

Abstract
Translation initiation generally occurs at AUG codons in eukaryotes, although it has been shown that non-AUG or noncanonical translation initiation can also occur. However, the evidence for noncanonical translation initiation sites (TISs) is largely indirect and based on ribosome profiling (Ribo-seq) studies. Here, using a strategy specifically designed to enrich N termini of proteins, we demonstrate that many human proteins are translated at noncanonical TISs. The large majority of TISs that mapped to 5′ untranslated regions were noncanonical and led to N-terminal extension of annotated proteins or translation of upstream small open reading frames (uORF). It has been controversial whether the amino acid corresponding to the start codon is incorporated at the TIS or methionine is still incorporated. We found that methionine was incorporated at almost all noncanonical TISs identified in this study. Comparison of the TISs determined through mass spectrometry with ribosome profiling data revealed that about two-thirds of the novel annotations were indeed supported by the available ribosome profiling data. Sequence conservation across species and a higher abundance of noncanonical TISs than canonical ones in some cases suggests that the noncanonical TISs can have biological functions. Overall, this study provides evidence of protein translation initiation at noncanonical TISs and argues that further studies are required for elucidation of functional implications of such noncanonical translation initiation.
Protein translation is one of the most critical steps in cellular homeostasis. Depending on the translation initiation site(s) on an mRNA sequence, different protein isoforms can be synthesized from the same mRNA (Wan and Qian 2014). Thus, accurate information regarding translation initiation sites (TISs) is important in understanding translational control in cells. Nevertheless, current annotations of human proteins generally lack direct evidence of TISs, with most annotations relying on the presence of AUG and the largest open reading frame (ORF) (Saeys et al. 2007). Recently, there have been a number of reports describing TISs using ribosome profiling (Ribo-seq) for inferring TISs based on mRNA protection by ribosomes at TISs (Ingolia et al. 2009, 2011, 2014). These studies have revealed that noncanonical TISs are more common than previously believed. Intriguingly, several such ribosome profiling studies have revealed that a large majority of TISs observed in 5′ untranslated regions (UTR) were noncanonical (Ingolia et al. 2011, 2014; Lee et al. 2012). Translation from a noncanonical TIS implies that translation begins at near-cognate AUG codons such as ACG, CUG, GUG, or UUG (Gerashchenko et al. 2010; Menschaert et al. 2013; Ingolia et al. 2014; Wan and Qian 2014; Gao et al. 2015). Although ribosome profiling studies have described noncanonical TISs, it should be noted that such annotations inferred by ribosome profiling depend on protection of mRNA sequences protected by the ribosome and still require protein level confirmation to validate those sites (Gerashchenko et al. 2010; Ingolia et al. 2011; Hinnebusch 2014).
In addition to the difficulty of ascertaining the TIS accurately, there is also uncertainty in determining which amino acid is incorporated when noncanonical TISs are utilized. Since Met-tRNAi is known to be incorporated into the translation initiation complex before encountering TISs, the noncanonical TISs could lead to incorporation of methionine, and the resulting peptide sequence should be able to establish the actual N-terminal amino acid used to encode the mature protein encoded by the gene (Hinnebusch 2014).
Tandem mass spectrometry allows facile determination of peptide sequences. However, annotation of TISs through identification of free protein N termini can still be ambiguous owing to post-translational proteolytic cleavages. Because almost 90% of intracellular proteins in humans are believed to be cotranslationally acetylated at their N termini, this acetylation can be utilized to obtain a signature modification that indicates the translation initiation site (Lange et al. 2014; Aksnes et al. 2015).
To study noncanonical TISs on a global scale, we focused on 5′ UTRs for two main reasons. First, 5′ UTRs have a lower frequency of AUG codons, and thus most N-terminally acetylated peptides that map to this region should be noncanonical TISs (Zur and Tuller 2013). Second, ribosome profiling studies already suggest that the majority of TISs at 5′ UTRs are noncanonical, making our approach more reliable for confirming true TISs (Gerashchenko et al. 2010; Ingolia et al. 2011; Hinnebusch 2014). However, identification of a large number of N-terminally acetylated peptides in routine proteomic surveys is not easy due to the large complexity of total peptides when digested by an enzyme. To circumvent this limitation, we applied terminal amine isotopic labeling of substrates (TAILS) to enrich protein N-terminal peptides through negative selection (Kleifeld et al. 2010). This approach specifically enriches for protein N-terminal peptides with high fidelity, including the key acetylated N-terminal peptides that are not captured by strategies based on positive selection. We chose two cell types, HEK293T cells and human umbilical vein endothelial cells (HUVEC), and two human tissues, colon and substantia nigra, for our analysis. We reasoned that, given the general nature of translational initiation, if overlapping results were observed across these cells/tissues, it would further attest to the reliability of the identified noncanonical TISs. Overall, our study provides evidence for noncanonical TISs in 5′ UTRs at the protein level and extends the data on putative TISs predicted by ribosome profiling. This is important in light of recent ribosome profiling studies that demonstrate that the majority of TISs in the 5′ UTR are noncanonical. Our studies lay the framework for future studies to study noncanonical TISs located within 5′ UTRs.
Results
In this study, our goal was to directly identify noncanonical TISs from human samples to validate TISs predicted by ribosome profiling studies. To do so, we enriched N-terminal peptides from proteins extracted from HEK293T cells, HUVEC, human colon tissue, and human substantia nigra tissue using the TAILS method. In this approach, all primary amine groups of proteins are first blocked by dimethylation at the protein level prior to trypsin digestion. The non-N-terminal peptides derived by trypsin are then depleted by capturing the peptides with a highly functionalized hyperbranched polyglycerol-aldehydes (HPG-ALD) polymer. The remaining N-terminal peptides can then be fractionated with high-pH reversed phase liquid chromatography before mass spectrometry analysis (Fig. 1A). We first searched the acquired mass spectra against annotated ORFs to remove spectra matching annotated proteins. To increase the possibility of identifying noncanonical TISs, ∼10 mg of protein lysate for each sample was used as a starting material. From this search, we identified ∼5600 N-terminally acetylated peptides with a false discovery rate of ≤1% that serve as the hallmark of translation initiation sites (Supplemental Table S1), and ∼4000 acetylated peptides were mapped to annotated TISs (Fig. 1B). Of the ∼4000 acetylated peptides that mapped to annotated TISs, ∼2800 (∼70%) were found in at least two samples and ∼2000 (∼50%) were observed in at least three samples, suggesting these acetylated peptides are true positive TISs (Fig. 1C).
Figure 1.

Identification of translation initiation sites. (A) A schematic diagram for enrichment of protein N-terminal peptides using the TAILS method. All primary amine groups (α- and ε-) on proteins were blocked by dimethylation followed by trypsin digestion. Non-N-terminal peptides displaying a free, trypsin-generated α-amine group were depleted using HPG-ALD polymer, and the remaining N-terminal peptides were analyzed in mass spectrometry after bRPLC fractionation. (B) The acetylated peptides mapping to annotated translation initiation sites were identified from HEK293T cells, HUVEC, human colon, and human substantia nigra using the TAILS method. (C) Overlap of the acetylated peptides mapping to annotated translation initiation sites from HEK293T cells, HUVEC, human colon, and human substantia nigra.
TISs mapped to 5′ UTRs were identified
Since the main goal of this study was to identify noncanonical translation initiation sites mapping to 5′ UTRs, we searched for TISs in 5′ UTRs with a three-frame translation database of 5′ UTRs. From the search, we observed that 90 N-terminally acetylated peptides mapped to 5′ UTRs, of which 41 were observed in at least two of the samples (Table 1). The majority (∼69%) of these led to an extension of the N termini of previously annotated proteins, whereas the remaining 31% of TISs originated from upstream small open reading frames (uORFs) located within the 5′ UTRs (Fig. 2A). Two TISs found in HDGF and FXR2 were reported previously (Menschaert et al. 2013). Notably, ∼92% of the TISs of N-terminal extension corresponded to a near-cognate AUG codon. Only one instance of N-terminal extension began with AUG (Fig. 2B). On the other hand, ∼33% and ∼11% of the TISs of uORFs began with near-cognate AUG and AUG, respectively (Fig. 2C). To investigate the sequence motif further, sequence logo analyses of mRNAs surrounding TISs was performed. For N-terminal extension forms, the sequence NUGGC was enriched at the initiation site (Fig. 2D). On the other hand, uORFs were enriched for (A/G)(C/U)GGC (Fig. 2E). The large majority of the acetylated peptides identified in this study presumably had the first amino acid removed, implying that most of acetylated peptides with the first amino acid-retained form were not identifiable in this database search. Considering that the first amino acid is encoded by the initiator tRNA and this initiator tRNA is predetermined before the preinitiation complex encounters TISs, the acetylated peptides with the first amino acid-retained form were expected to begin with Met and these peptides would not be identified by the database used because methionine was not encoded by the genomic sequence. Therefore, a more specialized database was required to identify the acetylated peptides retaining methionine at their N termini.
Table 1.
List of acetylated peptides mapped to 5′ UTRs identified from TAILS experiments
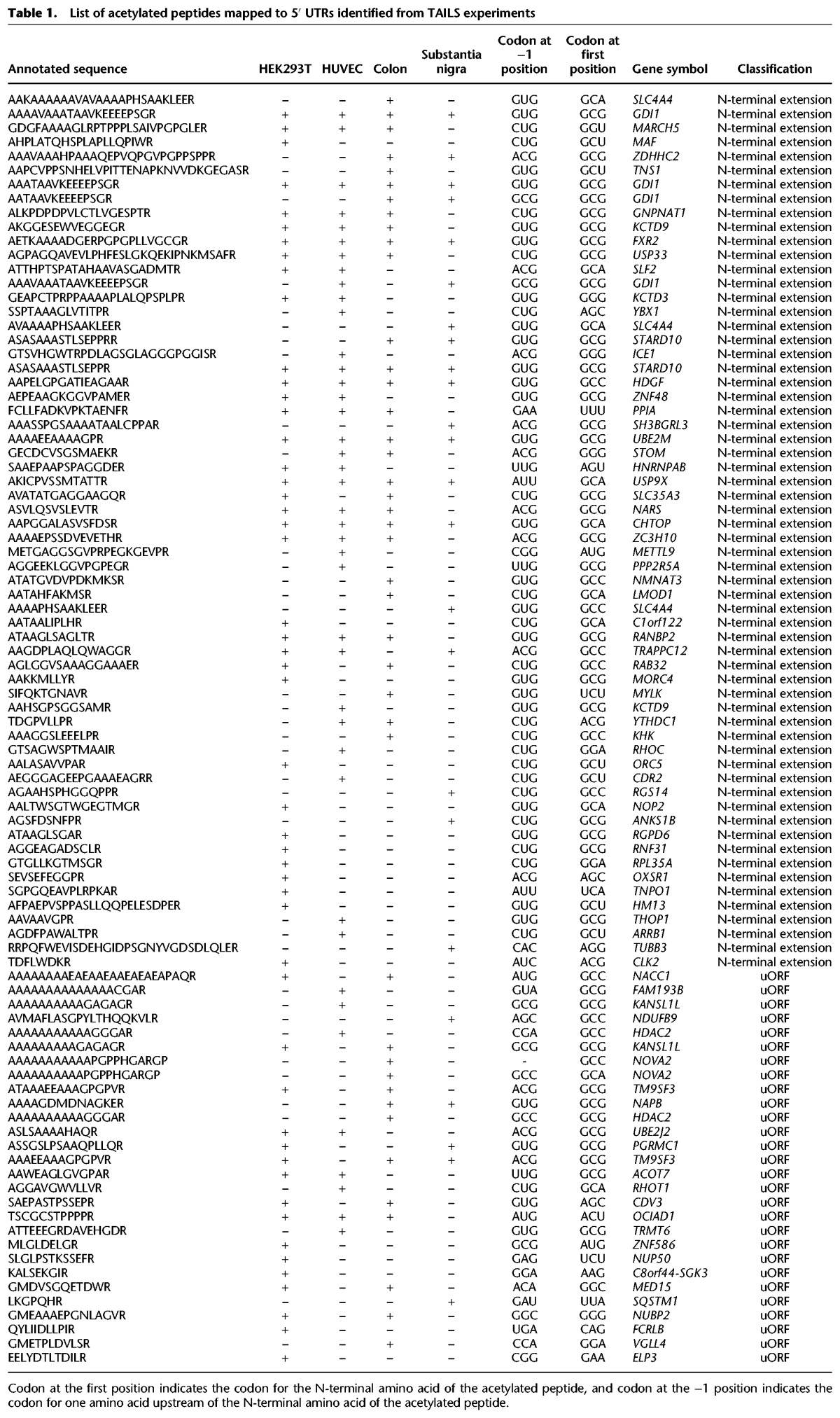
Figure 2.

Translation initiation sites mapping to 5′ UTRs. (A) Peptides acetylated at their N termini mapping to 5′ UTRs that were identified from HEK293T cells, HUVEC, human colon, and human substantia nigra as shown. The colors indicate whether the peptides correspond to uORFs or were N-terminal extensions of annotated proteins. (B) Codons for TISs corresponding to acetylated peptides mapping to 5′ UTRs which led to N-terminal extension of annotated proteins. (C) Codons for TISs corresponding to acetylated peptides mapping to 5′ UTRs which encode an uORF. (D) Sequence logo of nucleotides surrounding the TIS in cases where the TISs lead to N-terminal extension of annotated proteins. (E) Sequence logo of nucleotides surrounding the TIS in cases where the TISs are located in an uORF.
Near-cognate start codons specify methionine incorporation for TISs
Although it is well documented that near-cognate start codons can frequently be used as a TIS, exactly which amino acid is incorporated as the first amino acid in the protein is still not clear. One of the possibilities is that near-cognate start codons specify methionine. An alternative scenario is that near-cognate start codons lead to incorporation of the amino acids coded originally by those codons. If methionine is specified by those near-cognate codons, even if the N-terminally acetylated peptides contain methionine as the first amino acid, it would not be identified in a standard database search. To address this, we searched the data against peptides derived from a database in which near-cognate codons or four other non-near-cognate codons were substituted to AUG before three-frame translation (Fig. 3A). In all, 33 acetylated peptides that begin with methionine were identified from the four different samples. Thirty peptides corresponded to N-terminal extension, while the remaining three were derived from uORFs in the 5′ UTR (Fig. 3B). All identified peptides began with near-cognate start codons, strongly supporting that the corresponding TISs were bona fide TISs (Fig. 3C). Eight peptides were identified from two or more samples and the remaining 28 identified only from one sample (Table 2; Fig. 3B). Sequence logo analysis performed on TISs and their surrounding sequences revealed that the sequence NUGGA was enriched in the initiation sites (Fig. 3D). This result was quite similar to sequence motifs observed in N-terminal extended forms found in the search against databases without amino acid substitution. Methionine was incorporated for a large majority of the TISs that were coded by near-cognate codons. Strikingly, one TIS (Ac-(M/T)DFLWDKR) coded by ACG was discovered to be decoded into both Met and Thr (Table 1). Even though there have been reports that CUG can be decoded either by Met or Leu (Schwab et al. 2004; Starck et al. 2012), there has been no report that ACG can be decoded by Thr as well as Met. Interestingly, the penultimate amino acid of the peptide has a bulky side chain, consistent with a previously established rule that the first amino acid is not removed when the penultimate amino acid has a bulky side chain (Frottin et al. 2006; Xiao et al. 2010). These results suggest that almost all translation initiation commences with Met regardless of the codon for the amino acid at position 1, with some minor exceptions. In particular, it was notable that, in 22 out of 33 cases where methionine was retained at the N terminus, an acidic residue was observed at position 2 (Table 2). It has been previously shown that such acidic residues are N-end stabilizing, if acetylated (Lange et al. 2014).
Figure 3.

Translation initiation sites mapping to 5′ UTRs identified by a unique database search strategy. (A) Substitution of near-cognate initiation codons to AUG enables identification of acetylated peptides that begin with methionine by searching against a customized database as shown. (B) Peptides acetylated at their N termini mapping to 5′ UTRs that were identified from HEK293T cells, HUVEC, human colon, and human substantia nigra through a database search against a customized database that incorporates substitution of near-cognate initiation codons to AUGs. The colors indicate whether the peptides correspond to upstream open reading frames (ORFs) or were N-terminal extensions of annotated proteins. (C) Codons for TISs corresponding to acetylated peptides mapping to 5′ UTRs identified through a search against a customized database that incorporates substitution of near-cognate initiation codons to AUG. (D) Sequence logo of nucleotides surrounding the TISs identified by searching against the database with the substitution of near-cognate initiation codons to AUG.
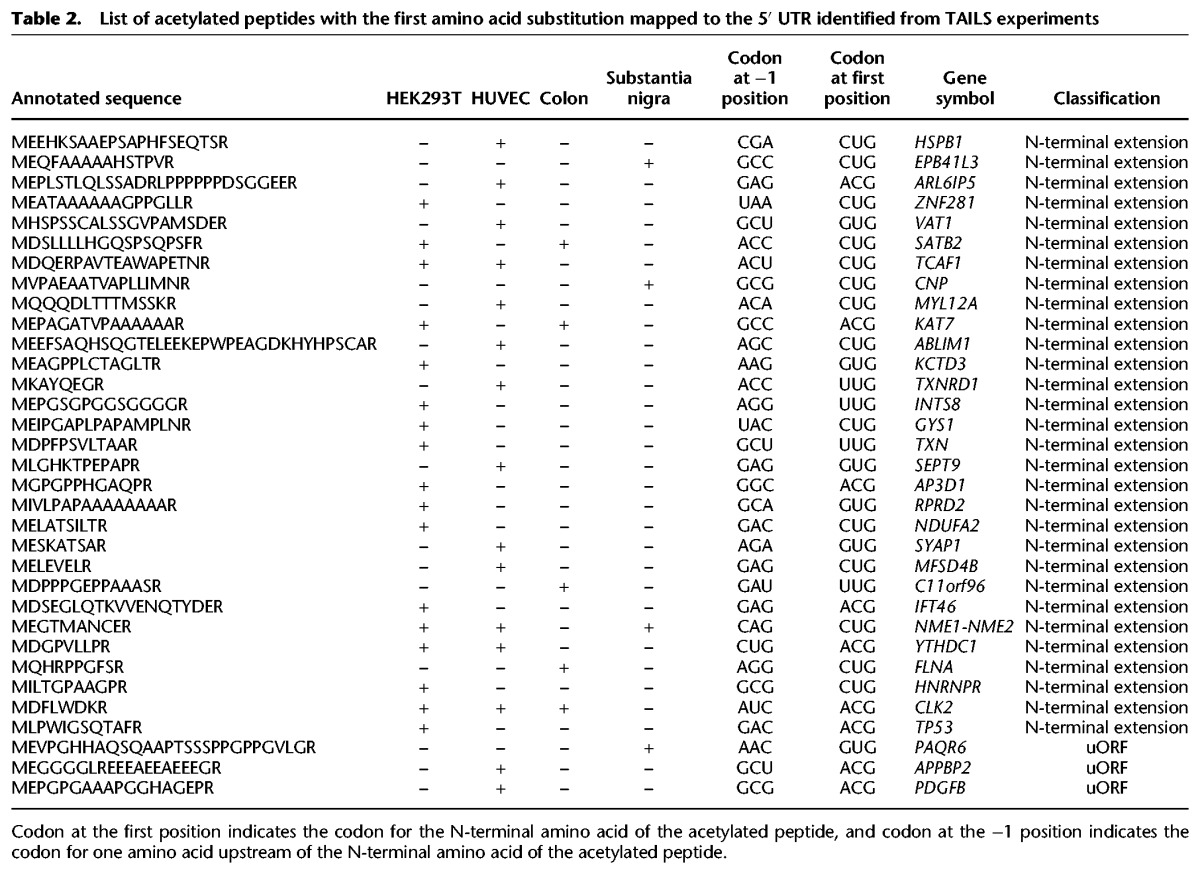
Table 2.
List of acetylated peptides with the first amino acid substitution mapped to the 5′ UTR identified from TAILS experiments
TISs mapping to 5′ UTRs are observed in multiple cells/tissues
To check whether translation initiation at noncanonical TISs is a general phenomenon, we examined if a similar pattern was also observed in a study in which 30 human cells/tissues were studied (Kim et al. 2014). Nineteen TISs mapping to 5′ UTRs were identified and eight TISs were already identified in the experiment performed using TAILS. Of the 19 TISs, 12 were from N-terminal extension and the remaining seven were encoded by uORFs. Two out of 12 TISs from N-terminal extension began with Met for near-cognate start codons (Supplemental Table S2). As observed in N-terminal enrichment for HEK293T, HUVEC, colon, and substantia nigra, the majority (∼70%) of TISs of N-terminal extension began with near-cognate start codons (Supplemental Fig. S1a). On the other hand, only 23% of TISs started with near-cognate initiation codons for uORFs (Supplemental Fig. S1b). Sequence logo analysis was performed for mRNA sequences at TISs. For the TISs of N-terminal extension, GUGGC was enriched at TISs, followed by high G or C enrichment at the downstream (Supplemental Fig. S1c). For TISs of uORFs, an obvious enrichment was not observed, but downstream showed higher enrichment for G or C (Supplemental Fig. S1d). These results imply that those noncanonical TISs are not limited to human cells/tissues that we analyzed with TAILS. If TAILS is applied to all tissues in a systematic fashion, we expect that additional noncanonical TISs would be identified from those samples as well.
Comparison with ribosome profiling data supports TISs located in 5′ UTRs
Since ribosome profiling studies have reported many putative TISs mapping to the 5′ UTR (Ingolia et al. 2009, 2011, 2014), we investigated how many TISs identified in this study corresponded to those reported in ribosome profiling studies. For this, we compared the acetylated peptides with the putative TISs annotated by ribosome profiling performed on HEK293 cells using the PROTEOFORMER pipeline (Lee et al. 2012; Crappe et al. 2015; Gao et al. 2015). Nearly one-third of the acetylated peptides matched putative TISs annotated by ribosome profiling (Supplemental Table S3). When we conducted a manual examination of the remaining acetylated peptides by aligning them against the ribosome profiling data, we were able to match nearly half of these peptides to ribosome profiling data (Supplemental Fig. S3). Overall, the fact that nearly two-thirds of identified acetylated peptides matched ribosome profiling data supports the notion that a large majority of TISs identified in this study are bona fide.
In the case of STARD10, two acetylated peptides were identified—one mapped to the 5′ UTR and the other one to the annotated initiator methionine. Both of these corresponded to TISs predicted by ribosome profiling data (Fig. 4A). The TIS predicted in the 5′ UTR of ZNF281 by ribosome profiling revealed the presence of CUG, although the acetylated peptide that mapped to this region contained methionine, which is not the amino acid specified by CUG (Fig. 4B). The TIS mapping to annotated initiator methionine of ZNF281 was not detected, likely because the corresponding N-terminal peptide generated by tryptic digest is too large (38 amino acids) for mass spectrometric detection. Also, the majority of N-terminal peptides mapped within a single nucleotide of the TISs predicted by ribosome profiling data.
Figure 4.

Acetylated peptides identified in 5′ UTRs map to TISs inferred using ribosome profiling data and were validated by synthetic peptides. (A) The acetylated peptide identified from HEK293T cells that was derived from the 5′ UTR of STARD10 along with the annotated TIS (the methionine “M” is marked in red) were aligned with ribosome profiling data from HEK293 cells. (B) The acetylated peptide identified from HEK293T cells that was derived from the 5′ UTR of ZNF281 was aligned with the ribosome profiling data from HEK293 cells. The methionine of the annotated TIS is marked as “M” in red. (C) The acetylated peptide positioned in the 5′ UTR of STARD10 was validated with synthetic peptides. The annotated mass spectrum derived from the sample (top) is aligned with the mass spectrum derived from a synthetic peptide (bottom). (D) The acetylated peptide positioned in the 5′ UTR of ZNF281 was validated with synthetic peptides. The annotated mass spectrum derived from the sample (top) is aligned with the mass spectrum derived from a synthetic peptide (bottom).
Because a database search identifies peptides by comparing spectra from a sample to theoretical spectra generated from peptide sequences in a database, the best practice to validate identified peptides is by comparing the experimentally obtained fragmentation spectra to spectra obtained from fragmentation of synthetic peptides. To validate the spectra for acetylated peptides mapping to 5′ UTRs, 56 randomly selected peptides were synthesized with the same modifications as the ones identified from the samples. We reasoned that if we randomly select about half of all identified TISs mapping to 5′ UTRs and if these peptides are proven to be true, then we could extrapolate the findings to the remaining peptides that were not validated by the synthetic peptides. Forty-nine out of 56 spectra perfectly matched those obtained from fragmentation of the synthetic peptides (Supplemental Fig. S2), six spectra matched with small differences, and only one spectrum matched poorly. Figure 4,C and D, illustrates how the mass spectra for peptides mapping to the 5′ UTR of STARD10 and ZNF281 match perfectly to the spectra from the corresponding synthetic acetylated peptides. These results suggest that the large majority of the identified acetylated peptides are true positives. Comparison of those acetylated peptides with ribosome profiling data and confirmation of the identified peptides with synthetic peptides support that the majority of the TISs mapping to 5′ UTRs are bona fide.
Sequence analysis reveals conservation of TISs mapped to 5′ UTRs
Although many protein N-terminal extensions mapping to 5′ UTRs were observed, there is a possibility that translation occurs randomly at the TISs that we identified. On the other hand, if these TISs have some functional importance, the sites are likely to be conserved across different species. To investigate the sequence conservation, protein sequences that were identified to have the N-terminal extension form of TISs at 5′ UTRs were aligned with proteins encoded by orthologous genes. Most TISs at 5′ UTRs were conserved, ranging from primates to zebrafish (Supplemental Fig. S4). In the case of STARD10, the protein sequence of the N-terminal extension was conserved to mouse. This conservancy disappeared from the fifth amino acid upstream of the noncanonical translation initiation site (Fig. 5A). In the case of ZNF281, the conservancy was not observed for cow, dog, and rat, but mouse showed conservancy (Fig. 5B). Since the studies for expressed RNA sequences for cow, dog, and rat were not as deep as that of mouse, we cannot rule out that those isoforms for the N-terminal extension form were not sequenced, while they are expressed in those species. Overall, these results suggest that the majority of TISs of the N-terminal extension form are conserved between closely related species and might have specific functions that are located in the conserved parts of the proteins that reside in the extended version of the proteins.
Figure 5.

Sequence conservation of acetylated peptides identified in 5′ UTRs across species. (A) Sequence conservation analysis for the 5′ UTR of the STARD10 gene between different species. (B) Sequence conservation analysis for the 5′ UTR of the ZNF281 gene between different species.
Relative abundance of noncanonical to canonical TISs
An indicator supporting the notion that noncanonical TISs have their own functions is if noncanonical TISs are more abundant or expressed at similar levels as their canonical counterparts. To investigate this, we used the number of peptide spectrum matches (PSMs) as a measure of abundance (Wong and Cagney 2010; Kim et al. 2014). The relative abundance of noncanonical TISs over canonical ones ranged from 0.01- to 3.6-fold. The majority of noncanonical TISs were less abundant than the canonical ones, although, in several cases, noncanonical TISs were found to be more abundant (Fig. 6A). As a next step, we investigated whether the relative abundance of noncanonical to canonical TISs for a given gene was the same across all cells and tissues. We observed that this showed a relatively wide distribution. Notably, the noncanonical TIS of RAB32 was about 2.5 times more abundant in HEK293T cells but one-third as abundant in colon. On the other hand, the noncanonical TIS of STARD10 was 2.5 times more abundant in colon but one-third as abundant in substantia nigra, confirming that some proteins that are translated from noncanonical TISs actually exist in higher abundance than the ones translated from canonical TISs. Moreover, the noncanonical to canonical TIS ratio is variable depending on the cell/tissue type. In light of recent reports that the translational apparatus is redirected toward unconventional upstream TISs during tumor initiation and that eukaryotic translation initiation factor 1 modulates the recognition of upstream TISs (Fijalkowska et al. 2017; Sendoel et al. 2017), our findings provide further evidence to support a biological function of noncanonical TISs.
Figure 6.

Relative abundance of noncanonical and canonical TISs. (A) The relative abundance was calculated by dividing the number of peptide spectrum matches (PSMs) corresponding to the noncanonical TIS of a gene by the number of PSMs of the corresponding canonical TIS observed in a sample. (B) The relative abundance is shown for the indicated genes across multiple samples.
Discussion
In this study, we identified ∼5600 TISs mapping to annotated ORF regions and 134 TISs mapping to 5′ UTRs using the TAILS N-terminomics approach. Strikingly, the majority of TISs mapping to 5′ UTRs were noncanonical. Although the existence of widespread putative noncanonical TISs has been suggested by a number of ribosome profiling studies, definitive data at the proteome level is lacking. Only a few small-scale proteome-level studies have been reported (Menschaert et al. 2013; Van Damme et al. 2014; Willems et al. 2017). There could be several possible explanations why those noncanonical TISs have not been identified at the proteome level, while ribosome profiling studies have reported so many putative noncanonical TISs. One possible explanation is that most of the noncanonical TISs reported by ribosome profiling studies were not true TISs. Another explanation could be that, given the noncanonical nature of TISs, the appropriate database searches were not conducted in previous studies. Our study was unique in several aspects. First, we considered that the starting material amount probably was not large enough to identify less abundant and less frequent noncanonical TISs. To overcome this limitation, we enriched protein N-terminal peptides by the TAILS method, with 10–20 times more starting material (∼10 mg) than common practice. Second, different human cells/tissues might use different noncanonical start sites, and thus it might be difficult to identify a large number of noncanonical TISs from a single human sample. To overcome this, we used three additional human samples—HUVEC, colon, and substantia nigra—to identify a larger repertoire of noncanonical TISs. Third, although there has been some effort to identify noncanonical TISs, all studies have relied on information from ribosome profiling studies, which will confine the TISs to those reported by ribosome profiling. To overcome this, we generated a customized database by translating 5′ UTRs into three frames independent of ribosome profiling data. Fourth, although three-frame translation of mRNAs from 5′ UTRs was used for database searches, the TIS peptides that start with Met at non-AUG codons cannot be identified, because most of the noncanonical TISs discovered in this study utilized Met-tRNAi for translation initiation. For this purpose, we generated a custom database in which near-cognate codons were replaced with AUG. Fifth, although N-terminal peptides were enriched, the amount was still not large enough for large-scale identification of noncanonical TISs. To overcome this, we used state-of-the-art mass spectrometry for identifying noncanonical TISs from the limited amounts of such low frequency N-terminal peptides.
Sequence logo analysis showed an enrichment of the UGGC motif when the first amino acid was removed and the UGGA motif when the first amino acid was retained. These sequences are followed by sequences slightly enriched for G or C downstream from the TISs. When the RNA sequence motif (G(G/C)(G/C)(A/G)NG(G/C)UGG(A/G)) for TISs was compared to Kozak sequence (GCC(A/G)CCAUGG), G at −6 position, A/G at −3 position, and G at +4 positions could be important for translation initiation at 5′ UTRs.
In this study, many TISs that start not only at near-cognate AUG codon such as ACG, CUG, GUG, and UUG but also those that start at non-cognate AUG codons such as GCG, GCC, and GGC were identified. When the TISs identified in this study were compared to the TISs annotated by ribosome profiling data, the majority (∼65%) had a corresponding match. This overlap in results indicates that some or many of the noncanonical start codons annotated by ribosome profiling experiments are indeed true TISs. On the other hand, an apparent discordance from ribosome profiling studies for the remaining one-third of TISs identified in this study could be caused by several possible reasons. The most likely cause is that, because the data sets are from two different studies, it is possible that the TISs identified in this study were not expressed in the cells used in the ribosome profiling study. This reasoning is also in agreement with previous observations (Menschaert et al. 2013). Another possible reason could be that TISs not supported by ribosome profiling are translated by an alternative translational mechanism. Finally, TISs determined through proteomics could represent false positives if they are generated by post-cleavage acetylation (Lange et al. 2014; Aksnes et al. 2015). Although the TAILS method permitted us to identify 134 TISs mapping to 5′ UTRs in addition to ∼6500 mapping to the ORF region, these numbers are likely to be underestimates as there are many other TISs that could not be identified by this approach because the N-terminal peptides generated by trypsin digestion were either too short or too long. To overcome this limitation, alternative enzymes need to be employed, which will enable us to identify TISs that were missed by using only trypsin for digestion.
The proteome-based discovery experiments performed in this study, independent of ribosome profiling experiments, validated that there are many noncanonical TISs that map to 5′ UTRs. The majority of those noncanonical TISs commence at near-cognate initiation codons such as ACG, CUG, GUG, and UUG. Notably, methionine is still incorporated at those noncanonical TISs. Moreover, sequence analysis of N-terminally extended regions revealed that there was conservation of some of the amino acid sequences across species. Although the abundance of peptides with noncanonical TISs was lower than the canonical TISs in the majority of cases, in some cases, it was dramatically more abundant. Further studies are clearly required for elucidation of functional implications of such noncanonical translation initiation, and our study provides a valuable resource for such future studies.
Methods
Cell culture and human sample acquisition
HEK293T cells were cultured in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum. HUVECs were cultured in EGM BulletKit Medium (Lonza) according to the manufacturer's instructions. Human colon tissue sample was collected during surgery, and the tissue was snap-frozen in liquid nitrogen. Human substantia nigra tissue sample was obtained from a rapid autopsy program. The tissues were evaluated by the pathologist involved, and microscopic observation showed minimal or no autolysis and putrefaction. Institutional ethical clearance was obtained for the use of the human samples from Johns Hopkins Medicine Institutional Review Board.
Protein extraction and N-terminal peptide enrichment by TAILS method
HEK293T cells, HUVEC, human colon, and substantia nigra tissues were sonicated for 5 min in 8 M guanidine hydrochloride, 50 mM HEPES, pH 7.0, 10 mM dithiotreitol in the presence of Halt Protease Inhibitor Cocktail (Thermo) followed by heating at 90°C for 3 min and subsequent centrifugation at 16,000g for 10 min. After cooling, the proteins were alkylated by treating with 30 mM iodoacetamide at room temperature for 15 min. All the primary amine groups of the proteins were dimethylated and non-N-terminal peptides of proteins were depleted as described previously (Kleifeld et al. 2011). Briefly, protein samples were diluted by adding the same volume of 50 mM HEPES, pH 7.0; formaldehyde and sodium cyanoborohydride were added to the final concentration of 40 and 20 mM, respectively. The pH was adjusted to 6.0–7.0 and incubated at 37°C overnight, followed by quenching the reaction by adding ammonium bicarbonate to a final concentration of 100 mM and incubation at 37°C for 4 h. After blocking all the primary amine groups by dimethylation, proteins were precipitated by adding eight volumes of ice-cold acetone and one volume of methanol at −80°C for 3 h. The proteins were collected by centrifuging at 14,000g at 4°C for 20 min and carefully decanting, followed by washing with ice-cold methanol twice. The tube with protein precipitant was dried at room temperature, and the proteins were resuspended with a small volume (500 µL for a high-speed centrifuge tube of 50-mL capacity) of 50 mM NaOH. Once the protein pellet was fully dissolved, NaOH was diluted with 50 mM HEPES, pH 8.0, followed by trypsin digestion with the enzyme-to-protein ratio of 1:100. The protein was digested at 37°C overnight. Protein non-N-terminal peptides were depleted by adding HPG-ALD polymer (http://flintbox.com/public/project/1948) at a protein-to-polymer ratio of 1:2, followed by addition of 20 mM NaBH3CN. The peptide and polymer mixture was incubated at 37°C overnight. Protein N-terminal peptides that were not captured to HPG-ALD polymer were collected by spin column with a 30-kDa molecular weight cutoff (Millipore). The collected N-terminal peptides were fractionated with high-pH reverse phase liquid chromatography from 10% to 35% of the gradient over a 95-min gradient with 0.3 mL/min flow rate (buffer A: 10 mM tetraethylamine ammonium bicarbonate, buffer B: 10 mM tetraethylamine ammonium bicarbonate /90% acetonitrile). The fractionated peptides were concatenated into 24 fractions and dried by SpeedVac.
LC-MS/MS
Peptide samples were reconstituted in 10% formic acid to keep the pH of the reconstituted peptide acidic because we sometimes observed that the high pH buffer after high-pH RPLC was not completely dry and 0.1% formic was not adequate to decrease the pH below 3. The peptides were analyzed on LTQ-Orbitrap Elite mass spectrometers (Thermo Electron) coupled with EASY-nLC II nanoflow liquid chromatography systems (Thermo Scientific), Orbitrap Fusion Tribrid mass spectrometer (Thermo Electron) coupled with EASY-nLC 1000 nanoflow liquid chromatography systems (Thermo Scientific), or Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Electron) coupled with EASY-nLC 1200 nanoflow liquid chromatography systems (Thermo Scientific). Peptides were resolved on an analytical column (75 µm × 60 cm) packed with 5-µm-sized C18 particles at a flow rate of 250 nL min−1 using a linear gradient of 7%–35% solvent B (0.1% formic acid in 90% acetonitrile) over 120 min for the EASY-nLC II system, and on an EASY-Spray analytical column (Thermo Scientific) (75 µm × 50 cm) packed with 2-µm-sized C18 particles at a flow rate of 250 nL min−1 using a linear gradient of 7%–35% solvent B (0.1% formic acid in 90% acetonitrile) over 120 min for EASY-nLC 1000 and EASY-nLC 1200 systems. Mass spectrometry (MS) spectra was acquired with full scans (300–1700 m/z) using an Orbitrap mass analyzer at a mass resolution of 120,000 at 400 m/z for LTQ-Orbitrap Elite and 120,000 at 200 m/z for Orbitrap Fusion Tribrid ETD. The ten most intense precursor ions from a survey scan were selected for MS/MS from each duty cycle. The selected precursor ions were fragmented by the higher-energy collision dissociation method and detected at a mass resolution of 30,000 at m/z of 400 in the Orbitrap analyzer for LTQ-Orbitrap Elite and 30,000 at m/z of 200 in the Orbitrap analyzer for Oribtrap Fusion Tribrid ETD. Dynamic exclusion was set for 60 sec with a 10-ppm mass window for both instruments. The automatic gain control for full FT MS was set to 1 million and for FT MS/MS was set to 0.05 million with maximum ion injection times of 100 and 300 msec, respectively. Internal calibration was carried out using lock-mass from ambient air (m/z 445.1200025).
Creation of customized databases
For the identification of the protein initiation sites positioned at annotated ORFs, all peptides commencing with M and ending with R from each protein of human RefSeq70 were extracted. After extraction of those peptides, the Met-removed forms of the peptides were generated as well to identify the Met-removed form. Those Met-retained and Met-removed forms of the peptides were concatenated for each protein to help proper decoy database generation. For the identification of the protein initiation sites positioned at 5′ UTRs, hg38 human gene sequence was downloaded from UCSC Genome Browser. The 5′ UTR region from the mRNA database was taken and translated into three different frames. For the database search of acetylated peptides with the first amino substituted to methionine, another customized database was generated by substituting near-cognate codons (ACG, CUG, GUG, and UUG) or control codons (CUA, GCC, GCG, and GUC) for the 5′ UTR of mRNA sequences to AUG. Each codon was substituted to AUG at a time, and this process was repeated for eight codons. The substituted mRNA sequences were translated into three frames. Those eight different databases were combined into one, and redundant sequences were removed.
Mass spectrometry data analysis
MS/MS data obtained from LC-MS analyses were searched against the RefSeq human protein database (version 70) for ORF regions or customized databases for 5′ UTRs with 248 common contaminant proteins using SEQUEST search algorithms through Proteome Discoverer 2.1 platform (Thermo Scientific). ArgC was used as the protease (since trypsin cleaves with ArgC specificity due to the dimethylation of lysines in the TAILS workflow) with a maximum of two missed cleavages allowed. Acetylation and dimethylation of peptide N termini and oxidation of methionine were included as variable modifications. Carbamidomethyl modification of cysteine and dimethyl modification of lysine were included as fixed modifications. The minimum peptide length was set to be 6 amino acids. The mass tolerances were set to 10 ppm and 0.02 Da for precursor and fragment ions, respectively. The matched spectra were filtered using the Percolator algorithm within the Proteome Discoverer suite. The identified peptides were further filtered by protein-level false discovery rate. Proteins were considered identified at a q value < 0.01. The unmatched spectra from the Human Proteome Map project were searched using the same parameters as those used for TAILS experiments except for nontryptic on N-terminal and tryptic on C-terminal, dynamic modification for peptide N-terminal acetylation, no fixed modification for Lys, and 0 missed-cleavage allowed. The acetylated peptides identified from the searches against 5′ UTRs were matched to the NCBI nonredundant human protein database, and only peptides that have at least 3 amino acids mismatch were retained.
Validation of acetylated peptides mapped to 5′ UTRs using synthetic peptides
Representative acetylated peptides were synthesized (JPT Peptide Technologies). Dried peptides were diluted using 10% formic acid, and approximately 1 pmol of peptides to the final concentration were mixed and subjected to LC-MS/MS analysis on the LTQ-Orbitrap Elite mass spectrometer (Thermo Fisher) or Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher). A database search was performed in the same manner as described in the mass spectrometry data analysis section with the exception of the database that is composed of synthetic peptides and 248 common contaminant proteins. Spectra of acetylated peptides from the samples were validated by aligning those from synthetic peptides using in-house generated software written in Python programming language.
Annotation of putative TISs using the ribosome profiling data
Ribosome profiling data for translation initiation profiling downloaded from the NCBI Sequence Read Archive (SRA) were searched for alternative translation initiation sites in 5′ untranslated regions (5′ UTRs) (Lee et al. 2012; Gao et al. 2015). The PROTEOFORMER pipeline was used to devise a list of alternative initiation sites that showed translation evidence based on the ribosome profiling data sets (Crappe et al. 2015). After clipping the adapter sequences using in-house software, Fastx, PROTEOFORMER starts by mapping the sequencing results of the two aforementioned data sets using the STAR aligner (Dobin et al. 2013). The mapping was executed nonuniquely with best rank selection (by using the “outSAMmultNmax” parameter of the STAR aligner). To pinpoint the alignments to their corresponding p-sites, offsets were calculated using the Plastid tool (Dunn and Weissman 2016). Transcript structures were built using the reference annotation from Ensembl version 87. The resulting read counts were then used to determine translated TISs with a minimal R-value of 0.05 and a minimum TIS count of 10. The R-value is defined as:
with Xk = number of reads on position x for data k, Nk = total number of reads on the transcript for data k, and k = initiation-seq (using lactimidomycin) or Ribo-seq (using cycloheximide).
Starting from these called TISs, open reading frames were constructed to check for complete translated sequences, as only TISs that lead to a valid ORF were used for validation. In this process, splicing and SNPs were taken into account. The alternative TISs pinpointed by the PROTEOFORMER ribosome profiling analysis were compared to the TISs annotated by the proteome analysis performed in this study by using a range of seven nucleotide positions up- and downstream around each TIS position. Sequences were also checked manually for sequence similarity.
Alignment of acetylated peptides with ribosome profiling data for the manual validation
Two different sets of ribosome profiling data performed for the annotation of TISs with HEK293 cells were downloaded from the GWIPS site, and the Ribo-seq read numbers were combined for each chromosome coordinate (Lee et al. 2012; Michel et al. 2014; Gao et al. 2015). The chromosome coordinates of the ribosome profiling data were lifted from hg19 to hg38 on the UCSC Genome Browser. The ribosome profiling data and acetylated peptides were aligned with in-house generated software written in Python programming language (Supplemental Scripts).
Sequence alignment and motif analysis
A homologous gene database was downloaded from Homologen of NCBI, and genes and mRNA accessions corresponding to the genes for the proteins with N-terminal extension were retrieved. Sequence from stop codon to stop codon was taken and translated. Sequence alignments were performed with Clustal Omega (Sievers et al. 2011). Sequence logo analysis was done with WebLogo (Crooks et al. 2004). DNA or protein sequences were taken and sequence logo was generated in probability mode.
Data access
All mass spectrometry data and search results from this study have been submitted to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD006633) via the PRIDE partner repository with the data set identifier PXD006633 and project name “Discovery of noncanonical translation initiation sites through mass spectrometric analysis of protein N-termini.”
Supplementary Material
Acknowledgments
This study was supported by a grant from the National Cancer Institute (CA184165 to A.P.) and NCI's Clinical Proteomic Tumor Analysis Consortium initiative (U24CA210985 to A.P.). We acknowledge support of a grant (to M.S.K.) from the International Research & Development Program of National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (Grant number: NRF-2016K1A3A1A47921601). G.M. is a fellow of the Research Foundation–Flanders (FWO-Vlaanderen, 12A7813N). We acknowledge support of the Center for Proteomics Discovery at Johns Hopkins and a shared instrumentation grant S10OD021844. We thank Dr. Rabih Jabbour of the US Army Edgewood Chemical Biological Center for access to the Orbitrap Fusion Tribrid mass spectrometer.
Author contributions: C.H.N. and A.P. designed research; C.H.N., M.A.B., M.S.K., O.P., and M.K.H. performed research; C.H.N., A.P., J.C.T., S.V., S.M.E., G.M., and C.M.O. analyzed data; C.H.N., A.P., C.M.O., S.V., G.M., M.K.H., and J.C.T. wrote the paper; A.P. supervised research.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.226050.117.
References
- Aksnes H, Hole K, Arnesen T. 2015. Molecular, cellular, and physiological significance of N-terminal acetylation. Int Rev Cell Mol Biol 316: 267–305. [DOI] [PubMed] [Google Scholar]
- Crappe J, Ndah E, Koch A, Steyaert S, Gawron D, De Keulenaer S, De Meester E, De Meyer T, Van Criekinge W, Van Damme P, et al. 2015. PROTEOFORMER: deep proteome coverage through ribosome profiling and MS integration. Nucleic Acids Res 43: e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, Weissman JS. 2016. Plastid: nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics 17: 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowska D, Verbruggen S, Ndah E, Jonckheere V, Menschaert G, Van Damme P. 2017. eIF1 modulates the recognition of suboptimal translation initiation sites and steers gene expression via uORFs. Nucleic Acids Res 45: 7997–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frottin F, Martinez A, Peynot P, Mitra S, Holz RC, Giglione C, Meinnel T. 2006. The proteomics of N-terminal methionine cleavage. Mol Cell Proteomics 5: 2336–2349. [DOI] [PubMed] [Google Scholar]
- Gao X, Wan J, Liu B, Ma M, Shen B, Qian SB. 2015. Quantitative profiling of initiating ribosomes in vivo. Nat Methods 12: 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko MV, Su D, Gladyshev VN. 2010. CUG start codon generates thioredoxin/glutathione reductase isoforms in mouse testes. J Biol Chem 285: 4595–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. 2014. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem 83: 779–812. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJ, Jackson SE, Wills MR, Weissman JS. 2014. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep 8: 1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. 2014. A draft map of the human proteome. Nature 509: 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleifeld O, Doucet A, auf dem Keller U, Prudova A, Schilling O, Kainthan RK, Starr AE, Foster LJ, Kizhakkedathu JN, Overall CM. 2010. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat Biotechnol 28: 281–288. [DOI] [PubMed] [Google Scholar]
- Kleifeld O, Doucet A, Prudova A, Keller UAD, Gioia M, Kizhakkedathu JN, Overall CM. 2011. Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat Protoc 6: 1578–1611. [DOI] [PubMed] [Google Scholar]
- Lange PF, Huesgen PF, Nguyen K, Overall CM. 2014. Annotating N termini for the human proteome project: N termini and Nα-acetylation status differentiate stable cleaved protein species from degradation remnants in the human erythrocyte proteome. J Proteome Res 13: 2028–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Liu B, Lee S, Huang SX, Shen B, Qian SB. 2012. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci 109: E2424–E2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menschaert G, Van Criekinge W, Notelaers T, Koch A, Crappe J, Gevaert K, Van Damme P. 2013. Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol Cell Proteomics 12: 1780–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AM, Fox G, M Kiran A, De Bo C, O'Connor PB, Heaphy SM, Mullan JP, Donohue CA, Higgins DG, Baranov PV. 2014. GWIPS-viz: development of a ribo-seq genome browser. Nucleic Acids Res 42: D859–D864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeys Y, Abeel T, Degroeve S, Van de Peer Y. 2007. Translation initiation site prediction on a genomic scale: beauty in simplicity. Bioinformatics 23: i418–i423. [DOI] [PubMed] [Google Scholar]
- Schwab SR, Shugart JA, Horng T, Malarkannan S, Shastri N. 2004. Unanticipated antigens: translation initiation at CUG with leucine. PLoS Biol 2: 1774–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendoel A, Dunn JG, Rodriguez EH, Naik S, Gomez NC, Hurwitz B, Levorse J, Dill BD, Schramek D, Molina H, et al. 2017. Translation from unconventional 5′ start sites drives tumour initiation. Nature 541: 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck SR, Jiang VV, Pavon-Eternod M, Prasad S, McCarthy B, Pan T, Shastri N. 2012. Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science 336: 1719–1723. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Gawron D, Van Criekinge W, Menschaert G. 2014. N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Mol Cell Proteomics 13: 1245–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Qian SB. 2014. TISdb: a database for alternative translation initiation in mammalian cells. Nucleic Acids Res 42: D845–D850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems P, Ndah E, Jonckheere V, Stael S, Sticker A, Martens L, Van Breusegem F, Gevaert K, Van Damme P. 2017. N-terminal proteomics assisted profiling of the unexplored translation initiation landscape in Arabidopsis thaliana. Mol Cell Proteomics 16: 1064–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JW, Cagney G. 2010. An overview of label-free quantitation methods in proteomics by mass spectrometry. Methods Mol Biol 604: 273–283. [DOI] [PubMed] [Google Scholar]
- Xiao Q, Zhang F, Nacev BA, Liu JO, Pei D. 2010. Protein N-terminal processing: substrate specificity of Escherichia coli and human methionine aminopeptidases. Biochemistry 49: 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H, Tuller T. 2013. New universal rules of eukaryotic translation initiation fidelity. PLoS Comput Biol 9: e1003136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.