

Abstract
We develop a partial charge-based Tightly Bound Ion (PCTBI) model for the ion effects in RNA folding. Based on the Monte Carlo Tightly Bound Ion (MCTBI) approach, the model can account for ion fluctuation and correlation effects, and can predict the ion distribution around the RNA. Furthermore, unlike the previous coarse-grained RNA charge models, where negative charges are placed on the phosphates only, the current new model considers the detailed all-atom partial charge distribution on the RNA. Thus, the model not only keeps the advantage of the MCTBI model but also has the potential to provide important detailed information unattainable by the previous MCTBI models. For example, the model predicts the reduction in ion binding upon protein binding and ion-induced conformational switches. For Hepatitis C virus (HCV) genomic RNA, the model predicts a Mg2+-induced stabilization of a kissing motif for a cis-acting regulatory element in the genomic RNA. Extensive theory-experiment comparisons support the reliability of the theoretical predictions. Therefore, the model may serve as a robust starting point for further development of an accurate method for ion effects in RNA conformational equilibrium and RNA-cofactor interactions.
Graphical abstract
1 INTRODUCTION
Metal ions play an important role in the stabilization of three-dimensional (3D) structures of nucleic acids (DNAs and RNAs), due to their ability to neutralize/screen the negative charges on nucleic acids backbones and specific ion-nucleic acid interactions.1, 2 Quantitative prediction for ion binding properties is essential for the understanding of nucleic acids structural formation and folding stabilities.3–20 However, despite tremendous theoretical and experimental efforts, accurate prediction of the ion effects at various solution conditions for different RNA structures remains one of main challenges in computational modeling of RNA structure and stability,21–25 especially for solutions involving multivalent ions such as Mg2+.
According to the different charges and sizes and the different ways of binding to the RNA, we can distinguish three types of ions.26–28 First, in one extreme, ions can be dehydrated and trapped at specific sites in RNA29 and ion-RNA binding is stabilized by specific interactions such as chelation with the electronegtive atomic groups of RNA. These ions are site-bound (or chelated). Second, in the other extreme, the majority of the bound ions can remain hydrated and bind to RNA diffusely. The large number of diffusely bound ions may dominate the ion electrostatic energy of the system.30 These ions “freely” diffuse at a distance from the RNA surface without any specific binding sites. These ions are called diffuse ions or diffusely bound (DB) ions. Third, there exist ions which are clustered around the RNA surface due to ion-RNA Coulomb attraction, resulting in high local concentration of ion. These (hydrated) ions are neither trapped at specific binding sites like chelated ions nor “free” to move around like diffuse ions. The high local concentration of the ion can cause strong coupling/correlation between the ions due to volume exclusion and Coulomb interaction. As a result, the local electric field for an ion is not only related to the position of the ion itself but also to the positions of the nearby ions. Such a correlation effect is more pronounced for multivalent ions such as Mg2+ due to the high ionic charge than for monovalent ions.13, 31–33 We call such ions, which involve the strong correlation, as the tightly bound (TB) ions.
A number of different methods have been used to investigate the ion effects in RNA folding and stability. Among these methods, molecular dynamics simulation34–37 has the advantage to provide insights into atomic details of the ion binding process, such as the site-specific ion binding in RNA kissing loop formation,38 the 5S rRNA loop E motif,39 SAM-I riboswitch,40 and many other systems.41–44 However, the accuracy of MD simulations is limited by the accuracy of the ion-dependent force field as well as the completeness of the sampling for the ions and water molecules,34 As a result, accurate calculations require significant computer resources and exceedingly long computer time even for small RNAs. The classical Counterion Condensation (CC) theory45 and the (nonlinear) Poisson-Boltzmann (NLPB) theory46–51 are simple yet useful theories for the prediction of the ion effects. However, both theories ignore ion correlation and ion fluctuations, two potentially important effects for ions around nucleic acids.31, 52 The CC theory treats ion binding with average (partially) neutralized charges while NLPB considers ions with an average continuous distribution. Recently, several new theories have been developed to account for the ion correlation effect for biomolecular systems.53–55 For example, the three-dimensional reference interaction site model (3D-RISM) was developed based on the Ornstein and Zernike integral equation theory,54 and a generalized counterion condensation theory was proposed to explicitly treat Mg2+ ions with ion-ion correlations.55
The tightly bound ion (TBI) model is another highly promising model that can accurately and efficiently predict the ion binding effects for RNAs.28, 52, 56–61 The key idea of the TBI model is to explicitly enumerate the many-body, correlated ion distributions for the TB ions while using a mean-field description (NLPB) for the DB ions. In this way, the model takes into account both the correlation and the fluctuation effects. Extensive comparisons between TBI predictions and experimental results support the validity and accuracy of the TBI model for predicting ion binding properties and ion-mediated nucleic acids stability for simple helices, pseudoknots, kissing complexes, and other more complex tertiary folds.28, 52, 56–61 However, because the original model is based on the explicit enumeration of ion distributions, which is time-consuming, the model is computationally inefficient. The low computational efficiency renders applications to medium (100–200 nts) and large RNA structures (> 200 nts) impractical.
Recently, we developed a new version of the TBI model, Monte Carlo tightly bound ion (MCTBI) model,62 which employs Monte Carlo Insertion Deletion (MCID) algorithm to sample the ion distributions of TB ions. In comparison with the original TBI model, the MCTBI model has two main advantages: more efficient computational speed and a higher resolution (3D coordinate-based) description for the ion distribution. The MCTBI model enables prediction of the highest probability regions for ion binding. However, similar to the original TBI model, the MCTBI model is based on a coarse-grained (CG) charge model for RNA, where the charges on a nucleotide are assumed to “collapse” into a point charge located at the center of the phosphate. In such a CG charge model, each phosphate carries an electronic charge −e and the rest atoms are assumed to be neutral.52, 60, 61 The coarse-grained charge distribution has the advantage to reduce the computational time. However, the over-simplification of the charge distribution prevents the model from applications to many biologically important problems which require information about realistic and detailed charge distributions on RNA. These problems include predictions of ions binding sites at higher accuracy and resolution63 and modeling of ligand-RNA and protein-RNA docking.64 These and many other ion-dependent RNA biology problems demand a new model that goes beyond the CG charge-based theory.
In this paper, we report a new model, PCTBI model, which uses all-atom partial charges for RNA atoms and can treat ion correlation and fluctuation effects. Tests against the previous CG charge-based model using the experimental data supports the validity and accuracy of this new model. Furthermore, compared with the CG charge model, the partial charge-based model reveals important details about the ion effects, including the influence of cofactor (protein) binding on ion distribution around the RNA. As an application of the model, we investigate the ion-dependent conformational equilibrium and conformational switches in Hepatitis C virus (HCV) genomic RNA.
Belonging to the Flaviviridae family, HCV is a Hepacivirus that causes the most common chronic blood-borne viral infection in the United States.65 Replication of HCV in human liver cells triggers and invades host immune responses, which lead to extensive damage to liver.66
Cell-culture based viral replication assays suggested a potential long-range RNA:RNA interaction in the 3′-end of the genomic RNA between 3′X and an upstream cis-acting regulatory element termed 5BSL3.2.67 The base pair complementarity between 5BSL3.2 and 3′X are phylogenetically conserved, and has been shown to be essential for HCV viral protein translation and RNA synthesis.68–70 Previous gel electrophoresis and surface plasmon resonance (SPR) studies have shown that in vitro transcribed 5BSL3.2 and 3′X can form an RNA:RNA complex and Mg2+ is required in the gel and running buffer in order to detect the RNA complex band.71, 72 We apply the PCTBI model developed in the present study to investigate the physical mechanism for the Mg2+-dependence of the folding stability and the shift of conformational equilibrium for the 5BSL3.2:3′X kissing complex. Our results indicate that Mg2+ ions can indeed stabilize the 5BSL3.2:3′X docked state over the undocked state and for 3′X, adding Mg2+ to the solution shifts the conformational equilibrium toward the kissing conformation.
2 THE PCTBI MODEL
2.1 RNA structure models
The development and test of the PCTBI model involve a number of RNA structures. The 80-nucleotide (nt) A-form RNA duplex and 48-nt B-form DNA helix are generated using the latest version (version 2.1) of the program X3DNA.73 Other RNA structures, such as the 76-nt yeast tRNAPhe (PDB ID: 1TRA74), 58-nt fragment of rRNA and rRNA-protein complex (PDB ID: 1HC875), and 72-nt Adenine riboswitch (A-riboswitch; PDB ID: 1Y2676), are generated from the Protein Data Bank coordinate files.77 For the 5BSL3.2:3X complex in the HCV genomic RNA, cell-based viral replication assays68–70 have suggested that the kissing complex is formed between 5BSL3.2 and the SL2 hairpin loop in the 3′X.67 We therefore focus on the two RNA segments 5BSL3.2 and SL2. The conformations are generated by the Vfold model combined with molecular dynamics (MD) simulations.78 In the MD simulations, we use the structure predicted by the Vfold model78 as the initial structure, and then apply a harmonic potential U(r) = k(r − r0)2 to constrain the base-pairs, where k = 1.0 kcal/mol and r0 = 3.5 Å. For each 2D structure (defined by the set of base pairs contained in the structure), totally more than 5000 different 3D conformations are generated in the MD simulation.
2.2 Partial charge assignment
The partial charges of the atoms in RNA are assigned using the “Dock Prep” module in Chimera.79 Before implementing the “Dock Prep” module, all the ions in the PDB are removed at first. Then, for a given structure, we run the “Dock Prep” module runs multiple times for solvent deletion, alternate locations deletion (keeping the highest occupancy), hydrogen addition, partial charges addition, and output with Mol2 format. Charges for standard residues in RNAs are taken from AMBER ff14SB80, 81 and charges for nonstandard residues, including ligand for the A-riboswitch, are calculated using the ANTECHAMBER module with the AM1-BCC charges.82, 83 In order to test the sensitivity of the different partial charge assignment methods, we also use PDB2PQR method84 to calculate the partial charges for the 58-nt fragment of rRNA with AMBER99,85 CHARMM22,86 or PARSE87 force fields.
2.3 Partial charge-based MCTBI model
For a given RNA with all-atom structure and partial charges assignment, we first run NLPB calculation to roughly compute the ion concentrations for each ion species around the RNA. In the NLPB computation, RNA is located in a large solution box, whose size is larger than six times of the Debye length in order to reduce the boundary effect.28 The mixed salt solution in the box can contain divalent cations Mg2+, monovalent cations K+ (or Na+), and monovalent anions Cl−. Their bulk concentrations , and satisfy the charge neutrality condition: .
To accelerate the NLPB calculation, we apply the three-step focusing process,88 where grid sizes 1.7 Å, 0.85 Å, and 0.425 Å for the three steps, respectively. From the ion concentration, we determine the average ion-ion distance and the ion correlation strength, which is defined as the ratio between the average ion-ion Coulomb energy and the thermal energy.28 Because the ion concentration is location-dependent, the ion correlation strength is coordinate-dependent. According to the spatial distribution of the correlation strength, we classify the region around RNA into TB region and DB region, for the TB ions (of strong correlation) and the DB ions (of weak correlation), respectively. For monovalent ions, which involve weaker Coulomb interactions than multivalent ions, the TB region (of strong correlation) is negligible and therefore, all the monovalent ions can be treated as DB ions.
Unlike monovalent ions, multivalent ions (such as Mg2+ ions), usually involve a significant TB region, which forms a thin layer of several Ås plus the ion radius from the RNA surface. Here we assume that ions are hydrated with radii rMg2+ = 4.5 Å, rK+ = 4.0 Å, rNa+ = 3.5 Å, and rCl− = 4.0 Å, respectively.58, 59 According to the above analysis, for a monovalent/divalent ion mixture solution, only for divalent ions (e.g., Mg2+) we need to distinguish TB ions from DB ions. The DB ions can be treated with the mean-field NLPB theory while the (strongly correlated) TB ions require sampling of many-body, discrete ion distributions.
For an RNA with N phosphates, we allow Nb, the number of the tightly bound Mg2+ ions (in the TB region), to vary from 0 to N. The total partition function of the many-body system can be calculated as the sum over Nb:
| (1) |
where Z(Nb) is the partition function for a given Nb (the number of Mg2+ ions in TB region):
| (2) |
Here Zid represents the ideal partition function without the RNA in the system (uniform ion solution). is the bulk concentration of Mg2+ ions and the factor Nb! accounts for the nondistinguishability of the Mg2+ ions. kB and T denote the Boltzmann constant and temperature. The statistical weight W (Nb) is equal to the configurational integral for all the Nb ions:
| (3) |
Here Ri denotes the coordinate of ion i in the TB region. ΔGb in Eq. 3 and ΔGd in Eq. 2 represent the interaction energy of all the charged particles in TB region (TB ions and atoms of RNA) and the free energy of DB ions, respectively. ΔGb includes volume exclusion, Coulombic interaction and dielectric polarization energies. In our calculation, the excluded volume effect is modeled by a Lennard-Jones (LJ) potential and the polarization energy is computed from the Generalized Born (GB) model:
| (4) |
The first term in the equation above is the Coulombic energy for the interaction between charges in the TB region. Zi(or j)e is the charge of particle i(or j), εR (= 20 in our calculation) is the dielectric constant of RNA, and rij is the distance between particles i and j. The second term is the LJ potential with uo (= 0.35) as the LJ constant and σij as the equilibrium distance between particles i and j. Here we set σij as the addition of the radius of the two particles. The third term above is the mutual polarization energy induced by other charges, εW (= 78) is the dielectric constant of water and Bi (or j) is the Born radius for particle i(or j). The forth and fifth terms represent the self-polarization energies of RNA (subscript R) and ions (subscript I). The free energy ΔGd of DB ions can be calculated from the effective single-particle ion distribution solved from NLPB:89, 90
| (5) |
Here α denotes the ion species. zαe is the charge of ion species α, ψ(r) and ψ′(r) are the electrical potentials at position r with and without ions in the solution, and cα(r) and represent the local (at r) and bulk concentrations, respectively.
We use a simple cubic lattice in TB region to configure the ion distributions for the TB ions. Assuming that the TB region contains Ns lattice sites with lattice grid length lb. Then the statistical weight in Eq. 3 can be computed from the following formula
| (6) |
where mi represents the number of all the available (empty) sites for TB ions. ΔGb is the interaction energy of all the charged particles in the TB region (see Eq. 4). The lattice size lb is adaptive in order to reach an optimal balance between the resolution of ion distribution and the efficiency of computation. For a low bulk concentration of Mg2+ ions, lb = 0.425 Å is determined from the grid size of the third focusing step in the NLPB calculation (for the DB ions). For a high ion bulk concentration, there is a larger TB region (sampling space) so that the sampling of the ion distribution could be time-consuming. Therefore, we increase the value of lb such that the number of lattice sites Ns inside the TB region is ≈ 200 × N. To circumvent the problem of the exceedingly long computational time for the exact enumeration of the ion distributions, we use the Rosenbluth-Rosenbluth (RR)-based Monte Carlo method91 and the Monte Carlo Insertion Deletion (MCID) algorithm,62 to sample the ion distributions in the TB region. The central idea of MCID algorithm is to sample ion distribution by inserting particles one by one according to the energy followed by a re-sampling for the ions by randomly removing high-energy ions. The purpose of the resampling/relaxation is to further enhance the sampling quality for the important (low-energy) ion distributions, we allow the multiple-ion system to relax.92 Specifically, we perform a re-sampling for the ions by randomly removing high-energy ions. The details of MCID algorithm has described in the Ref. 62.
3 Material and Methods for the HCV experiments
HCV-3′X and 5BSL3.2 stock solutions were created at a concentration of 25 μM in 10 mM Tris-HCL (pH 7.48). These starting stocks were boiled for 3 minutes, and then snap cooled at 4 °C. The 3′X and 5BSL3.2 samples were then mixed at 1:1 ratio in the presence of 1x incubation buffer (final concentration: 140 mM KCl, 10 mM NaCl, 2 mM MgCl2, 10 mM Tris-HCl, pH 7.5), and incubated at 37 °C for 60 minutes. The mixture was analyzed by electrophoresis in acrylamide gels containing 0, 1 and 2 mM MgCl2. The gels were run at 4 °C, and the RNA bands were stained by stains-all and imaged using a Kodak camera.
Purified 5BSL3.2 RNA samples were dissolved in 90% H2O, 10% D2O with 20 mM Tris-HCl, pH 6.5 at different MgCl2 concentrations. NMR spectra were acquired on a Bruker Avance III 800 MHz spectrometer equipped with TCI cryoprobe at the NMR core facility, University of Missouri, Columbia. For the assignment of imino protons, two-dimensional NOESY spectra were recorded for 5BSL3.2 with 0 and 5 mM MgCl2 at 15 °C. Data were processed with NMRPipe and NMR Draw,93 and analyzed with NMRViewJ (One Moon Scientific, NJ).
4 RESULTS AND DISCUSSION
4.1 Test of the PCTBI model
To test the accuracy of the new partial charge-based PCTBI model, we calculate the fraction of excess bound ions fα per nucleotide for ion species α, and compare the theoretical predictions with the experimental data. fα is given by the following formula:
| (7) |
where the partition functions Z(Nb) and Z in Eq. 7 are determined from Eqs. 1 and 2, respectively, and Γα(Nb) is the number of excess ions, which includes TB ions and excess DB ions:
| (8) |
In experiments, the excess number of bound ions can be measured by “Ion Counting” methods,94 such as fluorescence titration95–99 and buffer equilibration and atomic emission spectroscopy (BE-AES).31, 100 The fluorescence titration measurements take advantage of a fluorescent dye for an ion species such as Mg2+ to count the bound ions. Therefore, the method focuses on single ion species in each measurement. The BE-AES method that uses a filter to create a sample solution (with nucleic acids) and a reference solution (without nucleic acids) allows counting of the excess bound ions in the solution by atomic emission spectroscopy.31 The method has a lower detection limit100 and can also measure anion depletion. In the following calculations, unless otherwise stated, the partial charges of RNA atoms are calculated using the “Dock Prep” module in Chimera79 with AM1-BCC (or AM1 for short) charges.82, 83
Fig. 1 shows the binding fractions fMg2+ and fNa+ (or K+) (the average number of bound ions per nucleotide) of divalent and monovalent ions for the different RNAs as predicted by the original CG charge-based (MCTBI) model, where each phosphate group carries −e charge and other atoms are neutral, and the partial charge-based (PCTBI) model. The results for the binding fractions lead to three conclusions. First, our theoretical predictions are in accordance with the experimental data. Second, the PCTBI model gives slightly better predictions than the previous MCTBI model, such as the aRNA case at [Mg2+] = 0.1 mM. In order to make direct comparisons between the two models, we also show the prediction data in Table 1 for a 24-bp B-DNA, for which the experimental data is available.100 Indeed, the relative error ΔE (see table 1) shows that the PCTBI is (slightly) better than MCTBI. Third, fK+ of RNA-protein complex predicted by the PCTBI model is lower than that predicted by the original MCTBI model [see Fig. 1F]. The computational time increases with the RNA length (number of nucleotides) and the salt concentration. For example, for the 58-nt rRNA in a 20 mM K+ background solution shown in Fig. 1C, the computer times are about 12 and 30 minutes for 10−6 M and 10−2 M Mg2+, respectively. The computations were performed on a PC with an Intel i7-4790 processor and 16 GB RAM. On the same computer, for the 80-nt RNA shown in Fig. 1A, the computer times are about 30 and 60 minutes for 10−6 M and 10−2 M Mg2+, respectively.
Figure 1.

The [Mg2+]-dependence of the Mg2+ and Na+ (or K+) binding fractions per nucleotide for six different test systems: 80-nt RNA duplex (A-form helix) with 10mM Na+ (A), 76-nt yeast tRNAPhe (PDB ID: 1TRA74) with 32 mM Na+ (B), 58-nt fragment of rRNA (PDB ID: 1HC875) with 20 (C) and 60 mM K+ (D), respectively, adenine riboswitch with 50 mM K+ (E), and rRNA-protein complex with 60 mM K+ (F), respectively. The experimental data in A, B, C and D, E, and F are from the References 95, 96, 97, 98, and 99, respectively.
Table 1.
[Na+]-dependent Mg2+ Binding fractions for 24-bp B-DNA with [Mg2+] = 6 mM determined from the experiments,100 the PCTBI model, and the MCTBI model, respectively. DeltaE is the relative error between the theoretical prediction and the experimental result.
| Exp. data* | PCTBI | MCTBI | |||
|---|---|---|---|---|---|
| [Na+]/mM | fMg2+ | fMg2+ | ΔE** | fMg 2+ | ΔE |
| 0 | 0.467 ± 0.012 | 0.468 ± 0.37 ×10−4 | 0.17% | 0.469 ± 0.39 ×10−4 | 0.42% |
| 12 | 0.430 ± 0.004 | 0.413 ± 1.77 ×10−4 | 4.13% | 0.410 ± 1.50 ×10−4 | 4.81% |
| 18 | 0.400 ± 0.011 | 0.390 ± 2.49 ×10−4 | 2.59% | 0.386 ± 1.60 ×10−4 | 3.41% |
| 28 | 0.370 ± 0.002 | 0.355 ± 3.04 ×10−4 | 3.87% | 0.354 ± 1.82 ×10−4 | 4.10% |
| 48 | 0.304 ± 0.002 | 0.307 ± 3.10 ×10−4 | 0.93% | 0.300 ± 2.07 ×10−4 | 1.39% |
| 100 | 0.187 ± 0.004 | 0.195 ± 2.95 ×10−4 | 4.31% | 0.196 ± 1.73 ×10−4 | 4.85% |
The experiment data were provided in the Ref. 100.
The relative error ΔE = |(fpre − fexp)/fexp|, where fpre and fexp are the prediction and experiment data.
The original MCTBI model, unlike the current PCTBI model, cannot treat the partial charges on the protein, which can be critical for the RNA-protein docking. Specifically, the MCTBI model neglects the protein-induced change of Coulomb interactions as the residues of protein are assumed to be electrically neutral. In contrast, the PCTBI model can account for the detailed partial charge distributions not only on the RNA but also on the protein. Of particular importance is the charge distribution on the RNA-protein interface. The protein surface on the solvent side has positive partial charges, which tend to reduce cation binding. Therefore, the coarse-grained charge model, which cannot reflect such detailed charge distribution on the protein, would over-estimate the ion binding (see Fig. 1F).
To test the sensitivity of the predictions to the different partial charge models, we compute the ion binding fractions for the 58-nt rRNA fragment based on the different partial charge assignments, including the default method (“Dock Prep” with AM1),80–83 pdb2pqr84 with AMBER99 (AMB),85 CHARMM22 (CHA),86 and PARSE (PAR)87 force fields. Although the net charges on the 58-nt rRNA predicted by the above four methods are the same (−56e), the charge distributions are different. Using the partial charge assignment for a phosphate group in a standard nucleotide as an example (see Table 2), AM1 and AMB have the same charge assignment since they both use Parm9981 as the force field for a standard nucleotide. The charge assignments by PAR and CHA are different from the above two methods and the difference for CHA is more significant as shown by the net charge of the phosphate group (Table 2). As shown in Fig. 2, the partial charge models of AM1 (black line), AMB (red line), and PAR (green line) give nearly the same predictions, which are better than the predictions by CHA (blue line). Here, we note that for the case of the 58-nt rRNA with background salt of 20 mM K+, Mg2+ may be dehydrated and trapped at a specific binding site at high [Mg2+].75 Such a dehydration effect, however, is neglected in the current model. This lack of ion dehydration effect may contribute to the theory-experiment difference for [Mg2+] > 0.1 mM.
Table 2.
The partial charge assignments for a phosphate group in a standard nucleotide predicted by default method (AM1)80–83 pdb2pqr84 with AMBER99 (AMB),85 PARSE (PAR),87 and CHARMM22 (CHA),86 respectively.
Figure 2.

The [Mg2+]-dependence of the Mg2+ and Na+ (or K+) binding fractions per nucleotide for four different models of partial charge assignment.
One of the important applications of the PCTBI model is to predict the electrostatic free energies as a function of the ion concentrations and the ion-induced shift of the populational shift of the different conformations and corresponding conformational switches. As shown in Fig. 3 for six RNA systems, increasing the Mg2+ concentration results in a decreasing in the electrostatic free energy ΔG, which would contribute to the stabilization of the RNA structure. As shown in the result for the 58-nt rRNA fragment with 20 (blue line) and 60 mM K+ (cyan line), high concentration of monovalent cation can also help stabilize the RNA. However, at high concentration of divalent cation, the stabilization effect from the monovalent cation becomes negligible. Furthermore, the free energies of the rRNA (cyan line) and the rRNA-protein complex (magenta line) with 60 mM K+ indicate that the rRNA-protein complex is favored by the electrostatic interactions. It is important to note that such effects cannot be predicted by the original MCTBI model.
Figure 3.

The electrostatic free energy ΔG as a function of [Mg2+] for (from top to bottom) the rRNA with 20mM K+ (blue line), rRNA with 60 mM K+ (cyan line), adenine riboswitch with 50 mM K+ (green line), 76-nt yeast tRNAPhe with 32mM Na+ (red line), the 80-nt RNA duplex with 10 mM Na+ (black line), rRNA-protein complex with 60 mM K+ (magenta line), respectively.
With the use of the detailed partial charge distribution for RNA (and protein) and the sampling of ion distributions based on the 3D coordinates of the ions, the current new model enables more reliable predictions for the probable bound ion distributions and ion binding sites. The probability of finding a bound ion at position (grid point) k is given by
| (9) |
where n(Nb, k) is the number of Nb-ion distributions (out of the totally Nsample sampled distributions) with site k occupied by a TB ion.
For the tRNA (PDBID: 1TRA74) with 32 mM Na+ and 2 mM Mg2+ and the rRNA-protein complex (PDBID: 1HC875) with 60 mM K+ and 2 mM Mg2+ as examples, Fig. 4 shows the TB ion distributions of probability larger than 5% as predicted by the PCTBI model [Figs. 4A and C] and the original coarse-grained charge-based TBI model [see Figs. 4B and D]. The 3D structures of RNA used here were determined by X-ray diffraction, and the specific bound Mg2+ ions (cyan balls in Fig. 4) were determined from the electron density maps.74, 75 Comparison between the predicted high-probability ion distributions and the experimentally observed ion binding sites leads to two conclusions. First, the experimentally observed sites are in close proximity to our predicted “high probability” binding sites. We note that the predicted ion binding regions are usually close to the experimentally observed ion binding sites, however, the predicted binding sites may not agree exactly with the those shown in the crystallographic RNA structures. This is due to two reasons. First, the (MCTBI and PCTBI) models assume fully hydrated ions while experimentally observed specific site-bound ions often involve dehydration.29 Therefore, the (fully hydrated) ions in the models cannot enter the solvent-inaccessible pockets in RNAs. To best estimate the likelihood of ion binding, we use 5% as the cut-off probability for the probable ion binding sites. Second, ions bound to an RNA structure can be dynamic. Therefore, it is possible that the bound ions can distribute in a broad region instead of become fixed at a specific site shown in the crystallographic structure. Nevertheless, the results suggest that the theory may offer a low-resolution prediction for ion binding sites, which can be used as starting points for further refinements by including the ion dehydration effect. Second, the “high probability” locations predicted by the PCTBI model are more focused and accurate than those predicted by the original coarse grained charge-based TBI model (see the locations labeled by black and red cycles in Fig. 4). Because a Mg2+ ion has the same number of electrons as water molecules, it is possible that certain ion binding sites are not shown in the experimental structures.63, 76, 101, 102 Therefore, it might be possible that certain predicted highly probable binding sites are not explicitly shown in the crystallographic structures. Furthermore, as reported in a previous study,40 the predicted ion binding regions may also correspond to regions where ions have low diffusion.
Figure 4.

the predicted results of the high-probability binding positions for tRNA (PDB ID: 1TRA74) and rRNA-protein complex (PDB ID: 1HC875). RNA and protein are labeled by blue and yellow, cyan balls denote the site-bound ions, and the pink color denotes the predicted high-probability binding positions.
4.2 Mg2+ ion effects on the formation of the 5BSL3.2:3′X complex in HCV genomic RNA
In this section, by combining the experimental data and the theoretical predictions, we investigate the Mg2+ ions effect on the formation of the 5BSL3.2:3′X complex in HCV genomic RNA. Because the SL2 kissing loop is the main part in 3′X to form the kissing complex with 5BSL3.2, we focus on the 5BSL3.2 and the SL2 domains in our calculation.
In the experiment, the 5BSL3.2:3X complex was loaded to native polyacrylamide gels containing 0, 1, and 2 mM Mg2+. In agreement with the previous findings,71, 72 the 5BSL3.2:3′X complex band can be detected only in gels containing Mg2+ [see Fig. 5]. When Mg2+ was deprived in electrophoresis, the complex dissociated into free 5BSL3.2 and 3′X in the no Mg2+ gel, suggesting that Mg2+ is needed to maintain the complex. It is worth noting that the free 5BSL3.2 RNA migrated much faster in the Mg2+-free gel as its position was significantly lower than free 3′X, whereas in the 1 mM Mg2+ gel it was much closer to 3′X, and in the 2 mM Mg2+ gel it migrated to almost the same position as 3′X. These data suggest that Mg2+ is necessary to prevent dissociation of the 5BSL3.2:3′X complex, and Mg2+ might be involved in the folding of 5BSL3.2 due to the fact that 5BSL3.2 exhibited a pronounced decrease in migration rate in gels containing higher concentrations of Mg2+, whereas the migration rate of 3′X was relatively insensitive.
Figure 5.

Dependence of the 5BSL3.2:3′X complex formation on [Mg2+]. 5BSL3.2 and 3′X mixtures were analyzed by native polyacrylamide gels containing 0, 1 mM and 2 mM [Mg2+] (left, middle and right, respectively). Mg2+ in both gel and running buffer is necessary to maintain the RNA complex from dissociation during electrophoresis. Increasing [Mg2+] significantly slowed down the migration rate of 5BSL3.2 but had little impact on the migration rate of 3′X.
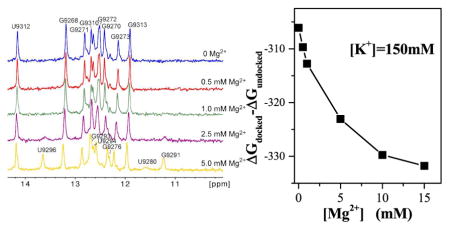
To confirm the sensitivity of 5BSL3.2 folding to Mg2+, one-dimensional imino proton spectra were collected for 5BSL3.2 with different Mg2+ concentrations. New signals were detected as the Mg2+ concentration increased, whereas the signals detected in the absence of Mg2+ remained unchanged. Assignment of these exchangeable imino protons on the two-dimensional NOESY spectra confirmed that formation of the kissing-loop conformation of the 5BSL3.2 relies on Mg2+. When the Mg2+ concentration was less than 1 mM, the imino proton signals observed were assigned to residues G9268–G9273 and G9310–G9313, demonstrating the formation of the bottom stem of 5BSL3.2. No signals indicative of a well-formed structure on the top stem were detected. The new imino signals detected at higher Mg2+ concentrations were assigned to residues of the 5BSL3.2 upper stem, demonstrating that the kissing conformation of 5BSL3.2 is stabilized by Mg2+ while the bottom stem remained unchanged [see Fig. 6a]. It is conceivable that the 5BSL3.2 upper stem residues are rapidly exchanging among various conformations in the absence of Mg2+ and cannot give rise to detectable imino signals, whereas the kissing conformation is favored in the presence of Mg2+ and thus afford new signals.
Figure 6.

(a) Imino proton spectra of 5BSL3.2 incubated with increasing [Mg2+]. (b) The electrostatic free energy change for the docking as a function of [Mg2+]. (c) The electrostatic free energy difference between the kissing conformation and non-kissing conformation for 5BSL3.2. (d) The electrostatic free energy of 5BSL3.2 (kissing conformation) and SL2, as a function of [Mg2+]. Here the unit of the free energy is kBT at the room temperature.
To quantitatively analyze the Mg2+ effects on the folding of 5BSL3.2, we use the Vfold method78 to predict 2D structures of 5BSL3.2 from the sequence. The 5BSL3.2 has two alternative 2D structures (shown in the insets of 6b): a stem-loop structure (kissing conformation), which can form the 5BSL3.2:3′X complex, and a three-way junction (non-kissing conformation), which cannot form the 5BSL3.2:3′X complex. For each 2D structure, we the use the Vfold3D method combined with molecular dynamics simulations78 to generate ensemble of 3D conformations for 5BSL3.2. We then apply the new partial charge-based model developed here to evaluate the electrostatics free energy for each conformation. For a given 2D structure, we predict its electrostatic free energy as the Boltzmann-weighted average electrostatic free energy over all the conformations. The predicted free energies for the two alternative 2D structures show that an increasing [Mg2+] would cause the free energy of the kissing conformation (of 5BSL3.2) to become lower than the non-kissing conformation [see Fig. 6b], indicating that a high [Mg2+] would favor the kissing structure over the non-kissing structure. Fig. 7 shows the “high probability” (> 5%) locations of Mg2+ ions for the kissing conformation (a representative 3D structure) of 5BSL3.2 with [K+] = 150 mM and [Mg2+] = 5 mM. The bulge pocket region in the kissing conformation (yellow) of 5BSL3.2 involves significant charge build-up of the RNA, resulting in a high probability for Mg2+ binding around the pocket region, especially in a solution of high [Mg2+]. Therefore, high [Mg2+] favors the kissing conformation of 5BSL3.2. As a caveat, we note that the stability of the 5BSL3.2 is determined by multiple factors, such as canonical and non-canonical hydrogen bonding, base-staking, and even the interactions with 3′X domain. Therefore, our results here only show the Mg2+-induced shift of the relative stability between given RNA structures.
Figure 7.

the predicted results of the high-probability binding positions around the kissing conformation of 5BSL3.2. The yellow part denotes the bulge of the kissing conformation for 5BSL3.2
In contrast to the sensitive Mg2+-dependence of the 5BSL3.2 folding, consistent with the gel electrophoresis results, no significant NMR signal changes were observed in the absence and presence of Mg2+, suggesting a weaker [Mg2+]-dependence of the 3′X folding. The quantitative effect of Mg2+ ions on the docking part of 3′X (SL2) is computed. Our Vfold2D model predicts that SL2 folds into a single 2D structure and the Vfold3D model gives the 3D structure of SL2. SL2 and the 5BSL3.2 kissing (stem-loop) conformation have different 3D structures and thus different [Mg2+]-dependence. The 5BSL3.2 kissing conformation involves a pocket structure in the bulge region (from 9298A to 9304G) and thus significant RNA backbone (local) charge build-up, which induces strong ion-binding. Therefore, 5BSL3.2 stability is highly sensitive to [Mg2+]. In contrast, the SL2 structure lacks such a ion-binding pocket and shows less [Mg2+] sensitivity. The Boltzmann-weighted average electrostatic free energy predicted by the PCTBI model for the Vfold3D-generated 3D conformational ensemble indeed shows a much weaker [Mg2+]-dependence (see Fig 6c). The result indicates that the predicted SL2 structure is stable over a wide range of [Mg2+] and the stability is not sensitive to Mg2+. The conclusion is in agreement with the experimental finding.
To understand the [Mg2+]-dependence of the 5BSL3.2-SL2 docking, we generate a docked 5BSL3.2:SL2 complex state [see the inset of Fig 6d] and an undocked state with separated 5BSL3.2 and SL2 structures. We find that the electrostatics free energy of the docked state is much lower than the undocked state, and the free energy gap increases with an increasing [Mg2+] (see 6d). Compared with the structure of undocked state, the docked state has a more compact structure and, meanwhile, create some extra TB regions for Mg2+ ions between the 5BSL3.2 and SL2. The Mg2+ ions in these TB region would subject to the attraction from the 5BSL3.2 as well as SL2. Therefore, the docked state could attract more TB Mg2+ ions, and these Mg2+ ions can indeed help stabilizing the 5BSL3.2-SL2 docked complex (over the undocked state).
5 CONCLUSION
In this paper, we describe a new partial charge-based model, PCTBI, to predict ion effects for RNA. Although both PCTBI and MCTBI models take into account the ion correlation and fluctuation effects, the current new model (PCTBI) has the advantage of accounting for atomistic charge distributions for RNAs. Unlike the previous coarse grained charge models, this new model enables calculations for the ion-mediated electrostatic interactions for RNAs at atomistic charge resolution. For a great variety of biologically significant problems, such as RNA-protein interactions, a detailed account of the charge distribution at the atomistic level is essential. Therefore, the new model would open up the possibility for many important applications such as RNA-protein docking and the kissing complex formation in HCV genomic RNA. The study described here leads to several main conclusions, as shown below.
Theory-experiment comparison shows that the PCTBI model can provide reliable predictions for the ion effects, especially for the divalent ions (Mg2+) in RNA folding. Moreover, comparison with the previous coarse grained charge model suggests that for the a protein-rRNA fragment docking, the protein prevents monovalent ion binding and adding Mg2+ ions can help stabilize the rRNA-protein docked complex over the undocked state.
The PCTBI model, compared with the previous coarse grained charge model, can predict the bound ion distributions and the probable ion binding sites with improved resolution and accuracy.
Experimental studies indicate that the stability of the 5BSL3.2:SL2 kissing complex in HCV genomic RNA is sensitive to [Mg2+] and Mg2+ ions stabilize the docked complex. By predicting the ion-dependent free energies and the bound ion distribution, the PCTBI model provides mechanistic insights into the experimental finding. Specifically, the ion electrostatic model, combined with the Vfold RNA structure prediction model, predicts that the SL2 domain forms a stable stem-loop structure in various [Mg2+]. In contrast, the 5BSL3.2 domain can form two alternative structures: a hairpin (the structure for the 5BSL3.2:SL2 docking) and a three-way junction (structure incompatible with the kissing complex), and Mg2+ ions promote the formation of the kissing complex by stabilizing the hairpin over the three-way junction structure.
From a computational point of view, the development of the PCTBI model involves two key ingredients. First, the consideration of ion correlation and fluctuation effects demands sampling of an ensemble of many-ion distributions. The use of atomistic partial charge model renders an increase in computational time. Second, the use of Monte Carlo sampling method significantly improves the computational efficiency for the sampling of ion distribution. The combination of the two strategies enables the PCTBI model to predict electrostatic free energy and ion binding properties with higher resolution and accuracy. Future development of the model involves consideration of ion dehydration effects and further improvement of the sampling method in order to treat larger RNAs.
Acknowledgments
This research was supported by NIH grants R01-GM117059 and R01-GM063732 (to SJC) and Zhejiang Provincial Natural Science Foundation China under grant LQ14A040004 (to LZS).
References
- 1.Brion P, Westhof E. Hierarchy and Dynamics of RNA Folding. Annu Rev Biophys Biomol Struct. 1997;26:113–137. doi: 10.1146/annurev.biophys.26.1.113. [DOI] [PubMed] [Google Scholar]
- 2.Tinoco I, Jr, Bustamante C. How RNA Folds. J Mol Biol. 1999;293:271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 3.Rook MS, Treiber DK, Williamson JR. An Optimal Mg2+ Concentration for Kinetic Folding of the Tetrahymena Ribozyme. Proc Natl Acad Sci U S A. 1999;96:12471–12476. doi: 10.1073/pnas.96.22.12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takamoto K, He Q, Brenowitz M. Monovalent Cations Mediate Formation of Native Tertiary Structure of the Tetrahymena Thermophila Ribozyme. Nat Struct Biol. 2002;9:928–933. doi: 10.1038/nsb871. [DOI] [PubMed] [Google Scholar]
- 5.Sosnick TR, Pan T. RNA Folding: Models and Perspectives. Curr Opin Struct Biol. 2003;13:309–316. doi: 10.1016/s0959-440x(03)00066-6. [DOI] [PubMed] [Google Scholar]
- 6.Woodson SA. Metal Ions and RNA Folding: A Highly Charged Topic with a Dynamic Future. Curr Opin Chem Biol. 2005;9:104–109. doi: 10.1016/j.cbpa.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Thirumalai D, Hyeon C. RNA and Protein Folding: Common Themes and Variations. Biochemistry. 2005;44:4957–4970. doi: 10.1021/bi047314+. [DOI] [PubMed] [Google Scholar]
- 8.Koculi E, Hyeon C, Woodson SA. Charge Density of Divalent Metal Cations Determines RNA Stability. J Am Chem Soc. 2007;129:2676–2682. doi: 10.1021/ja068027r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soto AM, Misra V, Draper DE. Tertiary Structure of an RNA Pseudoknot Is Stabilized by Diffuse Mg2+ Ions. Biochemistry. 2007;46:2973–2983. doi: 10.1021/bi0616753. [DOI] [PubMed] [Google Scholar]
- 10.Stellwagen E, Dong Q, Stellwagen NC. Quantitative Analysis of Monovalent Counterion Binding to Random-Sequence Doublestranded DNA Using the Replacement Ion Method. Biochemistry. 2007;46:2050–2058. doi: 10.1021/bi062132w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen AA, Pappu RV. Quantitative Characterization of Ion Pairing and Cluster Formation in Strong 1:1 Electrolytes. J Phys Chem B. 2007;111:6469–6478. doi: 10.1021/jp0708547. [DOI] [PubMed] [Google Scholar]
- 12.Chu VB, Herschlag D. Unwinding RNAs Secrets: Advances in the Biology, Physics, and Modeling of Complex RNAs. Curr Opin Struct Biol. 2008;18:305–314. doi: 10.1016/j.sbi.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Draper DE. RNA Folding: Thermodynamic and Molecular Descriptions of the Roles of Ions. Biophys J. 2008;95:5489–5495. doi: 10.1529/biophysj.108.131813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen SJ. RNA Folding: Conformational Statistics, Folding Kinetics, and Ion Electrostatics. Annu Rev Biophys. 2008;37:197–214. doi: 10.1146/annurev.biophys.37.032807.125957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu X, Andresen K, Pollack L. Abrupt Transition from a Free, Repulsive to a Condensed, Attractive DNA Phase, Induced by Multivalent Polyamine Cations. Phys Rev Lett. 2008;101:228101. doi: 10.1103/PhysRevLett.101.228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlatterer JC, Kwok LW, Pollack L. Hinge Stiffness Is a Barrier to RNA Folding. J Mol Biol. 2008;379:859–870. doi: 10.1016/j.jmb.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li PTX, Tinoco I., Jr Mechanical Unfolding of Two DIS RNA Kissing Comlexes from HIV-1. J Mol Biol. 2009;386:1343–1356. doi: 10.1016/j.jmb.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipfert J, Sim AY, Doniach S. Dissecting Electrostatic Screening, Specific Ion Binding, and Ligand Binding in an Energetic Model for Glycine Riboswitch Folding. RNA. 2010;16:708–719. doi: 10.1261/rna.1985110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye W, Yang J, Yu Q, Wang W, Hancy J, Luo R, Chen HF. Kink Turn sRNA Folding upon L7Ae Binding Using Molecular Dynamics Simulations. Phys Chem Chem Phys. 2013;15:18510–18522. doi: 10.1039/c3cp53145g. [DOI] [PubMed] [Google Scholar]
- 20.Xia Z, Bell DR, Shi Y, Ren P. RNA 3D Structure Prediction by Using a Coarse-Grained Model and Experimental Data. J Phys Chem B. 2013;117:3135–3144. doi: 10.1021/jp400751w. [DOI] [PubMed] [Google Scholar]
- 21.Serra MJ, Turner DH. Predicting Thermodynamic Properties of RNA. Methods Enzymol. 1995;259:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 22.SantaLucia J., Jr A Unified View of Polymer Dumbbell and Oligonucleotide DNA Nearest-Neighbor Thermodynamics. Proc Natl Acad Sci U S A. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathews DH, Sabina J, Turner DH. Expanded Sequence Dependence of Thermodynamic Parameters Improves Prediction of RNA Secondary Structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 24.Chen SJ, Dill KA. RNA Folding Energy Landscapes. Proc Natl Acad Sci U S A. 2000;97:646–651. doi: 10.1073/pnas.97.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuker M. MFold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cate JH, Doudna JA. Metal-Binding Sites in the Major Groove of a Large Ribozyme Domain. sturcture. 1996;15:1221–1229. doi: 10.1016/s0969-2126(96)00129-3. [DOI] [PubMed] [Google Scholar]
- 27.Draper DE, Grilley D, Soto AM. Ions and RNA Folding. Annu Rev Biophys Biomol Struct. 2005;34:221–243. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- 28.Tan ZJ, Chen SJ. Electrostatic Correlations and Fluctuations for Ion Binding to a Finite Length Polyelectrolyte. J Chem Phys. 2005;122:44903. doi: 10.1063/1.1842059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misra VK, Draper DE. A Thermodynamic Framework for Mg2+ Binding to RNA. Proc Natl Acad Sci U S A. 2001;98:12456–12461. doi: 10.1073/pnas.221234598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Misra VK, Draper DE. On the Role of Magnesium Ions in RNA Stability. Biopolymers. 1998;48:113–135. doi: 10.1002/(SICI)1097-0282(1998)48:2<113::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 31.Bai Y, Greenfeld M, Herschlag D. Quantitative and Comprehensive Decomposition of the Ion Atmosphere around Nucleic Acids. J Am Chem Soc. 2007;129:14981–14988. doi: 10.1021/ja075020g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grochowski P, Trylska J. Continuum Molecular Electrostatics, Salt Effects, and Counterion Binding–A Review of the Poisson-Boltzmann Theory and Its Modifications. Biopolymers. 2008;89:93113. doi: 10.1002/bip.20877. [DOI] [PubMed] [Google Scholar]
- 33.Wang K, Yu YX, Gao GH. Density Functional Study on the Structural and Thermodynamic Properties of Aqueous DNA-Electrolyte Solution in the Framework of Cell Model. J Chem Phys. 2008;128:185101. doi: 10.1063/1.2918342. [DOI] [PubMed] [Google Scholar]
- 34.Dong F, Olsen B, Baker NA. Computational Methods for Biomolecular Electrostatics. Methods Cell Biol. 2008;84:843–870. doi: 10.1016/S0091-679X(07)84026-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joung I, Cheatham TE. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model Specific Ion Parameters. J Phys Chem B. 2009;113:13279–13290. doi: 10.1021/jp902584c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuczera K, Jas G, Elber R. The Kinetics of Helix Unfolding: Molecular Dynamics Simulations with Milestoning. J Phys Chem A. 2009;113:7431–7473. doi: 10.1021/jp900407w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayes RL, Noel JK, Whitford PC, Mohanty U, Sanbonmatsu KY, Onuchic JN. Reduced Model Captures Mg2+-RNA Interaction Free Energy of Riboswitches. Biophys J. 2014;106:1508–1519. doi: 10.1016/j.bpj.2014.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen AA, Draper DE, Pappu RV. Molecular Simulation Studies of Monovalent Counterion- Mediated Interactions in a Model RNA Kissing Loop. J Mol Biol. 2009;390:805–819. doi: 10.1016/j.jmb.2009.05.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Auffinger P, Bielecki L, Westhof E. The Mg2+ Binding Sites of the 5S rRNA Loop E Motif as Investigated by Molecular Dynamics Simulations. Chem Biol. 2003;10:551–561. doi: 10.1016/s1074-5521(03)00121-2. [DOI] [PubMed] [Google Scholar]
- 40.Hayes RL, Noel JK, Mohanty U, Whitford PC, Hennelly SP, Onuchic J, Sanbonmatsu KY. Magnesium Fluctuations Modulate RNA Dynamics in the SAM-I Riboswitch. J Am Chem Soc. 2012;134:12043–12053. doi: 10.1021/ja301454u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krasovska MV, Sefcikova J, Sponer J. Cations and Hydration in Catalytic RNA: Molecular Dynamics of the Hepatitis Delta Virus Ribozyme. Biophys J. 2006;91:626–638. doi: 10.1529/biophysj.105.079368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen A, Marucho M, Baker NA, Pappu R. Simulations of RNA Interactions with Monovalent Ions. Methods Enzymol. 2009;469:411–432. doi: 10.1016/S0076-6879(09)69020-0. [DOI] [PubMed] [Google Scholar]
- 43.Sklenovsky P, Florova P, Banas P, Reblova K, Lankas F, Otyepka M, Sponer J. Understanding RNA Flexibility Using Explicit Solvent Simulations: The Ribosomal and Group I Intron Reverse Kink-Turn Motifs. J Chem Theory Comput. 2011;7:2963–2980. doi: 10.1021/ct200204t. [DOI] [PubMed] [Google Scholar]
- 44.Do TN, Ippoliti E, Parrinello M. Counterion Redistribution upon Binding of a Tat-Protein Mimic to HIV-1 TAR RNA. J Chem Theory Comput. 2012;8:688–694. doi: 10.1021/ct2005769. [DOI] [PubMed] [Google Scholar]
- 45.Manning GS. The Molecular Theory of Polyelectrolyte Solutions with Applications to the Electrostatic Properties of Polynucleotides. Q Rev Biophys. 1978;11:179–249. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 46.Zhou HX. Macromolecular Electrostatic Energy within the Nonlinear Poisson-Boltzmann Equation. J Chem Phys. 1994;100:3152–3162. [Google Scholar]
- 47.Misra V, Draper DE. The Interpretation of Mg2+ Binding Isotherms for Nucleic Acids Using Poisson-Boltzmann Theory. J Mol Biol. 1999;17:1135–1147. doi: 10.1006/jmbi.1999.3334. [DOI] [PubMed] [Google Scholar]
- 48.Baker NA, Sept D, McCammon JA. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker NA. Improving Implicit Solvent Simulations: A Poissoncentric View. Curr Opin Struct Biol. 2005;15:137–143. doi: 10.1016/j.sbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Tjong H, Zhou HX. The Dependence of Electrostatic Solvation Energy on Dielectric Constants in Poisson-Boltzmann Calculations. J Chem Phys. 2006;125:206101. doi: 10.1063/1.2393243. [DOI] [PubMed] [Google Scholar]
- 51.Tjong H, Zhou HX. GBr6NL: A Generalized Born Method for Accurately Reproducing Solvation Energy of the Nonlinear Poisson-Boltzmann Equation. J Chem Phys. 2007;126:195102. doi: 10.1063/1.2735322. [DOI] [PubMed] [Google Scholar]
- 52.Tan ZJ, Chen SJ. Predicting Ion Binding Properties for RNA Tertiary Structures. Biophys J. 2010;99:1–12. doi: 10.1016/j.bpj.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mak CH, Henke PS. Ions and RNAs: Free Energies of Counterion-Mediated RNA Fold Stabilities. J Chem Theory Comput. 2013;9:621–639. doi: 10.1021/ct300760y. [DOI] [PubMed] [Google Scholar]
- 54.Giambasu GM, Luchko T, Herschlag D, York DM, Case DA. Ion Counting from Explicit-Solvent Simulations and 3D-RISM. Biophys J. 2014;106:883–894. doi: 10.1016/j.bpj.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayes RL, Noel JK, Mandic A, Whitford PC, Sanbonmatsu KY, Mohanty U, Onuchic JN. Generalized Manning Condensation Model Captures the RNA Ion Atmosphere. Phys Rev Lett. 2015;114:258105. doi: 10.1103/PhysRevLett.114.258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan ZJ, Chen SJ. Ion-Mediated Nucleic Acid Helix-Helix Interactions. Biophys J. 2006;91:518–536. doi: 10.1529/biophysj.106.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan ZJ, Chen SJ. Electrostatic Free Energy Landscape for Nucleic Acid Helix Assembly. Nucleic Acids Res. 2006;34:6629–6639. doi: 10.1093/nar/gkl810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan ZJ, Chen SJ. RNA Helix Stability in Mixed Na+/Mg2+ Solution. Biophys J. 2007;92:3615–3632. doi: 10.1529/biophysj.106.100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan ZJ, Chen SJ. Salt Dependence of Nucleic Acid Hairpin Stability. Biophys J. 2008;95:738–752. doi: 10.1529/biophysj.108.131524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He Z, Chen SJ. Predicting Ion-Nucleic Acid Interactions by Energy Landscape-Guided Sampling. J Chem Theory Comput. 2012;8:2095–2102. doi: 10.1021/ct300227a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu Y, Chen SJ. Many-Body Effect in Ion Binding to RNA. J Chem Phys. 2014;141:055101. doi: 10.1063/1.4890656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun LZ, Chen SJ. Monte Carlo Tightly Bound Ion Model: Predicting Ion-Binding Properties of RNA with Ion Correlations and Fluctuations. J Chem Theory Comput. 2016;12:3370–3381. doi: 10.1021/acs.jctc.6b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Philips A, Milanowska K, Lach G, Boniecki M, Rother K, Bujnicki JM. Metal ion RNA: Computational Predictor of Metal-Binding Sites in RNA Structures. Bioinformatics. 2012;28:198–205. doi: 10.1093/bioinformatics/btr636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. DOCK 6: Combining Techniques to Model RNA-Small Molecule Complexes. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pestova TV, Shatsky IN, Fletcher SP, Jackson RJ, Hellen CU. A Prokaryotic-Like Mode of Cytoplasmic Eukaryotic Ribosome Binding to the Initiation Codon during Internal Translation Initiation of Hepatitis C and Classical Swine Fever Virus RNAs. Genes & development. 1998;12:67–83. doi: 10.1101/gad.12.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moradpour D, Penin F, Rice CM. Replication of Hepatitis C Virus. Nature reviews Microbiology. 2007;5:453–63. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 67.Cantero-Camacho A, Gallego J. The Conserved 3′X Terminal Domain of Hepatitis C Virus Genomic RNA Forms a Two-Stem Structure That Promotes Viral RNA Dimerization. Nucleic Acids Res. 2015;43:8529–39. doi: 10.1093/nar/gkv786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Friebe P, Boudet J, Simorre JP, Bartenschlager R. Kissing-Loop Interaction in the 3′ End of the Hepatitis C Virus Genome Essential for RNA Replication. J Virol. 2005;79:380–92. doi: 10.1128/JVI.79.1.380-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.You S, Rice CM. 3′ RNA Elements in Hepatitis C Virus Replication: Kissing Partners and Long Poly(U) J Virol. 2008;82:184–95. doi: 10.1128/JVI.01796-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.You S, Stump DD, Branch AD, Rice CM. A Cis-Acting Replication Element in the Sequence Encoding the NS5B RNA-Dependent RNA Polymerase Is Required for Hepatitis C Virus RNA Replication. J Virol. 2004;78:1352–66. doi: 10.1128/JVI.78.3.1352-1366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shetty S, Stefanovic S, Mihailescu MR. Hepatitis C Virus RNA: Molecular Switches Mediated by Long-Range RNA-RNA Interactions. Nucleic Acids Res. 2013;41:2526–40. doi: 10.1093/nar/gks1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palau W, Masante C, Ventura M, Di Primo C. Direct Evidence for RNA-RNA Interactions at the 3′ End of the Hepatitis C Virus Genome Using Surface Plasmon Resonance. RNA. 2013;19:982–91. doi: 10.1261/rna.037606.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Colasanti A, Lu XJ, Olson WK. Analyzing and Building Nucleic Acid Structures with 3DNA. J Vis Exp. 2013;74:e4401. doi: 10.3791/4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Westhof E, Sundaralingam M. Restrained Refinement of the Monoclinic Form of Yeast Phenylala-nine Transfer RNA. Temperature Factors and Dynamics, Coordinated Waters, and Base-Pair Propeller Twist Angles. Biochemistry. 1986;25:4868–4878. doi: 10.1021/bi00365a022. [DOI] [PubMed] [Google Scholar]
- 75.Conn GL, Gittis AG, Draper DE. A Compact RNA Tertiary Structure Contains a Buried Backbone-K+ Complex. J Mol Biol. 2002;318:963–973. doi: 10.1016/S0022-2836(02)00147-X. [DOI] [PubMed] [Google Scholar]
- 76.Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ. Structural Basis for Discriminative Regulation of Gene Expression by Adenine- and Guanine-Sensing mRNAs. Chem Biol. 2004;11:1729–1741. doi: 10.1016/j.chembiol.2004.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Website address: http://www.rcsb.org/pdb/home/home.do.
- 78.Xu XJ, Zhao PN, Chen SJ. Vfold: A Web Server for RNA Structure and Folding Thermodynamics Prediction. PLoS One. 2014;9:e107504. doi: 10.1371/journal.pone.0107504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera- Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 80.Cornell WD, Cieplak P, Baily CI, Gould IR, Merz KM, Ferguson DC, Jr, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins Nucleic Acids and Organic Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 81.Cheatham TE, III, Cieplak P, Kollman PA. A Modified Version of the Cornell et al. Force Field with Improved Sugar Pucker Phases and Helical Repeat. J Biomol Struct Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 82.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J Comput Chem. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 83.Wang J, Wang W, Kollman PA, Case DA. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J Mol Graph Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 84.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: An Automated Pipeline for the Setup, Execution, and Analysis of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang JM, Cieplak P, Kollman PA. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 86.MacKerell ADJ, Bashford D, Bellot M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 87.Sitkoff D, Sharp KA, Honig B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J Phys Chem. 1994;98:1978–1988. [Google Scholar]
- 88.Chen SW, Honig B. Monovalent and Divalent Salt Effects on Electrostatic Free Energies Defined by the Nonlinear PoissonBoltzmann Equation: Application to DNA Binding Reactions. J Phys Chem B. 1997;101:9113–9118. [Google Scholar]
- 89.Overbeek J, Th G. The Role of Energy and Entropy in the Electrical Double Layer. Colloids Surf. 1990;51:61–75. [Google Scholar]
- 90.Stigter D. Valuation of the Counterion Condensation Theory of Polyelectrolytes. Biophys J. 1995;69:380–388. doi: 10.1016/S0006-3495(95)79910-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rosenbluth MN, Rosenbluth AW. Monte Carlo Calculation of the Average Extension of Molecular Chains. J Chem Phys. 1955;23:365–369. [Google Scholar]
- 92.Li Z, Scheraga HA. Monte Carlo-Minimization Approach to the Multiple-Minima Problem in Protein Folding. Proc Natl Acad Sci U S A. 1987;84:6611–6615. doi: 10.1073/pnas.84.19.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 94.Lipfert J, Doniach S, Das R, Herschlag D. Understanding Nucleic Acid-Ion Interaction. Annu Rev Biochem. 2014;83:813–841. doi: 10.1146/annurev-biochem-060409-092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krakauer H. The Binding of Mg2+ Ions to Polyadenylate, Polyuridylate, and Their Complexes. Biopolymers. 1971;10:2459–2490. doi: 10.1002/bip.360101209. [DOI] [PubMed] [Google Scholar]
- 96.Romer R, Hach R. tRNA Conformation and Magnesium Binding. A Study of a Yeast Phenylalanine-Specific tRNA by a Fluorescent Indicator and Differential Melting Curves. Eur J Biochem. 1975;55:271–284. doi: 10.1111/j.1432-1033.1975.tb02160.x. [DOI] [PubMed] [Google Scholar]
- 97.Grilley D, Misra V, Draper DE. Importance of Partially Unfolded Conformations for Mg2+-Induced Folding of RNA Tertiary Structure: Structural Models and Free Energies of Mg2+ Interactions. Biochemistry. 2007;46:10266–10278. doi: 10.1021/bi062284r. [DOI] [PubMed] [Google Scholar]
- 98.Leipply D, Draper DE. Effects of Mg2+ on the Free Energy Landscape for Folding a Purine Riboswitch RNA. Biochemistry. 2011;50:2790–2799. doi: 10.1021/bi101948k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leipply D, Draper DE. Evidence for a Thermodynamically Distinct Mg2+ Ion Associated with Formation of an RNA Tertiary Structure. J Am Chem Soc. 2011;133:13397–13405. doi: 10.1021/ja2020923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gebala M, Bonilla S, Bisaria N, Herschlag D. Does Cation Size Affect Occupancy and Electrostatic Screening of the Nucleic Acid Ion Atmosphere? 2016;138:10925–10934. doi: 10.1021/jacs.6b04289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang J, Ferré-DAmaré AR. Dramatic Improvement of Crystals of Large RNAs by Cation Replacement and Dehydration. Structure. 2014;22:1363–71. doi: 10.1016/j.str.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sun LZ, Zhang D, Chen SJ. Theory and Modeling of RNA Structure and Interactions with Metal Ions and Small Molecules. Annu Rev Biophys. 2017;46:227–46. doi: 10.1146/annurev-biophys-070816-033920. [DOI] [PMC free article] [PubMed] [Google Scholar]