

Abstract
Apolipoprotein (apo)A-I is an organizing scaffold protein that is critical to high density lipoprotein (HDL) structure and metabolism, likely mediating many of its cardioprotective properties. However, HDL biogenesis is poorly understood as lipid-free apoA-I has been notoriously resistant to high resolution structural study. Published models from low resolution techniques share certain features but vary considerably in shape and secondary structure. To tackle this central issue in lipoprotein biology, we assembled an unprecedented team of lipoprotein structural biologists and set out to build a consensus model of monomeric lipid-free human apoA-I. Combining novel and published cross-link constraints, small angle X-ray scattering (SAXS), hydrogen-deuterium exchange (H-DX) and crystallography data, we propose a time averaged model consistent with much of the experimental data published over the last 40 years. The model provides a long sought platform for understanding and testing details of HDL biogenesis, structure and function.
Keywords: apolipoprotein A-I, structure, cross-linking, mass spectrometry, lipid-free, oligomerization
Apolipoprotein (apo)A-I is the most abundant protein in high density lipoprotein (HDL). Although HDL contains up to 95 ‘accessory’ proteins (http://homepages.uc.edu/∼davidswm/HDLproteome.html) that modulate particle function, apoA-I is central to mediating HDL's structure, interactions and function. Indeed, it is a cofactor for lecithin:cholesterol acyl transferase, mediating HDL maturation via the esterification of free cholesterol in plasma1. Lipid-free (poor) apoA-I is also critical for HDL biogenesis via the cell surface transporter ATP-binding cassette A1 (ABCA1)2. Thus, there has been a sustained effort to understand apoA-I structure in both its lipid-free and -bound states. Lipid-free monomeric apoA-I represents the protein in its most nascent form. As such, it is a logical starting point for the investigation into the structural steps of HDL biogenesis.
Unfortunately, apoA-I is difficult to study by traditional protein structural analyses like X-ray crystallography or nuclear magnetic resonance (NMR). It has a tendency to self-associate, bind hydrophobic substances, and exceptional structural flexibility. Indeed, multiple laboratories have been trying to crystalize the full-length protein for decades without success. However, two groups published crystal structures of tetrameric3 and dimeric4 truncation mutants of lipid-free apoA-I. The former, lacking the N-terminal 44 amino acids, was widely thought to mimic apoA-I configuration on a nascent, discoidal HDL particle. The latter structure lacked the C-terminal 58 amino acids and may be more reflective of lipid-free apoA-I. Independent studies of the same truncation mutant showed that most of the structural features of the crystal structure were borne out in solution5. These reports have provided critical insights into structural details of lipid-free apoA-I. However, they must still be extrapolated to the in vivo condition where i) the N- and C-termini are both present and ii) the protein is in aqueous solution.
The difficulty of crystallizing full-length apoA-I has led to hundreds of studies using clever applications of lower resolution techniques such as circular dichroism (CD)6, analytical ultracentrifugation7, NMR8, fluorescence spectroscopy9, molecular dynamics10, chemical cross-linking11, hydrogen-deuterium exchange12, electron paramagnetic resonance spectroscopy (EPR)13,14, and others15. Cross-linking has been particularly useful for 3-D models because it provides distance constraint data under physiological conditions in solution. To date, there have been eight models proposed for full-length, monomeric apoA-I from four laboratories10,11,16,17. Examples are shown in Fig. 1. They all convey the generally held view that lipid-free apoA-I is a four-helix bundle in a pseudo-stable state poised to interact with lipid. However, looking more deeply, the models differ widely in shape, secondary structure placement, and the location of the N- and C-termini. Thus, the field still lacks the structural detail to understand how apoA-I binds lipids, interacts with ABCA1 to form mature HDL, exchanges between HDL, and mediates particle interactions with plasma-borne remodeling factors.
Figure 1. Published models of full-length lipid-free human apoA-I.
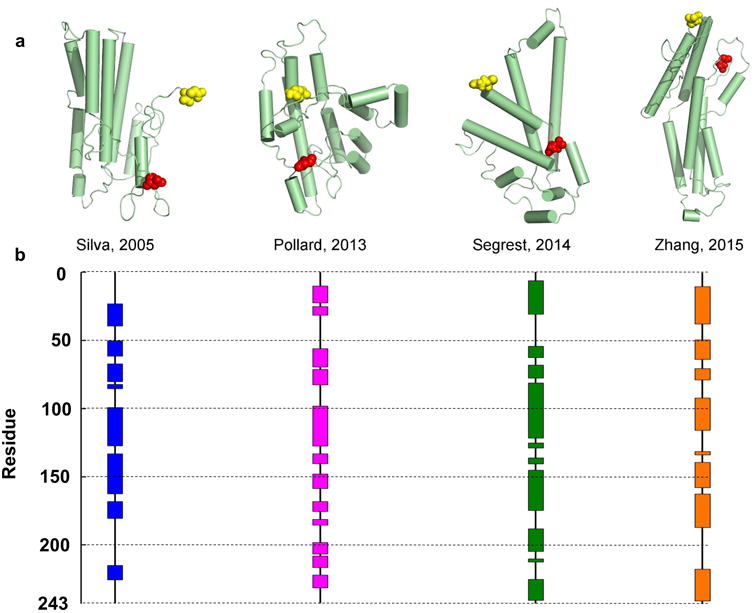
There are 8 published models of lipid-free monomeric apoA-I that have coordinates available. Silva et al.11 proposed a model based on chemical cross-linking and homology sequence threading techniques. Pollard et al.16 generated their model using chemical cross-linking and cysteine point mutations. Segrest et al.10 used chemical cross-linking and molecular dynamics simulations using information from the crystal structures of apoA-I truncation mutants as templates. Note: this paper also proposed 3 other models that differed in terms of length of simulation (one of which is shown here, 15 ns). Zhang et al.17 produced molecular simulations using information from the crystal structures as initial templates. The models are shown in cartoon form with cylinders representing helical domains in panel (a). The N-terminal amino acids are shown space-filled in red while the C-terminal amino acids are in yellow. The alpha helical content of each model (as assessed by Pymol) is shown on the linear diagram for each model in panel (b) with each colored block representing helical domains and black lines representing random coil or loop regions.
In the absence of a high-resolution structure of lipid-free apoA-I but with the benefit of nearly 5 decades of spectroscopic and cross-linking data, we assembled a group of leading apoA-I structural investigators, the ApoA-I Working Group, to derive a consensus model that consolidates the apoA-I structural literature. In addition, we report new cross-linking data to round out a universal list of 95 distance constraints from 3 laboratories, utilizing multiple cross-linking reagents. We also included new molecular shape data from small-angle x-ray scattering (SAXS) and incorporated secondary structural information from hydrogen-deuterium exchange (H-DX)12. Using the crystal structure of dimeric apoA-I4 as a template, we applied computer modeling and simulation techniques to derive the new model.
Results
Cross-linking
We used the homo-bifunctional cross-linkers CBDPS (Cyanur-biotin-dimercapto-propionyl-sulfo-succinimide) and BS3 (bis-sulfosuccinimidyl suberate), both NHS esters which cross-link lysine and, to a lesser extent, serine residues18,19. Wild-type (14NN) and isotopically labeled (15N) apoA-I were mixed 1:1 under denaturing conditions. This allowed i) unambiguous peptide identification and ii) confirmation that cross-links were not due to oligomerization5. After cross-linking, monomeric apoA-I was isolated by size exclusion chromatography (SEC). We routinely obtained >95% purity of cross-linked monomeric apoA-I as shown by SDS-PAGE (Supplementary Fig. 1). Supplementary Tables 1 and 2 list the 65 cross-linked peptide pairs identified (see Methods for criteria). All cross-links exhibited a dual peak pattern indicating intramolecular span5,20. 45 were identified in both BS3 and CBDPS treated samples with 15 unique to BS3 and 5 unique to CBDPS.
Small-Angle X-ray Scattering (SAXS)
We used SAXS to assess the shape of monomeric apoA-I in solution. ApoA-I's propensity for concentration dependent self-association posed a challenge since reliable SAXS data collection requires concentrations up to 4 mg/ml. We circumvented this by performing SAXS on apoA-I that had been locked into the monomeric state by cross-linking with BS3 or CBDPS (see Methods). Careful evaluation by SEC showed that this prevented further oligomerization. The SAXS parameters are shown in Supplementary Table 3. For both cross-linking reagents, scattering intensity increased proportionally with sample concentration and the Guinier range was linear at low scattering angles (not shown), indicating no concentration dependent effects and good data quality. No significant differences were observed in Rg (Guinier and real space), Dmax, and comparable molecular volumes were found across all concentrations. Additionally, scattering profiles and pairwise distribution plots for both samples were highly related (Fig. 2a and Fig. 2b). There may be small differences in the flexibility of differentially cross-linked apoA-I species. ApoA-I cross-linked with CBDPS shows a dip in q3·I(q) plot (Fig. 2c) and a plateau in the q4·I(q) plot (Fig. 2d) indicative of a folded, rigid structure. The BS3 treated sample exhibited a plateau in the q3·I(q) plot and lack of plateau in the q4·I(q) plot indicating a possible flexible domain. The deviation in dynamics is likely due to differences in cross-linking efficiency as much less CBDPS is required for linking oligomers vs. BS3. The Rg of both samples were similar (25.35 ± 0.15 Å and 25.34 ± 0.17 Å for CBDPS and BS3, respectively) and, taken with the pairwise distribution plots, suggested apoA-I had characteristics of a globular protein. Twenty-three independent envelope reconstructions were performed and averaged using DAMMIF21 to generate a composite. Both DAMMIF reconstructions had normalized spatial discrepancies (NSD) between 0.5 and 0.7 (Supplementary Table 3) indicating good convergence of the independent reconstructions. The envelopes (Fig. 2e and Fig. 2f) serve as a low-resolution representation of the overall shape of the molecule which can be used as a rough tool for visualizing the fit of a model. ApoA-I cross-linked with CBDPS had a calculated volume of ∼69,400 Å3 compared to ∼79,500 Å3 for that cross-linked with BS3, consistent with the more flexible nature of the BS3 sample. Taken together, the data suggest minimal structural differences between the samples.
Figure 2. Molecular envelopes of monomeric apoA-I using SAXS.
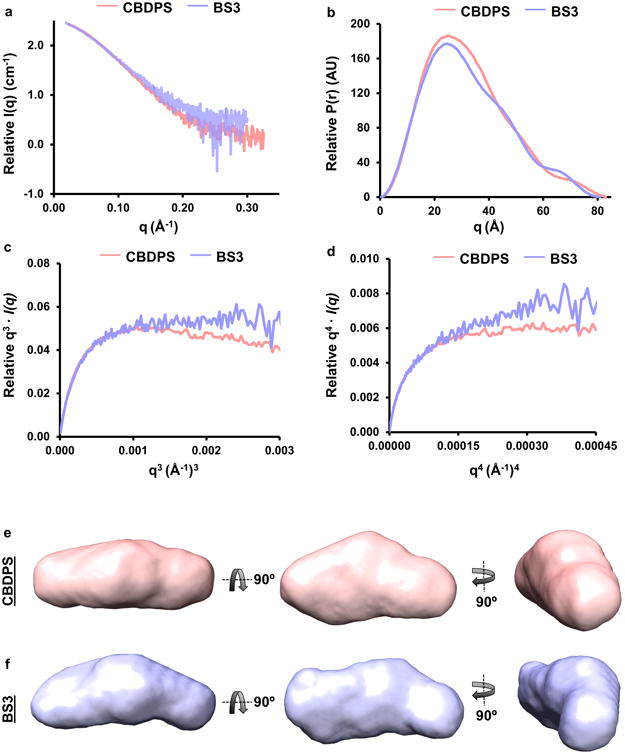
Plasma and recombinant apoA-I were cross-linked with CBDPS and BS3 and monomeric species were isolated by gel filtration chromatography and subjected to SAXS. Panel (a): Scattering profiles of monomeric apoA-I cross-linked with CBDPS and BS3. Panel (b): P(r) curves of monomeric apoA-I cross-linked with CBDPS and BS3. Panels (c and d): Flexibility plots of monomeric apoA-I cross-linked with CBDPS and BS3. Panels (e and f): DAMMIF ab initio molecular envelopes of recombinant apoA-I cross-linked with CBDPS and BS3, respectively.
Generation and Evaluation of the Model
The starting construct of the model was derived from the crystal structure of apoA-I1-184 as shown in Supplementary Figure 2. As Mei and Atkinson4 proposed, an inflection point was first placed in H5, near the center of the long helix shared by two protomers (A and B) in the dimer. However, guided by cross-linking constraints and SAXS, the inflection point was shifted more C-terminal to residue 139 in helix 5. The C-terminal portion of monomer A was then folded back resulting in the juxtaposition of H6(A) with H5(A) (Supplementary Figure 2b). Using Pymol and Modeller v9.14, the missing residues 185-243 were threaded in and positioned guided by the relevant cross-linking constraints10,11,16 in Supplementary Table 4. This construct was used as a base model for further refinement. Secondary structural data derived exclusively from H-DX12 was next implemented onto the base model. This required conversion of helical segments observed in the crystal structure to random coil (and vice versa in some cases). Next, the entire model was simultaneously refined for fit to the SAXS scattering curves and the cross-linking data, while holding the secondary structure assignments as constant as possible. Lastly, the model was subjected to energy minimization using YASARA22 and side chain rotamers were manually refined in COOT23. Please see Methods for more detail on each of these steps.
The finalized model (Fig. 3) depicts monomeric lipid-free apoA-I as a helical bundle composed of three main helices: H1 (residues 8-35), H5 (81-115), and H6 (148-179). Three shorter helical regions are interspersed between these: H2 (37-45), H3 (54-64), and H4 (68-78). The remainder is random coil with two main stretches spanning residues 116-147 and the entire C-terminus 180-243. The C-terminus forms a globular domain that sits on the bundle at the N-terminal end of H5 and the C-terminal end of H6. The N- and C-termini are located near each other at the base of the bundle and opposite the bulk of the C-terminal globular domain. Proline residues at positions 7 and 66 reside in regions of transition between helix and random coil with residue 66 clearly mediating a turn structure. While most of the other prolines are located in random coil regions, prolines 99 and 165 occur in the middle of helix 5 and 6, respectively, without apparent disruption.
Figure 3. All atom model of full-length, lipid-free monomeric apoA-I.
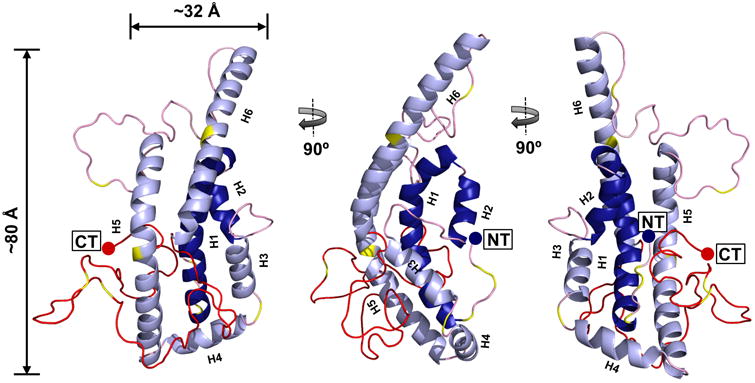
The final time-averaged model is shown where dark blue represents N-terminal helical α-helical domains H1 and H2, light blue represents α-helical domains H3, H4, H5 and H6, pink represents random coil, red represents the C-terminal 59 residues absent in the crystal structure and yellow shows the position of prolines on each model.
Compatibility of the model with experimental data old and new
A contact plot of the final structure is shown in Fig. 4a with experimental cross-links superimposed in blue and violations in red. Overall, 89 of 95 cross-links (both previously published and newly reported here) were consistent with the model. It is worth noting that this is 3-5x the number of constraints used in previous modeling attempts, significantly reducing the conformational possibilities that can fit the data. The six violations were, on average, within ∼2 Å of the allowable span. Fig. 4b shows the model agreement with the H-DX data of Chetty et al12. The new model has 235 of 243 residues (97%) assigned in keeping with the H-DX data. The small differences resulted from tradeoffs made for optimal cross-link and SAXS data fitting. These occurred near residue 36 which is predicted to form the minor H2 helix (also observed in the crystal structure4). The remaining differences involved 1-2 amino acids bookending α-helical regions. Fig. 4c shows the model superimposed onto both DAMMIF ab initio molecular envelopes generated from recombinant apoA-I cross-linked with CBDPS and BS3. The primary measure of agreement to SAXS was the model fit to the scattering curves. Fig. 4d and 4e shows that the model was in good agreement with both SAXS scattering curves (χ=1.13 and χ=0.81 for CBDPS and BS3, respectively). As a point of reference, our previous work compared a crystal structure of a truncation mutant of apoA-IV with similarly obtained SAXS data with a χ=0.97.
Figure 4. Evaluation of experimental cross-links, secondary structure, and SAXS data with respect to the model.
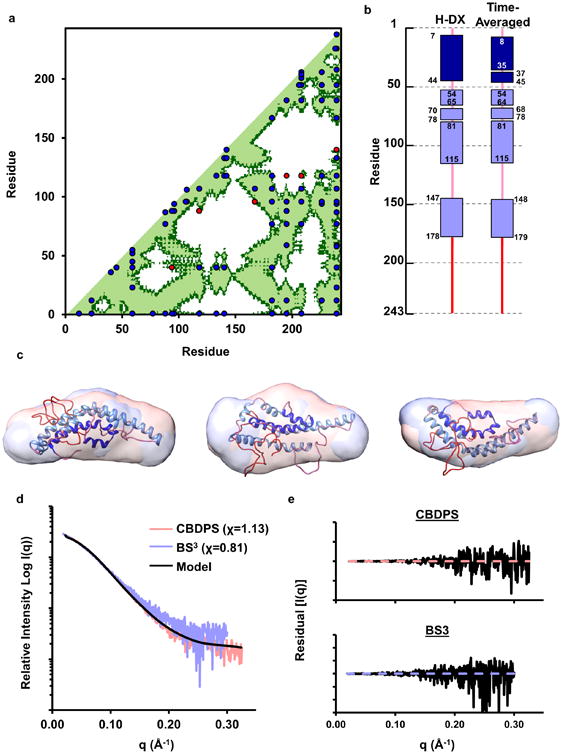
Panel (a): Cross-linking data Supplementary Table 4 containing all published cross-links reported by Melchior, Silva11, Pollard16, and Segrest10. The X and Y axis of the plots indicate the residue number of apoA-I from 1-243. The diagonal line bisects the figure and the bottom/right shows intramolecular interactions. Dark green represents residues within 28 Å (upper limit for alpha-carbons cross-linked with CBDPS or BS3) and light green represents residues within 23 Å (upper limit for alpha-carbons cross-linked with MDA). Blue dots represent experimental cross-links that fall within the upper-limit distance constraint and red dots represent show those that exceed the allowable distance on the time-averaged model. Panel (b): Comparison of the secondary structure reported in the H-DX study by Chetty et. al.12 and the time-averaged model. Rectangles represent α-helical segments and the lines represent random coil. Panel (c): Superposition of the model on molecular envelopes derived from monomeric apoA-I cross-linked with CBDPS and BS3. Panel (d): The theoretical scattering curve of the proposed model (black line) superimposed on the experimental scattering profile of apoA-I cross-linked with CBDPS and BS3. The fit to the respective scattering profiles are shown in parenthesis. Panel (e): The residual plot of the model (black) to experimental scattering data from apoA-I cross-linked with CBDPS and BS3.
We also reviewed the literature for additional structural data for lipid-free apoA-I collected under conditions where it was monomeric. Supplementary Table 5 summarizes numerous studies that used far UV circular dichroism (CD) to estimate α-helix and random coil contents. α-helical content ranged from 40% to 68% across 27 studies for an average of 52 ± 6 %, or 126 ± 14 residues. The new model, driven primarily by the H-DX restraints, matched exactly with 126 α-helical residues. Another technique for identifying areas of low structural organization is limited proteolysis. Supplementary Table 6 summarizes two studies that applied this technique to primarily monomeric apoA-I. Proteolytically susceptible sites (highlighted on the new model in Figure 5a) tended to occur in random coil areas near the molecule surface. The solvent accessibility of these sites in the new model, as assessed by VADAR24, was highly consistent with the proteolysis data with only two of 13 reported cleavage sites (Tyr115, Phe57) showing solvent accessible areas of less than 20 Å2.
Figure 5. Predictions of the new model with respect to proteolytic susceptibility and stability.
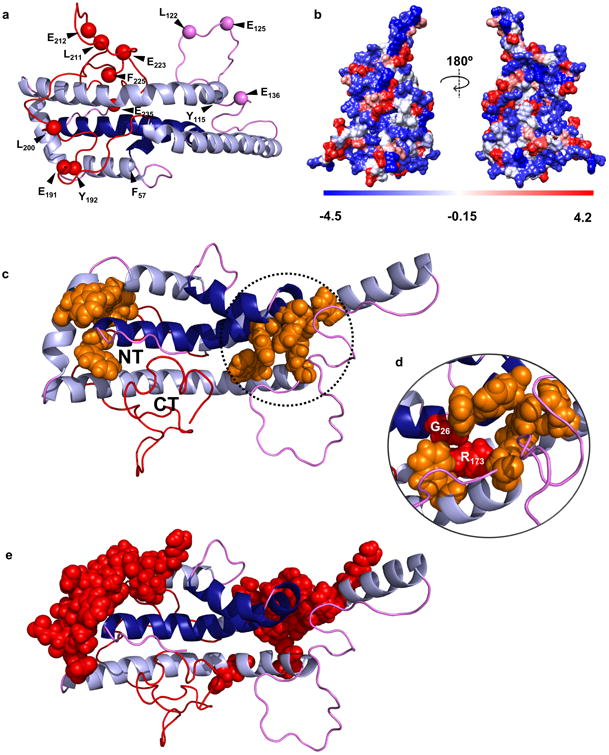
Panel (a) shows the proteolytic cleavage sites from Supplementary Table 6. Panel (b) shows a space-filled rendering of the model by residue hydrophobicity with hydrophilic residues in blue and hydrophobic residues in red. Panel (c) highlights aromatic clusters (orange) that may participate in molecular stabilization and the proximity of the N- and C-termini in the time-averaged model. Panel (d) shows the position of natural mutations in apoA-IIOWA (Gly 26) and apoA-IMilano (Arg 173) within one aromatic cluster. Panel (e) shows critical stabilizing residues mapped by Gorshkova et. al. for lipid-free apoA-I33,42.
Supplementary Figure 3 summarizes how the new model compares to the 8 published models of monomeric apoA-I with respect to the experimental data outlined above. In most cases, the new model showed considerably improved compatibility with the data.
Discussion
Lipid-free apoA-I is of direct physiological relevance because it and its related forms (i.e. preβ1-HDL) are essential to de novo HDL biogenesis via ABCA1-mediated efflux of phospholipid and cholesterol, a critical anti-atherogenic pathway in human plasma25. This involves direct interaction of apoA-I with both ABCA1 and specialized domains in the plasma membrane 26,27. The concentration of total apoA-I in human plasma is about 130 mg/dL (∼50 μM). At 5-10% of total apoA-I28, lipid-free apoA-I in plasma is around 2 μM and should be mostly monomeric. In peripheral lymph (interstitial fluid) it is about 10-fold less concentrated (∼0.2 μM or about 5 μg/ml) corresponding to the Km for lipid efflux via ABCA129. Thus, a detailed understanding of apoA-I in its most “nascent” biophysical state (lipid-free and monomeric) will aid in our understanding of HDL biogenesis and its eventual interaction with critical HDL mediators such as plasma lecithin:cholesterol acyl transferase. This problem has been pursued intensively for decades but a high resolution structural model of apoA-I's native form has remained elusive. The model described here represents a comprehensive multi-technique attempt to address this issue given apoA-I's recalcitrance to traditional high resolution structural techniques. We first describe the features of the model, touch on its implications for understanding HDL biology and then discuss its limitations.
Figures 3 and 5 show that the model is consistent with previous proposals of a two-domain structure with a relatively organized N-terminal helical bundle and a mostly random coil C-terminal domain from residues 180-243. With respect to the C-terminal domain, we point out that our methodology does not fully preclude the existence of some helical structure, particularly highly unstable helices that cannot be not detected at the timescale of the H-DX experiments. Indeed, truncation of the C-terminus from residue 243 to residues 231 and 221 reduced CD-detectable helix content by 7 and 14 amino acid residues, respectively30,31. On the basis of such observations, Mei and Atkinson31 suggested that the segment spanning residues 231-241 contains α-helical structure. Such a C-terminal helix is further supported by preliminary molecular dynamics simulations of the new model (Segrest et al, unpublished observation). Overall, the C-terminal domain sits on N-terminal α-helical domains H1 and H2 (dark blue) (Fig. 5) with the termini residing close to each other. This theme was also observed in the solution structure for apoA-IV20 and may explain structural and energetic studies indicating that the N- and C-terminal domains of apoA-I act cooperatively to modulate its stability32 and lipidation kinetics5,33-36. The space-filling illustration in Fig. 5b shows that the model is well packed, particularly in the N-terminal domain, with hydrophobic faces of the helices oriented toward the bundle core. However, there are clear instances of solvent exposed hydrophobic residues (red), possibly contributing to the protein's low thermodynamic stability37. Another intriguing feature is two potential networks of aromatic residues (orange) (Fig 5c). Aromatic residues play important roles in protein stability, protein-protein interactions, and tertiary folding38. Indeed, natural mutations such as G26R in apoA-IIOWA39 and the R173C mutation in apoA-IMILANO40 both result in substantial decreases in free energy of denaturation of lipid-free apoA-I41 and both are central to an aromatic cluster that potentially stabilizes helices 1, 5, and 6 (Fig. 5d). Lastly, residues reported by Gorshova and colleagues33,42 to be critical for apoA-I stability (Fig 5e, space-filled in red) exhibit remarkable co-localization with the aromatic clusters highlighted in Fig. 5c. The model predicts that additional mutations in these areas should affect stability and likely the propensity to oligomerize and interact with lipid.
We considered the new model in light of apoA-I's propensity to self-associate. Fig. 6 compares the consensus model to the crystal structure of dimeric apoA-I1-184 and its monomer as postulated by Mei et al 4. Comparing Fig. 6b and 6c, the full-length model shares numerous features with the postulated monomer. Both models show an N-terminal helix between residues 7 and 36 (blue) situated alongside another helix composed of residues 80-116 (green/yellow). Though these helices are bent more acutely in the full-length model, the inter-helical interactions are similar. Also, the N-termini of both models are in close proximity to a short helix of residues ∼66-76. The full-length model shows a large random coil region between residues 116-147 that acts as a turn allowing H6 (148-178, red) to double back and participate in the helical bundle. Although more ordered in the crystal model, a similarly centered turn (near residue 129) generates an intramolecular helical bundle. The main difference is that, in the full-length model, the position of H6 is shifted relative to the bundle, perhaps by the presence residues 185-243 that sit against the bundle (note how the red helix in Fig. 6c is shifted to the right vs 6b). Given the relative agreement with the monomeric model derived from the crystal structure, it follows that the full length protein may dimerize similarly as the crystal structure dimer. Fig. 6d and 6e show how this might occur. By placing a hinge point near residue 129, the C-terminal portion of full-length apoA-I can be extended to interact with a similarly extended apoA-I partner. Aside from the increased random coil structure in the consensus model dimer, many of the intermolecular alignments seen in the apoA-I1-184 dimer are highly plausible. Thus, we propose that the dimer/monomer conversion scheme proposed by Mei et al. for apoA-I1-184 is compatible with the new consensus model and may describe how the full-length protein dimerizes. Further experimental work will be required to test this hypothesis.
Figure 6. Comparison of the consensus apoA-I model to the apoA-I1-184 crystal structure.
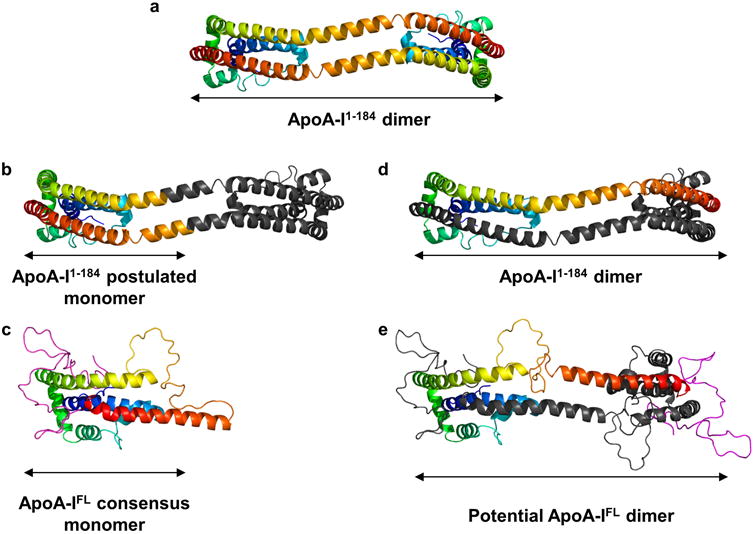
Panel (a): The dimeric apoA-I1-184 crystal structure is shown in cartoon rendering. For each protomer, the N-terminus is blue and gradates through cyan, green, yellow, and orange to the C-terminus (residue 184 in this case) in red. Panel (b) A postulated monomeric form of apoA-I1-184 as proposed by Mei et al.4. An inflection point was placed near the center of the long helix shared by two protomers (A and B) at about residue 126. Monomer A is rendered with the same color gradient as panel (a) until residue 126, at which point all remaining residues are colored gray. With its N-terminal portion colored gray, the C-terminal portion of monomer B was colored in a continuation of the gradient as though it was a single molecule with a turn introduced as indicated by the arrow. Panel (c): The consensus model of full-length apoA-I colored the same way as panel (a) with residue 182 being red. The residues that are missing from the crystal structure are colored in magenta. Panel (d): The apoA-I1-184 crystal structure rendered with all of molecule B in gray. Panel (e): Postulated model of dimeric full-length apoA-I. Molecule A is colored as for the monomer in panel (c) with residues 183-243 colored magenta. Molecule B is entirely gray.
We also considered the implications of the new model for apoA-I lipid-binding and HDL biogenesis. There is a general consensus that lipid-binding is initiated when the relatively unstable C-terminal domain comes in contact with a lipid surface31,43-45, either via a packing defect46 or an ABCA1 mediated mechanism26. It is easy to visualize the periodic movement of the C-terminus away from the helical bundle in the consensus model. If it encounters lipid, the formation and/or stabilization of C-terminal helices provides a favorable ΔG30 that drives further lipid association. There is also evidence that the N-terminus may play a role in initial lipid contact35,47. Freed of the C-terminal domain, the bundle helices can then unpack, reorienting the hydrophobic faces of the central part of apoA-I to penetrate the lipid surface35. One of the main benefits of having the consensus structure is that it gives a ‘road map’ to the initial position of the helices and offers a structural basis for how they may unpack during this transition. For example, Saito et al. speculated that H1 and H2, based on their similar low stabilities, may be in close opposition and unfold as a unit30 early on in the lipid binding process. Our model shows that H2 (in our case, residues 50-80) forms a bent helix that actually wraps around H1, supporting this idea.
There is considerable debate as to the exact subsequent steps involved in the creation of a nascent HDL particle. Key questions remain as to the role and timing of apoA-I dimerization (i.e. does it happen prior to, during or after lipidation?) and the exact mechanism and role of ABCA1 (does it play a role in apoA-I dimer formation and how does it physically load lipid onto apoA-I?). A recent cryo-EM structure of ABCA1 offers interesting possibilities48, but a consensus on how this works has yet to materialize. In Figure 7, we show a simplified scheme consistent with that proposed by Mei et. al.31 of how the consensus model could be unpacked to generate a ring-like conformation as proposed for the double belt structure of apoA-I in discoidal HDL particles49. We caution that this is only one of many possibilities and the role of apoA-I dimerization and ABCA1 was intentionally not addressed. Now that we have good ideas of the structures of both apoA-I and ABCA1, we look forward to exciting new work on this critical mechanism.
Figure 7. Hypothesis on lipid-binding and relationship to apoA-I arrangement on discoidal HDL.
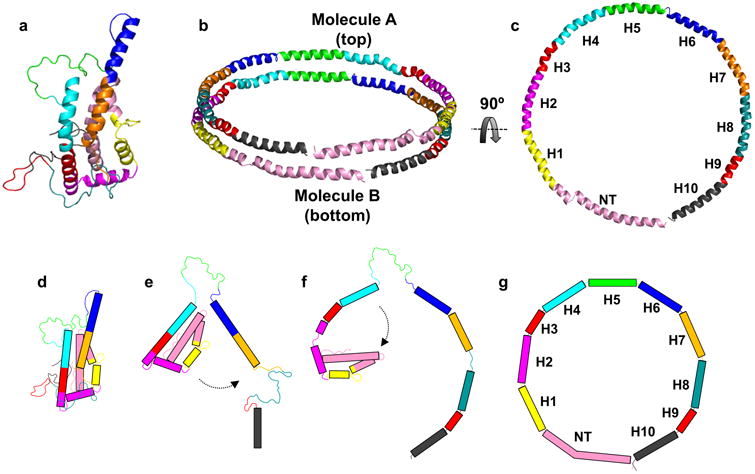
Panel (a): The consensus model of full-length apoA-I. Panel (b): Proposed arrangement of two molecules of apoA-I on discoidal HDL; the “double-belt” model. Panel (c): Molecule B from the double-belt model rotated 90° (top view). Panel (d): Cartoon representation of the time-averaged model in its folded state. Panel (e): Dissociation of helix 6 and C-terminus from the lipid-free model and formation of a helical region upon interaction of the CT with lipid. Panel (f): formation of additional alpha-helical segments upon lipid filling and unfurling of the N-terminal bundle. Panel (g): Endpoint as a belt on an HDL particle. The model and cartoon is colored based on alpha-helical repeats identified and reported by Segrest et. al. 49. Rectangles in panels (d-g) represent alpha-helical segments.
Limitations of the model
Despite its consistency with much of the known structural data, we note that the model is still limited in resolution compared to NMR or X-ray crystallography. The general backbone configuration is likely correct, but more refined molecular interactions such as salt-bridging and hydrogen bonding remain unclear. Additionally, one might question whether it is possible to represent a highly dynamic protein with a single, time-averaged model. ApoA-I has ΔG of helix stabilization ranging from ∼3-5 kcal/mol12. At neutral pH and room temperature, H-DX studies have demonstrated complete hydrogen exchange into apoA-I α-helical segments occurring in ∼10 minutes (Supplementary Figure 4); i.e. all apoA-I helical segments have unfolded and reformed at least once in this timeframe. To put this in context, a stable globular protein like cytochrome C has a ΔG of helix stabilization of 10 kcal/mol with complete hydrogen exchange requiring ∼10 weeks. Given this degree of dynamics, it seems likely that, at physiological temperatures, apoA-I probably exists in an ensemble of related structures at any point in time. Indeed, the fact that some cross-links failed to fit the model (6 were close, but not strictly allowed) directly suggests differential conformations. Our model is a time-averaged structure derived from experimental data obtained on a longer time scale than typical secondary structure oscillations. For this reason, we think of it as a base model upon which hypothesized dynamics and conformational alterations can be further modeled and tested. However, because the majority of cross-links did fit the model and the SAXS patterns showed consistent molecular shapes, we argue that i) the consensus model likely reflects a majority of the molecules existing at a given time or ii) any conformers exhibit related shapes and structures.
Another issue relates to the notion of solvent accessibility of the cross-linking reagents. While most cross-links fit the model in terms of Euclidian distance (‘as the crow flies’), many are impeded by some obstruction like a side chain from a non-participating residue or an adjacent helical domain. Given that the model represents a time-average of an ensemble of related apoA-I structures, cross-links that appear sterically hindered or solvent inaccessible could occur on an alternate conformation within the boundaries of the experimental system. Additional studies are needed to better define these boundaries and the extent of rearrangement apoA-I can achieve in vivo and in vitro. Also, the constant unfolding of helical domains over the course of the experiment could allow cross-links that might not be expected in a static structure. Nevertheless, previous reports have shown excellent consistency between observed cross-links in solution with crystal structures of apoA-I1-184 (5) and apoA-IV20 and non-apolipoproteins50-53 validating the approach. Importantly, instances of steric cross-linker hindrance were also routinely observed in these studies. For a more detailed discussion of potential limitations of the model, see the online supplement.
In summary, we propose a new time-averaged model of the structure of apoA-I in solution that is, by and large, consistent with decades worth of experimental data. While still lacking amino acid level resolution, it provides a much-needed starting point upon which to test structural hypotheses with regard to loss-of-function mutations, interaction with cell-surface proteins, and HDL formation.
Online Methods
Protein expression and purification
Recombinant apoA-I was expressed and purified as previously described54 with minor modifications. Briefly, a pET30 vector (Novagen) containing mature apoA-I (sequence confirmed by the Cincinnati Children's Hospital Sequencing Core on an Applied Biosystems 3730 DNA Analyzer) was transformed into BL-21 E. coli cells. Cells were grown at 37°C in Luria-Bertani (LB) culture media containing kanamycin to select transformants. Protein expression was induced by addition of isopropyl β-d-thiogalactopyranoside (IPTG) at 0.1 mM followed by shaking at 225 RPM for 2 h at 37°C. Cells were pelleted by centrifugation and stored at -20°C. Cells were thawed on ice, resuspended in binding buffer (5 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl, pH 7.9) and lysed at 4°C by probe sonication. The insoluble cell components were pelleted by centrifugation and supernatant was applied to His-bind columns (Novagen), which were extensively washed and eluted with elution buffer (1 M imidazole, 0.5 M NaCl, 20 mM Tris pH 7.9). The His-tag was cleaved by tobacco etch virus (TEV) protease at a ratio of 20:1 for 2 h at room temperature. The sample was reapplied to the His-binding resin to remove uncut protein. Fractions with purified protein were pooled, dialyzed into 10 mM ammonium bicarbonate pH = 8.1 and lyophilized for storage at -80°C until ready for use. Lyophilized protein was solubilized in a Tris buffer containing 3M guanidine HCl at concentrations below 0.1 mg/ml for 2 h at 4°C and then exhaustively dialyzed into appropriate buffer for experimentation. Our recombinant protein contains an N-terminal glycine which does not affect any protein structural or functional index that we have examined54. The 15N-labeled version of apoA-I was expressed and purified with minor modifications to the system as previously described20. The experimental molecular weight of 15N apoA-I was 28,483.7 Da determined by direct injection mass spectrometry. This translated to 342/348 successfully labeled nitrogen atoms which was >98% labeling efficiency.
Mixing of wild-type and isotopically labeled protein
Recombinant 14N and 15N apoA-I were solubilized separately in 3M guanidine in Tris-HCl (1.5 M NaCl, 100 mM Tris-HCl, 10 mM EDTA, 0.2% Azide), pH = 8.0 at a concentration below 0.1 mg/ml to ensure complete unfolding and dissociation of higher order oligomers. The species were then mixed at a 1:1 molar ratio and dialyzed against PBS (140 mM NaCl, 2.7 mM KCl, 10.1 mM Na2HPO4, 1.8 mM KH2PO4, pH=7.4) to refold the protein. The 14N/15N recombinant apoA-I was further purified using size exclusion chromatography using a Superdex 200 gel filtration column (10/300 GL; GE Healthcare) on a ӒKTA FPLC system (GE Healthcare) in PBS. Fractions containing pure apoA-I were pooled and concentrated to between 1-2 mg/ml for cross-linking. Protein concentration was determined by the Markwell modified Lowry assay55. Purity was routinely >99% as determined by SDS-PAGE and mass spectrometry.
Cross-linking and Isolation of Monomeric ApoA-I
ApoA-I was cross-linked with bis-(sulfosuccinimidyl) suberate (BS3) (Thermo Scientific) or Cyanur-biotin-dimercapto-propionyl-sulfo-succinimide (CBDPS), a homo-bifunctional, biotin-tagged cross-linking agent (Creative Molecules Inc.)56. Samples were cross-linked at molar ratios of 50:1 (BS3) or 20:1 (CBDPS) which were determined based on optimal formation of oligomeric species. The reactions proceeded for 12 h at 4°C and were quenched by excess Tris-HCl. Monomeric species of apoA-I were isolated by size exclusion chromatography on three Superdex 200 gel filtration columns (10/300 GL; GE Healthcare) in series on a ӒKTA FPLC system (GE Healthcare) in PBS at a flowrate of 0.3 ml/min. 0.25 ml fractions were collected and analyzed by SDS PAGE. Fractions containing purified monomeric apoA-I were pooled, concentrated and quantified. Monomers undergoing MS analysis were dialyzed into 10 mM ammonium bicarbonate pH 8.1. 50 μg aliquots were digested with sequencing grade trypsin (Promega) at 1:20 mass ratio of trypsin to protein for 16 h at 37°C. Peptides were lyophilized to dryness and stored at -20°C until analysis by MS.
Peptide Purification with CBDPS
We utilized the biotin tag on CBDPS to isolate peptides that had been modified by the cross-linker56. ApoA-I was cross-linked with CBDPS-H8/D8 at a molar ratio of 20:1 cross-linker to protein. Spin cups were loaded with 150 μg of Ultralink streptavidin resin (Pierce, 53117) and washed 5× with 300 μl PBS by centrifuging at 4000 × g for 1 min at RT. 100 μg of digested peptides per 150 μl of slurry were incubated together on an orbital shaker for 1 h at RT. Unlabeled peptides were removed by centrifugation at 4000 × g for 1 min at RT. The resin was washed once with PBS and then eluted with two aliquots of 300 μl of 50% acetonitrile, 0.4% trifluoroacetic acid. The peptides were then lyophilized to dryness for MS analysis.
Mass Spectrometry and Identification of Cross-linked Peptides
Mass spectrometry analyses were performed as previously described20. Briefly, Nano-LC-MS/MS analyses were performed on a TripleTOF® 5600+ (AB Sciex, Toronto, Canada) coupled to an Eksigent (Dublin, CA) NanoLC-Ultra® nanoflow system. Dried samples were reconstituted in formic acid/H2O 0.1/99.9 (v/v), and 5 μl (∼1-2 μg of digest) was loaded onto C18 IntegraFrit™ trap column (New Objective, Inc.) at 2 μl/min in FA/H2O 0.1/99.9 (v/v) for 15 min to desalt and concentrate the samples. For the chromatographic separation, the trap-column was switched to align with the analytical column, Acclaim® PepMap100 (Dionex-Thermo Fisher Scientific). Peptides were eluted at 300 nL/min using a mobile phase gradient from 95% phase A (FA/H2O 0.1/99.9, v/v) to 40% phase B (FA/ACN 0.1/99.9 v/v) for 35 min (1% per min), then from 40% B to 85% B in 5 min with re-equilibration. Effluent was introduced to the mass spectrometer using a NANOSpray® III Source (AB Sciex, Toronto, Canada). The instrument was operated in positive ion mode for 65 min, where each cycle consisted of one TOF-MS scan (0.25 s accumulation time, in a 350 to 1500 m/z window) followed by 30 information dependent acquisition mode MS/MS-scans on the most intense candidate ions selected from initially performed TOF-MS scan during each cycle. Each product ion scan was operated under vendor-specified high-sensitivity mode with an accumulation time of 0.075 s and CE of 43 with an 8 unit scan range. The .wiff files were converted to Mascot generic files using PeakView® v1.2.0.3 software (AB Sciex). Cross-links were identified using SimXL57 as previously described5. A user-defined error of 20 ppm was used for mass detection in all experiments. Two independent experiments were performed for apoA-I cross-linked with CBDPS and three independent experiments were performed for apoA-I cross-linked with BS3. Positive identification of cross-linked peptides required confirmation from the MS/MS fragmentation pattern and visual identification of mass peaks independently by two experienced analysts. Additionally, a unique cross-link had to appear in a minimum of two experiments for inclusion.
Small Angle X-ray Scattering
SAXS data were collected using the SIBYLS beamline (Berkeley, CA)58. Cross-linked monomeric apoA-I samples were shipped overnight at 4°C within 48 h of isolation to minimize aggregation artifacts. The cross-linked molecules were sampled at 4 mg/ml, 2 mg/ml, and 1 mg/ml at 10°C with four exposure times; 0.5, 1.0, 2.0, and 5.0 s. Scattering profiles showing evidence of radiation damage or particle repulsion were discarded. ScÅtter (SIBYLS) and ATSAS program suite (EMBL) were used for data analysis. 23 independent ab initio molecular envelopes were generated, averaged, and superimposed using DAMMIF (ATSAS, EMBL-Hamburg)21. The averaged molecular envelope graphics were rendered using UCSF Chimera.
Iterative Model Generation and Evaluation
A 3-D composite homology model of monomeric mutant apoA-I1-184 was generated from the dimeric crystal structure4,59 using Modeller v9.1460 as previously described5. The C-terminal 59 residues were modeled in incremental segments guided by experimental cross-linking data for respective regions using Modeller v9.14. Models were constrained with previously published cross-links10,11,16 and newly identified cross-links using an upper bound of 23.0 ± 0.001 Å or 28.0 ± 0.001 Å from C-α to C-α for cross-links identified with MDA and BS3, respectively. The models primary and secondary structure were adapted and refined iteratively guided by hydrogen-deuterium exchange (H-DX) data published by Chetty and colleagues12. Models were evaluated against experimental data obtained from SAXS using the online server FoxS61,62. The final model was subjected to energy minimization using the YASARA server22 and rotamers were manually refined using COOT23. Given the lack of a density map, rotamer position was determined using the most probable position without resulting in clashes. The reliability of the model was determined using MolProbity63 and solvent accessibility to individual amino acids was determined using VADAR24.
Data Availability
The .pdb file for the model is available for download from the online supplement or from the Davidson Lab homepage: (http://homepages.uc.edu/∼davidswm/structures.html). All other source data supporting the findings in this study are available upon request. A Life Sciences Reporting Summary for this article is available.
Supplementary Material
Acknowledgments
This work was supported by an American Heart Association postdoctoral fellowship grant (16POST27710016 to J. Melchior.), National Institutes of Health Heart Lung and Blood Institute funded predoctoral fellowship to M. Castleberry (HL125204-03), R01 HL127649 to W Davidson, R01 HL112276 to M Sorci-Thomas, P01 HL026335 and R01 HL116518 to D Atkinson, P01 HL12803 to W. Davidson, J. Segrest and J. Heinecke. The mass spectrometry data was acquired in the University of Cincinnati Proteomics Laboratory under the direction of K Greis on a mass spectrometer funded in part through an NIH S10 shared instrumentation grant (RR027015 Gries-PI).
Footnotes
Author Contributions: Conceived and designed new experiments reported in this paper: JTM, WSD, Performed experiments: JTM, RGW, ALC, JM, MC, HDS, Analyzed data: JTM, RGW, MC, TBT, MKJ, HDS, JPS, MCP, WSD. Derived the model: JTM, MC, TBT, MKJ, HDS, KAR, MNO, MGST, MJT, JWH, XM, DA, JPS, SLK, MCP, WSD. Wrote the manuscript: JTM, TBT, KAR, MNO, MGST, MJT, JWH, DA, JPS, SLK, MCP, WSD.
Competing Financial Interests: The authors report no competing financial interests with respect to this work.
References
- 1.Soutar AK, et al. Effect of the human plasma apolipoproteins and phosphatidylcholine acyl donor on the activity of lecithin: cholesterol acyltransferase. Biochemistry. 1975;14:3057–64. doi: 10.1021/bi00685a003. [DOI] [PubMed] [Google Scholar]
- 2.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289:24020–9. doi: 10.1074/jbc.R114.583658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borhani DW, Engler JA, Brouillette CG. Crystallization of truncated human apolipoprotein A-I in a novel conformation. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 9):1578–1583. doi: 10.1107/s0907444999008914. [DOI] [PubMed] [Google Scholar]
- 4.Mei X, Atkinson D. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem. 2011;286:38570–82. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melchior JT, et al. An Evaluation of the Crystal Structure of C-terminal Truncated Apolipoprotein A-I in Solution Reveals Structural Dynamics Related to Lipid Binding. J Biol Chem. 2016;291:5439–51. doi: 10.1074/jbc.M115.706093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nolte RT, Atkinson D. Conformational analysis of apolipoprotein A-I and E-3 based on primary sequence and circular dichroism. Biophys J. 1992;63:1221–39. doi: 10.1016/S0006-3495(92)81698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbeau DL, Jonas A, Teng T, Scanu AM. Asymmetry of apolipoprotein A-I in solution as assessed from ultracentrifugal, viscometric, and fluorescence polarization studies. Biochemistry. 1979;18:362–9. doi: 10.1021/bi00569a021. [DOI] [PubMed] [Google Scholar]
- 8.Okon M, Frank PG, Marcel YL, Cushley RJ. Heteronuclear NMR studies of human serum apolipoprotein A-I. Part I. Secondary structure in lipid-mimetic solution. FEBS Lett. 2002;517:139–43. doi: 10.1016/s0014-5793(02)02600-5. [DOI] [PubMed] [Google Scholar]
- 9.Davidson WS, et al. Structural organization of the N-terminal domain of apolipoprotein A-I: studies of tryptophan mutants. Biochemistry. 1999;38:14387–95. doi: 10.1021/bi991428h. [DOI] [PubMed] [Google Scholar]
- 10.Segrest JP, Jones MK, Shao B, Heinecke JW. An experimentally robust model of monomeric apolipoprotein A-I created from a chimera of two X-ray structures and molecular dynamics simulations. Biochemistry. 2014;53:7625–40. doi: 10.1021/bi501111j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silva RA, Hilliard GM, Fang J, Macha S, Davidson WS. A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry. 2005;44:2759–69. doi: 10.1021/bi047717+. [DOI] [PubMed] [Google Scholar]
- 12.Chetty PS, et al. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci U S A. 2009;106:19005–19010. doi: 10.1073/pnas.0909708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagerstedt JO, et al. The “beta-clasp” model of apolipoprotein A-I--a lipid-free solution structure determined by electron paramagnetic resonance spectroscopy. Biochim Biophys Acta. 2012;1821:448–55. doi: 10.1016/j.bbalip.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oda MN, Forte TM, Ryan RO, Voss JC. The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nat Struct Biol. 2003;10:455–60. doi: 10.1038/nsb931. [DOI] [PubMed] [Google Scholar]
- 15.Phillips MC. Thematic Review Series: High Density Lipoprotein Structure, Function, and Metabolism New insights into the determination of HDL structure by apolipoproteins. Journal of Lipid Research. 2013;54:2034–2048. doi: 10.1194/jlr.R034025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pollard RD, Fulp B, Samuel MP, Sorci-Thomas MG, Thomas MJ. The conformation of lipid-free human apolipoprotein A-I in solution. Biochemistry. 2013;52:9470–81. doi: 10.1021/bi401080k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X, Lei D, Zhang L, Rames M, Zhang S. A model of lipid-free apolipoprotein A-I revealed by iterative molecular dynamics simulation. PLoS One. 2015;10:e0120233. doi: 10.1371/journal.pone.0120233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swaim CL, Smith JB, Smith DL. Unexpected products from the reaction of the synthetic cross-linker 3,3′-dithiobis(sulfosuccinimidyl propionate), DTSSP with peptides. J Am Soc Mass Spectrom. 2004;15:736–749. doi: 10.1016/j.jasms.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 19.Leavell MD, Novak P, Behrens CR, Schoeniger JS, Kruppa GH. Strategy for selective chemical cross-linking of tyrosine and lysine residues. J Am Soc Mass Spectrom. 2004;15:1604–11. doi: 10.1016/j.jasms.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 20.Walker RG, et al. The Structure of Human Apolipoprotein A-IV as Revealed by Stable Isotope-assisted Cross-linking, Molecular Dynamics, and Small Angle X-ray Scattering. Journal of Biological Chemistry. 2014;289:5596–5608. doi: 10.1074/jbc.M113.541037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krieger E, et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77(Suppl 9):114–22. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willard L, et al. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003;31:3316–9. doi: 10.1093/nar/gkg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de la Llera-Moya M, et al. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vedhachalam C, et al. ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler Thromb Vasc Biol. 2007;27:1603–1609. doi: 10.1161/ATVBAHA.107.145789. [DOI] [PubMed] [Google Scholar]
- 27.Hassan HH, et al. Identification of an ABCA1-dependent phospholipid-rich plasma membrane apolipoprotein A-I binding site for nascent HDL formation: implications for current models of HDL biogenesis. Journal of Lipid Research. 2007;48:2428–2442. doi: 10.1194/jlr.M700206-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Rye KA, Barter PJ. Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2004;24:421–428. doi: 10.1161/01.ATV.0000104029.74961.f5. [DOI] [PubMed] [Google Scholar]
- 29.Vedhachalam C, et al. Influence of ApoA-I structure on the ABCA1-mediated efflux of cellular lipids. Journal of Biological Chemistry. 2004;279:49931–49939. doi: 10.1074/jbc.M406924200. [DOI] [PubMed] [Google Scholar]
- 30.Saito H, et al. Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. J Biol Chem. 2003;278:23227–32. doi: 10.1074/jbc.M303365200. [DOI] [PubMed] [Google Scholar]
- 31.Mei X, Liu M, Herscovitz H, Atkinson D. Probing the C-terminal domain of lipid-free apoA-I demonstrates the vital role of the H10B sequence repeat in HDL formation. J Lipid Res. 2016;57:1507–17. doi: 10.1194/jlr.M068874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koyama M, et al. Interaction between the N- and C-terminal domains modulates the stability and lipid binding of apolipoprotein A-I. Biochemistry. 2009;48:2529–2537. doi: 10.1021/bi802317v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorshkova IN, Liadaki K, Gursky O, Atkinson D, Zannis VI. Probing the lipid-free structure and stability of apolipoprotein A-I by mutation. Biochemistry. 2000;39:15910–15919. doi: 10.1021/bi0014406. [DOI] [PubMed] [Google Scholar]
- 34.Gross E, Peng DQ, Hazen SL, Smith JD. A novel folding intermediate state for apolipoprotein A-I: role of the amino and carboxy termini. Biophysical Journal. 2006;90:1362–1370. doi: 10.1529/biophysj.105.075069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollard RD, Fulp B, Sorci-Thomas MG, Thomas MJ. High-Density Lipoprotein Biogenesis: Defining the Domains Involved in Human Apolipoprotein A-I Lipidation. Biochemistry. 2016;55:4971–81. doi: 10.1021/acs.biochem.6b00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Panagotopulos SE, et al. The role of apolipoprotein A-I helix 10 in apolipoprotein-mediated cholesterol efflux via the ATP-binding cassette transporter ABCA1. J Biol Chem. 2002;277:39477–84. doi: 10.1074/jbc.M207005200. [DOI] [PubMed] [Google Scholar]
- 37.Brouillette CG, et al. Forster resonance energy transfer measurements are consistent with a helical bundle model for lipid-free apolipoprotein A-I. Biochemistry. 2005;44:16413–25. doi: 10.1021/bi051018v. [DOI] [PubMed] [Google Scholar]
- 38.Puchkaev AV, Koo LS, Ortiz de Montellano PR. Aromatic stacking as a determinant of the thermal stability of CYP119 from Sulfolobus solfataricus. Arch Biochem Biophys. 2003;409:52–8. doi: 10.1016/s0003-9861(02)00402-2. [DOI] [PubMed] [Google Scholar]
- 39.Nichols WC, Dwulet FE, Liepnieks J, Benson MD. Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem Biophys Res Commun. 1988;156:762–8. doi: 10.1016/s0006-291x(88)80909-4. [DOI] [PubMed] [Google Scholar]
- 40.Franceschini G, Sirtori CR, Capurso A, Weisgraber KH, Mahley RW. A-Imilano Apoprotein - Decreased High-Density Lipoprotein Cholesterol Levels with Significant Lipoprotein Modifications and without Clinical Atherosclerosis in an Italian Family. Journal of Clinical Investigation. 1980;66:892–900. doi: 10.1172/JCI109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chetty PS, et al. Effects of the Iowa and Milano mutations on apolipoprotein A-I structure and dynamics determined by hydrogen exchange and mass spectrometry. Biochemistry. 2012;51:8993–9001. doi: 10.1021/bi300926j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorshkova IN, et al. Structure and stability of apolipoprotein a-I in solution and in discoidal high-density lipoprotein probed by double charge ablation and deletion mutation. Biochemistry. 2006;45:1242–54. doi: 10.1021/bi051669r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davidson WS, Hazlett T, Mantulin WW, Jonas A. The role of apolipoprotein AI domains in lipid binding. Proc Natl Acad Sci U S A. 1996;93:13605–10. doi: 10.1073/pnas.93.24.13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saito H, et al. alpha-Helix formation is required for high affinity binding of human apolipoprotein A-I to lipids. Journal of Biological Chemistry. 2004;279:20974–20981. doi: 10.1074/jbc.M402043200. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka M, et al. Influence of N-terminal helix bundle stability on the lipid-binding properties of human apolipoprotein A-I. Biochim Biophys Acta. 2011;1811:25–30. doi: 10.1016/j.bbalip.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pownall HJ, Massey JB, Kusserow SK, Gotto AM., Jr Kinetics of lipid--protein interactions: interaction of apolipoprotein A-I from human plasma high density lipoproteins with phosphatidylcholines. Biochemistry. 1978;17:1183–1188. doi: 10.1021/bi00600a008. [DOI] [PubMed] [Google Scholar]
- 47.Palgunachari MN, et al. Only the two end helixes of eight tandem amphipathic helical domains of human apo A-I have significant lipid affinity - Implications for HDL assembly. Arteriosclerosis Thrombosis and Vascular Biology. 1996;16:328–338. doi: 10.1161/01.atv.16.2.328. [DOI] [PubMed] [Google Scholar]
- 48.Qian H, et al. Structure of the Human Lipid Exporter ABCA1. Cell. 2017;169:1228–1239 e10. doi: 10.1016/j.cell.2017.05.020. [DOI] [PubMed] [Google Scholar]
- 49.Segrest JP, et al. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. Journal of Biological Chemistry. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 50.Huang BX, Kim HY, Dass C. Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J Am Soc Mass Spectrom. 2004;15:1237–47. doi: 10.1016/j.jasms.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 51.Jacobsen RB, et al. Structure and dynamics of dark-state bovine rhodopsin revealed by chemical cross-linking and high-resolution mass spectrometry. Protein Sci. 2006;15:1303–17. doi: 10.1110/ps.052040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Young MM, et al. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc Natl Acad Sci U S A. 2000;97:5802–6. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng L, Rasmussen MI, Chailyan A, Houen G, Hojrup P. Probing the structure of human protein disulfide isomerase by chemical cross-linking combined with mass spectrometry. J Proteomics. 2014;108:1–16. doi: 10.1016/j.jprot.2014.04.037. [DOI] [PubMed] [Google Scholar]
- 54.Tubb MR, Smith LE, Davidson WS. Purification of recombinant apolipoproteins A-I and A-IV and efficient affinity tag cleavage by tobacco etch virus protease. J Lipid Res. 2009;50:1497–504. doi: 10.1194/jlr.D900003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Markwell MA, Haas SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Analytical Biochemistry. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 56.Petrotchenko EV, Serpa JJ, Borchers CH. An isotopically coded CID-cleavable biotinylated cross-linker for structural proteomics. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.001420. M110 001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lima DB, et al. SIM-XL: A powerful and user-friendly tool for peptide cross-linking analysis. J Proteomics. 2015;129:51–5. doi: 10.1016/j.jprot.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 58.Dyer KN, et al. High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol Biol. 2014;1091:245–58. doi: 10.1007/978-1-62703-691-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci U S A. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 61.Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–4. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J. 2013;105:962–74. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The .pdb file for the model is available for download from the online supplement or from the Davidson Lab homepage: (http://homepages.uc.edu/∼davidswm/structures.html). All other source data supporting the findings in this study are available upon request. A Life Sciences Reporting Summary for this article is available.