

Abstract
FDA-approved BRAF inhibitors produce high response rates and improve overall survival in patients with BRAF V600E/K mutant melanoma, but are linked to pathologies associated with paradoxical ERK1/2 activation in wild-type BRAF cells. To overcome this limitation, a next-generation paradox breaking RAF inhibitor (PLX8394) has been designed. Here we show that by using a quantitative reporter assay, PLX8394 rapidly suppressed ERK1/2 reporter activity and growth of mutant BRAF melanoma xenografts. Ex vivo treatment of xenografts and use of a patient-derived explant system (PDeX) revealed that PLX8394 suppressed ERK1/2 signaling and elicited apoptosis more effectively than the FDA-approved BRAF inhibitor, vemurafenib. Furthermore, PLX8394 was efficacious against vemurafenib-resistant BRAF splice-variant expressing tumors and reduced splice-variant homodimerization. Importantly, PLX8394 did not induce paradoxical activation of ERK1/2 in wild-type BRAF cell lines or PDeX. Continued in vivo dosing of xenografts with PLX8394 led to the development of acquired resistance via ERK1/2 reactivation through heterogeneous mechanisms; however, resistant cells were found to have differential sensitivity to ERK1/2 inhibitor. These findings highlight the efficacy of a paradox-breaking selective BRAF inhibitor and the use of PDeX system to test efficacy of therapeutic agents.
Keywords: precision medicine, melanoma
INTRODUCTION
Melanoma is the most aggressive form of cutaneous malignancy with a short time to metastasis and high mortality rate. Enhanced MEK-ERK1/2 signaling occurs in most, if not all, cutaneous melanomas and is frequently activated by a valine to glutamic acid mutation at residue 600 (V600E) in the v-Raf murine sarcoma viral oncogene homolog B (BRAF) protein (1). Recent targeted therapies have focused on selectively targeting BRAF V600E in mutant BRAF-harboring melanomas. Selective BRAF inhibitors, vemurafenib (PLX4032) and dabrafenib (GSK’436), have high response rates and provide remarkable improvements in patients with mutant BRAF melanoma; however, the majority of patients develop resistance within one year (2,3). In addition, a frequent side effect of vemurafenib and dabrafenib is the induction of squamous cell carcinomas (SCCs) and keratoacanthomas (KAs), which generally require surgical removal (4,5). While vemurafenib favors the V600E form of BRAF (6), its binding to wild-type (WT) BRAF induces heterodimerization with CRAF and ERK1/2 activation (7,8). This “paradoxical activation” of ERK1/2 likely mediates vemurafenib induction of SCCs and KAs (4,5), leukemia (9,10) and mutant KRAS pancreatic adenocarcinoma (11). Vertical targeting of the ERK1/2 pathway in melanoma with BRAF plus MEK inhibitor combinations achieves a 64–76% response rate, extends median progression free survival to over 9 months and reduces the adverse events associated with paradoxical ERK1/2 activation (12–14). However, the BRAF plus MEK inhibitor combination does not prevent relapse and can cause significant toxicities that may result in treatment discontinuation (15). Checkpoint inhibitor agents, such as ipilimumab, nivolumab, and pembrolizumab, act to relieve immunosuppressive signals and often elicit durable responses; however they do not elicit response rates as high as targeted small molecule inhibitors (11%–57.6% vs. 48%–69.6% respectively) (16). Furthermore, immunotherapy approaches are generally not suitable for patients with bulky disease that require rapid intervention (16), and an initial clinical trial combining vemurafenib with ipilimumab (a CTLA-4 targeting agent) resulted in significant hepatotoxicity (17).
New targeted therapies that efficiently inhibit the ERK1/2 pathway with fewer and less serious side effects would be clinically beneficial. Recently, next generation mutant BRAF inhibitors have been designed that elicit strong efficacy in mutant BRAF melanoma cells but do not elicit paradoxical ERK1/2 activation in mutant RAS-expressing keratinocytes (18–24). Further examination of PLX8394 as a targeted agent is warranted as this agent enters clinical trials since it may elicit fewer high grade toxicities than previous generations of mutant selective BRAF inhibitors and the combination of BRAF plus MEK inhibitors. Targeted inhibitors produce heterogeneous effects in mutant BRAF patients due to intrinsic mechanisms of resistance and adaptive drug responses. There is an important need for targeted agents to be tested in a personalized manner. Patient-derived xenograft models have been developed but typically take several months to be propagated in mice (25–27). Here, we describe the use of a patient-derived melanoma biopsy explant system (PDeX) and in vivo ERK1/2 reporter models to show that PLX8394 is a potent BRAF inhibitor and does not elicit paradoxical activation of ERK1/2 in vivo and ex vivo.
MATERIALS AND METHODS
Ion Torrent sequencing
Vehicle treated and PLX8394 resistant tumors were harvested for genomic DNA with Wizard Genomic DNA purification kit (Promega, Madison, WI). Samples were barcoded, and sequenced using the Ion PGM 200 Sequencing Kit (Life Technologies). Full details see Supplemental Data.
Western blot analysis
Western blot analysis was performed as in (28) with volumetric analysis in Quantity One (Bio-Rad; Hercules, CA). Antibodies were purchased from Cell Signaling Technology (Danvers, MA), Santa Cruz Biotechnology Inc., Biosciences Inc., Enzo (Farmingdale, NY), and Sigma-Aldrich Co. (St. Louis, MO). Full details are in Supplemental Data.
Inhibitors
Vemurafenib, dabrafenib, and trametinib (GSK’212) were purchased from Selleck Chemicals LLC (Houston, TX). PLX8394, was provided by Dr. Gideon Bollag (Plexxikon Inc., Berkeley, CA). PLX8394 for in vivo experiments was sent to Research Diets Inc. (New Brunswick, NJ) for the production of chow.
Cell culture
1205LuTR GAL4-ELK1 reporter cells (Modified cell line – the parental was a gift from Dr. Meenhard Herlyn (2005), PRT #3 (26), PBRT #15 and #16 cells (in vivo derived resistant cells of 1205LuTR GAL4-ELK1 (2013)) were grown in MCDB 153 medium containing 20% Leibovitz-L15 medium, 2% FBS, 0.2% sodium bicarbonate, and 5 μg/mL insulin. Additionally, PRT #3 cells were cultured in 1 μM PLX4720, and PBRT #15 and #16 cells were cultured in 0.5 μM PLX8394. BOWES cells (Gift from Dr. Mark Bracke (2013)) were grown in MEM containing 10% FBS, 1% non-essential amino acids, 1% sodium pyruvate, and 1% HEPES buffer. B6, MeWo, (Gifts from Dr. Barbara Bedogni (2013)) and CHL-1 cells (Purchased from ATCC in 2013) were cultured in DMEM with 10% FBS. Pen/strep (1%) was added to all media. All cells were grown at 37°C in a humidified incubator supplemented with 5% CO2. Cells are routinely assayed for mycoplasma contamination with MycoScope kit (Genlantis, San Diego, CA). Cells were assayed in April, May, and September 2016. Cell line authentication via STR analysis was completed in April 2015 for BOWES, MeWo, B6, and CHL-1, and in February 2017 for 1205LuTR GAL4-ELK1 reporter cells and PBRTs. B6 cells produced a unique profile, while all other cells matched to known profiles.
Immunohistochemistry
Tissue was fixed in formalin and paraffin embedded. Sections were stained for ERK1/2 phosphorylation (Thr202/Tyr204, #4370, Cell Signaling Technology), Staining was scored using the digital Aperio ScanScope GL system in a blinded fashion by a pathologist (A. Goldberg).
Colony formation assays
Cells (1.4 × 104) were seeded in individual wells of 6-welled plates in regular culture medium (containing 0.5 μM PLX8394 for PBRTs). The next day, plates were washed and medium was replaced with medium supplemented with drugs of interest. Medium and drugs were changed every 2 days. After 9 days, cells were fixed in buffered formalin with 0.2% crystal violet. Plates were then scanned for quantitation via ImageJ.
Viability assays
Cells (2 × 103) were seeded in triplicate in wells of a 96-welled plate in regular culture medium (containing 0.5 μM PLX8394 for PBRTs). On the next day, cells were washed twice with PBS and drug laced media added. After 4 days (including one medium change), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Sigma-Aldrich Co.) was added for 3 hours. Solubilized formazan was analyzed at 450 nM in a Multiskan Spectrum spectrophotometer (Thermo Scientific, Chicago, IL). Results are normalized to DMSO conditions and are a composite of three independent experiments.
Statistical analysis
Unless noted otherwise, significant values (indicated by an asterisk) were considered to have a p value of ≤ 0.05 as determined by a two-tailed student’s T-test assuming unequal variance and error bars are −/+ SEM. The effects of drug treatment on BRAF homodimers was modeled by considering the treatment and experimental replicate (N=4) as predictors of log(Myc/FLAG). ANOVA analysis was then performed with these considerations. IC50 calculations for ERK1/2 phosphorylation were performed using GraphPad Prism.
S-phase entry analysis
Cells (2.0 × 105) were seeded in 6-well plates. Cells were treated with drug of interest for 48 hours. The thymidine analog, EdU was added at a final concentration of 10 μMol/L for the final 16 hours. EdU incorporation was measured using the Click-it EdU Alexa Flour 647 Flow Cytometry Assay Kit and was utilized as per manufacturer’s instructions (Molecular Probes). EdU staining was quantified on BD FacsCalibur and data were analyzed with FlowJo software. Data points are shown as averages of three experimental replicates.
Ex-vivo explant system
Tumors were collected following informed patient consent at Thomas Jefferson University Hospital under an IRB-approved protocol (#10D.341). Less than 16 hours post-surgery, excess adipose and stromal tissue was removed and tumors were cut into 1 mm3 pieces. Vetspon absorbable hemostatic gelatin 1 cm3 sponges (Novartis; Basel, Switzerland) were pre-soaked in 12-welled plates for 15 minutes at 37°C in 500 μL of DMEM/10% FBS containing drugs or DMSO as a vehicle control. To avoid concerns of intratumoral heterogeneity, up to three 1 mm3 pieces from different locations of the original tumor were placed per sponge per treatment condition. Similarly, xenograft tumors were dissected into 1 mm3 pieces and placed on medium/drug-soaked sponges. Medium was replaced every 24 hours. Tumor pieces for western blotting were homogenized in modified RPPA lysis buffer (29) with phosphatase and inhibitors (PhosSTOP and cOmplete tablets Roche, Basel, Switzerland). Laemmli sample buffer was added and samples were heated at ≥95°C for 5 minutes. For IHC analysis, tumor pieces were fixed in formalin for 24 hours. Two of the samples (TJU-MEL-27A and TJU-MEL-27B) were different lesions from the same patient and combo treatment was not assayed for TJU-MEL-30.
In vivo experiments
Seven-week old female nude mice (Jackson Laboratory, Bar Harbor, ME stock# 007850) were injected with 1 × 106 1205LuTR GAL4-ELK1 reporter cells. Tumors were allowed to form to ~100 mm3 at which point the mice were randomly divided into 2 cohorts and fed either vehicle or PLX8394 laced chow. Tumor volumes and ERK1/2 reporter activity via firefly luciferase measurements were recorded every 3–4 days. All mouse experiments were performed at Thomas Jefferson University (Association for Assessment and Accreditation of Laboratory Animal Care-accredited) and approved by the Institutional Animal Care and Use Committee (IACUC). For full details see Supplemental Data.
Reverse Phase Protein Array (RPPA) analysis
1205LuTR GAL4-ELK1 parental reporter cells and PB-resistant tumor (PBRT) #15 and #16 cells (2.5 × 105/per condition) were seeded in 6-well plates in normal growth media (containing 500 nM PLX8394 for PBRTs). The next day, cells were treated with either DMSO or 0.5 μM PLX8394 for 24 hrs. Lysates from three independent experiments were processed and analyzed as previously described (29), producing triplicates for each. For analysis details see Supplemental Data section.
Immunoprecipitation assays
1205LuTR cells expressing both Myc and FLAG tagged BRAF V600E ΔEx 2–8 were seeded (1.0 × 106) on 10cm plates overnight. Cells were then dosed with 100 ng/mL doxycycline to induce both splice variant expression for 48 hours. Plates were treated with DMSO, PLX4720, or PLX8394 for an additional 4 hours. Cells were PBS washed, and lysed in an NP40 based lysis buffer. 20 μL of pre-washed anti-FLAG Affinity Gel (#A2220 Sigma-Aldrich, Saint Louis, MO) was used to immunoprecipitate FLAG tagged target during an overnight incubation at 4°C. The affinity gel was then washed 3× with cold TBS, resuspended in Laemmli sample buffer and boiled for 5 minutes. Equal volume was loaded on acrylamide gels for western analysis.
RESULTS
PLX8394 suppresses ERK1/2 signaling and tumor growth in vivo
PLX8394 is a mutant BRAF selective inhibitor, which potently blocks ERK1/2 signaling in BRAF V600E/D-harboring melanoma cells in vitro (18,19,24). The structure of PLX8394 has been previously published (18). As an initial assessment of the cellular response to PLX8394, we performed reverse phase protein array (RPPA) analysis on 1205LuTR GAL4-ELK1 reporter cells (28) (Figure 1A). RPPA allows for quantitative assessment of >200 targets involved in growth factor signaling, cell cycle progression, apoptosis and histone modification (29). In order to allow for cell cycle and apoptotic changes to take place, we assayed samples at a 24 hour time point compared to an acute time point which would most likely only affect signaling. PLX8394 treatment significantly (p<0.05 and fold change ≥1.5) altered 13 targets including down-regulation of phosphorylated MEK and ERK1/2, up-regulation of the pro-apoptotic protein BIM, and down-regulation of cyclin B1 (Figure 1B). We also observed up-regulation of the growth factor receptor, ERBB3, consistent with previous findings with vemurafenib (30,31). Phosphorylation of Raf-1/CRAF and Src, which are implicated in paradoxical ERK1/2 signaling and are suppressed by pan-RAF inhibitors, remained unaffected by PLX8394 treatment (Supplemental Figure 1). To quantitatively measure the effects of PLX8394 in vivo, we utilized xenografts from BRAF V600E melanoma cells expressing an ERK1/2 luciferase-based reporter. This model permits quantitative and temporal analysis in a non-invasive manner (28). ERK1/2 reporter luciferase levels (adjusted for tumor volume) were significantly reduced within 7 days of PLX8394 treatment compared to vehicle controls (Figures 1C & 1D). PLX8394 also significantly reduced tumor growth compared to vehicle-treated mice (Figure 1E). Together these results show that PLX8394 inhibits ERK1/2 signaling in vitro, in vivo, and reduces tumor growth in mutant BRAF melanoma xenografts.
Figure 1. PLX8394 effectively reduces ERK1/2 signaling and tumor volume in vivo.
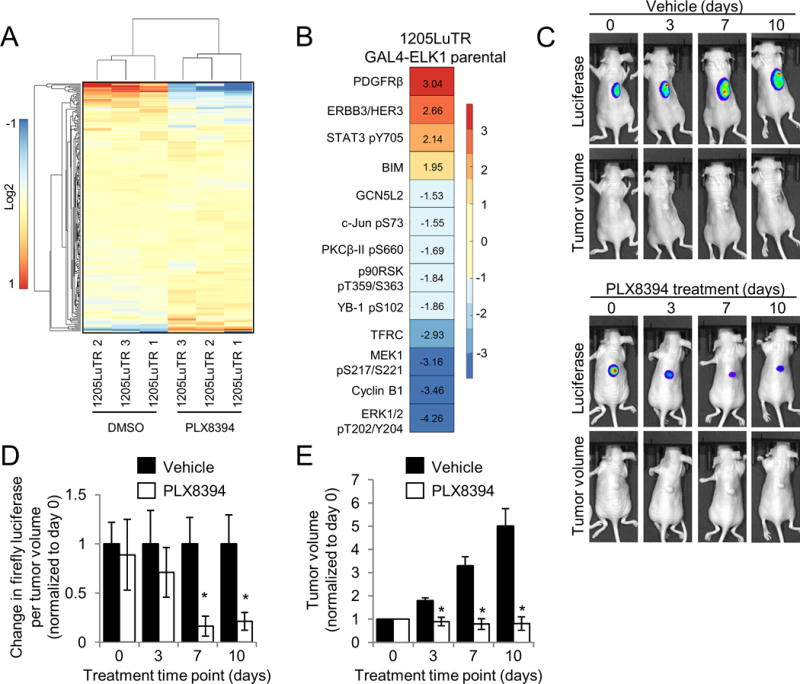
A. 1205LuTR GAL4-ELK1 cells were treated for 24 hours with either DMSO or 0.5 μM PLX8394. Lysates were obtained from three independent experiments and processed for RPPA analysis. A heat map was generated using median-centered data across each protein measurement for each sample. B. Proteins with a p value ≤ 0.01 and a fold change of ≥ 1.5 that were significantly altered following PLX8394 treatment. C. Mice bearing 1205LuTR GAL4-ELK1 xenografts were fed either vehicle chow or PLX8394 laced chow. Representative images of a vehicle and PLX8394 treated mouse with overlaid luciferase output across 10 days of treatment are shown. D. Quantification of firefly luciferase. Graph depicts fold change in luciferase output per tumor volume compared to vehicle for each day of treatment. E. Average fold change in tumor volume between mice fed vehicle chow and PLX8394-laced chow.
PLX8394 suppresses phospho-ERK1/2 and elicits apoptotic markers in patient samples as efficiently as combo treatment
An ex vivo explant model has been previously utilized in prostate cancer (32,33). These systems are advantageous for preclinical testing as they contain a stromal component and, thus, more closely mimic the tumor microenvironment. We established and validated this model in melanoma, using explants derived from xenograft tumors of 1205LuTR cells (partially sensitive to PLX4720 - the tool compound for vemurafenib) and 1205LuTR-PRT #3 cells which express a BRAF V600E splice variant and are resistant to PLX4720 (28,34). Tumor tissue was treated ex vivo with vemurafenib at 1 μM, a standard concentration for in vitro experiments (6,7,34) and 3D melanoma systems (35) or with 1 μM dabrafenib/16 nM trametinib combination (combo) (Supplemental Figure 2A–2D). The dabrafenib and trametinib combination at the given concentration is a clinically relevant molar ratio of the two drugs that was found to have significant effect on downstream signaling (Supplemental Figure 2F); this treatment served as a positive control for ERK1/2 pathway suppression to demonstrate the range of response in the ex vivo explant system. These results provided proof of concept for the Patient Derived eXplant (PDeX) system. Next, we extended PDeX analysis to fresh human melanomas. Sequence-validated, mutant BRAF V600E melanoma biopsy explants (Supplemental Figure 3A; Supplemental Table 1) were treated either with vemurafenib, dabrafenib/trametinib combo, or PLX8394 for 48–72 hours. By immunohistochemical staining of paraffin-embedded tumor sections and quantitative analyses, vemurafenib inhibited ERK1/2 phosphorylation but inhibition was partial and variable (Figure 2A & 2B). This observation is consistent with others who report a modest response to vemurafenib in 3D tumor systems (35), and in stroma/melanoma co-culture settings (36–38). By contrast, both combo and PLX8394 treatment consistently and significantly inhibited ERK1/2 phosphorylation in PDeX (Figure 2A & 2B). By western blot analysis, vemurafenib inhibition of ERK1/2 phosphorylation was again variable, but statistically significant compared to vehicle treatment (Figure 2C & 2D; Supplemental Figure 3C).
Figure 2. PLX8394 effectively inhibits ERK1/2 signaling in patient tumors comparable to dabrafenib/trametinib treatment.
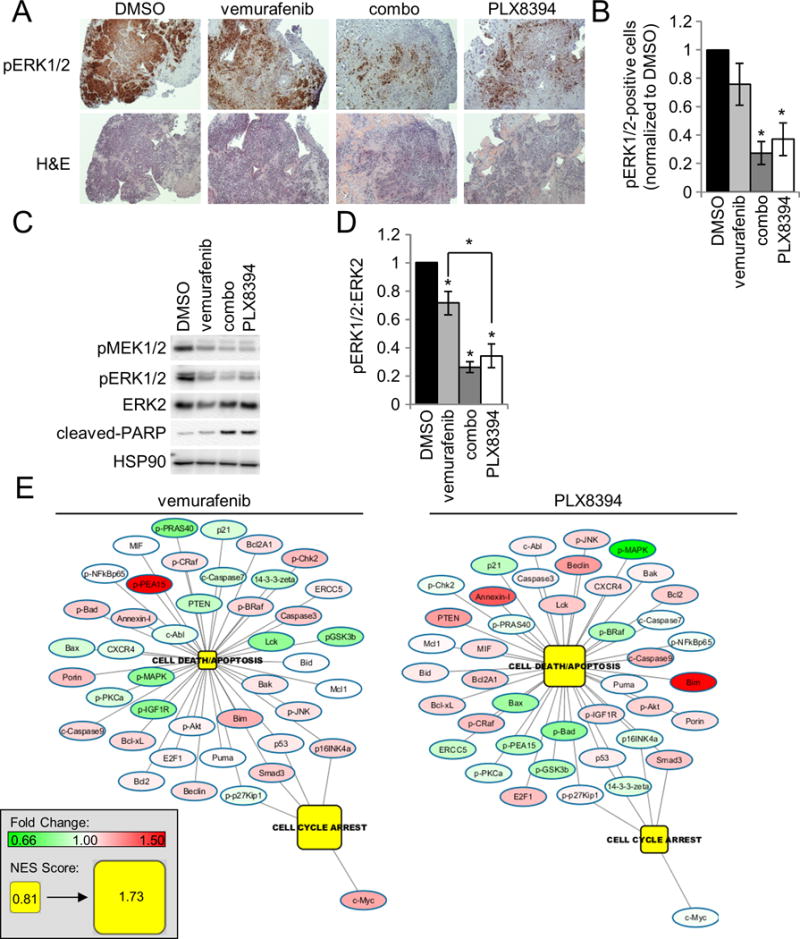
A. H&E and IHC analysis of pERK1/2 staining from a representative mutant BRAF patient sample (TJU-MEL-27A) treated with either DMSO, vemurafenib (1 μM), combo (1 μM dabrafenib/16 nM trametinib) or PLX8394 (0.5 μM). B. Quantitation of A across a panel of 6 different mutant BRAF melanoma patient samples. C. Western blot analysis of ERK1/2 signaling and PARP cleavage from a representative patient sample (TJU-MEL-27A). D. Western blot quantitation of the normalized pERK1/2 to ERK2 signal from 5 patient samples. E. RPPA data from mutant BRAF patient samples were analyzed via GSEA. Patient explants treated with vemurafenib (left) and PLX8394 (right), were grouped and compared to DMSO treated samples. Enrichment of the Programmed Cell Death/Apoptosis and Cell Cycle Arrest GO pathways and corresponding changes in RPPA determined protein levels compared to DMSO are shown. Pathway nodes and protein levels for all treatments are on the same scale (bottom left).
Importantly, both PLX8394 and the dabrafenib/trametinib combination inhibited ERK1/2 phosphorylation by >60% (Figure 2B & 2D). Furthermore, PARP cleavage is associated with ERK1/2 inhibition following combo and PLX8394 treatments (Figure 2C and Supplemental Figure 3). To better understand pathway alterations, RPPA analysis was performed on BRAF V600E human melanoma PDeX treated with targeted inhibitors. Pathway analysis of the Programmed Cell Death/Apoptosis and Cell Cycle Arrest Gene Ontology pathways by Gene Set Enrichment Analysis (GSEA) showed that vemurafenib predominantly enriched a cell cycle arrest response, whereas PLX8394 and combo treatment induced an apoptotic/cell death response (Figure 2E and Supplemental Figure 3F). In a tumor with sufficient sample to assay the effects of a dose response to PLX8394, we observed a dose dependent increase of PARP cleavage, as well as suppression of ERK1/2 phosphorylation. RPPA analysis of this sample demonstrated a dose dependent decrease of ERK1/2 pathway targets, and increase in the pro-apoptotic protein BIM (Supplemental Figure 3D and 3E). Taken together, these data suggest that PLX8394 is a potent inhibitor of ERK1/2 phosphorylation in human BRAF V600E melanomas and elicits effects comparable to the current FDA-approved dabrafenib/trametinib combination in an explant model.
Figure 3. PLX8394 suppresses MEK/ERK signaling in mutant BRAF splice variant expressing cells and is associated with reduction of splice variant homodimerization.
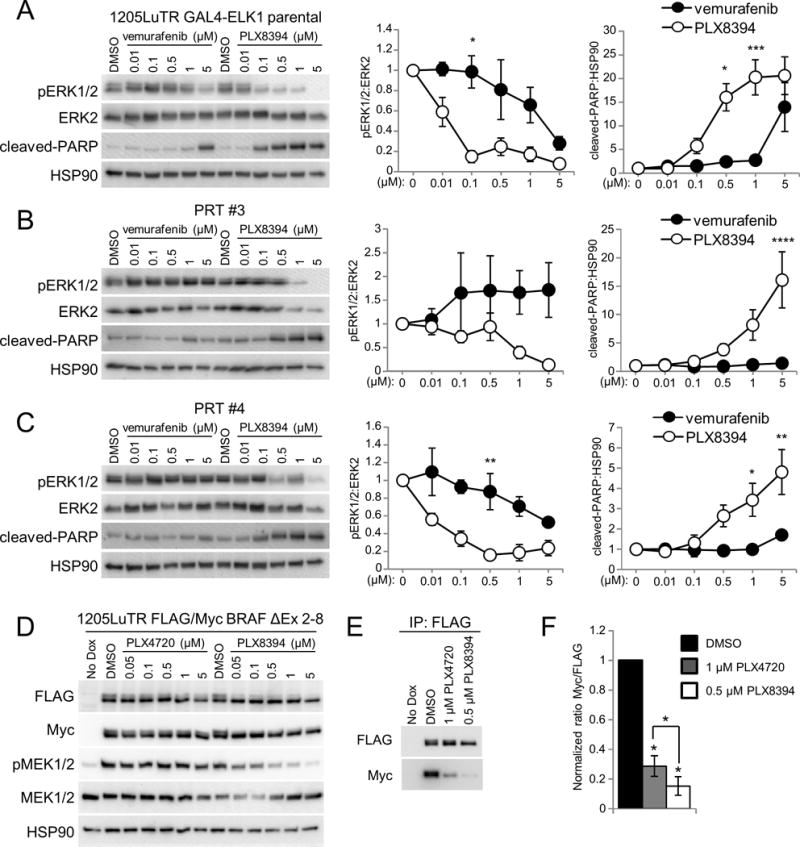
A. 1205LuTR GAL4-ELK1 cells were used to generate xenograft tumors that were harvested and dissected into ~1 mm3 pieces for use in explant system. After 48 hours of treatment, lysates were collected and analyzed by western blotting. Using densitometry, the normalized ratio of phospho ERK1/2 to ERK2 levels and cleaved PARP to HSP90 for each cohort was quantified and graphed. Data was analyzed with a two-way ANOVA corrected for multiple comparisons with Tukey analysis. Error bars are +/− SEM, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. B. Similar to A except PRT#3 cell line was used to generate xenograft tumors. C. Similar to A but PRT#4 xenograft tumors. D. Western blots of whole cell lysates from 1205LuTR FLAG/Myc BRAF ΔEx 2-8 cells after 48 hours of doxycycline induced splice variant expression and an additional 4 hours of PLX4720 or PLX8394 treatment at the indicated concentration. E. Parallel lysates from D were used to immunoprecipitate the FLAG tagged mutant BRAF splice variant and western blots reveal its associated Myc tagged binding partner. F. Quantification of splice variant homodimerization after treatment with 1 μM PLX4720 and 0.5 μM PLX8394 (N=4).
PLX8394 is more potent in suppressing ERK1/2 phosphorylation than vemurafenib and is efficacious against constitutively dimerized BRAF splice variants
To determine the efficacy of PLX8394 compared to vemurafenib in the explant system, xenografts were generated with 1205LuTR GAL4-ELK1 parental cells and two different in vivo derived RAF inhibitor resistant lines, PRT #3 and PRT #4 (28). Xenografts tumors were excised and used in the explant system to assay dose responses. After 48 hours of treatment, western blotting was used to measure ERK1/2 phosphorylation and PARP cleavage. We found that PLX8394 more efficiently suppressed ERK1/2 phosphorylation (IC50 0.01 uM vs. 1.39 uM) and elicited PARP cleavage compared to vemurafenib in parental 1205LuTR (Figure 3A, and Supplemental Figure 4A). Importantly, while vemurafenib treatment was largely ineffective (ERK1/2 phosphorylation IC50 is undefined for PRT #3 and 4.05 mM for PRT #4), PLX8394 inhibited ERK1/2 phosphorylation (0.97 μM and 0.096 μM for PRT #3 and #4, respectively) and induced PARP cleavage in BRAF splice-variant expressing tumors, PRT #3 and PRT #4 (Figure 3B, 3C & Supplemental Figure 4B, 4C). It is noteworthy that while the PRT tumors were sensitive to PLX8394, both required a higher dose of PLX8394 than parental cells to suppress ERK1/2 phosphorylation. This result is consistent with other RAF inhibitor-resistant cells treated with potential second-line RAS-RAF-MEK-ERK pathway targeting agents (34,39). Since constitutive BRAF splice variant homodimerization has been linked to vemurafenib resistance (39), we investigated if PLX8394 affects homodimerization of BRAF splice variants. 1205Lu cell lines were created to inducibly co-express both Myc-tagged and FLAG-tagged versions of BRAF splice variants lacking exons two through eight (1205LuTR FLAG/Myc BRAF ΔEx 2-8). BRAF ΔEx 2-8 is equivalent to the BRAF splice variant expressed in PRT #4. Similar to the PRT tumors, this cell line demonstrated a dose dependent reduction of ERK1/2 pathway signaling from PLX8394 but not PLX4720 treatment (Figure 3D). Parallel lysates were then used to query homodimerization by immunoprecipitation of the FLAG-tagged BRAF splice variant and probing for the association of its Myc-tagged binding partner. While both drugs impaired homodimerization, PLX8394 treatment elicited a more profound reduction than PLX4720 (Figure 3E and 3F). Interestingly, PLX8394 dosing had little effect on homodimerization status (Supplemental Figure 4D), yet higher doses of PLX8394 were able to inhibit MEK phosphorylation. These observations suggest that while PLX8394 blocks dimerization and ERK1/2 pathway more efficiently than PLX4720, the extent of BRAF splice variant homodimerization itself may not be wholly responsible for vemurafenib resistance. Together, these data demonstrate superior efficacy of PLX8394 as a single agent RAF inhibitor in comparison to vemurafenib and that PLX8394 can overcome mutant BRAF splice variants, a common mechanism of BRAF inhibitor resistance.
PLX8394 attenuates the paradoxical ERK1/2 activation in WT/WT melanoma tissue
An important goal in the design of PLX8394 is to reduce hyper-activation of ERK1/2 in WT BRAF-containing tissues. To demonstrate the “paradox breaking” ability of PLX8394, we treated WT BRAF/WT NRAS (WT/WT) melanoma cell lines with vemurafenib, PLX8394 and trametinib in 2D culture conditions. Vemurafenib significantly increased ERK1/2 signaling in CHL-1, BOWES, MeWo, and B6 WT/WT melanoma cell lines compared to DMSO control (Figures 4A & 4B). In contrast, PLX8394 treatment did not produce a statistically significant increase in paradoxical activation and treatment with trametinib strongly reduced ERK1/2 phosphorylation. Extending these studies into explants derived from xenografts from the WT/WT BOWES and B6 cells, vemurafenib enhanced ERK1/2 activation in WT/WT xenograft explants similar to experiments in 2D (Figure 4C). In comparison to vemurafenib, PLX8394 did not induce strong paradoxical ERK1/2 activation in these samples. Furthermore, we tested paradoxical activation by RAF inhibitors using the PDeX system in a WT/WT patient sample (Supplemental Figure 3B). Western blot analysis demonstrated a strong paradoxical phosphorylation of ERK1/2 induced by vemurafenib, but comparatively weak ERK1/2 phosphorylation in response to PLX8394 (Figure 4D). As expected, the MEK inhibitor trametinib suppressed ERK1/2 signaling (Figure 4D). Taken together, these data show that when using doses effective in suppressing ERK1/2 signaling in mutant BRAF tumor, PLX8394 does not elicit strong paradoxical signaling in WT BRAF tissue, representing an improvement over the previous generation of BRAF inhibitors.
Figure 4. PLX8394 prevents significant paradoxical ERK1/2 activation in WT BRAF melanomas.
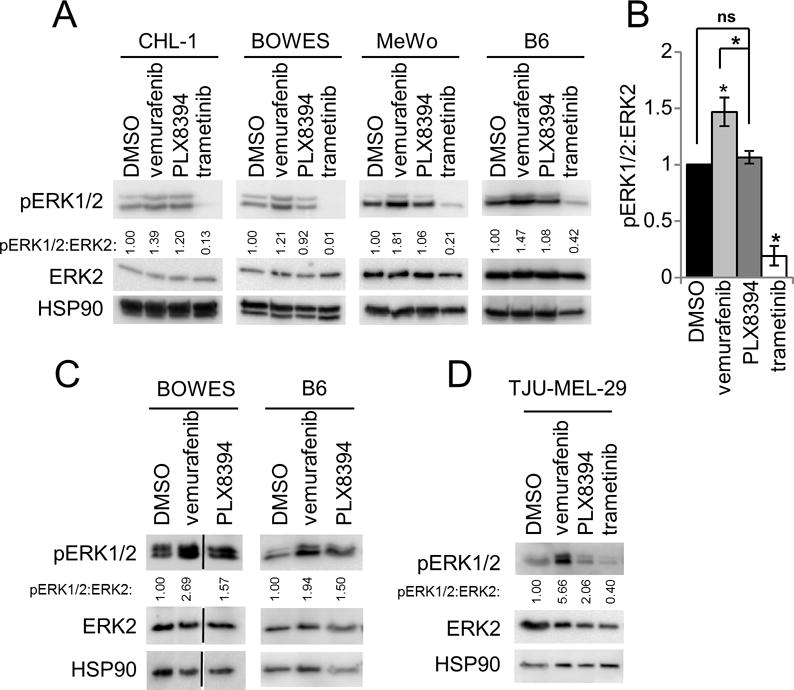
A. Western blot analysis of WT/WT melanoma cell lines after treatment with DMSO, vemurafenib (1 μM), PLX8394 (0.5 μM) or trametinib (50 nM) for 48 hours. B. Quantitation of the normalized pERK1/2 signal from A. C. As in Fig 2B, except that two different WT/WT melanoma cells were used to form xenografts and were processed/treated in the ex vivo explant system. Western blot analysis of lysates prepared from explants treated with DMSO, vemurafenib or PLX8394 after 48 hours. Densitometry results of pERK1/2 to ERK2 are indicated. D. WT/WT patient sample explants (TJU-MEL-29) were treated as in A for 48 hours. Densitometry values are shown.
Acquired resistance to PLX8394 is associated with ERK1/2 reactivation and deregulation of ERK1/2-independent pathways
Treatment with targeted therapies is invariably associated with acquired resistance; therefore, we investigated the duration of PLX8394 effects on BRAF V600E melanomas in vivo. Mice bearing mutant BRAF xenografts were continued on PLX8394 treatment until progression (≥1000mm3 tumor size or displayed signs of ulceration). Progressing tumors were excised and two Paradox Breaker Resistant Tumor (PBRT) cell lines, #15 and #16 were propagated. PBRT #15 and PBRT #16 were isolated at day 45 and 35 post drug treatment, respectively. In 2D colony formation assays, PLX8394 suppressed the growth of 1205LuTR parental cells in a dose dependent manner (Figure 5A & 5B). Conversely, PBRT #15 maintained growth in PLX8394 and PBRT #16 exhibited addiction to PLX8394, similar to a phenomenon observed in vemurafenib-resistant cells (34). In MTT assays, PLX8394 potently inhibited the viability of parental cells but both PBRT #15 and #16 cell lines were highly resistant to the inhibitor (Supplemental Figure 5A).
Figure 5. In vivo acquired resistance of 1205LuTR GAL4-ELK1 xenografts to PLX8394.
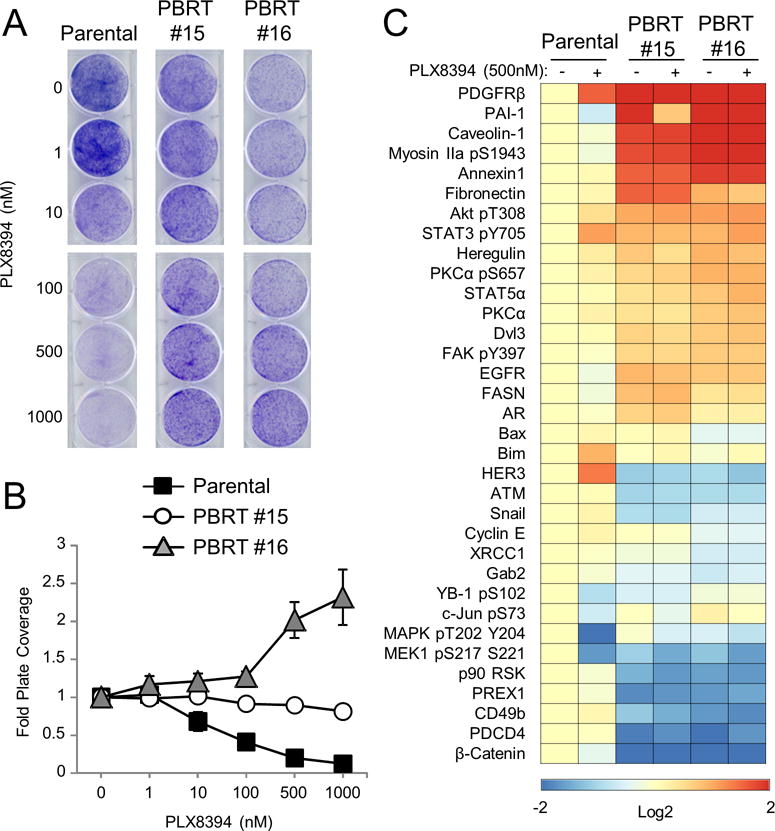
A. Progressing xenografts were harvested and used to generate PLX8394 resistant cell lines (PBRTs). Crystal violet staining of PBRT #15 and #16 cells treated with increasing doses of PLX8394 compared to parental cells is shown. B. Quantified results of three independent experiments as in B. C. RPPA analysis of PBRT #15 and PBRT #16 cells compared to parental cells treated with either DMSO or PLX8394 (0.5 μM) for 24 hours. Heatmap showing proteins with a p value ≤ 0.01 and a fold change of ≥ 1.5 found to be significantly altered in PBRT #15 or #16 cells when compared to parental cells.
To understand pathway alterations associated with resistance to PLX8394, we performed RPPA analysis on PBRT #15 and #16 compared to parental 1205LuTR cells. Compared to parental 1205LuTR cells, the levels of 34 proteins significantly changed (1.5 fold; p≤ 0.01) in either the DMSO control or PLX8394 conditions (Figure 5C). Western blotting confirmed the RPPA results showing maintenance of ERK1/2 phosphorylation in PBRT #15 and #16 cells treated with PLX8394 (Supplemental Figure 5B & 5C). Additionally, PBRT cells treated with PLX8394 exhibited significantly higher levels of Rb phosphorylation than PLX8394-treated parental cells (Supplemental Figure 5D), reflecting the ability of PBRT cells to overcome PLX8394-mediated cell cycle inhibition. Interestingly, both PBRT cell lines displayed increased AKT phosphorylation, enhanced PDGFR levels and reduced β-catenin expression compared to 1205LuTR parental cells (Supplemental Figure 5B and 5E–G). These alterations have been previously implicated in resistance to vemurafenib (40–44), but did not appear to be primary drivers of resistance in the PBRTs (Supplemental Figure 6). Furthermore, BRAF V600E splice variants, which drive resistance to vemurafenib, were not detected in PBRT cell lines (Supplemental Figure 7A). Thus, as with other RAF inhibitors and MEK-ERK1/2 regimens, prolonged exposure to PLX8394 results in acquired resistance associated with ERK1/2 pathway re-activation and compensatory pathway alterations.
PBRT cell lines have differential sensitivities to ERK1/2 pathway inhibition
We tested whether vertical targeting of the ERK1/2 pathway would overcome the acquired resistance to PLX8394, as it does in other resistant melanoma models (27,39). Individual treatments of PLX8394, vemurafenib, trametinib, and the ERK1/2 inhibitor SCH772984 (SCH772) suppressed ERK1/2 signaling and reduced Rb phosphorylation in parental cells (Figure 6A). While trametinib reduced phospho-ERK1/2 in both PBRT #15 and #16, Rb phosphorylation was only affected in PBRT #15 (Figure 6A). Similarly, SCH772 treatment reduced Rb phosphorylation in PBRT #15 but not PBRT #16 (Figure 6A). Dose escalation of SCH772 was associated with an increase in PARP cleavage and BIM levels, as well as a reduction of both Rb phosphorylation and cyclin A expression in PBRT #15 (Supplemental Figure 7B). However, these changes were not observed in PBRT #16 (Supplemental Figure 7B). Similarly, crystal violet growth and EdU incorporation assays demonstrated that both parental and PBRT #15 cell lines were more sensitive to ERK1/2 inhibitor treatment than PBRT #16 (Figure 6B–6C and Supplemental Figure 7C).
Figure 6. PBRTs have differential responses to second-line therapies.
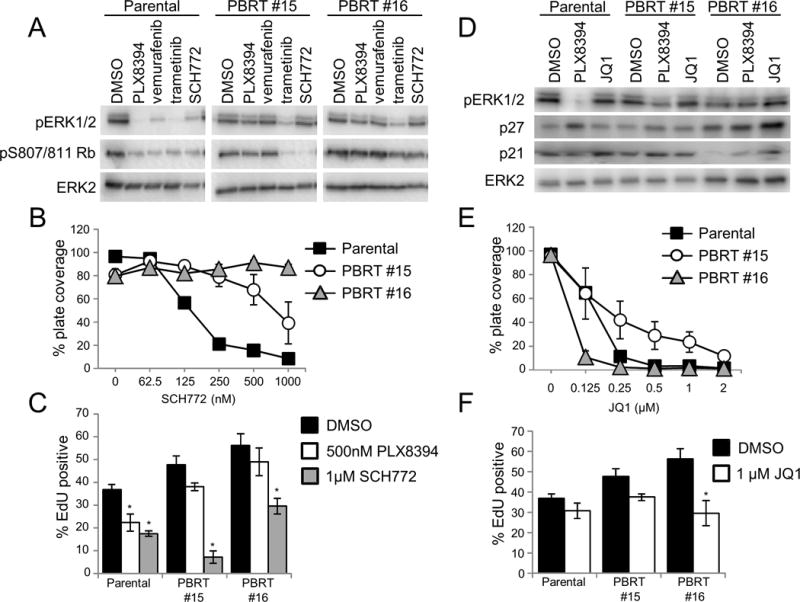
A. 1205LuTR GAL4-ELK1, PBRT #15, and PBRT #16 were seeded in 6 well plates overnight then cells were washed and media was replaced and supplemented with DMSO, 0.5 μM PLX8394, 1 μM vemurafenib, 50 nM trametinib, or 1 μM SCH772. After 24hours, lysates were harvested and samples were analyzed by western blot. B. Quantification of crystal violet 2D growth assays for 1205LuTR GAL4-ELK1, PBRT #15 and PBRT #16 in the presence of increasing SCH772. Data points represent the average percent plate coverage of at least three independent experiments. Error bars are SEM. C. S-phase entry of 1205LuTR GAL4-ELK1, PBRT #15 and PBRT #16 cells were assayed by EdU incorporation. Graph is the average EdU positivity from at least three experimental replicates. Error bars are SEM, * indicates p value < 0.05 compared to each cell line’s DMSO condition using a two-way student’s T-test assuming unequal variance. D. Western blot analysis of 1205LuTR GAL4-ELK1, PBRT #15 and PBRT #16 cells after 24 hour drug treatment of DMSO, 0.5 μM PLX8394, or 1 μM JQ1. E–F. Similar to B-C except cells were treated with JQ1.
Ion torrent sequencing results of PBRT #15 and PBRT #16 did not yield any missense mutations that would be indicative of ERK1/2 inhibitor resistance (Supplemental Tables 2 and 3). Consequently, we postulated that transcriptional alterations may contribute to resistance. Therefore, we utilized epigenetic agents, the bromodomain and extra-terminal domain (BET) bromodomain (BRD) inhibitor, JQ1. Western blot analysis demonstrated that JQ1 treatment increased levels of the cyclin-dependent kinase inhibitors, p21 and p27, in PBRT #16 (Figure 6D). This correlated with increased sensitivity of PBRT #16 to BET/BRD inhibitors in crystal violet growth assays and EdU incorporation (Figure 6E–6F and Supplemental Figure 7D). While PBRT #16 was more sensitive to BET/BRD inhibitor treatment, these agents also suppressed growth of PBRT #15 suggesting a potential universal second-line therapy option (Figure 6E and Supplemental Figure 7D).
DISCUSSION
FDA-approved mutant BRAF selective inhibitors have markedly improved the treatment options and outcomes for BRAF V600E/K melanoma patients but are limited by the occurrence of adverse events associated with paradoxical ERK1/2 activation. In this study, we show the efficacy of the next-generation “paradox breaking” BRAF inhibitor, PLX8394. Overall, our data support that PLX8394 is a viable treatment option to efficiently inhibit ERK1/2 signaling while limiting paradoxical effects in WT BRAF cells and highlight the use of tumor explants to rapidly assess the utility of targeted agents.
Using an in vivo GAL4-ELK1 reporter system to quantitatively and temporally measure ERK1/2 signaling in melanoma xenografts (28), we show that PLX8394 effectively inhibits ERK1/2 signaling and reduces mutant BRAF melanoma growth. This approach is complemented by an ex vivo explant system that demonstrates PLX8394 effectively inhibits ERK1/2 phosphorylation in patient tumors. Vemurafenib treatment only elicited a ~20% reduction in ERK1/2 phosphorylation in explants consistent with others’ observations of minimal vemurafenib efficacy in 3D tumor mimics (35), and stroma/melanoma co-culture systems (36–38). Variability in the response of patient tumors to vemurafenib may also be due to different treatment histories of the patients (Supplemental Table 1) and/or a high stromal component, which displays paradoxical ERK1/2 activation (35). By contrast, PLX8394 consistently inhibited ERK1/2 signaling in all patient biopsies independent of treatment history, and was comparable to dabrafenib/trametinib combination therapy.
PLX8394 demonstrated enhanced efficacy when directly compared to vemurafenib in parallel ex vivo dosing of xenograft tissue. This may not be surprising as the PLX8394 IC50 for ERK1/2 phosphorylation is ~10 fold lower than vemurafenib (18). By contrast, in the explant system, the dose required to reach ERK1/2 phosphorylation IC50 of parental mutant BRAF tumor tissue treated with vemurafenib was much higher (~44x IC50) compared to PLX8394 (~3x IC50) (Figure 3A). One explanation for the larger difference of vemurafenib effectiveness is the potential paradoxical activation of ERK1/2 signaling in the stromal component present in the explant tissue which is not present in 2D culture systems. Vemurafenib has been shown to paradoxically elicit tumor protective responses from stromal components in ex vivo systems, as well as stimulate production of mitogenic growth factors from wild type BRAF tumors in vivo (18,35), a phenomenon that should be attenuated in PLX8394 treatment. The moderate increase in ERK1/2 phosphorylation IC50 for PLX8394 treatment in the explant system, compared to 2D culture, may reflect ERK1/2 signaling present in stromal cells that should not be inhibited or paradoxically activated.
PLX8394 was effective in suppressing ERK1/2 signaling and eliciting PARP cleavage in BRAF splice variant-expressing, vemurafenib-resistant samples. This may be in part due to PLX8394’s ability to better suppress splice variant homodimerization, thereby facilitating efficient inhibition of monomeric mutant BRAF kinase. Homodimerization of mutant BRAF splice variant expressing cells has been linked to RAF inhibitor resistance (28,39); however, the dimerization status of these splice variants in the presence of RAF inhibitors has not been tested. Vemurafenib and PLX4720 have previously been shown to destabilize homodimerization of full length mutant BRAF (45) and heterodimerization of wild-type BRAF kinase domain with CRAF (46). On the other hand, reports indicate that these drugs may enhance BRAF/CRAF heterodimers (23). In the present study, we utilize differentially tagged V600E BRAF splice variants to measure homodimerization status during drug treatment to model the setting in which splice variants are expressed. We found that while both PLX4720 and PLX8394 reduced homodimerization of V600E BRAF splice variants, PLX8394 was significantly more effective. It is possible that the residual dimerized splice variant in PLX4720 treated cells may be adequate for drug resistance as further blocking of homodimerization by PLX8394 is associated with ERK1/2 pathway inhibition. Alternatively, these data may indicate that although splice variant homodimerization contributes to signaling in the presence of vemurafenib, it may not be wholly responsible for RAF inhibitor resistance.
The explant system was also used to show that vemurafenib, but not PLX8394, induces a strong paradoxical activation of ERK1/2 in WT/WT melanoma. Other groups have reported on pan-RAF/Src inhibitors that do not elicit paradoxical activation properties (47); however, the mutant BRAF selectivity of PLX8394 may afford a higher therapeutic index than agents that broadly inhibit RAF kinases. Furthermore, the mutant BRAF specific targeting properties of PLX8394 may enable its use in combinatorial regimens with immune therapies. Since suppression of ERK1/2 signaling is associated with increased melanoma antigen presentation (48), it is advantageous to use targeted inhibitors as an adjuvant to improve immunotherapy efficacy. However, there are conflicting reports of how systemic pathway inhibition (i.e. MEK inhibitor treatment) affects the anti-tumor immune response (49–51). Our results indicate that at doses that would inhibit mutant BRAF tumors, PLX8394 minimally affects ERK1/2 status in WT/WT cells, suggesting it will not alter normal T-cell activation. Thus, PLX8394 may be an appropriate partner with immune-based therapies such as CTLA-4 and PD-1/PDL-1 inhibitors (52).
With an increasing number of available therapies for the treatment of mutant BRAF melanomas, identifying the best therapy for an individual patient is increasingly important. Patient-derived tumor xenograft (PDX) models accurately reproduce a patient’s response to therapy (53); however, these models are associated with long generation times and high cost. As an alternative, we describe a patient derived explant system, PDeX, to test multiple treatment strategies using a single patient biopsy that accounts for intratumoral heterogeneity by assaying multiple sample pieces from different parts of the lesion. The ability of PDeX to test the efficacy of small molecule inhibitors and monoclonal antibodies in a short time period offers an inexpensive and rapid assay that is individualized and may inform patient treatment options.
As with other targeted therapies, acquired resistance to PLX8394 eventually occurs in our pre-clinical studies. Phospho-proteomic analysis implicated well-known BRAF inhibitor resistance markers in the PLX8394-resistant cell lines (40,43,54). However, initial experiments suggest that enhanced AKT activity and up-regulation of PDGFR are not sole drivers of resistance in these cells (Supplemental Figure 6). Rather, it is likely that they work in co-ordination with re-activation of the ERK1/2 pathway. It is possible that resistance mechanisms to PLX8394 will be unique from vemurafenib and dabrafenib and is underscored by the finding that PBRT #16 is resistant to vertical targeting of the ERK1/2 signaling pathway but is sensitive to BET/BRD inhibitor treatment. Since enrollment for PLX8394 phase 1/2a study (ClinicalTrials.gov #NCT02428712) has only recently started, resistance mechanisms in patients remain unknown.
In summary, PLX8394 is a promising next-generation mutant BRAF selective inhibitor that does not elicit strong paradoxical ERK1/2 activation in non-mutant BRAF cells. PLX8394 monotherapy is entering clinical trials with hope that it will prevent side effects associated with paradoxical ERK1/2 activation while simultaneously reducing grade 4 toxicities and permanent discontinuations associated with dual inhibitor therapies (14). Our parallel dosing experiments of tumor tissue suggest that PLX8394 is more effective than vemurafenib at suppressing ERK1/2 signaling even when considering vemurafenib’s lower biochemical potency (18). Additionally, the PDeX system utilized in this study provides a rapid and quantitative method to determine the efficacy of PLX8394 (and other targeted therapies) in patient tissues. As a result, our data suggest that PLX8394 is a promising new therapy for the treatment of mutant BRAF melanomas refractive to vemurafenib, and that the PDeX system can potentially be used to guide the treatment of patients in a personalized manner.
Supplementary Material
Acknowledgments
We are grateful to Sheera Rosenbaum for generating the WT/WT melanoma xenografts, Dr. Kevin Basile for help with mutant BRAF xenografts, Dr. Meenhard Herlyn (Wistar Institute, Philadelphia, PA) for the WM melanoma cell lines and Dr. Barbara Bedogni (Case Western Reserve University, Cleveland, OH) for the B6, and MeWo cells.
References
- 1.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 4.Belum VR, Rosen AC, Jaimes N, Dranitsaris G, Pulitzer MP, Busam KJ, et al. Clinico-morphological features of BRAF inhibition-induced proliferative skin lesions in cancer patients. Cancer. 2015;121:60–8. doi: 10.1002/cncr.28980. [DOI] [PubMed] [Google Scholar]
- 5.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–15. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JT, Li L, Brafford PA, van den Eijnden M, Halloran MB, Sproesser K, et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res. 2010;23:820–7. doi: 10.1111/j.1755-148X.2010.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yaktapour N, Meiss F, Mastroianni J, Zenz T, Andrlova H, Mathew NR, et al. BRAF inhibitor-associated ERK activation drives development of chronic lymphocytic leukemia. J Clin Invest. 2014;124:5074–84. doi: 10.1172/JCI76539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367:2316–21. doi: 10.1056/NEJMoa1208958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlino MS, Kwan V, Miller DK, Saunders CA, Yip D, Nagrial AM, et al. New RAS-mutant pancreatic adenocarcinoma with combined BRAF and MEK inhibition for metastatic melanoma. J Clin Oncol. 2015;33:e52–6. doi: 10.1200/JCO.2013.51.5783. [DOI] [PubMed] [Google Scholar]
- 12.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 13.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 15.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–88. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 16.Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017;14:463–82. doi: 10.1038/nrclinonc.2017.43. [DOI] [PubMed] [Google Scholar]
- 17.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 18.Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–6. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- 19.Le K, Blomain ES, Rodeck U, Aplin AE. Selective RAF inhibitor impairs ERK1/2 phosphorylation and growth in mutant NRAS, vemurafenib-resistant melanoma cells. Pigment Cell Melanoma Res. 2013;26:509–17. doi: 10.1111/pcmr.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi J, Landrette SF, Wang T, Evans P, Bacchiocchi A, Bjornson R, et al. Identification of PLX4032-resistance mechanisms and implications for novel RAF inhibitors. Pigment Cell Melanoma Res. 2014;27:253–62. doi: 10.1111/pcmr.12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A. 2013;110:5957–62. doi: 10.1073/pnas.1219232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karoulia Z, Wu Y, Ahmed TA, Xin Q, Bollard J, Krepler C, et al. An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell. 2016;30:485–98. doi: 10.1016/j.ccell.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basile KJ, Le K, Hartsough EJ, Aplin AE. Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigment Cell Melanoma Res. 2014;27:479–84. doi: 10.1111/pcmr.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Girotti MR, Gremel G, Lee R, Galvani E, Rothwell D, Viros A, et al. Application of sequencing, liquid biopsies, and patient-derived xenografts for personalized medicine in melanoma. Cancer Discov. 2016;6:286–99. doi: 10.1158/2159-8290.CD-15-1336. [DOI] [PubMed] [Google Scholar]
- 26.Kemper K, Krijgsman O, Kong X, Cornelissen-Steijger P, Shahrabi A, Weeber F, et al. BRAF(V600E) kinase domain duplication identified in therapy-refractory melanoma patient-derived xenografts. Cell Rep. 2016;16:263–77. doi: 10.1016/j.celrep.2016.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krepler C, Xiao M, Sproesser K, Brafford PA, Shannan B, Beqiri M, et al. Personalized preclinical trials in BRAF inhibitor-resistant patient-derived xenograft models identify second-line combination therapies. Clin Cancer Res. 2016;22:1592–602. doi: 10.1158/1078-0432.CCR-15-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basile KJ, Abel EV, Dadpey N, Hartsough EJ, Fortina P, Aplin AE. In vivo MAPK reporting reveals the heterogeneity in tumoral selection of resistance to RAF inhibitors. Cancer Res. 2013;73:7101–10. doi: 10.1158/0008-5472.CAN-13-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–21. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 30.Abel EV, Basile KJ, Kugel CH, 3rd, Witkiewicz AK, Le K, Amaravadi RK, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J Clin Invest. 2013;123:2155–68. doi: 10.1172/JCI65780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kugel CH, 3rd, Hartsough EJ, Davies MA, Setiady YY, Aplin AE. Function-blocking ERBB3 antibody inhibits the adaptive response to RAF inhibitor. Cancer Res. 2014;74:4122–32. doi: 10.1158/0008-5472.CAN-14-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Centenera MM, Gillis JL, Hanson AR, Jindal S, Taylor RA, Risbridger GP, et al. Evidence for efficacy of new Hsp90 inhibitors revealed by ex vivo culture of human prostate tumors. Clin Cancer Res. 2012;18:3562–70. doi: 10.1158/1078-0432.CCR-12-0782. [DOI] [PubMed] [Google Scholar]
- 33.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–49. doi: 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartsough EJ, Basile KJ, Aplin AE. Beneficial effects of RAF inhibitor in mutant BRAF splice variant-expressing melanoma. Mol Cancer Res. 2014;12:795–802. doi: 10.1158/1541-7786.MCR-13-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, et al. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell. 2015;27:574–88. doi: 10.1016/j.ccell.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fedorenko IV, Wargo JA, Flaherty KT, Messina JL, Smalley KS. BRAF Inhibition Generates a Host-Tumor Niche that Mediates Therapeutic Escape. J Invest Dermatol. 2015;135:3115–24. doi: 10.1038/jid.2015.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Capparelli C, Rosenbaum S, Berger AC, Aplin AE. Fibroblast-derived neuregulin 1 promotes compensatory ErbB3 receptor signaling in mutant BRAF melanoma. J Biol Chem. 2015;290:24267–77. doi: 10.1074/jbc.M115.657270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70:6670–81. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–60. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biechele TL, Kulikauskas RM, Toroni RA, Lucero OM, Swift RD, James RG, et al. Wnt/beta-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci Signal. 2012;5:ra3. doi: 10.1126/scisignal.2002274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chien AJ, Haydu LE, Biechele TL, Kulikauskas RM, Rizos H, Kefford RF, et al. Targeted BRAF inhibition impacts survival in melanoma patients with high levels of Wnt/beta-catenin signaling. PLoS One. 2014;9:e94748. doi: 10.1371/journal.pone.0094748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, et al. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol. 2015;22:37–43. doi: 10.1038/nsmb.2924. [DOI] [PubMed] [Google Scholar]
- 46.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 47.Girotti MR, Lopes F, Preece N, Niculescu-Duvaz D, Zambon A, Davies L, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell. 2015;27:85–96. doi: 10.1016/j.ccell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res. 2006;4:779–92. doi: 10.1158/1541-7786.MCR-06-0077. [DOI] [PubMed] [Google Scholar]
- 49.Ebert PJ, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity. 2016;44:609–21. doi: 10.1016/j.immuni.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 50.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 51.Allegrezza MJ, Rutkowski MR, Stephen TL, Svoronos N, Tesone AJ, Perales-Puchalt A, et al. IL15 Agonists Overcome the Immunosuppressive Effects of MEK Inhibitors. Cancer Res. 2016;76:2561–72. doi: 10.1158/0008-5472.CAN-15-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu-Lieskovan S, Robert L, Homet Moreno B, Ribas A. Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challenges. J Clin Oncol. 2014;32:2248–54. doi: 10.1200/JCO.2013.52.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sabbatino F, Wang Y, Wang X, Flaherty KT, Yu L, Pepin D, et al. PDGFRalpha up-regulation mediated by sonic hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget. 2014;5:1926–41. doi: 10.18632/oncotarget.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.