

ABSTRACT
Cell surface molecules of the B7/CD28 family play an important role in T-cell activation and tolerance. The relevance of the PD-1/PD-L1 pathway in cancer has been extensively studied whereas PD-L2 has received less attention. However, recently the expression of PD-L2 was described to be independently associated with clinical response in anti-PD1-treated cancer patients. Here, we investigated whether PD-L2 might represent a natural target that induces specific T cells. We identified spontaneous specific T-cell reactivity against two epitopes located in the signal peptide of PD-L2 from samples from patients with cancer as well as healthy individuals ex vivo. We characterized both CD8+ and CD4+ PD-L2-specific T cells. Interestingly, the epitope in PD-L2 that elicited the strongest response was equivalent to a potent HLA-A2-restricted epitope in PD-L1. Importantly, PD-L1-specific and PD-L2-specific T cells did not cross-react; therefore, they represent different T-cell antigens. Moreover, PD-L2-specific T cells reacted to autologous target cells depending on PD-L2 expression. These results suggested that activating PD-L2 specific T cells (e.g., by vaccination) might be an attractive strategy for anti-cancer immunotherapy. Accordingly, PD-L2 specific T cells can directly support anti-cancer immunity by killing of target cells, as well as, indirectly, by releasing pro-inflammatory cytokines at the microenvironment in response to PD-L2-expressing immune supressive cells.
KEYWORDS: Anti-cancer immunity, CD4+ T cells, CD8+ T cells, Immune checkpoint regulator, PD-L2
Introduction
The B7/CD28 pathways provide positive co-stimulatory signals to sustain T-cell activity, and they contribute inhibitory signals that modulate the magnitude of T-cell responses. Inhibitory molecules of the B7/CD28 family play key roles in inducing immune tolerance in the tumor microenvironment.1 The programmed death-1 receptor (PD-1, CD279) and its ligands, PD-L1 (CD274, B7-H1) and PD-L2 (CD273, B7-DC), constitute one such inhibitory pathway. The relevance of the PD-1/PD-L1 pathway in cancer has been extensively studied. Therapeutic approaches that target PD-1 (e.g., nivolumab or pembrolizumab) and PD-L1 (e.g., avelumab or atezolizumab) have been developed and approved for several different indications of cancer, including lymphoma. Thus, blocking PD-1 has shown remarkable clinical effects in Hodgkin lymphoma (HL). Recently, the FDA approved the anti-PD-1 antibody, nivolumab (Opdivo®), for treating classic HL. However, PD-L2 has not received as much attention, and its role in modulating tumor immunity is not clear. Studies at the protein level have been limited by the lack of reliable, commercially available anti-PD-L2 antibodies. However, in a recent analysis of more than 400 archival tumor samples,2 PD-L2 expression was observed in several tumor types and was described to be expressed in the absence of PD-L1 in subsets of patients. This has also been reported elsewhere.3 Moreover, PD-L2 expression was independently associated with clinical response in pembrolizumab-treated patients. Thus, PD-L2 expression may play an important role in response to PD-1 axis targeted therapies.
Studies that characterized these two ligands reported that PD-L2 could bind to PD-1 with 2-6-fold higher affinity than PD-L1.4 PD-L2 is expressed by antigen-presenting cells, such as macrophages and dendritic cells (DCs), but its expression can be induced in other immune and non-immune cells, mainly through Th2-associated cytokines. PD-L2 expression has been detected in samples from patients with different solid cancers. In addition, PD-L2 seems to play a pivotal role in hematologic malignancies. In classical Hodgkin lymphoma (HL), both PD-L1 and PD-L2 expression were considered a defining feature; expression levels increased with 9p24.1 alterations in 97% of cases.5 Expression studies in NHL have shown highly variable results, potentially due to differences in staining antibodies and cut-off thresholds. However, a gene expression study in follicular lymphoma (FL) demonstrated higher expression of PD-L2 than PD-L1, which was confirmed with immunohistochemistry. They showed that PD-L2 was expressed in FL cells in 75% of cases, and PD-L1 was only found on environmental cells.6 In myeloid malignancies, PD-L2 was upregulated in CD34+ stem cells. Likewise, PD-L2 expression was reported to be elevated in acute myeloid leukemia (AML) cells.7 The same study found that myeloid-derived suppressor cells (MDSCs) expressed higher levels of PD-L2 than CD34+ cells. MDSCs are known to suppress anti-tumor immunity and promote tumor growth in a variety of cancers, including hematological malignancies.8 Hence, it is important to consider PD-L2 expression in tumor cells as well as in the microenvironment. The presence of PD-L2 in the microenvironment makes this ligand attractive, both as a checkpoint molecule and as a tumor antigen for therapeutic targeting.
Recent studies showed that the immune system has an anti-cancer mechanism that works via PD-L1-specific effector T cells.9–13 Our previous studies was the first to describe spontaneous CD8+ and CD4+ T-cell reactivity against PD-L1 in peripheral blood of both healthy donors and patients with various cancers. Those results suggested that PD-L1-specific T cells might modulate adaptive immune reactions. In the present study, we aimed to identify PD-L2 as a novel T-cell target. We investigated different PD-L2-derived epitopes that might elicit T cell reactivity, and we tested spontaneous T-cell mediated reactivity against PD-L2 in samples from both healthy donors and patients with different cancers. Finally, we determined whether PD-L2-specific T cells could recognzie target cells expressing PD-L2.
Results
Natural T cell reactivity against PD-L2
The amino acid sequence of the PD-L2 protein was screened with the “SYFPEITHI” database18 to predict the best HLA-A2 peptide epitopes. The algorithm identified 22 peptides that were top candidates, based on predictive scores in the range of 22–29. We selected 9 peptides for synthesis and further study, based on their location in the PD-L2 protein. At least part of the peptide had to be located in the signal peptide or the transmembrane domain. These nine peptides were used with the IFN-γ ELISPOT in vitro assay to test for the presence of specific T-cell responses in PBMCs from HLA-A2+ patients with malignant melanoma. We detected immune responses against PD-L215 (PD-L2234-243), and in particular, against PD-L201 (PD-L24-12) and PD-L205 (PD-L216-25) (Figure 1).
Figure 1.
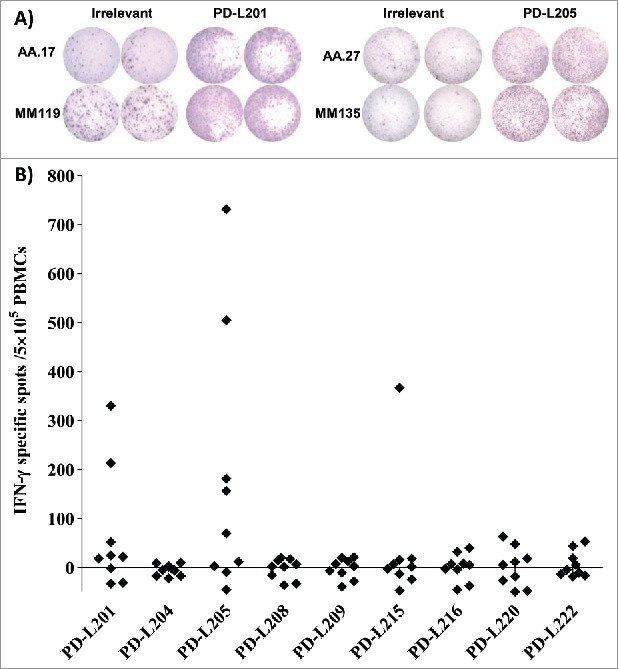
Screening for T-cell responses towards minimal peptides derived from PD-L2. (A) Examples of ELISPOT results for PBMCs isolated from patients with malignant melanoma (AA and MM), in response to PD-L201 (PD-L24-12; LLLMLSLEL) and PD-L205 (PD-L216-25; QIAALFTVTV). (B) In-vitro IFN-γ ELISPOT results. PBMCs from 9 patients with malignant melanoma were stimulated once in vitro with each peptide. Then, the PBMCs were exposed to the peptides, and IFN-γ secretion was measured with ELISPOT. The response was calculated as the number of peptide-specific spots, minus the number of spots that reacted to an irrelevant peptide (HIV/HLA-A2; pol476-484; ILKEPVHGV), per5 × 105 PBMCs.
Next, we utilized both the IFN-γ and TNF-α ELISPOT assays to examine 5 selected PBMCs for immune responses against PD-L201 (PD-L24-12) and PD-L205 (PD-L216-25) (Figure 2A and 2B). All IFN-γ and TNF-α responses were statistically significant, according to the DFR test (Figure 2A and 2B). In addition, the IFN-γ responses and one TNF-α response were statistically significant according to the DFR × 2 rule (Figure 2A and 2B). Next, we tested PBMCs from healthy donors for immune responses against both PD-L2-derived epitopes with the IFN-γ ELISPOT assay. We detected strong immune responses against PD-L205 (PD-L216-25), and weaker responses against PD-L201 (PD-L24-12) in healthy individuals (Figure 2C). In general, PD-L205 (PD-L216-25) appeared to be the dominant epitope for eliciting immune responses. Next, we tested PBMCs from four donors, directly ex vivo (without prior in vitro peptide stimulation), for responses against PD-L205 (PD-L216-25) with the IFN-γ ELISPOT assay (Figure 2D). The PBMCs from one of these donors showed an ex vivo IFN-γ response that was statistically significant, according to the DFR rule (Figure 2D).
Figure 2.
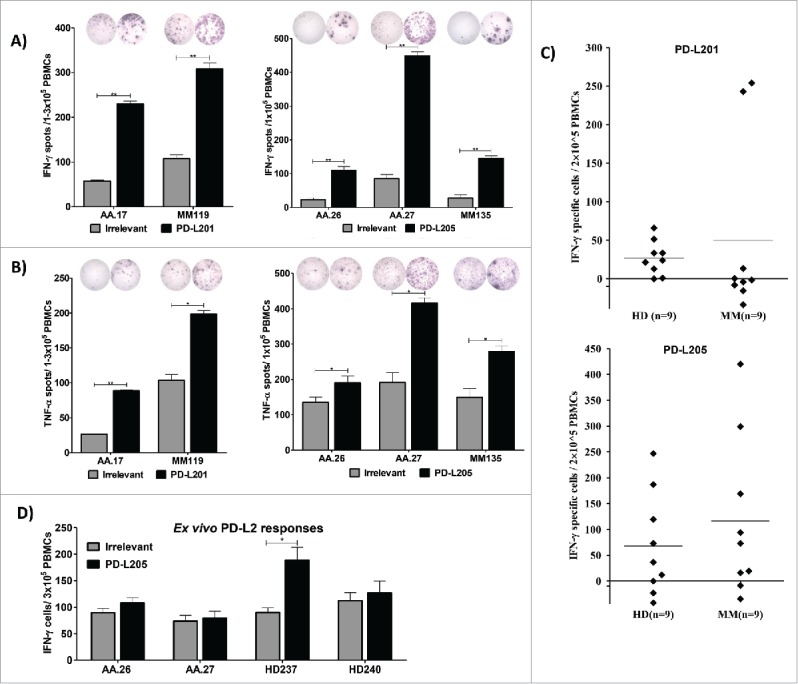
Natural T-cell responses towards two minimal PD-L2-derived epitopes in both patients with cancer and healthy donors. (A) Examples of IFN-γ responses against PD-L201 (PD-L24-12) and PD-L205 (PD-L216-25)(black bars) or irrelevant peptide (grey bars) in PBMCs from patients with malignant melanoma (AA and MM). All experiments were performed in triplicate, ** significant according to the DFR and DFR × 2. (B) Examples of TNF-α responses against PD-L201 (PD-L24-12) and PD-L205 (PD-L216-25) (black bars) or irrelevant peptide (grey bars) in PBMCs from patients with malignant melanoma (AA and MM), ** significant according to the DFR and DFR × 2; * significant according to only the DFR. (C) In-vitro IFN-γ ELISPOT results. PBMCs from 9 patients with malignant melanoma and 9 healthy donors were stimulated once in vitro with PD-L201 (PD-L24-12) or PD-L205 (PD-L216-25). Then, PBMCs were exposed to the peptides, and IFN-γ secretion was measured with ELISPOT. The average number of peptide-specific spots (after subtracting the number of spots without added peptide) was calculated per 2–5 × 105 PBMCs. (D) Ex vivo IFN-γ ELISPOT results. PD-L205 (PD-L216-25)(black bars) or the irrelevant peptide (grey bars) elicited responses in PBMCs from two patients with malignant melanoma (AA) and in PBMCs from two healthy donors (HD).
Finally, to test for spontaneous, PD-L2-specific, CD4+ T-cell responses, we synthesized two longer PD-L2 peptides. One of these, PD-L2long1 (PD-L29-29; SLELQLHQIAALFTVTVPKEL), included the PD-L205 (PD-L216-25) epitope; and the other, PD-L2long2 (PD-L21-25; MIFLLLMLSLELQLHQIAALFTVTV), included both the PD-L201 (PD-L24-12) and PD-L205 (PD-L216-25) epitopes. We tested PBMCs from 11 patients with malignant melanoma and 11 healthy donors for the presence of CD4+ T cell responses against these long peptides with the IFN-γ ELISPOT assay. We detected frequent but moderate responses (Figure 3A). Next, we isolated PBMCs from four non-hodgkin lymphoma patients and screened for IFN-γ responses towards PD-L2long2 (PD-L21-25) in an in vitro ELISPOT assay. All four patients seemed to have a spontaneous immune response towards the long peptide. Indeed, the responses were significant according to the DFR rule in three of the patients, and the last could not be calculated since this experiment were only performed in duplicates (Figure 3B and 3C). Tumor infiltrating T- lymphocytes (TILs) from two melanoma patients also elicited IFN-γ CD4+ T-cell responces towards PD-L2long2 (PD-L21-25) measured by using intracellular cytokine staining (Figure 3D). TILs from both patients additionally elicited a weak IFN-γ and TNF-α CD8+ T-cell response towards PD-L2long2 (PD-L21-25) (Figure 1S).
Figure 3.
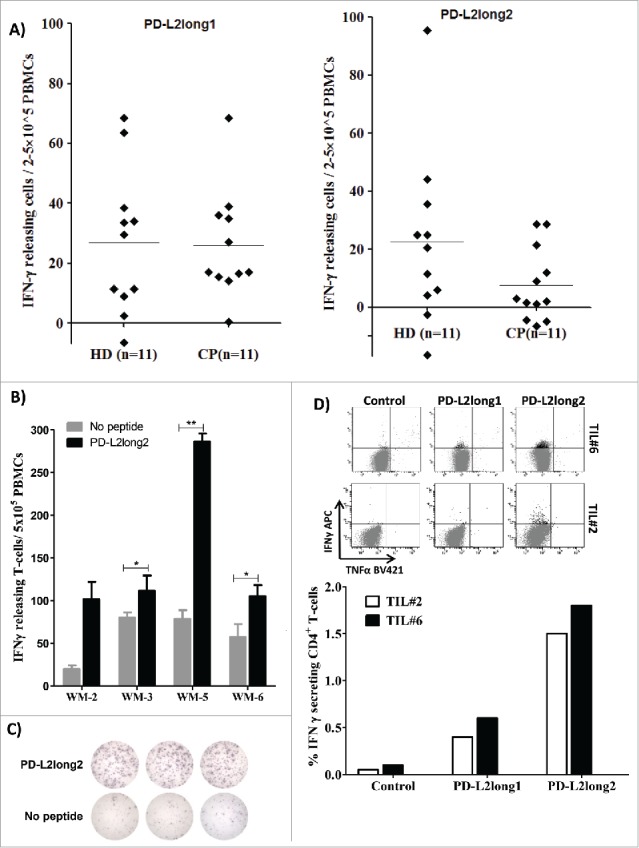
Reactivity towards long PD-L2 peptides spanning the signal peptide part of the PD-L2 sequence. (A) In vitro IFN-γ ELISPOT results. PBMCs from 11 patients with malignant melanoma and 11 healthy donors were stimulated with PD-L2long1 (PD-L29-29; SLELQLHQIAALFTVTVPKEL) or PD-L2long2 (PD-L21-25; MIFLLLMLSLELQLHQIAALFTVTV) and screened for IFNγ responses, by measuring IFNγ release in an in vitro ELISPOT assay. (B) PBMCs from four non-hodgkin lymphoma patients (WM) screened for IFN-γ responses towards PD-L2long2 (PD-L21-25) in an in vitro ELISPOT assay. All assays were made in triplicates with 3*10^6 cells per well, except one which were made in duplicates (WM-2). ** denotes as significant according to the DFR and DFR × 2; * denotes significant according to only the DFR. (C) Examples of ELISPOT well images for WM-5 patient in response to PD-L2long2. (D) Intracellular cytokine staining of tumor infiltrating T-lymphocytes (TILs) from two melanoma patients (TIL2, white bars and TIL6, black bars) shows CD4+ T cell release of IFN-Υ, upon exposure to PD-L2long1 (PD-L29-29), PD-L2long2 (PD-L21-25) and a control HIV peptide (HIV-1 pol476-484).
PD-L2-specific T cells were effector cells releasing pro-inflammatory cytokines
To characterize the immune response elicited by PD-L2, we isolated and expanded PD-L205-specific T cells from a patient with melanoma (AA26) either using anti-CD137 (PD-L2T cell culture-A) or by TNF-α enrichment (PD-L2T cell culture-B). These specific T cells were analyzed for specificity against PD-L205 (PD-L216-25) with intracellular cytokine staining (Figure 4A and 4B), ELISPOT (Figure 4C) and cytotoxicity assays (Figure 4D). We detected TNF-α release in response to PD-L205(PD-L216-25) in about 4.5% and 1% of CD4+ and CD8+ T cells, respectively, from PD-L2T cell culture-A; and about 7% and 2% of CD4+ and CD8+ T cells, respectively, from PD-L2T cell culture-B (Figure 4A). Additonally, we detected TNF-α release in response to autologous DCs in around 3% and 2.8% of CD4+ T cells from PD-L2T cell culture-A and -B respectively (Figure 4B). Furthermore, IFN-Υ and TNF-α T cell responses were observed towards PD-L205(PD-L216-25) peptide and autologous DCs in both PD-L2 specific cultures (Figure 4C). Both expanded cultures also recognized and lysed T2 cells that had been pulsed with PDL205 (PD-L216-25) in conventional 51chromium-release assays, but they did not recognize T2 cells pulsed with an irrelevant HIV peptide (HIV-1 pol476-484) (Figure 4D).
Figure 4.
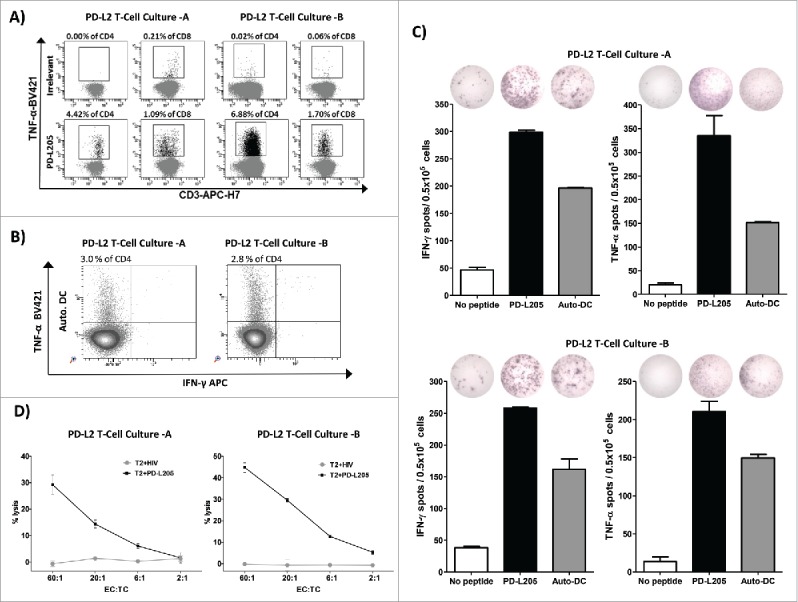
PD-L2-specific T cells are effector T cells. (A) Intracellular cytokine staining showing CD4+ and CD8+ T cells that release TNF-α in response to either an irrelevant control peptide HIV peptide (HIV-1 pol476-484) or PD-L205 (PD-L216-25) in cultures of PD-L2T cells-A (left,) and PD-L2T cells-B (right). (B) Intracellular TNF-α and IFN- γ cytokine staining of PD-L2T-cells culture-A (left) and PD-L2T-cells culture-B (right) in response to 5 hours stimulation with autologous DCs. (C) IFN-γ and TNF-α secretion by PD-L2T-cell culture-A (top) and PD-L2T-cell culture-B (bottom) towards PD-L205 (PD-L216-25) peptide (black bars) and autologous DCs when cultured at ratio 1:5 (grey bars) as measured by ELISPOT assay. (D) T2 cells pulsed either with PD-L205 (PD-L216-25) or a control HIV peptide (HIV-1 pol476-484) as recognized by by PD-L2T-cell culture-A (left) and PD-L2T-cell culture-B (right) in a standard 51Cr-release assay.
PD-L2 dependent reactivity in response to PD-L2 expressing DCs
PD-L2 can be induced in immune cells. Thus, as the next and very more important step, we addressed the question whether PD-L2-expressing DCs would also be recognized by PD-L2-reactive T cells. To test this notion, we generated autologous DCs; and transfected these with PD-L2 siRNA. We first examined PD-L2 protein expression on the matured siRNA transfected DCs (Figure 5A). PD-L2 expression was down regulated on DCs transfected with PD-L2 siRNA compared to a negative control siRNA, 48 hours after electroporation (Figure 5A). Next, we examined reactivity of PD-L2T-cell cultures in response to the transfected DCs using intracellular cytokine staining (Figure 5B) and ELISPOT assay (Figure 5C). Both PD-L2T cell cultures show reduced CD4+ and CD8+ T-cell cytokine response towards DCs transfected with PD-L2 siRNA compared to a negative control siRNA (Figure 5B). Similarly in both cultures, the number of TNF-α releasing T cells were significantly reduced in response to DCs transfected with PD-L2 siRNA compared to a negative control siRNA in an TNF-α ELISPOT assay (Figure 5C). These results confirmed that the reactivity of target cells were dependent on PD-L2 expression.
Figure 5.
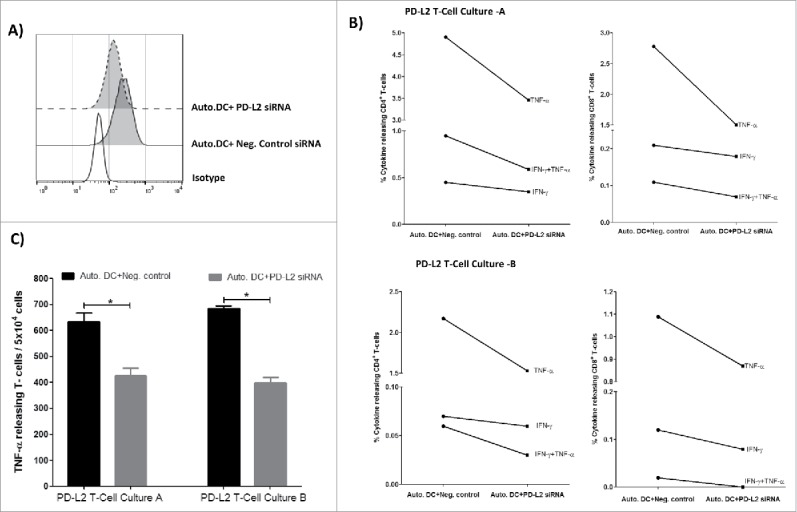
PD-L2 dependent reactivity towards DCs. (A) Flow cytometric analysis showing profile of PD-L2 surface expression on autologous DCs transfected with either PD-L2 siRNA or negative control siRNA, 48hr after electroporation. (B) PD-L2T-cells culture-A (top) and PD-L2T-cells culture-B (bottom) were stimulated with autologous DCs transfected PD-L2 siRNA or negative control siRNA for 5 hours at a ratio of 1:5 (DC:T-cell). Percentage of cytokine releasing CD4+ T cells (left) and CD8+ T cells (right) was measured using intracellular cytokine staining. (C) Number of TNF-α releasing T cells in PD-L2 cultures in response to autologous DCs transfected with either a negative control siRNA (black bars) or PD-L2 siRNA (grey bars) measured at 48 hours after electroporation using ELISPOT assay. The assay was performed in triplicate and * denotes significant according to the DFR.
PD-L1-specific and PD-L2-specific T cells did not cross-react
The dominant PD-L2 epitope in eliciting a T cell response was PD-L205 (PD-L216-25; QIAALFTVTV). Interestingly, we previously described another HLA-A2 restricted epitope in PD-L1 (termed PD-L101 (PDL115-23; LLNAFTVTV),9,10,12 which elicited a strong T cell response.. These two dominant T-cell epitopes were located in almost the same position in the PD-L1 and PD-L2 proteins (Figure 6A). Moreover, these two dominant epitopes shared five amino acids (FTVTV), due to sequence similarities between PD-L1 and PD-L2 (Figure 6A). Thus, we tested for a potential cross reactivity between PD-L205 (PD-L216-25)-specific and PD-L101 (PDL115-23)-specific T cells.
Figure 6.
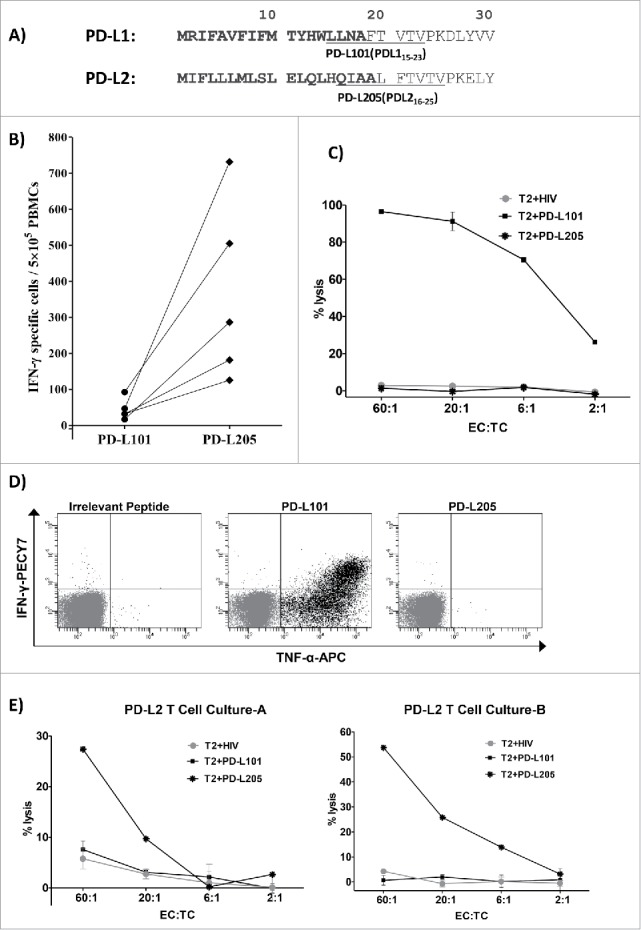
No cross-reactivity between PD-L1-specific and PD-L2-specific T cells. (A) The first 30 amino acid sequences of PD-L1 and PD-L2 and the location of the peptides PD-L101 (PDL115-23; LLNAFTVTV) and PD-L205 (PD-L216-25; QIAALFTVTV) in the signal peptide part of the proteins are marked in bold. (B) In vitro IFN- γ ELISPOT results show responses of T cells from five patients with cancer towards PD-L101 (PDL115-23) and PD-L205 (PD-L216-25) peptides. (C) 51Cr-release assay results show percent lysis of T2 cells pulsed with PD-L101 (PDL115-23), PD-L205 (PD-L216-25), or an irrelevant HIV peptide (HIV-1 pol476-484) when exposed to PD-L101-specific T-cells (CTLs) at different effector-to-target ratios. (D) Intracellular cytokine staining of cultured PD-L101-specific T-cells shows CD8+ T cell release of TNF-α, upon exposure to PD-L101 (PDL115-23), PDL205 (PD-L216-25), or an irrelevant HIV peptide (HIV-1 pol476-484). (D) Percent lysis of T2-cells, pulsed with PDL205 (PD-L216-25), PD-L101 peptide (PDL115-23), or an irrelevant HIV peptide (HIV-1 pol476-484), after exposure to PD-L2T-cell culture-A (left) or PD-L2T-cell culture-B (right).
First, we isolated PBMCs from patients with cancer that responded to PD-L205 (PD-L216-25). When we examined whether these PBMCs showed spontaneous T-cell responses against PD-L101(PDL115-23), we did not detect any immune response with the IFN-γ ELISPOT test (Figure 6B). Next, we examined the potential cross-reactivity of a PD-L101-specific T-cell culture.9 We used standard 51Cr release assays with TAP-deficient T2 cells as target cells. The target cells were loaded with PD-L101(PDL115-23), PD-L205(PD-L216-25), or an irrelevant control peptide from HIV (HIV-1 pol476-484). Figure 6C illustrates that the PD-L101-specific T cells only lysed T2-cells pulsed with PD-L101(PDL115-23), and no cytotoxicity was observed against T2-cells pulsed with either PD-L205(PD-L216-25), or the irrelevant HIV peptide. Next, we performed an intracellular cytokine staining assay to analyze cytokine release (IFN-γ and TNF-α) from PD-L101-specific CD8+ T cells, in response to PD-L101(PDL115-23), PD-L205(PD-L216-25), or the control HIV peptide (Figure 5C). Again, we found that PD-L101-specific T cells released cytokines only in response to PD-L101(PDL115-23) and not in response to PD-L205(PD-L216-25) or the irrelevant HIV peptide (Figure 6D).
Finally, we examined whether PD-L205-reactive T cells could specifically recognize PD-L205(PD-L216-25), but not PD-L101(PDL115-23). We examined the two established PD-L205-reactive T-cell cultures with the standard 51Cr release assays, with TAP-deficient T2 cells as target cells. The target cells were loaded with PD-L205(PD-L216-25), PD-L101(PDL115-23), or the control HIV peptide (HIV-1 pol476-484). The PD-L205-specific T cells could indeed recognize T2 cells pulsed with PD-L205(PD-L216-25),, but they did not kill T2 cells pulsed with PD-L101(PDL115-23), or with the control HIV peptide (Figure 6E).
Discussion
Regulatory feedback mechanisms, such as the upregulation of PD-L1 and PD-L2, are essential for limiting the strength and magnitude of immune responses that might otherwise harm the host. However, immune evasion is detrimental in the framework of cancer immunotherapy. Thus, by design, all effective immune therapies aim to induce immunological activation. Therapeutic blockade of immune checkpoint pathways, particular the PD-1 pathway, has become a paradigm-shifting treatment in solid tumor oncology. Many solid malignancies, including melanoma and NCSLC, are known to be immune-sensitive, and therefore, they are also promising clinically exploitable targets for this type of treatment. In addition, PD-1 blockade in HL showed remarkable results, and last year, the FDA approved the use of nivolumab (Opdivo®)―an anti-PD-1 antibody―for treating classical HL. Several clinical trials on blocking checkpoints in hematological malignancies have raised hope that this approach may also alter the treatment landscape in leukemia and lymphomas, in general.
We recently described an alternative approach for targeting the PD1/PD-L1 pathway with specific T-cells.19,20 In the present study, we examined PD-L2 as a target for specific T cells, and we determined that PD-L2-specific T cells are spontaneously present in patients with different cancers including NHL. In general, the strongest responses were elicited by the peptide, PD-L205(PD-L216-25). Surprisingly, we observed both CD8+ and CD4+ T cells that specifically recognized the minimal epitope in PD-L205(PD-L216-25), selected for its HLA-A2 peptide binding motif. Interestingly, the PD-L205(PD-L216-25) epitope was located similarly in the protein sequence as the main HLA-A2 restricted epitope that we identified previously in PD-L1 (PDL115-23). PD-L1 and PD-L2 share the same receptor PD-1 and share some amino acid identity. Thus, these two dominant epitopes indeed shared five amino acids (FTVTV), due to sequence similarities between PD-L1 and PD-L2. However, importantly, PD-L1- and PD-L2-specific T cells did not cross react; therefore, they should also be considered different T-cell antigens. Our results further showed that PD-L2-specific T cells specifically recognize PD-L2+ target cells. Hence, PD-L2-specific T cells recognize target cells in response to their PD-L2 expression levels. We therefore suggest that inducing/boosting T-cell responses against PD-L2 (e.g., by vaccination) represents an attractive strategy for treating hematologic malignancies, including NHL. We believe that these findings justify clinical testing to evaluate the efficacy of a PD-L2-based vaccination. Consequently, we are currently initiating the first PD-L2 vaccine study in humans at Herlev Hospital (Denmark). In that study, the long PD-L2 epitope described in this study will be administered to high-risk patients with NHL that are in remission after second-line chemotherapy. This approach represents a major difference from employing monoclonal antibodies to target the PD1/PDL pathway. Indeed, in addition to reducing the direct immunoregulatory effects of PD-L2, these PD-L2-specific T cells might also inhibit other routes of immune suppression that are mediated by PD-L2+ target cells.19
As importantly, the induction or activation of PDL2-specific T cells may induce or increase Th1 inflammation at the sites of tumors that are otherwise excluded due to infiltration of PD-L2 expressing immune suppressive cells. Thus, PD-L2-specific T cells might indirectly affect anti-cancer immunity by releasing pro-inflammatory cytokines at the site of the tumor. Hence, we demonstrated that PD-L2-specific T-cells released both IFN-γ and TNF-α. Notably, we found PD-L2 specific T-cell responses directly ex vivo underlining the immunogenicity of this antigen.21 Accordingly, PD-L2-based vaccines should be viewed as complementary, rather than competitive, to other forms of immunotherapy. For example, combining a PD-L2 vaccination with a checkpoint blocker could be an effective therapy. A checkpoint blockade could boost vaccine-activated PD-L2-specific T cells by preventing their inhibition at the tumor site. Likewise, the vaccine-induced upregulation of checkpoint molecules, due to the release of pro-inflammatory cytokines, would also be blocked by the checkpoint inhibitors. Finally, we found that T cells spontaneously reacted to PD-L2-derived epitopes located in the signal peptide region of PD-L2. Hence, the PD-L2 epitopes recognized by T cells are therefore different than any epitope recognized by anti-PDL2 blocking antibodies. In general, cancer vaccines represent a way to eliminate minimal residual disease without inducing significant toxicity and secondary malignancies. However, to date, they have largely failed to demonstrate a significant improvement in patient outcome.22 This failure probably reflects the ability of malignant cells to suppress the function of the induced immune cells. The addition of PD-L2 epitopes to current cancer vaccine strategies is likely to be highly beneficial, and it would be easy to implement. Furthermore, unlike tumor cells, stromal cell types in the tumor microenvironment are genetically stable, and thus, they represent attractive therapeutic targets with reduced risk of resistance and tumor recurrence.
In conclusion, this study described naturally-occurring, PD-L2-specific T cells in patients with cancer. PD-L2 may thus serve as a highly accessible target for immunotherapeutic strategies.
Materials and methods
Patient material
We collected blood samples from 18 melanoma, 4 non-hodgkin lymphoma patients, and 18healthy individuels. Samples were collected a minimum of four weeks after the termination of any kind of anti-cancer therapy. Peripheral blood mononuclear cells (PBMCs) were isolatedusing Lymphoprep™ (Alere AS, cat. 1114547) separation, HLA-typed and frozen in FCS with 10% DMSO (Sigma-Aldrich, cat. D5879-100ML). Tumor Infiltrating lymphocytes (TIL) from lesions from two melanoma patients were expanded using high dose IL-2 (6000 units/ml). The protocol was approved by the Scientific Ethics Committee for The Capital Region of Denmark and conducted in accordance with the provisions of the Declaration of Helsinki. Before study entry, a written informed consent from the patients was obtained.
Peptides
We identified 22 HLA-A2 restricted, 9–10 amino acid-long peptides in the human PD-L2 protein with an online epitope prediction database, SYFPEITHI (www.syfpeithi.de). Of these 22 peptides, we selected 9, from either the signal sequence or the transmembrane domain of PD-L2, and had them synthesized by TAG Copenhagen (Copenhagen, Denmark). These peptides were: PD-L201 (PD-L24-12;LLLMLSLEL), PD-L204 (PD-L2231-240; IIAFIFIATV), PD-L205 (PD-L216-25; QIAALFTVTV), PD-L208 (PD-L26-14; LMLSLELQL), PD-L209 (PD-L29-17; SLELQLHQI), PD-L215 (PD-L2234-243; FIFIATVIAL), PD-L216 (PD-L211-20; ELQLHQIAAL), PD-L220 (PD-L21-10; MIFLLLMLSL), and PD-L222 (PD-L2226-235; FIPFCIIAFI). We also analyzed two peptides with 21–25 amino acids: PD-L2long1 (PD-L29-29; SLELQLHQIAALFTVTVPKEL) and PD-L2long2 (PD-L21-25; MIFLLLMLSLELQLHQIAALFTVTV). The HLA-A2 high affinity binding epitope, HIV-1 pol476-484 (ILKEPVHGV), was used as an irrelevant control. A previously described PD-L1 peptide named as PD-L101 (PDL115-23; LLNAFTVTV) was used in cross reactivity experiments.9 All peptides were dissolved in either DMSO or sterile water before the experiments.
ELISPOT assay
The IFN-γ and TNF-α ELISPOT technique was performed as described previously.14 We performed the assays according to the guidelines provided by the cancer immunotherapy immunoguiding program (CIP; http://cimt.eu/cimt/files/dl/cip_guidelines.pdf). Unless stated otherwise, PBMCs were stimulated once in vitro with peptide prior to analysis to extend the sensitivity of the assay. To measure T-cell reactivity, nitrocellulose-bottomed 96-well plates (MultiScreen MSIPN4W; Millipore) were coated overnight at room temperature or two days at 4°C with the relevant antibodies. The wells were washed and blocked with X-vivo medium for 2h. The PBMCs were added at different cell concentrations in triplicate wells, with PD-L2 peptide or with control peptide, and incubated overnight. The following day, the wells were washed, and the relevant biotinylated secondary antibody (Mabtech) was added, followed by the avidin-enzyme conjugate (AP-Avidin; Calbiochem/Invitrogen Life Technologies); finally, we added the enzyme substrate, NBT/BCIP (Invitrogen Life Technologies) for visualization. The spots on the developed ELISPOT plates were analyzed on a CTL ImmunoSpot S6 Ultimate-V analyzer with Immunospot software, v5.1.
Generation of PD-L2-specific T-cell cultures
PBMCs from a patient with melanoma were stimulated with irradiated (30Gy) autologous DCs, which had been pulsed with PD-L205 (PD-L216-25) peptide (PBL:DC ratio 10:1) and IL-7 (40 U/ml) were added (PeproTech, London, UK). The next day, IL-12 (20 U/ml) was added (PeproTech, London, UK). At weekly intervals, we performed three identical stimulations, with irradiated autologous DCs loaded with PDL205 (PD-L216-25), and IL-12 (20 U/ml) added the next day. Then, at weekly intervals, we stimulated the culture three more times with irradiated autologous PBLs loaded with PDL205 (PD-L216-25) (culture:PBL ratio 1:1), but the next day, we added IL-2 (120 U/ml; Proleukin, Novartis).
The culture was enriched for specific T cells, either by staining with an anti-CD137-PE antibody (BD-Biosciences) or by employing a TNF-α-enrichment and detection kit (according to the procedure by Miltenyi Biotec). Next, we performed magnetic cell isolation with a MACs microbead column (according to the procedure by Miltenyi Biotec). The sorted cells were rapidly expanded by incubating with 0.6µg anti-CD3 antibody (eBioscience, clone OKT3) and a high dose of IL-2 (Proleukin, Novartis).
Intracellular cytokine staining
To detect cell subpopulations that produced cytokines, we stimulated a PD-L1-specific T-cell culture (previously described9) and a PD-L2-specific T-cell culture with 5µg/ml of relevant or irrelevant peptide, and incubated the cells for 5h at 37°C with 5% CO2. After 1h of incubation, we added GolgiPlug (BD), diluted at 1:200. After 4h, cells were washed twice with PBS, stained with fluorochrome-conjugated antibodies specific for surface markers (CD3-APC-H7, CD4-PerCP/FITC, CD8-Pacific Blue/PerCP, and Horizon Fixable Viability Stain 510, all from BD). Cells were washed, fixed, and permeabilized with Fixation/Permeabilization and Permeabilization Buffer (eBioscience), according to the manufacturer's instructions. Cells were subsequently stained with fluorochrome-conjugated antibodies to visualize intracellular cytokines. The following antibody-fluorochrome combinations were used: IFNγ-PE-CY7/APC (eBioscience) and TNFα-APC/BV421 (eBioscience). Relevant isotype controls were used to enable correct compensation and confirm antibody specificity. Stained cells were analyzed with a BD FACSCanto II flow cytometer. Analysis was performed with BD FACSDiva Software.
Cytotoxicity assay
PD-L1-specific and PD-L2-specific T-cell-mediated cytotoxicity was measured with conventional 51chromium-release assays, as previously described.15 Target cells were TAP deficient T2 cells, pulsed with HIV-1 pol476-484 (ILKEPVHGV), PD-L101 (PD-L115-23), or PD-L205 (PD-L216-25).
siRNA-mediated PD-L2 silencing of DCs
All Silencer® Select siRNA duplexes for targeted silencing of PD-L2 and Silencer® Select siRNA negative control duplex for medium GC content were obtained from Ambion® by Life Technology.
The PD-L2 siRNA duplexes consisted of three transcripts: 1. (sence) 5´-CAUCCUAAAGGUUCCAGAAtt-3´, (antisense) 5´-UU CUGGAACCUUUAGGAUGtg3´- (siRNA ID#s37285), 2. (sense) 5´-CCUAAGGAACUGUACAUAAtt -3´, (antisense) 5´-UUAUGUACAGUUCCUUAGGga 3´- (siRNA ID#s37286) and 3. (sence) 5´-GAAAACAACUCUGUCAAAAtt-3´, (antisense) 5´-UUUUGACAGAGUUGUUUUCtt 3´- (siRNA ID#s37287). The siRNA duplexes were dissolved in RNase-free water to a final concentration of 100 μM and subsequently stored at −80ºC.
Autologous CD14+ monocytes were enriched using MACS CD14 MicroBeads (Miltenyi Biotech). The enriched monocytes were cultured using CellGro (CellGenix), and supplemented with GM-CSF (1000 U/ml) and IL-4 (250 U/ml) (both PeproTech). The next day, the cells were harvested and transfected with either PD-L2 siRNA or negative control siRNA. The transfection procedure and electroporation parameters were used as previously described16 For electroporation, cells were resuspended at a concentration of 2 × 106 per 200ul of Opti-MEM medium (Invitrogen). Cells were kept on ice and added with 0.25nmol of each PD-L2 siRNA duplexes. Subsequently, cells were transferred into a 2mm kuvette and were electroporated with a single pulse at 250 Volts for 2 milli seconds using a BTX 830 square-wave electroporator (Harvard Apparatus, Holliston MA, USA).
Immediately after electroporation, monocytes were transferred to prewarmed CellGro medium containing DC-maturation cocktail: IL-β(1,000 U/mL), IL-6 (1,000 U/mL), TNF-a (1,000 U/mL), and PGE2 (1mg/mL) (all from PeproTech). After 48 hours incubation, the transfected matured DCs were used for experimental analysis. PD-L2 surface expression on the DCs transfected with PD-L2 siRNA and negative control siRNA was analyzed using anti-human PD-L2 -PE (BD biosciences). Functionality of PD-L2-specific T-cell cultures towards transfected autologous DCs was analysed using ICS and ELISPOT assay as described above. For ICS, T-cell cultures were stimulated with either PD-L2 siRNA or negative control transfected DCs for 5 hours with ratio of 1:5. In EliSpot assay T-cell cultures were stimulated with the DCs for 24hours with ratio of 1:5. Statistical analysis
An ELISPOT response was defined, based on the guidelines and recommendations provided by CIP and Moodie et al.17 The non-parametric distribution-free resampling (DFR) and more conservative DFRx2 statistical test were used for a formal comparison between antigen-stimulated wells and negative-control wells. The ELISPOT assays were performed at least in triplicate.
Supplementary Material
Conflict of interest statement
The authors declare no competing financial interests. It should be noted, however, that Mads Hald Andersen is an author of a filed patent application based on the use of PD-L2 for vaccination. The rights of the patent applications have been transferred to Copenhagen University Hospital, Herlev, according to Danish Law of Public Inventions at Public Research Institutions.
Acknowledgements
We thank Merete Jonassen and Tina Seremet for their excellent technical assistance, and Per thor Straten and Morten Hansen for scientific discussions. Supported by the Danish Cancer Society, the Danish Council for Independent Research, and Herlev Hospital.
References
- 1.Andersen MH. The targeting of immunosuppressive mechanisms in hematological malignancies. Leukemia. 2014;28:1784–1792. doi: 10.1038/leu.2014.108. PMID:24691076 [DOI] [PubMed] [Google Scholar]
- 2.Yearley JH, Gibson C, Yu N, Moon C, Murphy E, Juco J et al.. PD-L2 expression in human tumors: Relevance to Anti-PD-1 therapy in cancer. Clin Cancer Res. 2017;23:3158–3167. doi: 10.1158/1078-0432.CCR-16-1761. PMID:28619999 [DOI] [PubMed] [Google Scholar]
- 3.Danilova L, Wang H, Sunshine J, Kaunitz GJ, Cottrell TR, Xu H et al.. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc Natl Acad Sci U S A. 2016;113:E7769–E7777. doi: 10.1073/pnas.1607836113. PMID:27837027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. PMID:18173375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H et al.. PD-L1 and PD-L2 genetic alterations define classical Hodgkin Lymphoma and predict outcome. J Clin Oncol. 2016;34:2690–2697. doi: 10.1200/JCO.2016.66.4482. PMID:27069084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laurent C, Charmpi K, Gravelle P, Tosolini M, Franchet C, Ysebaert L et al.. Several immune escape patterns in non-Hodgkin's lymphomas. Oncoimmunology. 2015;4:e1026530. doi: 10.1080/2162402X.2015.1026530. PMID:26405585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Bueso-Ramos C, Dinardo C, Estecio MR, Davanlou M, Geng QR et al.. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28:1280–1288. doi: 10.1038/leu.2013.355. PMID:24270737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solito S, Pinton L, Mandruzzato S. In Brief: Myeloid-derived suppressor cells in cancer. J Pathol. 2017;242(1):7–9. doi: 10.1002/path.4876. PMID:28097660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munir S, Andersen GH, Met O, Donia M, Frosig TM, Larsen SK et al.. HLA-restricted cytotoxic T cells that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Research. 2013;73:1674–1776. doi: 10.1158/0008-5472.CAN-12-3507. [DOI] [PubMed] [Google Scholar]
- 10.Munir S, Andersen GH, Woetmann A, Odum N, Becker JC, Andersen MH. Cutaneous T cell lymphoma cells are targets for immune checkpoint ligand PD-L1-specific, cytotoxic T cells. Leukemia. 2013;27:2251–2253. doi: 10.1038/leu.2013.118. PMID:23660624 [DOI] [PubMed] [Google Scholar]
- 11.Munir S, Andersen GH, Svane IM, Andersen MH. The immune checkpoint regulator PD-L1 is a specific target for naturally occurring CD4+ T cells. Oncoimmunology. 2013;2:e23991. doi: 10.4161/onci.23991. PMID:23734334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmad SM, Larsen SK, Svane IM, Andersen MH. Harnessing PD-L1-specific cytotoxic T cells for anti-leukemia immunotherapy to defeat mechanisms of immune escape mediated by the PD-1 pathway. Leukemia. 2014;28:236–238. doi: 10.1038/leu.2013.261. PMID:24091833 [DOI] [PubMed] [Google Scholar]
- 13.Ahmad SM, Svane IM, Andersen MH. The stimulation of PD-L1-specific cytotoxic T lymphocytes can both directly and indirectly enhance antileukemic immunity. Blood Cancer J. 2014;4:230–233. doi: 10.1038/bcj.2014.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larsen SK, Munir S, Woetmann A, Froesig TM, Odum N, Svane IM et al.. Functional characterization of Foxp3-specific spontaneous immune responses. Leukemia. 2013;27:2332–2340. doi: 10.1038/leu.2013.196. PMID:23812418 [DOI] [PubMed] [Google Scholar]
- 15.Martinenaite E, Ahmad SM, Hansen M, Met O, Westergaard MW, Larsen SK et al.. CCL22-specific T cells: Modulating the immunosuppressive tumor microenvironment. Oncoimmunology. 2016;5:e1238541. doi: 10.1080/2162402X.2016.1238541. PMID:27999757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Met O, Balslev E, Flyger H, Svane IM. High immunogenic potential of p53 mRNA-transfected dendritic cells in patients with primary breast cancer. Breast Cancer Res Treat. 2011;125:395–406. doi: 10.1007/s10549-010-0844-9. PMID:20336365 [DOI] [PubMed] [Google Scholar]
- 17.Moodie Z, Price L, Janetzki S, Britten CM. Response determination criteria for ELISPOT: toward a standard that can be applied across laboratories. Methods Mol Biol. 2012;792:185–196. doi: 10.1007/978-1-61779-325-7_15. PMID:21956511 [DOI] [PubMed] [Google Scholar]
- 18.Rammensee HG, Falk K, Roetzschke O. MHC molecules as peptide receptors. Curr Biol. 1995;5:35–44. PMID:769734427677755 [DOI] [PubMed] [Google Scholar]
- 19.Andersen MH. Immune regulation by self-recognition: Novel possibilities for anticancer immunotherapy. J Natl Cancer Inst. 2015;107:154. doi: 10.1093/jnci/djv154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen MH. Anti-regulatory T cells. Semin Immunopathol. 2016;39(3):317–326. doi: 10.1007/s00281-016-0593-x.Review. PMID:27677755 [DOI] [PubMed] [Google Scholar]
- 21.Keilholz U, Weber J, Finke JH, Gabrilovich DI, Kast WM, Disis ML et al.. Immunologic monitoring of cancer vaccine therapy: Results of a workshop sponsored by the Society for Biological Therapy. J Immunother. 2002;25:97–138. doi: 10.1097/00002371-200203000-00001. PMID:12074049 [DOI] [PubMed] [Google Scholar]
- 22.Rhee F. Idiotype vaccination strategies in myeloma: how to overcome a dysfunctional immune system. Clin Cancer Res. 2007;13:1353–1355. doi: 10.1158/1078-0432.CCR-06-2650. PMID:17332275 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.