2013;34:E109–119. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.