

ABSTRACT
Ectoine and hydroxyectoine are widely synthesized by members of the Bacteria and a few members of the Archaea as potent osmostress protectants. We have studied the salient features of the osmostress-responsive promoter directing the transcription of the ectoine/hydroxyectoine biosynthetic gene cluster from the plant-root-associated bacterium Pseudomonas stutzeri by transferring it into Escherichia coli, an enterobacterium that does not produce ectoines naturally. Using ect-lacZ reporter fusions, we found that the heterologous ect promoter reacted with exquisite sensitivity in its transcriptional profile to graded increases in sustained high salinity, responded to a true osmotic signal, and required the buildup of an osmotically effective gradient across the cytoplasmic membrane for its induction. The involvement of the −10, −35, and spacer regions of the sigma-70-type ect promoter in setting promoter strength and response to osmotic stress was assessed through site-directed mutagenesis. Moderate changes in the ect promoter sequence that increase its resemblance to housekeeping sigma-70-type promoters of E. coli afforded substantially enhanced expression, both in the absence and in the presence of osmotic stress. Building on this set of ect promoter mutants, we engineered an E. coli chassis strain for the heterologous production of ectoines. This synthetic cell factory lacks the genes for the osmostress-responsive synthesis of trehalose and the compatible solute importers ProP and ProU, and it continuously excretes ectoines into the growth medium. By combining appropriate host strains and different plasmid variants, excretion of ectoine, hydroxyectoine, or a mixture of both compounds was achieved under mild osmotic stress conditions.
IMPORTANCE Ectoines are compatible solutes, organic osmolytes that are used by microorganisms to fend off the negative consequences of high environmental osmolarity on cellular physiology. An understanding of the salient features of osmostress-responsive promoters directing the expression of the ectoine/hydroxyectoine biosynthetic gene clusters is lacking. We exploited the ect promoter from an ectoine/hydroxyectoine-producing soil bacterium for such a study by transferring it into a surrogate bacterial host. Despite the fact that E. coli does not synthesize ectoines naturally, the ect promoter retained its exquisitely sensitive osmotic control, indicating that osmoregulation of ect transcription is an inherent feature of the promoter and its flanking sequences. These sequences were narrowed to a 116-bp DNA fragment. Ectoines have interesting commercial applications. Building on data from a site-directed mutagenesis study of the ect promoter, we designed a synthetic cell factory that secretes ectoine, hydroxyectoine, or a mixture of both compounds into the growth medium.
KEYWORDS: osmoregulation, promoters, compatible solutes, chemical chaperones, excretion
INTRODUCTION
Ectoine [(S)-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid] and its derivative 5-hydroxyectoine [(4S,5S)-5-hydroxy-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid] (1) are representatives of a special class of organic osmolytes, the compatible solutes (2). These compounds are widely synthesized as osmostress protectants by members of the Bacteria (3) and some members of the Archaea (4) and might also be produced by a few obligatory halophilic protists (5). Ectoines are well suited for this physiologically demanding task, since their high water solubility and physicochemical attributes make them compliant with cellular biochemistry and the functionality of macromolecular structures (6–11). The function-preserving characteristics of ectoines allow their high-level cellular accumulation, a process which in turn raises the osmotic potential of the cytoplasm and thereby allows the cell to counteract the high-osmolarity-instigated efflux of water to preserve vital turgor (12, 13). At the same time, the composition and solvent properties of the cytoplasm are optimized for biochemical reactions (13) so that growth of microorganisms can occur under osmotically unfavorable circumstances (2, 14, 15).
The amassing of ectoines by osmotically stressed microbial cells can be accomplished either through uptake from environmental sources via osmotically induced transporters or through de novo synthesis. Synthesis of ectoine proceeds from l-aspartate-β-semialdehyde (Fig. 1A), a central hub in bacterial amino acid and cell wall synthesis (16). A considerable number of microbial ectoine producers possess a specialized aspartokinase (Ask_Ect) (Fig. 1A) to ensure a sufficient supply of this precursor when the cellular demand for ectoine synthesis is high (17–19). The EctB protein (l-2,4-diaminobutyrate transaminase) converts l-aspartate-β-semialdehyde into l-2,4-diaminobutyrate, which is then transformed by EctA (l-2,4-diaminobutyrate acetyltransferase) into N-γ-acetyl-l-2,4-diaminobutyric acid, a metabolite that is cyclized by ectoine synthase (EctC) to form ectoine (Fig. 1A) (20). In a substantial subgroup of ectoine producers (3), ectoine can be further transformed into 5-hydroxyectoine through a position- and stereospecific enzyme reaction catalyzed by the ectoine hydroxylase (EctD) (21, 22) (Fig. 1A).
FIG 1.

Pathway for the synthesis of ectoine and hydroxyectoine and design of a recombinant cell factory for the production of ectoines. (A) Biosynthetic route for ectoine and its derivative 5-hydroxyectoine from l-aspartate. The relevant data for the enzymes and metabolites involved in this process were compiled from the literature (17, 20, 21, 86). (B) The E. coli cell factory carries a low-copy-number plasmid harboring the ectABCD-ask_ect gene cluster from P. stutzeri A1501 (67) under the control of its authentic and osmotically inducible promoter (P_ect) (17). This plasmid (pLC68) is a derivative of the cloning vector pHSG575 (89), which carries a lac promoter (P_lac) that is constitutively expressed in all the E. coli strains used in this study, as they carry a deletion of the entire lac operon, including that of the lacI regulatory gene. ProP and ProU are osmotically inducible transport systems for osmostress protectants; ProP is a member of the MFS family (88), and ProU is a binding-protein-dependent ABC transporter (80). The presumed ectoine/hydroxyectoine efflux system is shown as a yellow box; its molecular identity is unknown. The trimeric OmpC and OmpF proteins (represented here as monomers) function as general porins that are inserted into the E. coli outer membrane. IM, inner membrane; OM, outer membrane.
The ectoine biosynthetic genes are typically organized as an operon (ectABC), which may also comprise the ectoine hydroxylase gene (ectD) and the gene (ask_ect) for the specialized Ask_Ect aspartokinase (1, 4, 17, 23). Increases in the environmental osmolarity/salinity usually trigger enhanced levels of ect expression (21, 23–26). These genes are transcribed in some microorganisms from a single osmotically stimulated promoter (21, 24); however, a more complex arrangement of promoters driving ect expression also exists (26–28). The repressor CosR controls ectABC transcription in the human pathogen Vibrio cholerae, but this MarR-type regulator regulates not only compatible solute synthesis and uptake but also motility and biofilm formation (29). Likewise, ectABCD expression in the soil bacterium Streptomyces coelicolor is negatively controlled by a regulatory protein (GlnR) which serves as a globally acting transcription factor for nitrogen metabolism in many actinomycetes (30, 31). Since the CosR and GlnR repressors also control cellular processes other than ectoine/hydroxyectoine synthesis, their involvement in the production of ectoines seems to be restricted to the mentioned species and their closest relatives. In contrast, in a substantial subgroup of ectoine/hydroxyectoine producers, a regulatory gene (ectR) has been found in close physical association with the corresponding biosynthetic ect gene clusters (in 107 out of 440 inspected genome sequences) (3). EctR is a member of the MarR family of transcriptional regulators and was initially discovered in Methylomicrobium alcaliphilum, where its operator sequence overlaps with one of the two promoters driving ectABC-ask_ect transcription (28). Disruption of the ectR gene derepresses ect expression but this does not abolish osmotic control of this biosynthetic gene cluster (28); unfortunately, the environmental or cellular cue(s) to which the EctR repressor responses is not known. Despite these interesting findings, osmoregulation of ect gene expression is in general not well understood, in particular since many gene clusters for the synthesis of ectoines are not associated with a regulatory gene (3).
In addition to their function as osmostress protectants (1, 32), ectoines also afford cytoprotection against extremes in high and low growth temperatures in some bacteria (31, 33–35). The physicochemical characteristics of ectoines (36, 37) bestow them, both in vivo and in vitro, with excellent function-preserving attributes, e.g., enhancement of protein folding and stability, preservation of membrane structures and membrane proteins, and protection of the integrity of isolated molecules and even of entire cells (38–47). Although the difference between ectoine and 5-hydroxyectoine seems minor from a chemical point of view, hydroxylation of ectoine endows the newly formed 5-hydroxyectoine with substantially enhanced, or additional, stress-protective and function-preserving attributes (e.g., in particular against heat and desiccation stress) (31, 34, 39, 42, 43, 45).
The excellent stress-protective, function-preserving, and anti-inflammatory properties of ectoines attract considerable interest in the biotechnological use of these compounds as protein stabilizers, as well as for skin care and cosmetics, and potential medical applications are also actively pursued (1, 32, 48, 49). The financially rewarding practical applications of ectoines drove the development of an industrial-scale production process which harnesses the highly salt-tolerant ectoine/hydroxyectoine producer Halomonas elongata (26) as a natural microbial cell factory (1, 32, 48). In this process, high-level synthesis of ectoines is triggered through the growth of H. elongata in high-salinity media. The newly produced ectoines (along with other low-molecular-weight compounds) are then released from the producer cells through the transient opening of mechanosensitive channels (50), an activity that is triggered by a severe osmotic downshock of the cells (1, 32). Since these mechanosensitive safety valves close again once osmotic equilibrium has been achieved (50), the osmotically downshocked H. elongata cells survive this harsh treatment. Hence, the already formed biomass can be recycled into the fermentation vessel, while the released ectoines are recovered and purified via downstream processes (1, 32). This bacterial milking procedure (51) was subsequently modified by using a genetically engineered H. elongata strain lacking ectoine catabolic genes (26, 52) and in which the osmotically controlled ectoine/hydroxyectoine-specific tripartite ATP-independent periplasmic (TRAP) transport system TeaABC (53) was deleted. Overall, this strain excretes considerable amounts of ectoines into the growth medium and allows their recovery in a highly purified form on the scale of several tons per annum (32).
The very high salinity used to cultivate the ectoine/hydroxyectoine producer organism H. elongata in the established industrial production process is harsh on the fermentation and downstream processing equipment (32). Therefore, there has been considerable interest in developing alternative fermentation processes (54, 55), in exploiting additional natural ectoine/hydroxyectoine producers (56, 57) (including a bacterium that secrets them naturally into the medium [58]), and in engineering natural and synthetic microbial production hosts for enhanced recovery of these high-value compounds (1, 32). With respect to the latter approaches, ectoine/hydroxyectoine biosynthetic genes have been cloned into plasmids under the transcriptional control of either natural ect or synthetic promoters and transferred to Escherichia coli or Corynebacterium glutamicum host strains (59–62) or integrated into the chromosomes of biotechnological workhorses such as C. glutamicum (63) or the methylotrophic yeast Hansenula polymorpha (64). In some cases, advanced pathway engineering approaches were also used to enhance the synthetic production of ectoines in homologous and heterologous hosts (62, 63, 65, 66).
Since most of the ectoine/hydroxyectoine biosynthetic gene clusters are not associated with a dedicated regulatory gene (3), their osmostress-responsive induction might be an intrinsic property of the ect promoter elements and their immediate flanking regions. However, the salient features of ect type promoters have so far not been genetically analyzed at any level of detail. We focused here on the promoter driving the osmotically induced transcription of the Pseudomonas stutzeri A1501 ectABCD-ask_ect operon (17) through reporter gene studies and site-directed mutagenesis. Building on the knowledge gained through engineering of the P. stutzeri A1501 ect promoter, we designed ectoine and/or hydroxyectoine synthetic E. coli cell factories that allow the production and efficient secretion of these compounds into the growth medium.
RESULTS
Osmotic control of the P. stutzeri A1501 ect promoter in the non-ectoine-producing E. coli surrogate host.
The activity of the promoter(s) driving transcription of the ectABCD-ask_ect gene cluster from the plant-root-associated bacterium P. stutzeri A1501 (67) is induced by high salinity of the growth medium (17). However, the molecular identity of this promoter and the details of its transcriptional regulation are not known. We wondered if one could transfer the osmotically inducible pattern of P. stutzeri A1501 ect gene transcription into E. coli, a surrogate host that can import but cannot synthesize ectoines (68). To this end, we constructed an ect-lacZ reporter plasmid (pGJK4) that carries 264 bp of genomic DNA upstream of the start codon of ectA, the entire ectA gene, and a fusion junction to the promoterless lacZ gene within codon 87 of ectB. We observed a 6.3-fold increase in lacZ reporter activity when the MC4100(pGJK4) E. coli strain was grown in a chemically defined minimal medium (MMA) in the presence of 0.4 M NaCl in comparison with cells that were grown in MMA (Fig. 2A).
FIG 2.

Transcriptional activity of the ect promoter in response to increases in osmolarity and presence of compatible solutes. (A) E. coli strain MC4100 carrying the ectB-lacZ gene fusion plasmid pGJK4 was grown in MMA in the absence or presence of various compounds to increase the osmolarity of the medium. The solute concentration was chosen such that all the media possessed an equivalent osmolarity [1.000 mosmol (kg H2O)−1]. (B) Cultures of strain MC4100(pGJK4) were grown in MMA with increasing NaCl concentrations until they reached approximately the same optical density (OD578 of about 1.8), whereupon they were harvested and processed for β-galactosidase reporter enzyme activity assays. (C) Cultures of strain MC4100(pGJK4) were grown in MMA with 0.4 NaCl in the absence or presence of 1 mM concentrations the indicated compatible solutes or Casamino Acids. When the cultures reached an OD578 of about 1.8, the cells were harvested and assayed for β-galactosidase reporter enzyme activity. The data shown were derived from four independently grown cultures, and each enzyme assay was performed at least twice. β-Galactosidase enzyme activity is given in Miller units (MU) (108).
We then asked whether the observed increase in ect promoter activity occurred only in response to an increase in the external NaCl concentration or whether its transcriptional activity is responsive to a true osmotic cue. To test this, we grew the MC4100(pGJK4) reporter strain in the presence of both ionic and nonionic osmolytes and with glycerol, a membrane-permeative alcohol that cannot establish an osmotically effective gradient across the cytoplasmic membrane of E. coli when provided at high external concentrations. For these experiments, we added the solutes to the growth media in concentrations so that they had about the same osmolarity [1.000 mosmol (kg H2O)−1]. Both ionic (NaCl and KCl) and nonionic (sucrose and lactose) solutes triggered enhanced transcription of the ect promoter and activated it to approximately the same degree (Fig. 2A). In contrast, glycerol was unable to stimulate ect promoter activity (Fig. 2A).
After we had established that the activity of the ect promoter was under true osmotic control, we asked if this was an all-or-none transcriptional response. We therefore monitored ect-lacZ promoter activity in cultures of strain MC4100(pGJK4) that were grown in MMA with steadily and sustained increased NaCl concentrations over a wide range of salinities. This experiment revealed an exquisitely sensitive response of the ect promoter to the degree of the osmotic stress imposed onto the E. coli cells, since its transcriptional activity increased linearly concomitant with the increase in the NaCl content of the growth medium (Fig. 2B). In this experiment, we tested a substantial concentration range of salinities (from 0 M NaCl to 0.5 M NaCl); salinities higher than 0.5 M NaCl severely impair growth of the E. coli cells and were therefore not tested to avoid indirect effects on the transcriptional profile of the ect promoter.
Compatible solutes not only are effective osmostress protectants (2, 14, 15) but also frequently downregulate osmotically induced promoters since their accumulation alleviates the degree of osmotic stress perceived by the bacterial cell (18, 69, 70). To test whether the ect promoter also responded in its transcriptional activity to an external supply of compatible solutes, we propagated the MC4100(pGJK4) reporter strain at increased salinity (MMA with 0.4 M NaCl) in the absence or the presence (1 mM) of the osmostress protectants ectoine, hydroxyectoine, glycine betaine, and carnitine. Each of these compounds downregulated the activity of the ect promoter to approximately the same degree (Fig. 2C). The components of rich media contain compatible solutes or their precursors; e.g., glycine betaine is present in yeast extract, carnitine is present in meat extracts, and proline or proline-containing peptides are present in Casamino Acids. Indeed, the addition of 1 mM Casamino Acids to high-salinity-grown cultures of the MC4100(pGJK4) reporter strain significantly reduced the activity of the ect-lacZ reporter fusion (Fig. 2C).
RpoS, H-NS, OmpR, and cAMP/CRP do not participate in osmoregulation of ect expression in E. coli.
A number of regulatory genes have previously been implicated in controlling the transcription of osmostress-responsive genes in E. coli. Many osmotically regulated promoters in E. coli can be driven by forms of RNA polymerase complexed with the alternative sigma factor RpoS, the master regulator of the general stress response system (71, 72). Likewise, the nucleoid-associated H-NS protein not only affects gene expression on a global scale but also targets various osmotically controlled promoter regions, e.g., that of the proU operon encoding a glycine betaine/proline betaine ABC-type transporter (73). The EnvZ/OmpR two-component regulatory system is a key determinant for the reciprocal osmoregulation of the ompC and ompF porin genes (74). Furthermore, the cAMP/CRP complex has been implicated in osmoregulation (75, 76), e.g., by controlling the induction of the proP-P1 promoter (77). Although this promoter (5′-TTGATC-X17-TAGGGT-3′) for the osmolyte importer ProP bears a striking resemblance in its −10 region to the ect promoter of P. stutzeri A1501 (Fig. 3) (see below), it is transcriptionally active only upon a sudden osmotic upshock (77), while the ect promoter operates continuously under sustained high-osmolarity growth conditions (Fig. 2B). We tested an isogenic set of gene disruption mutations for potential effects of the RpoS, H-NS, EnvZ/OmpR, and cAMP/CRP regulatory proteins/complexes on ect-lacZ expression under both osmotically noninduced and osmotically induced growth conditions. None of these regulatory proteins had any effect on the ect promoter activity (Table 1).
FIG 3.

Deletion analysis of the ect promoter region. (A) Plasmid pGJK4 carries an ectB-lacZ reporter fusion that is expressed from the ect promoter present upstream of the ectA gene. The P. stutzeri A1501 genomic DNA located in front of the ectA start codon has a length of 264 bp. This genomic segment was successively shortened from its 5′ end, and the resulting E. coli reporter strains were assayed for β-galactosidase enzyme activity along with the wild-type plasmid-bearing strain. Cultures were grown in MMA or MMA containing 0.4 M NaCl and were harvested and processed for β-galactosidase enzyme activity when they reached an optical density (OD578) of about 1.8. The data shown were derived from four independently grown cultures, and each enzyme assay was performed at least twice. β-Galactosidase enzyme activity is given in Miller units (MU) (108). (B) DNA consensus sequence of Sig70-type E. coli promoters (78), of the predicted ect promoter, and of a hybrid promoter created through deletion analysis of the ect regulatory region (deletion junction K7) that generated plasmid pNST40. The −10 and −35 elements are highlighted, and the spacer length typical for Sig70-type E. coli promoters is indicated.
TABLE 1.
Influence on ect-lacZ expression of regulatory systems that had previously been implicated in osmotically controlled gene transcription in E. colia
| Strain | Gene disruption | LacZ activity (MU) with: |
|
|---|---|---|---|
| 0 M NaCl | 0.4 M NaCl | ||
| MC4100(pBBR1MCS-2-lacZ) | 0.3 ± 0.1 | 0.3 ± 0.1 | |
| MC4100(pGJK4) | 29 ± 0.7 | 141 ± 1.6 | |
| PD32(pGJK4) | hns | 28 ± 1.6 | 148 ± 4 |
| RH90(pGJK4) | rpoS | 24 ± 0.3 | 149 ± 3.7 |
| RH76(pGJK4) | cya | 29 ± 1.2 | 143 ± 4.3 |
| LC30(pGJK4) | ompR | 24 ± 1.9 | 140 ± 4.9 |
Transcriptional activity of the ectB-lacZ reporter fusion present on plasmid pGJK4 in response to increased salinity. The reporter plasmid was introduced by DNA transformation into an isogenic set of strains harboring different gene disruption mutations: MC4100 (wild type), PD32 (hns-206::Apr), RH90 (rpoS359::Tn10), RH76 (Δcya-851), and LC30 (ompR::Tn10). These strains are all derivatives of the ΔlacIZYA strain MC4100. The vector, pBBR1MCS-2-lacZ, used for the construction of the ect-lacZ reporter fusion was used as the control. All strains were grown in MMA in the absence or presence of 0.4 M NaCl until they reached an OD578 of approximately 1.8; they were then harvested by centrifugation and assayed for β-galactosidase reporter enzyme activity. The data shown (means ± standard deviations) were derived from four independently grown cultures, and each enzyme assay was performed at least twice. β-Galactosidase enzyme activity is given in Miller units (MU).
Pinpointing the osmotically regulated ect promoter through deletion analysis.
The ect-lacZ reporter plasmid (pGJK4) carries 264 bp of genomic DNA upstream of the start codon of ectA, a region that will contain the osmotic-stress-responsive ect promoter. To delineate its approximate position, we successively shortened the 5′ region of this genomic segment while preserving the original ectB-lacZ fusion junction in codon 88 of ectB (Fig. 3A). The DNA sequences of the various deletion endpoints are summarized in Fig. S1 in the supplemental material and are graphically represented in Fig. 3A. The removal of 47 bp from the 5′ end of the original genomic segment preceding the ectA start codon (plasmid pGJK4) had only very modest effects on both the noninduced and salt-stress-induced levels of the lacZ reporter fusion (up to deletion endpoint K6) (Fig. 3A). By shortening the K6 deletion junction (plasmid pNST29) further by 40 bp (deletion K7; plasmid pNST40), we observed a drastic fall in promoter activity. However, the remaining reporter enzyme activity was still osmotically inducible (Fig. 3A). An additional deletion of 70 bp then abolished the transcriptional activity of the ect-lacZ reporter fusion (deletion K8; plasmid pNST41) (Fig. 3A). Since the 5′ end of the K8 deletion construct lies in codon four of the ectA coding region (Fig. S1), one can infer that no promoter internal to the ect gene cluster is present between the K8 deletion endpoint in ectA and the fusion junction in codon 88 of ectB (Fig. 3A and S1).
We were particularly interested in the striking drop of ect-lacZ promoter activity found in strains carrying plasmid pNST40 with the K7 deletion endpoint (Fig. 3A), since it might provide potential clues about the position of either the ect promoter or regulatory regions required for its full activity. We therefore inspected the ect DNA sequence around the 5′ end of the K7 deletion junction more closely and found through sequence gazing potential −10 and −35 elements of sigma-70 (Sig70)-type promoters (78) that were separated from each other by 18 bp (Fig. 3B). While the potential −35 region of the ect promoter possesses a good fit to the consensus sequence with just a single-base-pair deviation, the putative −10 region diverges very strongly from the AT-rich consensus sequence (78), since it is very GC rich (Fig. 3B). Furthermore, the spacer length of 18 bp is suboptimal for Sig70-type promoters (78). Interestingly, the K7 deletion endpoint removes the −35 region of the putative ect promoter and also 2 bp from the spacer region (Fig. 3B). However, DNA sequences derived from the cloning vector with resemblance to a potential −35 region replaced the authentic ect −35 DNA sequence while maintaining a spacer length of 18 bp of the hybrid promoter (Fig. 3B). The fortuitously generated −35 region of the K7 deletion junction deviates more strongly from the consensus sequence of −35 regions from Sig70-type promoters (78) than the authentic −35 region of the putative ect promoter (Fig. 3B). These DNA sequence features of the fortuitously generated hybrid promoter could thus potentially explain the significant drop in ect-lacZ reporter activity observed in strain MC4100(pNST40) (Fig. 3A).
Defining a minimal DNA fragment required for osmoregulation of ect transcription.
Building on the knowledge gained through the deletion analysis of the 5′ segment of the ect regulatory region, we constructed an ect-lacZ reporter fusion containing a minimal DNA fragment still allowing osmoregulation. To this end, we fused a 116-bp DNA fragment to the lacZ reporter gene (Fig. 4A); it contains the −35 and −10 elements of the ect promoter, 28 bp upstream of the −35 region, and a fusion junction to lacZ within codon seven of ectA (Fig. 4B). This ectA-lacZ reporter construct (present on plasmid pASTI11) provided approximately the same level of gene expression and osmotic inducibility as the parent ectB-lacZ reporter fusion (present on plasmid pGJK4) (Fig. 4C). Hence, all elements required in cis for osmoregulation of ect expression are contained in this 116-bp DNA region. This ectA-lacZ reporter construct is a gene fusion that leads to the formation of an EctA-LacZ hybrid protein; as a consequence, its translation depends on the ectA ribosome-binding site.
FIG 4.
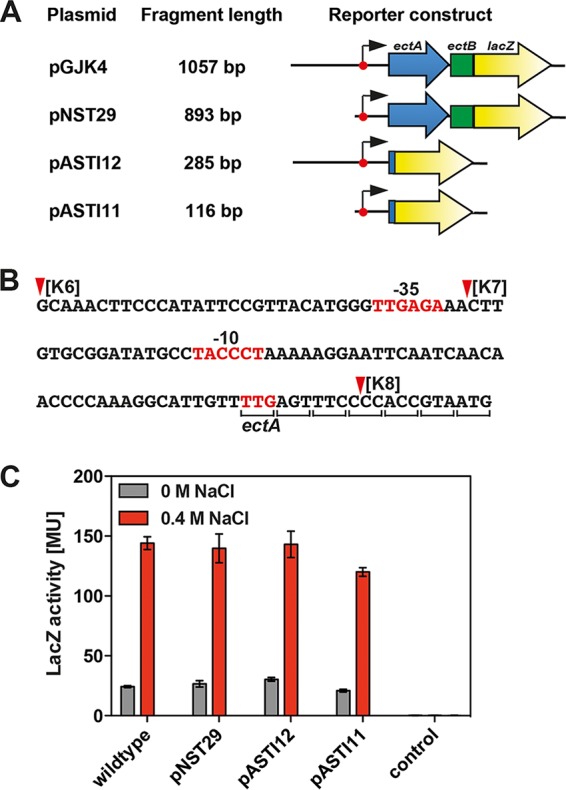
A minimal DNA fragment directing osmoregulated ect transcription. (A) Physical structures of ect-lacZ reporter constructs. (B) DNA sequence of the DNA fragment present in plasmid pASTI11. The deletion endpoints K6, K7, and K8 defined in the experiments for Fig. 3A are indicated, and the −35 and −10 elements of the ect promoter are highlighted. The fusion junction within ectA to the lacZ reporter gene lies in codon seven. (C) β-Galactosidase reporter enzyme activity in cells of strain MC4100 carrying the plasmids depicted in panel A. Cells of MC4100 carrying the vector (pBBR1MCS-2-lacZ) used to construct plasmids pGJK4, pNST29, pASTI12, and pASTI11 were used as the control. Cultures were grown in MMA or MMA containing 0.4 M NaCl and were harvested and processed for β-galactosidase enzyme activity when they reached an optical density (OD578) of about 1.8. The data shown were derived from four independently grown cultures, and each enzyme assay was performed at least twice. β-Galactosidase enzyme activity is given in Miller units (MU) (108).
Site-directed mutagenesis of the ect promoter reveals major features of its osmotic control.
To provide further evidence that the putative −35 and −10 sequences described above (Fig. 3B and 4B) are actually part of the ect promoter, we targeted by site-directed mutagenesis base pairs in both the −35 and −10 elements that are highly conserved in Sig70-type promoters and crucially contribute to promoter activity and strength (78). To this end, we replaced the highly conserved TTG sequence at the 5′ end of the −35 region with a GGG string of base pairs, and in the −10 region we replaced the so-called invariant T with a GC pair (Fig. 5A). Both promoter variants (Mut 1 and Mut 2) abolished ect-lacZ reporter activity down to a basal level (Fig. 5B), thereby providing compelling evidence that the region we deemed to be the ect promoter (Fig. 3B and 4B) actually represents this element for the transcriptional control of the ectoine/hydroxyectoine biosynthetic operon from P. stutzeri A1501.
FIG 5.

Site-directed mutagenesis of the ect promoter and assessment of the transcriptional activities of the promoter variants. (A) DNA sequences of the wild-type ect promoter and its mutant derivatives. The base pairs changed through site-directed mutagenesis are highlighted in red. (B) β-Galactosidase reporter enzyme activity of E. coli strains (MC4100) harboring the wild-type ectB-lacZ fusion or its mutant derivatives. Cultures were grown in MMA or MMA containing 0.3 M NaCl and were harvested and processed for β-galactosidase enzyme activity when they reached an optical density (OD578) of about 1.8. The data shown were derived from four independently grown cultures, and each enzyme assay was performed at least twice. β-Galactosidase enzyme activity is given in Miller units (MU) (108).
To glean information about the features of the ect promoter that contribute to its osmotic control, we first targeted the spacer region. Shortening the spacer to a suboptimal length of 16 bp rendered the promoter constitutive (Mut 3) at an expression level corresponding to that exhibited by the wild-type ect promoter in E. coli cells grown in minimal medium in the presence of 0.3 M NaCl (Fig. 5B). Conversely, when we extended the spacer sequence of the ect promoter to a suboptimal length of 19 bp (Mut 6) for Sig70-type promoters (78), ect-lacZ reporter activity was abolished (Fig. 5A and B). Interesting effects were seen when we decreased the spacer length of the authentic ect promoter from 18 bp to the optimal spacer length of 17 bp for the activity of Sig70-type promoters (78) by deleting single base pairs at two different positions in the ect promoter sequence. In the corresponding mutants (Mut 4 and Mut 5) (Fig. 5A), the noninduced level of ect-lacZ promoter activity rose substantially but both mutant promoters still permitted osmotic induction of transcription (Fig. 5B). Sig70/SigA-type promoters from E. coli and Bacillus subtilis often possess a TG motif at position −16 which contributes to promoter strength (78, 79). We generated through a deletion of one base pair in the ect spacer a promoter variant (Mut 18) that generated a TG motif at the −16 position (Fig. 5A). This promoter variant remained osmotically inducible at a level of transcription similar to that observed for the Mut 4 and Mut 5 17-bp spacer variants (Fig. 5B). The properties of the Mut 18 variant indicate that the artificially generated −16 motif is not crucial for either the strength of the ect promoter or its response to osmotic stress.
We then targeted the unusual GC-rich −10 region of the ect promoter by single- and double-base-pair changes that collectively bring the authentic sequence closer to the A/T-rich consensus sequence of Sig70-type promoters (78). In all of these mutants, strong osmotic induction occurred (Fig. 5A and B). Particularly notable was the property of the Mut 9 promoter variant, in which one of the three CG base pairs was replaced by a TA base pair (Fig. 5A). It allowed a very strong salt induction, 18.9-fold in the Mut 9 variant versus 5.2-fold for the wild-type promoter (Fig. 5B).
As noted above, the −35 region of the ect promoter deviates at just a single position from the consensus Sig70-type promoter sequence (Fig. 3B). When we altered the −35 region to a fully consensus-like sequence (Mut 12), a drastic effect on the strength of promoter activity was observed: the basal promoter activity rose by 33.4-fold compared to that of the wild type, and its salt-stress-induced level of activity exceeded that of the wild-type promoter by 16.2-fold (Fig. 5B). Strong increases in transcriptional activity were also seen when we optimized both the −10 and −35 regions and spacer length of the ect promoter (Fig. 5A and B). In all these cases, the basal level of ect promoter activity was strongly increased in the absence of osmotic stress while still allowing increased promoter activity at high salinity (Fig. 5B). As a result of this profile in promoter activity, the extent of osmotic induction was reduced. Particularly noteworthy are the properties of the ect promoter variants (Mut 15 and Mut 16) that we mutated such that they matched in their −10 and −35 regions, and with their spacer length (17 bp), the features of a perfect consensus sequence of Sig70-type promoters (78) (Fig. 5A and B). The transcriptional activities of these mutant promoters occurred at high basal levels but still retained osmotic inducibility, by about 1.8-fold for Mut 15 and 1.7-fold for Mut 16. For a comparison, the osmotic inducibility of the wild-type ect promoter under the same experimental conditions was 5.2-fold (Fig. 5A and B).
We noted the presence of an inverted-repeat DNA sequence (5′-CCCAT-N10-ATGGG-3′) just upstream of the ect −35 region (see Fig. S2A in the supplemental material). Since this inverted repeat could potentially form the operator sequence for a regulatory protein, we changed the left side of the repeat through site-directed mutagenesis to 5′-GGGTA-3′, thereby destroying the dyad symmetry (Fig. S2A). This mutation had no influence on ect-lacZ reporter gene expression (Fig. S2B).
As a note of caution, in conducting this comprehensive set of ect-lacZ reporter fusion experiments with mutant ect promoters, we initially experienced some experimental difficulties. We repeatedly observed that the β-galactosidase reporter enzyme activity varied strongly between parallel-grown cultures of E. coli strains carrying the same ect-lacZ reporter plasmid when they were grown in MMA with 0.4 M NaCl. Substantial amounts of β-galactosidase protein were present in both cultures (as assessed by Western blotting); however, one culture possessed very high β-galactosidase reporter enzyme activity, while the other culture showed greatly diminished reporter enzyme activity. We therefore took care that the data included in this communication are all derived from experiments where the parallel-grown cultures yielded consistent β-galactosidase reporter enzyme activities. We observed the phenomenon of substantially differing β-galactosidase values only with mutant ect promoters exhibiting high transcriptional activity and only when the salinity of the growth medium exceeded that of 0.3 M NaCl. An example of this phenomenon is documented for strain MC4100(pPH11) harboring the perfect Sig70-promoter driving ect-lacZ expression when it was grown at various salinities (see Fig. S3 and S4 in the supplemental material). We were unable to define the apparently subtle physiological differences in the growth behavior of the cultures incubated in parallel that led to the irregular behavior of the β-galactosidase reporter enzyme. However, it is known from previous studies that high-level expression of hybrid proteins (e.g., ProV-LacZ; ProV is the ATPase of the ProU ABC transporter [80]) can lead to protein aggregation under high-salinity growth conditions and therefore obscure the physiologically correct regulatory pattern of the osmoregulated promoter under study (81). Since the reporter fusion present on plasmid pGJK4 and its derivatives carrying mutant ect promoters fuses 88 codons of ectB to the lacZ reading frame, the resulting EctB-LacZ hybrid protein might be prone to aggregation in a high-osmotic-strength cytoplasm.
Details of the transcriptional responses of selected ect promoter variants with high levels of transcriptional activity under sustained osmotic stress.
Since we intended to use the wild-type promoter of the ectABCD-ask_ect gene cluster from P. stutzeri A1501 (67) and some of its promoter up-mutations for the establishment of a heterologous ectoine/hydroxyectoine cell factory (see below), we studied the promoter activities of Mut 12 (with a perfect −35 region) (Fig. 5A) and of Mut 15 (with a consensus Sig70-type promoter) (Fig. 5A) in somewhat greater detail. We grew the strains carrying the corresponding ect-lacZ reporter plasmids, pPH8 (Mut 12) and pPH11 (Mut 15), under sustained osmotic stress condition in cultures in which we systematically increased the salinity from 0 M NaCl to 0.4 M NaCl (Fig. 6A, B, and C). The β-galactosidase reporter enzyme activities of strains MC4100(pPH8) and MC4100(pPH11) increased in a finely tuned manner in response to corresponding increases in the external salinity. Their pattern of transcriptional activation mirrored that of the wild-type ect-lacZ reporter strain MC4100(pGJK4) (Fig. 6). Hence, as noted above (Fig. 5B), the two mutant ect promoters had not lost their osmotic control when they were tested in a wider range of salinities. However, the basal activity of the reporter fusion in the absence of osmotic stress (cells grown in MMA) was very substantially increased; that of strain MC4100(pPH8) was raised by 42.3-fold (Fig. 6B) and that of strain MC4100(pPH11) (Fig. 6C) by 47.2-fold relative to that with the wild-type plasmid pGJK4 (Fig. 6A). The osmotically induced levels of β-galactosidase reporter enzyme activity in the ect-lacZ reporter fusion constructs exceeded greatly the activity of the reporter enzyme in the wild type ect-lacZ reporter fusion, by 14.2-fold, and 16.8-fold, respectively (Fig. 6A, B, and C).
FIG 6.

Transcriptional activity of the ect wild-type promoter and two of its mutant derivatives in response to sustained osmotic stress. (A to C) E. coli strains (MC4100) harboring either the wild-type ectB-lacZ reporter fusion plasmid pGJK4 (A), a plasmid (pPH8) carrying a point mutation (Mut 12) (Fig. 4A) in the ect −35 region (B), or a plasmid (pPH11) carrying a mutant ect promoter (Mut 15) (Fig. 4A) that was changed in its −10 region, −35 region, and spacer length to the consensus sequence of Sig70-type E. coli promoters (78) (C) were grown at various salinities. (D) Cells of strain MC4100 or FF4169 (otsA1::Tn10) carrying the wild-type plasmid pGJK4, pPH8, or pPH11 were grown in MMA with 0.3 M NaCl. Cultures were harvested and processed for β-galactosidase enzyme activity when they reached an optical density (OD578) of about 1.8. The data shown were derived from four independently grown cultures, and each enzyme assay was performed at least twice. β-Galactosidase enzyme activity is given in Miller units (MU) (108).
Genetic design of a heterologous ectoine/hydroxyectoine cell factory.
E. coli synthesizes large amounts of the compatible solute trehalose under osmotic stress conditions as a cytoprotectant (82, 83). It partially releases the newly produced trehalose also into the periplasmic space, where this disaccharide is hydrolyzed by an osmotically inducible trehalase (TreA), and the glucose monomers are then recovered again by the E. coli cells through a phosphotransferase transport system (PTS) for their use as carbon sources (84). To avoid the contamination of newly produced ectoine/hydroxyectoine by trehalose and to provide an incentive to the osmotically stressed cells to enhance the production of ectoines in response to increased salinity to provide physiologically adequate osmostress protection for E. coli (2), we used for the setup of the cell factory a strain that is defective in trehalose synthesis (FF4169; otsA1::Tn10) (83, 85). The use of such a host strain proved to be beneficial for the expression of the ect-lacZ reporter fusions carrying either the wild-type ect promoter or its Mut 12 and Mut 15 derivatives in comparison with its otsBA+ parent strain, MC4100 (Fig. 6D). We therefore constructed derivatives of the wild-type ectABCD-ask_ect plasmid pLC68 harboring either the Mut 12 (plasmid pASTI1) or Mut 15 (plasmid pASTI9) variant in their promoters.
We assessed the production performance of strain MC4100 (otsAB+) and its otsA1::Tn10 derivative, strain FF4169, for ectoines when the cells carried plasmid pLC68. In MC4100 cultures, a mixture of ectoine and hydroxyectoine was found intracellularly, while the supernatant contained exclusively hydroxyectoine (Fig. 7A and B). Use of strain FF4169 for such an experiment yielded higher production levels of ectoines, with hydroxyectoine being again exclusively accumulated in the growth medium (Fig. 7A and B). In both strains, the external concentration of hydroxyectoine substantially exceeded that of the internal pool size of ectoines (Fig. 7A and B).
FIG 7.
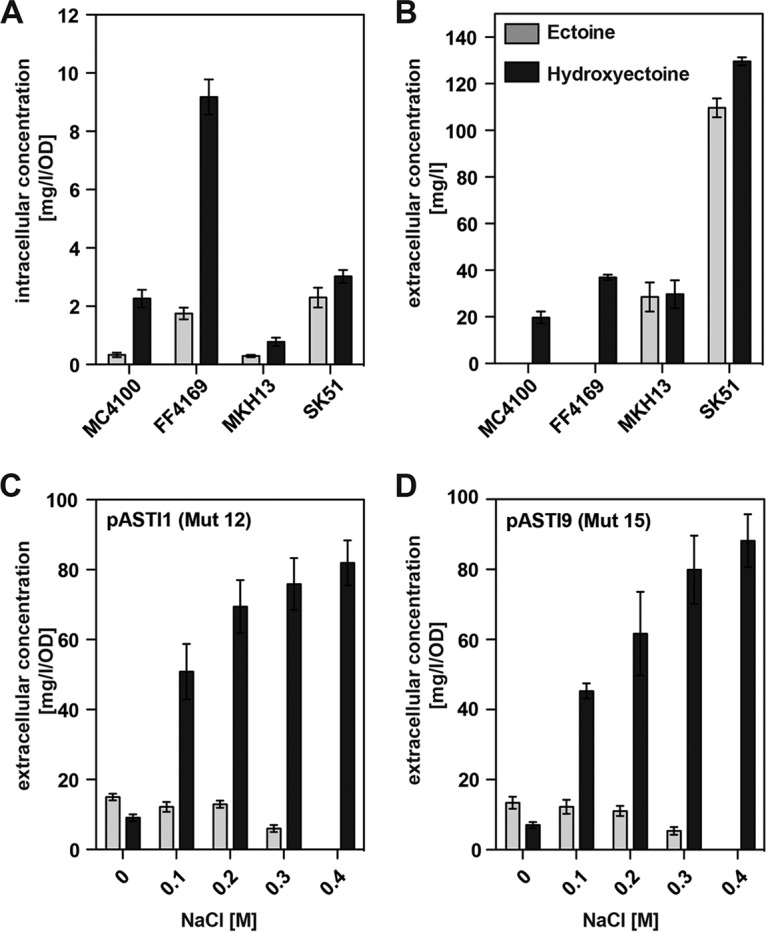
Comparison of ectoine and hydroxyectoine production in different E. coli mutant strains. (A and B) Cells of the E. coli wild-type strain MC4100 and its mutant derivatives FF4169 (otsA1::Tn10), MKH13 [Δ(proP)2 Δ(proU::spc)608], and SK51 [Δ(proP)2 Δ(proU::spc)608 otsA1::Tn10] carrying plasmid pLC68 (with a wild-type ect promoter) were cultivated in MMA with 0.4 M NaCl. After incubation for 48 h, cells were harvested and assayed for their intracellular (A) and extracellular (B) concentrations of ectoine (gray bars) and hydroxyectoine (black bars) via HPLC analysis. (C and D) Comparison of ectoine and hydroxyectoine production in strains carrying mutant ect promoters in response to graded increases in the external salinity. E. coli strain FF4169 (otsA1::Tn10) carrying either plasmid pASTI1 (Mut 12; point mutation in the −35 region) (Fig. 3A) (C) or plasmid pASTI9 (Mut 15; consensus Sig70-type promoter variant) (Fig. 3A) (D) was cultivated in MMA containing various concentrations of NaCl. The cultures were grown for 24 h, and the extracellular ectoine/hydroxyectoine content was then determined by HPLC analysis. The data shown were derived from four independently grown cultures, and each assessment of their ectoine/hydroxyectoine content was performed at least twice. Since the concentration of NaCl in the medium significantly influences cell growth, the ectoine/hydroxyectoine content of the supernatant was normalized to an OD568 of 1 and is reported here as mg/liter/OD unit.
Aspartate is the biosynthetic precursor of ectoine (20, 86) (Fig. 1A), and its addition to the growth medium has already been proven beneficial for the recombinant production of ectoines (87). In exploratory experiments, we observed that the addition of 25 mM aspartate to our Ots− E. coli cell factory harboring plasmid pLC68 substantially enhanced hydroxyectoine production (see Fig. S5A in the supplemental material). However, increases in the aspartate concentration successively reduced the amount of the produced hydroxyectoine, while this phenomenon was not observed in strains harboring the ectABCD-ask_ect gene cluster carrying promoter up-mutations (plasmids pASTI1 [Mut 12] and pASTI9 [Mut 15]) (Fig. S5). We did not further investigate the drop in hydroxyectoine production of the Ots− E. coli host strain FF4169(pLC68), but we suspect that it might be related to the notable increase in medium osmolarity caused by the addition of high concentrations of aspartate (see Fig. S6 in the supplemental material). Furthermore, the limited ability of the wild-type ectABCD-ask_ect plasmid to provide adequate levels of osmostress protection to the host strain lacking trehalose (82, 83) might also be a contributing factor.
ect promoter mutations increase ectoine/hydroxyectoine production by the E. coli cell factory.
To further increase ectoine/hydroxyectoine production titers, we separately introduced plasmids pASTI1 (Mut 12, a point mutant of the −35 region in the ect promoter) (Fig. 5A) and pASTI9 (Mut 15, a mutant ect promoter altered to the Sig70-type consensus sequence) (Fig. 5A) into the Ots− E. coli mutant strain FF4169. Consistent with the ect-lacZ reporter fusion assays (Fig. 6D), the presence of either plasmid leads to substantially increased production titers in comparison with those in strain FF4169 carrying the wild-type plasmid pLC68 (Fig. 7A and B) in an osmostress-dependent manner, with hydroxyectoine being the predominant compatible solute in the culture supernatant (Fig. 7C and D).
Hypersecretion of ectoine and hydroxyectoine by E. coli strains lacking the osmolyte importers ProP and ProU.
The osmotically regulated ABC transporter ProU and the major facilitator superfamily (MFS)-type transporter ProP serve as the major uptake systems for compatible solutes in E. coli (80, 88) and are also responsible for the uptake of ectoine under osmotic stress conditions (68). We noted that the secretion of both ectoine and hydroxyectoine was particularly pronounced in strain MKH13 (Ots+ ProP− ProU−) and in strain SK51, which is simultaneously defective in trehalose synthesis and the ProU and ProP transport systems (Fig. 7A and B). Apparently, E. coli continuously secretes substantial amounts of the heterologously produced ectoines into the growth medium that subsequently cannot be recovered again by the cells due to the defects in the ProU and ProP importers (Fig. 7B).
To study this phenomenon in more detail, we constructed an isogenic set of strains that lacked the ability to synthesize trehalose and possessed only either the ProU (strain LC6) or ProP (strain LC7) transporter. As long as one of these ectoine/hydroxyectoine importers was intact, the external pool of hydroxyectoine matched that produced by the ProP+ ProU+ parent strain FF4169. Notably, ectoine was a very minor component of the supernatant of the osmotically stressed cells (Fig. 8B), while in the supernatant of the non-osmotically challenged cultures ectoine was present (Fig. 8A). This picture changed substantially when we analyzed ectoine/hydroxyectoine excretion in the ProP− ProU− strain SK51. Supernatants of cultures of the SK51 cell factory contained both ectoine and hydroxyectoine, with hydroxyectoine being the dominant solute again (Fig. 8B).
FIG 8.
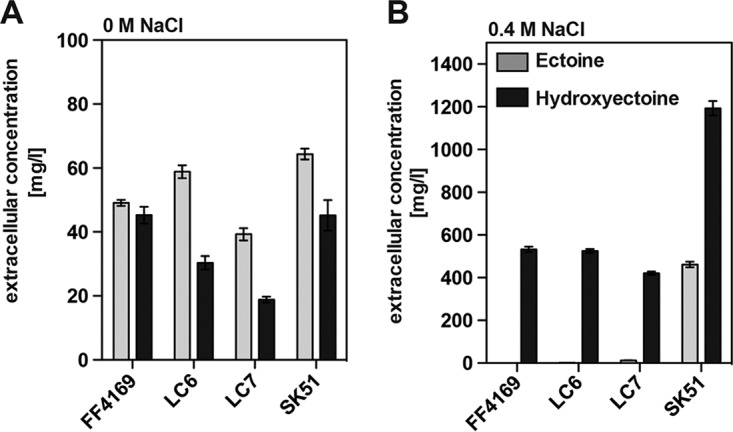
Influence of the ProP and ProU ectoine uptake systems on the extracellular amounts of ectoine and hydroxyectoine. E. coli strains FF4169, LC6, LC7, and SK51 carrying plasmid pASTI1 (Mut 12; point mutation in the −35 region) (Fig. 3A) were cultivated in MMA containing either 0 M NaCl (A) or 0.4 M NaCl (B) for 48 h, and the extracellular ectoine/hydroxyectoine content was then determined by HPLC analysis. The data shown were derived from four independently grown cultures, and each assessment of their ectoine/hydroxyectoine content was performed at least twice. The relevant genotypes of the E. coli strains used are as follows: FF4169, otsA1::Tn10 proP+ proU+; LC6, otsA1::Tn10 proP proU+; LC7, otsA1::Tn10 proP+ proU; and SK51, otsA1::Tn10 proP proU.
Deletion of ectD results in a recombinant cell factory secreting exclusively ectoine.
In strain SK51 (Ots− ProP− ProU−) harboring the ectABCD-ask_ect wild-type plasmid pLC68, ectoine and hydroxyectoine are present in an almost a 1:1 mixture (Fig. 7B), while in the supernatant of strain FF4169 (Ots− ProP+ ProU+) carrying the ect promoter mutant plasmids pASTI1 and pASTI9, hydroxyectoine predominates (Fig. 7C and D). To construct a recombinant cell factory producing exclusively high levels of ectoine, we deleted the ectD gene from the ectABCD-ask_ect gene cluster present on plasmid pASTI1. When we introduced the resulting plasmid, pLC75, into strain SK51, only ectoine was found, both intracellularly and extracellularly (Fig. 9A and B). This strain yields an ectoine/hydroxyectoine production titer of 1.5 g liter−1 per 24 h. The strain harboring the corresponding ectD+ plasmid pASTI1 had a practically identical productivity but yielded a 2:1 mixture of hydroxyectoine to ectoine in the supernatant (Fig. 9B). Only approximately 8.7% of the ectoines produced were retained by cells of strain SK51(pASTI1 [ectD+]), whereas the corresponding value for cells of strain SK51(pLC75 [ΔectD]) was 4.2% (Fig. 9A and B). Hence, these recombinant cell factories secrete the vast majority of the newly produced ectoines into the growth medium. The ca. 1.5 g ectoine produced per liter of culture of strain SK51(pLC75) within 24 h corresponds to a yield of 0.3 g ectoine per g glucose, values that can also be described as 1.3 g ectoine produced per 1.24 g cell dry weight.
FIG 9.
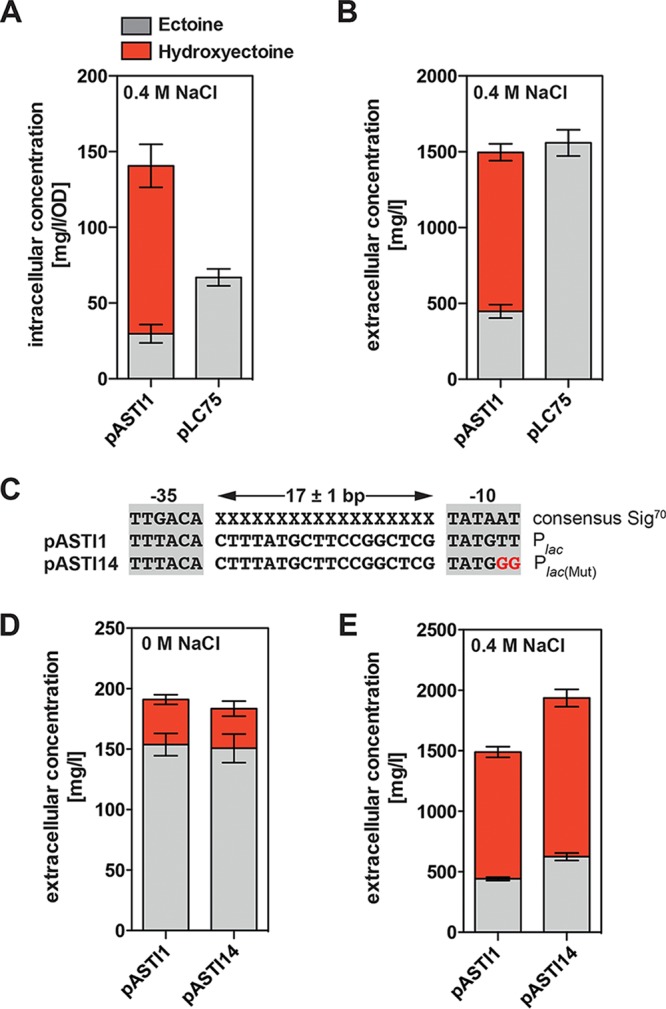
Influence of an ectD deletion on ectoine/hydroxyectoine production and secretion. (A and B) E. coli strain SK51 (otsA1::Tn10 proP proU) harboring either plasmid pASTI1 (ectABCD-ask_ect) or plasmid pLC75 [ectABC(ΔectD)-ask_ect] carrying a point mutation in the −35 region of the ect promoter was grown in MMA containing 0.4 M NaCl for 24 h, and the intracellular (A) and extracellular (B) ectoine/hydroxyectoine content was then determined by HPLC analysis. (C) Comparison of the lac promoter sequence present in the cloning vector pHSG575 (89) and its mutant derivative present in plasmid pASTI14 (ectABCD-ask_ect). (D and E) Cells of strain SK51 carrying either pASTI1 or pASTI14 were grown either in MMA (D) or MMA with 0.4 M NaCl (E) for 24 h, and the extracellular ectoine and hydroxyectoine concentrations were then determined by HPLC analysis. The data shown were derived from four independently grown cultures, and each assessment of their ectoine/hydroxyectoine content was performed at least twice.
Quantitative analysis of the performance of the ectoine/hydroxyectoine synthetic cell factory.
To study the salient features of the ectoine/hydroxyectoine synthetic cell factory further, we performed an experiment in which we not only monitored the secretion of ectoines but also quantitated the consumption of glucose and that of the ectoine precursor aspartate under both inducing and noninducing osmotic conditions in baffled shake flask cultures. For these experiments, we used strain SK51(pASTI1) as an example (Table 2). After 24 h of cultivation of this strain in MMA with 0.4 M NaCl, we found ectoine and hydroxyectoine in the growth medium in concentrations similar to that in the experiment for Fig. 9B. Since pASTI1 carries a ect promoter up-mutation (in its −35 sequence; Mut 12) (Fig. 5A), we also detected ectoine and hydroxyectoine in the supernatant of the cultures grown in MMA. While the corresponding value for ectoine represented about a third of that detected under osmotic stress conditions, the production of hydroxyectoine was greatly stimulated under high-osmolarity conditions (by about 52-fold) (Table 2). Together, this corresponded to 1.3 g ectoines produced per liter of culture.
TABLE 2.
Analysis of glucose, aspartate, and the content of ectoines before and after cultivation of the heterologous E. coli cell factory SK51(pASTI1)a
| Compound | Concn (mg/liter) in supernatant |
|||
|---|---|---|---|---|
| Before cultivation |
After 24 h of cultivation |
|||
| Without NaCl | With 0.4 M NaCl | Without NaCl | With 0.4 M NaCl | |
| Ectoine | 1.5 ± 0.3b | 1.4 ± 0.06b | 144.1 ± 4.8 | 371.8 ± 11.6 |
| Hydroxyectoine | NDc | ND | 18.1 ± 2.4 | 936.7 ± 48.5 |
| Aspartate | 4,246.6 ± 197.6 | 4,358.8 ± 149.9 | 2.2 ± 0.4 | 979.1 ± 94.2 |
| Glucose | 4,606.6 ± 200.6 | 4,627.2 ± 168.4 | 3.7 ± 0.5 | 2.2 ± 0.5 |
E. coli strain SK51 (otsA1::Tn10 proP proU) harboring plasmid pASTI1 (ectABCD-ask_ect) was grown in 200 ml MMA (in a 2-liter baffled Erlenmeyer flask) containing either 0 or 0.4 M NaCl. Cells of preculture grown in MMA overnight were used to inoculate the main cultures to an OD578 of about 0.15; the cells were then cultivated for 24 h at 37°C on an aerial orbital shaker. Immediately after inoculation and at the end of the experiment after 24 h, 2-ml samples were withdrawn and used to determine the glucose, aspartate, ectoine, and hydroxyectoine contents of the cultures. The data shown (means ± standard deviations) were derived from four independently grown cultures, and each assessment of their ectoine, hydroxyectoine, aspartate, and glucose contents was performed at least twice.
This small amount of ectoine results from carryover from the inoculum.
ND, not detected.
By the time the experiment was terminated (after 24 h), both the osmotically nonstressed and the osmotically stressed cells had consumed practically all the glucose that was added to the growth medium (Table 2). In contrast, the cells had consumed essentially all the added aspartate under nonstressed conditions, while substantial amounts of this amino acid were still present in the supernatants of cultures grown under osmotic stress conditions (Table 2). It is thus apparent from this analysis that the SK51(pASTI1) synthetic cell factory is carbon source limited under the studied growth conditions in baffled shake flasks. Furthermore, since both the nonstressed and osmotically stressed cultures reached essentially the same optical density within the 24-h time frame (see Fig. S7 in the supplemental material), it appears that under osmotic nonstressed conditions, the cells funnel aspartate primarily into the formation of biomass, while this route of aspartate consumption is substantially reduced under osmotic stress conditions (Table 2).
By monitoring the growth of the cells, we found the interesting phenomenon that ectoine/hydroxyectoine production was beneficial under osmotic stress conditions, while the growth of the E. coli cells was not notably altered under nonstressed conditions (Fig. S7). The improved growth of the ectoine/hydroxyectoine-producing cells at high salinity can readily be understood since the internally amassed ectoines will provide osmoprotection to the otherwise osmotically sensitive host strain SK51, which lacks the ability to synthesize the compatible solute trehalose (82, 83).
A lac promoter-directed ect antisense transcript limits production of ectoines in plasmid-carrying cells.
The low-copy-number vector (pHSG575) used for the cloning of the ectABCD-ask_ect operon (plasmid pLC68) carries a lac promoter (89) that is constitutively expressed in the ΔlacIZYA host strain MC4100 and its derivatives that were used in our study. We obtained in our experiments involving cloning of the ect gene cluster only pHSG575-derived ect+ plasmids in which the lac promoter will direct an antisense RNA transcript relative to the ectABCD-ask_ect mRNA, which is driven by the transcriptional activity of the ect promoter (Fig. 1B). This antisense transcript could potentially either destabilize the ectABCD-ask_ect mRNA or impair its effective translation. We therefore constructed a derivative of pASTI1 in which we inactivated the lac promoter through site-directed mutagenesis (Fig. 9C). When we tested the performance of the resulting plasmid, pASTI14, for ectoine/hydroxyectoine production in comparison with the parent plasmid pASTI1, the extracellular levels of ectoines increased from about 1.5 g liter−1 in strain SK51(pASTI1) to about 1.9 g liter−1 in strain SK51(pASTI14) under high-salinity growth conditions (Fig. 9E). This corresponded to 1,749 mg ectoines per g (dry weight) of the producing cells. Hence, the lac promoter-directed antisense transcript (Fig. 1B) limits the production of ectoines in our cell factory to a notable degree (by approximately 21%).
DISCUSSION
When challenged by high-osmolarity environments, many microorganisms synthesize copious amounts of ectoines as cytoprotectants (1, 3, 4, 32). However, the mechanism(s) through which the cell perceives increases in the environmental osmolarity, how it processes this information, and how it transmits it to the transcriptional apparatus to drive enhanced transcription of ectoine/hydroxyectoine biosynthetic genes are rather incompletely understood. CosR, a regulator of ectoine biosynthetic genes in V. cholerae, has been proposed to detect changes in the external osmolarity through the ensuing increases in the ionic strength of the cytoplasm (29). In a recently reported study (3), about a quarter of the 440 inspected ectoine/hydroxyectoine gene clusters were associated with a gene that encodes the MarR-type regulator EctR. However, even in the absence of EctR, osmoregulation of ect gene expression persisted in the single microorganism (M. alcaliphilum) in which the interaction of this repressor with the ect regulatory region has been studied (28). In addition, since the majority of ectoine/hydroxyectoine gene clusters are not associated with a regulatory gene (3), these observations imply that the promoter elements and their flanking region itself probably harbor critical information for osmotic control of ect transcription.
Consistent with this concept, we were able to transfer the trait of osmoregulated expression of the ect gene cluster from the ectoine/hydroxyectoine producer P. stutzeri A1501 (17), an organism that lacks CosR and EctR (3, 67), into the surrogate host E. coli, which does not synthesize ectoines naturally (68). While osmoregulation of several heterologous ect gene clusters in E. coli has already been reported (18, 23, 59), no detailed molecular analysis of this remarkable process has so far been pursued. We were able to show that the ect promoter from P. stutzeri A1501 responds to a true osmotic signal and requires the buildup of an osmotically effective gradient across the cytoplasmic membrane for enhanced transcriptional activity (Fig. 2A). We ruled out the involvement of the general stress-responsive alternative sigma factor RpoS, of the nucleoid-associated protein H-NS, of the two-component regulatory system EnvZ/OmpR, and of the cAMP/CRP complex, regulatory systems that target various osmotically controlled genes of E. coli (71–77), in the osmotic control of the P. stutzeri A1501 ect expression (Table 1).
Perception of the osmotic stress signal by E. coli is finely tuned, because graded increases in sustained high osmolarity led to corresponding linear increases in the lasting transcriptional activity of the ect promoter over a wide range of salinities (from 0 M NaCl to 0.5 M NaCl) (Fig. 2B). This finding implies that the E. coli host strain can sensitively perceive small increases in sustained high environmental osmolarity and is able to process this information genetically/physiologically such that a precise setting of the transcriptional activity of the ect promoter will ensue (Fig. 2B). We consider it highly unlikely that the non-ectoine/hydroxyectoine-producing enterobacterium E. coli possesses specific protein- or RNA-based osmoregulatory circuits that would allow it to control the promoter of a heterologous ect biosynthetic gene cluster derived from a plant-root-associated soil bacterium in such a finely tuned fashion. As a working hypothesis, our data therefore suggest that osmoregulation of the ect promoter in the heterologous E. coli host is an inherent feature of the promoter elements and their flanking regions (Fig. 4). It remains to be determined how RNA polymerase, perhaps with contributions of the intracellular ion and compatible solute pools (Fig. 2C) (90, 91) and increases in global and local DNA supercoiling (92, 93), can afford the exquisitely sensitive transcriptional response to high osmolarity that we observed for the ect promoter (Fig. 2B).
It is striking how moderate changes in the ect promoter sequence that increase its resemblance to housekeeping Sig70-type promoters of E. coli (78) cause substantially enhanced expression, both in the absence and in the presence of osmotic stress (Fig. 5A and B and 6A, B, and C). Deviations from the consensus Sig70 promoter sequence thus serve to keep ect transcription low under osmotic conditions where the cells do not rely on the synthesis of compatible solutes for growth while simultaneously allowing induction of these types of genes when the cell has to physiologically cope with high environmental osmolarity (94). Essentially all of the ect promoter variants that we have constructed retained osmotic control, at least to a certain degree (Fig. 5B). Surprisingly, even an ect promoter variant in which we adjusted the −35, and −10 regions, and also the spacer length, to those of corresponding elements present in the consensus Sig70-type promoter (78) (e.g., Mut 15) (Fig. 5A) still remains responsive to high salinity (Fig. 6C). It therefore follows from our analysis that DNA sequence determinants for osmotic control of ect gene expression reside not only in the canonical promoter elements. Through deletion analysis and the study of reporter fusions, we have delineated all DNA sequences required in cis for osmoregulation of the ect promoter to a 116-bp DNA fragment. It encompasses the canonical −35 and −10 elements, contains 28 bp upstream of the −35 region, and extends from the beginning of the −10 region 58 bp down to codon seven of ectA, the first gene in the ectABCD-ask_ect gene cluster (Fig. 4B). DNA sequences relevant for osmotic control of the ect operon are certainly not present upstream of the −35 element, since their replacement with a fortuitously fused plasmid-derived DNA sequence somewhat related to canonical Sig70-type −35 regions still allows osmotic induction of ect transcription (deletion endpoint K7) (Fig. 3A), albeit at a substantially reduced level of promoter activity. Currently, our data cannot exclude the possibility that elements important for osmoregulation are contained in the 5′ region of the ect transcript or that a small antisense RNA targets the region around the translation initiation site of ectA (Fig. 4B). In any event, the identification of a small DNA fragment (116 bp) directing osmoregulated ect gene expression should allow the development of genetic screens to search for variants of the ect regulatory region that no longer respond to the osmotic cue but that do not simply destroy the promoter.
It is apparent from our mutational analysis of the P. stutzeri A1501 ect promoter that there is a complex interplay between different promoter elements that allow the cell to respond differently in transcriptional activity to the intracellular cue generated though an increase in the environmental osmolarity (Fig. 5). This proposal is reminiscent of data reported by Borowiec and Gralla (95) on the lac promoter from E. coli, where point mutations in the −10, the −35, or the spacer region alter the transcriptional response to DNA supercoiling in a distinct fashion but where the promoter elements then act as a whole to facilitate unwinding of the DNA during open complex formation and thereby set promoter activity. The strength of the intracellular cue that triggers enhanced ect expression is modulated by the accumulation of various types of compatible solutes. Since this also includes ectoine and hydroxyectoine (Fig. 2C), the ectoine/hydroxyectoine producer bacterium is provided with a homeostatic system allowing it to tune down the activity of the ect promoter once a physiologically appropriate cellular adjustment to the external osmolarity has been attained.
A particular intriguing feature of the P. stutzeri A1501 ect promoter is the string of three consecutive CG base pairs in the −10 region, whereas the canonical counterpart of this element in Sig70-type promoters lacks CG pairs altogether (Fig. 3B) (78). Such unusual −10 regions have also been noted in several osmostress-responsive genes in other microorganisms (77, 94). For an understanding of the role possibly played by the three consecutive CG base pairs in the ect −10 region (Fig. 3B), we find it useful to consider the properties of the leu-500 allele of Salmonella enterica serovar Typhimurium. In this allele, a single A-to-G point mutation is introduced into the −10 region of the promoter driving expression of the leu operon. This mutation renders the already unusual −10 element particularly GC rich (depending on the assumed spacer length of the leu promoter, the −10 region is either TGCCAC or GCCACT), strongly impairs leu transcription, and thereby causes leucine auxotrophy (96). Suppressors restoring leucine prototrophy map in topA, the structural gene for DNA topoisomerase I, a key player in setting the overall level of DNA supercoiling in the cell. Furthermore, local changes in negative superhelicity also stimulate leu-500 promoter activity strongly (93, 97). It is thus thought that increases in negative DNA superhelicity aid the RNA polymerase in overcoming the increased energy barrier posed by the GC-rich −10 region of the leu-500 promoter to allow DNA strand separation during open complex formation (78, 93, 97). The degree of DNA supercoiling is an important determinant of bacterial gene expression and growth, and it fluctuates in response to cellular and environmental changes (98, 99). High-osmolarity-instigated increases in negative DNA superhelicity (92) are thus likely to contribute to the unwinding of the CG-rich −10 element of the ectC promoter (Fig. 3B and 4B), thereby stimulating its activity under osmotically unfavorable environmental circumstances (2, 12, 14, 15).
There has been a growing interest in ectoines as stabilizers of macromolecules and cellular structures for various practical applications, and both natural and synthetic microbial cell factories for their production have been developed (1, 32, 48, 49, 59, 66). We designed an ectoine/hydroxyectoine-producing synthetic cell factory by combining optimized ect promoters with an engineered E. coli chassis strain that lacks the ability to synthesize the osmostress protectant trehalose (82, 83) and to import ectoines via the ProP and ProU transport systems (68, 80, 88). By flexibly choosing plasmids with combinations of ectABCD-ask_ect or ectABC(ΔectD)-ask_ect gene clusters and different E. coli host strains, we were able to achieve production and secretion of either pure ectoine (Fig. 9B) or pure hydroxyectoine (Fig. 7C and D and 8B) or a mixture of both compounds (Fig. 9B, D, and E). Our best-performing cell factory yields a total ectoine/hydroxyectoine production titer of about 1.9 g liter−1 within 24 h in baffled shake flask cultures (Fig. 9E). This is probably not the maximal productivity of our synthetic cell factory, since under the evaluated conditions (Table 2), the growth/productivity of the culture is limited by glucose availability but not by the added amount (25 mM) of the ectoine biosynthetic precursor aspartate (Fig. 1A).
Our recombinant cell factory also allows the synthesis of ectoine under nonstressed conditions, but since it is dependent on osmotically stimulated ect promoters, its ectoine/hydroxyectoine production yields strongly increase (by about 10-fold) when the cells are osmotically challenged (Table 2; Fig. 7C and D and 9D and E). However, in comparison with natural ectoine/hydroxyectoine-producing microorganisms, which demand very high salinities (between 1 and 2.5 M NaCl) (1) for good production levels, the cell factory that we describe here requires rather moderate salinities (Fig. 2A and 9D and E), conditions that might be more favorable for fermentation vessels and downstream processing equipment (1, 32). Furthermore, recombinant ectoine/hydroxyectoine cell factories that rely on synthetic promoters to drive the expression of the ect biosynthetic genes in the absence of osmotic stress often require costly inducers, while the osmotic stimulation of ect promoter activity requires only a commodity compound. Another significant advantage of our cell factory is that it does not require osmotic downshifts for the release of the ectoines, and it is designed such that the excreted ectoines cannot be reacquired by the producer cells (Fig. 1B). Others have already described various synthetic ectoine or hydroxyectoine cell factories (for summaries, see references 1, 32, 59, and 66), but we find that the reported performances of these cell factories are rather difficult to compare since the growth and production conditions differ substantially (Table 3).
TABLE 3.
Comparison of the productivities of different recombinant ectoine/hydroxyectoine cell factories
| Compound and host | Origin of ect biosynthetic genes | Titer of ectoines (g liter−1) | Productivity (g liter−1 h−1) | Production yielda | Production system | Reference |
|---|---|---|---|---|---|---|
| Ectoine | ||||||
| C. glutamicum LYS-1 | P. stutzeri A1501 | 6.7 | 0.28 | 36.1 μmol (g DCWb)−1 [5.12 mg (g DCW)−1] | Fed batch, in a fermentor | 63 |
| C. glutamicum | C. salexigens | 22 | 0.32 | NRc | Fed batch, in a fermentor | 62 |
| E. coli DH5α | C. salexigens | 6 | 0.04 | 5 mg (g DCW)−1 | Batch, in a fermentor | 61 |
| E. coli BW25113 | H. elongata | 25.1 | 1.05 | 4,048 mg (g DCW)−1 | Whole-cell catalysis, in a fermentor | 87 |
| E. coli W3110 | H. elongata | 25.1 | 0.84 | 800 mg (g DCW)−1 | Fed batch, in a fermentor | 65 |
| E. coli SK51(pASTI14) | P. stutzeri A1501 | 1.9d | 0.08 | 1,749 mg (g DCW)−1 | Shake flasks | This study |
| 5-Hydroxyectoine | ||||||
| E. coli DH5α | P. stutzeri DSM5190 | NR | NR | 500 μmol (g DCW)−1 [79.1 mg (g DCW)−1] | Shake flasks | 59 |
| H. polymorpha | H. elongata | 2.8 | 365 μmol (g DCW)−1 [57.7 mg (g DCW)−1] | Fed batch, in a fermentor | 64 |
Values in brackets were recalculated by us on the basis of the molecular mass of ectoine/hydroxyectoine.
DCW, dry cell weight.
NR, not reported.
Mixture of ectoine and hydroxyectoine (see Fig. 9E).
The recombinant cell factory described here secretes most of the newly synthesized ectoines into the growth medium (Fig. 9B and E). It shares this feature with other synthetic cell factories that rely on microbial chassis strains that do not produce ectoines naturally (61–65, 87). Investigations into the release of hydroxyectoine from a recombinant E. coli strain expressing the ectD gene from P. stutzeri A1501 under the control of the tet promoter (85) demonstrated that this process was not mediated, in the absence of an osmotic downshock, by any of the mechanosensitive channels (MscL, MscS, and MscM) operating in E. coli (100). In our recombinant cell factory, ectoines were efficiently secreted in strains lacking the ectoine/hydroxyectoine uptake systems ProU and ProP (68, 80, 88) (Fig. 8B), thereby firmly ruling out the possibility that these importers are somehow involved in the release of ectoines as well. Data on the release/excretion of different types of compatible solutes have been reported for cells when the natural producer bacteria lack the corresponding import systems. Examples are the release of glycine betaine synthesized from the precursor choline by E. coli (101), glucosylglycerol from Synechocystis sp. strain PCC6803 (102), ectoine from H. elongata (53), and proline by Bacillus subtilis (103). A cycle of synthesis, release, and recapture might thus help osmotically stressed cells to fine-tune the steady-state concentration of compatible solutes, and hence of turgor, in response to cellular and environmentally imposed temporary constraints and cellular imbalances during growth and cell division (103, 104).
Efflux systems for various compounds (105), in particular for amino acids, are well known to exist in microorganisms (106). While passive diffusion of ectoine and hydroxyectoine across the cytoplasmic membrane remains a possibility to explain the accumulation of ectoines in the growth medium of our cell factory (Fig. 9B and E), data reported by Jebbar et al. (68) point to the existence of a yet-unrecognized ectoine efflux system(s) in E. coli. Such a system may also be used for the release of types of compatible solutes that E. coli actually synthesizes naturally, e.g., glycine betaine from the precursor choline or trehalose (84, 101). Previously published data (53, 103) and our findings on the secretion of substantial amounts of ectoines from an E. coli cell factory lacking the ProU and ProP osmolyte importers (Fig. 9B and E) might aid in the setup of a genetic screening procedure to identify potential efflux systems for ectoines.
MATERIALS AND METHODS
Chemicals and reagents.
Ectoine and 5-hydroxyectoine that were used as standards in high-performance liquid chromatography (HPLC) analysis were kindly provided by bitop AG (Witten, Germany). Acetonitrile (HPLC grade) was obtained from VWR International GmbH (Darmstadt, Germany). All other chemicals were purchased from Serva Electrophoresis GmbH (Heidelberg, Germany), Sigma-Aldrich (St. Louis, MO, USA), and Carl Roth GmbH (Karlsruhe, Germany). All compatible solutes used in this study were from laboratory stocks (107). The colorimetric substrate for β-galactosidase enzyme activity assay, o-nitrophenyl-β-d-galactopyranoside (ONPG), was obtained from Serva Electrophoresis GmbH (Heidelberg, Germany).
Media and growth conditions.
All E. coli strains and P. stutzeri A1501 (67) were routinely maintained on Luria-Bertani (LB) agar plates and propagated in liquid LB medium (108). When the E. coli strains used contained a recombinant plasmid, chloramphenicol (30 μg ml−1) or kanamycin (50 μg ml−1) was added to the growth medium. For experiments involving the production of ectoine and hydroxyectoine or the measurement of β-galactosidase activity, E. coli strains were grown in minimal medium A (MMA) (108) supplemented with 0.5% (wt/vol) glucose as the carbon source, 1 mM MgSO4, and 3 mM thiamine. When indicated, 25 mM sodium aspartate was added to the modified MMA to provide extra resources of the precursor for the biosynthesis of ectoines (17, 18, 20). The osmolarity of the growth medium was routinely adjusted by adding various concentrations of NaCl from a 5 M stock solution, as specified for the individual experiments. The osmolarity values of these media were determined with a vapor pressure osmometer (Vapor Pressure 5500; Wescor, Inc., UT). Shake flask cultures of E. coli strains were incubated at 37°C in a shaking water bath set to 220 rpm. Typically, a 100-ml baffled Erlenmeyer flask filled with 20 ml of growth medium was used to propagate the cultures.
Growth of E. coli strain SK51 harboring different plasmids (pASTI1 or pASTI9) leading to the synthesis of ectoines and the empty cloning vector (pHSG575) used for their construction was monitored within a time frame of 24 h in a microplate reader (Biotek Epoch2, Winooski, VT, USA) using a 48-well microplate. Cultures (500 μl) were inoculated with a preculture of strain SK51 harboring various plasmids that had been grown in MMA overnight at 37°C to an optical density at 578 nm (OD578) of 0.15 and were then incubated in MMA or MMA with 0.4 M NaCl for 24 h with double orbital shaking (800 circles min−1) at 37°C. Optical densities of the cultures were measured once per hour.
To determine the dry weight of the formed biomass, 200-ml cultures of E. coli strain SK51(pASTI1) (in 2-liter baffled Erlenmeyer flasks) were inoculated with a preculture grown in MMA overnight at 37°C to an OD578 of 0.15; the cultures were grown in MMA or in MMA containing 0.4 M NaCl for 24 h in an aerial shaker (set to 200 rpm) at 37°C. Fifty-milliliter samples of the cultures were collected in weighed Falcon tubes, and the cells were then harvested by centrifugation (16,000 × g for 10 min) in an Eppendorf tabletop centrifuge. The supernatant was discarded, and the cells were washed once with 20 ml distilled water. The cell pellet was dried overnight at 70°C. Falcon tubes with dried cell pellets were weighed and a dry cell weight (DCW) of ∼0.25 g per OD578 unit was calculated.
Bacterial strains and genetic construction of E. coli mutants.
P. stutzeri strain A1501 (67) was kindly provided by C. Elmerich (Institute Pasteur, Paris, France). E. coli K-12 strain FF4169 [(otsA::Tn10)1] is a derivative of strain MC4100 (109) and is deficient in the synthesis of the stress protectant trehalose (83). MC4100 is also the parent of strains BK32 [Δ(proP)2 proU+], MKH17 [proP+ Δ(proU::spc)608 (Spcr)], and MKH13 [Δ(proP)2 Δ(proU::spc)608 (Spcr)], which carry in various combinations defects in the genes encoding the ProP or ProU compatible solute uptake systems (80, 110). Strains RH76 (Δcya-851) (111), RH90 (rpoS359::Tn10) (112), PD32 (hns-206::Apr) (113), and LC30 (ompR::Tn10) (from T. J. Silhavy) are also derivatives of MC4100. The genotypes of these E. coli strains are listed in Table 4.
TABLE 4.
E. coli strains used in this study
| Straina | Genotype | Reference or source |
|---|---|---|
| MC4100 | F− Δ(argF-lac)U169 araD139 rpsL150 relA1 flbB5301 deoC1 ptsF25 rbsR | 116 |
| RH90 | F− MC4100 rpoS359::Tn10 | 112 |
| PD32 | F− MC4100 hns-206::Apr | 113 |
| RH76 | F− MC4100 Δcya-851 | 111 |
| LC30 | F− MC4100 ompR::Tn10 | T. J. Silhavy |
| FF4169 | F− MC4100 otsA1::Tn10 | 83 |
| MKH17 | F− MC4100 Δ(proU::spc)608 | 110 |
| BK32 | F− MC4100 Δ(putPA)101 Δ(proP)2 | 110 |
| MKH13 | F− MC4100 Δ(putPA)101 Δ(proP)2 Δ(proU::spc)608 | 110 |
| SK51 | F− MC4100 Δ(putPA)101 Δ(proP)2 Δ(proU::spc)608 otsA1::Tn10 | This study |
| LC6 | F− MC4100 Δ(putPA)101 Δ(proP)2 otsA1::Tn10 | This study |
| LC7 | F− MC4100 Δ(putPA)101 Δ(proU::spc)608 otsA1::Tn10 | This study |
Strains SK51, LC6, and LC7 were constructed by transducing the otsA1::Tn10 gene disruption mutation with P1vir into strains MKH13, BK32, and MKH17, respectively.
Standard genetic methods such as phage P1vir-mediated transduction were performed as described previously (108). For the construction of E. coli strains SK51, LC6, and LC7, a P1vir phage lysate was prepared on cells of strain FF4169 [(otsA::Tn10)1] and was then used to transduce the defective trehalose synthesis gene into the otsBA+ E. coli strains MKH13, BK32, and MKH17 (Table 4) by selecting for tetracycline-resistant colonies on LB agar plates containing 15 μg ml−1 of the antibiotic. Representative colonies were picked and purified by restreaking several times on tetracycline-containing LB agar plates; the cells were then tested for their inability to produce trehalose in response to osmotic stress (83) using a kit purchased from Megazyme (Wicklow, Ireland).
Construction of recombinant plasmids.
To monitor the transcription of the ectoine biosynthetic operon (ectABCD-ask_ect) of P. stutzeri A1501 (17, 67), a lacZ reporter gene fusion was constructed. To this end, a 1,057-bp DNA fragment starting upstream of ectA and carrying 396 bp of the ectB coding region (Fig. 3A) was amplified from chromosomal DNA of P. stutzeri A1501 using the DNA primers EctABCD_ask_fw(SpeI) and LacZectB_rev(HindIII) (Table 5). Both the PCR fragment and the broad-host-range plasmid pBBR1MCS-2-lacZ (114) were digested with SpeI and HindIII prior to ligation. The resulting ect-lacZ reporter plasmid was pGJK4 (Table 6). In a similar fashion, a series of ect-lacZ reporter plasmids was constructed by systematically shortening the 5′ region in front of ectA using a set of custom-synthesized DNA primers (Table 5) and the LacZectB_rev(HindIII) DNA primer to provide the same ectB/lacZ fusion junction present in plasmid pGJK4; the resulting plasmids are listed in Table 6, and their physical structures are shown in Fig. 3A and 4A. All these plasmids carry the same fusion junction between the ectB and lacZ genes (in codon 88 of ectB), and this leads to the production of a hybrid EctB-LacZ protein whose translation is dependent on the ectB ribosome-binding site. The ectB and lacZ genetic material in this gene fusion is connected by a 48-base-pair DNA region derived from the polylinker of the pBBR1MCS-2-lacZ cloning vector that inserts a linker peptide of 16 amino acids between the N-terminal EctB′ and C-terminal LacZ proteins in the EctB-LacZ hybrid. To shorten the ectA and ectB DNA sequences present in the ectB-lacZ reporter plasmids, we constructed plasmids pASTI11 and pASTI12 (Fig. 4A), in which we fused codon seven of ectA to the lacZ coding region, thereby causing the synthesis of a hybrid EctA-LacZ protein. Again, a 48-base-pair region derived from the polylinker of the pBBR1MCS-2-lacZ cloning vector inserts a 16-amino-acid peptide between the N-terminal EctA′ and C-terminal LacZ proteins in the EctA-LacZ hybrid. The translation of the hybrid ectA-lacZ gene fusion is dependent on the ectA ribosome-binding site.
TABLE 5.
DNA primers used in this study
| Plasmid | Primer name | Primer sequencea |
|---|---|---|
| pGJK4 | EctABCD_ask_fw(SpeI) | AAAAAACTAGTCCGTCAGTCAGAGAACTTCTGACG |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pNST22 (K1) | LacZ_ectB_K2(BamHI) | AAAAGGATCCCTGAATTCAAATTGCATTTCGGTG |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pNST21 (K2) | LacZ_ectB_K1(BamHI) | AAAAGGATCCGGCAAGCTGACCACCGCAATAC |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pNST26 (K3) | LacZ_ectB_K6(BamHI) | AAAAGGATCCCACAGAAACATTCTGCGCGC |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pNST27 (K4) | LacZ_ectB_K7(BamHI) | AAAAGGATCCCTGCGCGCCAGCATAGTTATC |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pNST28 (K5) | LacZ_ectB_K8(BamHI) | AAAAGGATCCGCGGGTTTCAGCGGCATATAC |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pNST29 (K6) | LacZ_ectB_K9(BamHI) | AAAAGGATCCCGCAAACTTCCCATATTCCGTTAC |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pNST40 (K7) | LacZ_ectB_K11(BamHI) | AAAAGGATCCACTTGTGCGGATATGCCTACCCT |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pNST41 (K8) | LacZ_ectB_K13(BamHI) | AAAAGGATCCCCACCGTAATGCTCCGTCGC |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pNST39 (K9) | LacZ_ectB_K10(BamHI) | AAAAGGATCCCGTTACATGGAAACCACCATCTCG |
| LacZectB_rev(HindIII) | AAAAAAGCTTGAAGGTTTCGAGGAAACGCTC | |
| pASTI11 | KlacZ-for | AAGCTTATCGATACCGTCGACCTC |
| K11ectAshort-rev | CATTACGGTGGGGAAACTCAAAACAAT | |
| pASTI12 | KlacZ-for | AAGCTTATCGATACCGTCGACCTC |
| K11ectAshort-rev | CATTACGGTGGGGAAACTCAAAACAAT | |
| pPH1 (Mut 1) | Mut_35_TTzuGG_for | GTTACATGGGggGAGAAACTTGTGC |
| Mut_35_TTzuGG_rev | GGAATATGGGAAGTTTGC | |
| pPH7 (Mut2) | Mut_10_TzuC_for | ATGCCTACCCcAAAAAGGAATTC |
| Mut_10_TzuC_rev | ATCCGCACAAGTTTCTCAAC | |
| pPH4 (Mut3) | Mut_Spacer_Del3_for | GGATATGCCTACCCTAAAAAG |
| Mut_Spacer_Del3_rev | ACAAGTTTCTCAACCCATG | |
| pPH2 (Mut4) | Mut_Spacer_Del1_for | CGGATATGCCTACCCTAAAAAG |
| Mut_Spacer_Del1_rev | ACAAGTTTCTCAACCCATGTAAC | |
| pPH3 (Mut5) | Mut_Spacer_Del2_for | TGCCTACCCTAAAAAGGAATTC |
| Mut_Spacer_Del2_rev | ATCCGCACAAGTTTCTCAAC | |
| pPH17 (Mut6) | Mut_Spacer_In1_for | tATATGCCTACCCTAAAAAG |
| Mut_Spacer_In1_rev | CCGCACAAGTTTCTCAAC | |
| pPH5 (Mut7) | Mut_10_CzuT_for | GATATGCCTAtCCTAAAAAGGAATTC |
| Mut_10_CzuT_rev | CGCACAAGTTTCTCAACC | |
| pPH14 (Mut8) | Mut_10_1. CzuA_for | GATATGCCTAaCCTAAAAAGG |
| Mut_10_1. CzuA_rev | CGCACAAGTTTCTCAACC | |
| pPH15 (Mut9) | Mut_10_2. CzuT_for | ATATGCCTACtCTAAAAAGGAATTC |
| Mut_10_2. CzuT_rev | CCGCACAAGTTTCTCAAC | |
| pPH16 (Mut10) | Mut_10_3. CzuT_for | TATGCCTACCtTAAAAAGGAATTC |
| Mut_10_3. CzuT_rev | TCCGCACAAGTTTCTCAAC | |
| pPH6 (Mut11) | Mut_10_CCzuAA_for | ATATGCCTACaaTAAAAAGGAATTCAATCAAC |
| Mut_10_CCzuAA_rev | CCGCACAAGTTTCTCAAC | |
| pPH8 (Mut12) | Mut_35_perfekt_for | CATGGGTTGAcAAACTTGTGC |
| Mut_35_perfekt_rev | TAACGGAATATGGGAAGTTTG | |
| pPH9 (Mut13) | Mut_10_perfekt_for | GATATGCCTAtaaTAAAAAGGAATTCAATCAACAAC |
| Mut_10_perfekt_rev | CGCACAAGTTTCTCAACC | |
| pPH10 (Mut14) | Mut_10_perfekt_for | GATATGCCTAtaaTAAAAAGGAATTCAATCAACAAC |
| Mut_10_perfekt_rev | CGCACAAGTTTCTCAACC | |
| pPH11 (Mut15) | Mut_Spacer_Del1_for | CGGATATGCCTACCCTAAAAAG |
| Mut_Spacer_Del1_rev | ACAAGTTTCTCAACCCATGTAAC | |
| pPH12 (Mut16) | Mut_Spacer_Del2_for | TGCCTACCCTAAAAAGGAATTC |
| Mut_Spacer_Del2_rev | ATCCGCACAAGTTTCTCAAC | |
| pPH13 (Mut17) | Mut_Spacer_Del3_for | GGATATGCCTACCCTAAAAAG |
| Mut_Spacer_Del3_rev | ACAAGTTTCTCAACCCATG | |
| pASTI16 (Mut18) | Del_TG_for | CTACCCTAAAAAGGAATTC |
| Del_TG_rev | CATATCCGCACAAGTTTC | |
| pASTI17 (Mut19) | pGJK4_GGGTA_for | ACGCAAACTTgggtaATTCCGTTACATGGGTTGAGAA |
| pGJK4_GGGTA_rev | ACTTGTGCTGTATATGCCGCTGA | |
| pLC68 | Pstutz_EctClu_Blunt_for | CCGCAATACACAGAAACATTCTG |
| Pstutz_EctClu_Blunt_rev | CTTGCAGCCATGGTCCTC | |
| pASTI1 | Mut_35_perfekt_for | CATGGGTTGAcAAACTTGTGC |
| Mut_35_perfekt_rev | TAACGGAATATGGGAAGTTTG | |
| pLC75 | EctD_KO_for | CAGTATCTCTGAGGCGATGG |
| EctD_KO_rev | GTCGGCTTGCATAGGGTTC | |
| pASTI9 | Mut_35_perfekt_for | CATGGGTTGAcAAACTTGTGC |
| Mut_35_perfekt_rev | TAACGGAATATGGGAAGTTTG | |
| pASTI14 | plac_kaputt_for | AATTCCACACccCATACGAGCC |
| plac_kaputt_rev | GTGAGCGGATAACAATTTC |
Mutations are indicated with bold lowercase letters.
TABLE 6.
Plasmids used in this study
| Plasmid | Description and mutationsa | Reference or source |
|---|---|---|
| pBBR1MCS-2-lacZ | Broad-host-range cloning vector for lacZ reporter gene fusions; Kanr | 114 |
| pHSG575 | Low-copy-number cloning vector; Catr | 89 |
| pGJK4 | pBBR1MCS-2-lacZ with 1,057-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pNST22 (K1) | pGJK4 derivative with 1,010-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pNST21 (K2) | pGJK4 derivative with 970-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pNST26 (K3) | pGJK4 derivative with 953-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pNST27 (K4) | pGJK4 derivative with 942-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pNST28 (K5) | pGJK4 derivative with 918-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pNST29 (K6) | pGJK4 derivative with 892-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pNST40 (K7) | pGJK4 derivative with 856-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pNST41 (K8) | pGJK4 derivative with 786-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pNST39 (K9) | pGJK4 derivative with 472-bp fragment starting upstream of ectA and including ectA and 396 bp of ectB; Kanr | This study |
| pASTI11 | pNST29 derivative with 116-bp fragment starting upstream of ectA and including 18 bp of ectA; Kanr | This study |
| pASTI12 | pGJK4 derivative with 285-bp fragment starting upstream of ectA and including 18 bp of ectA; Kanr | This study |
| pPH1 (Mut 1) | pGJK4 derivative with a mutation in the −35 region of the ect promoter; TTGAGA→GGGAGA; Kanr | This study |
| pPH7 (Mut2) | pGJK4 derivative with a mutation in the −10 region of the ect promoter, TACCCT→TACCCC; Kanr | This study |
| pPH4 (Mut3) | pGJK4 derivative with a 2-bp (CG) deletion in the spacer of the ect promoter; Kanr | This study |
| pPH2 (Mut4) | pGJK4 derivative with a 1-bp (G) deletion in the spacer of the ect promoter; Kanr | This study |
| pPH3 (Mut5) | pGJK4 derivative with a 1-bp (A) deletion in the spacer of the ect promoter; Kanr | This study |
| pPH17 (Mut6) | pGJK4 derivative with a 1-bp (T) insertion in the spacer of the ect promoter; Kanr | This study |
| pPH5 (Mut7) | pGJK4 derivative with a mutation in the −10 region of the ect promoter, TACCCT→TATCCT; Kanr | This study |
| pPH14 (Mut8) | pGJK4 derivative with a mutation in the −10 region of the ect promoter, TACCCT→TAACCT; Kanr | This study |
| pPH15 (Mut9) | pGJK4 derivative with a mutation in the −10 region of the ect promoter, TACCCT→TACTCT; Kanr | This study |
| pPH16 (Mut10) | pGJK4 derivative with a mutation in the −10 region of the ect promoter, TACCCT→TACCTT; Kanr | This study |
| pPH6 (Mut11) | pGJK4 derivative with a mutation in the −10 region of the ect promoter, TACCCT→TACAAT; Kanr | This study |
| pPH8 (Mut12) | pGJK4 derivative with a mutation in the −35 region of the ect promoter, TTGAGA→TTGACA; Kanr | This study |
| pPH9 (Mut13) | pGJK4 derivative with a mutation in the −10 region of the ect promoter, TACCCT→TATAAT; Kanr | This study |
| pPH10 (Mut14) | pPH8 derivative with a mutation in the −10 region of the ect promoter, TACCCT→TATAAT; Kanr | This study |
| pPH11 (Mut15) | pPH10 derivative with a 1-bp deletion (G) in the spacer of the ect promoter leading to a perfect Sig70-type promoter; Kanr | This study |
| pPH12 (Mut16) | pPH10 derivative with a 1 bp deletion (A) in the spacer of the ect promoter leading to a perfect Sig70-type promoter; Kanr | This study |
| pPH13 (Mut17) | pPH10 derivative with a 2-bp deletion (GC) in the spacer of the ect promoter leading to a 16-bp spacer; Kanr | This study |
| pASTI16 (Mut18) | pGJK4 derivative with a 1-bp (C) deletion in the spacer of the ect promoter leading to a TG motif at position −16 at the ect promoter; Kanr | This study |
| pASTI17 (Mut19) | pGJK4 derivative with a mutation in the inverted repeat upstream of the ect promoter, CCCAT→GGGTA; Kanr | This study |
| pLC68 | pHSG575 containing a 5,065-bp fragment of the ectoine gene cluster from P. stutzeri A1501; Catr | This study |
| pASTI1 | pLC68 derivative with a mutation in the −35 region of the ect promoter, TTGAGA→TTGACA; Catr | This study |
| pLC75 | Derivative of plasmid pASTI1 carrying an 885-bp in-frame deletion of ectD; Catr | This study |
| pASTI9 | Derivative of plasmid pASTI1 carrying a mutant ect promoter changed to the consensus sequence of a Sig70-type promoter; Catr | This study |
| pASTI14 | a-derivative of plasmid pASTI1 with an inactivated lac promoter, TATGTT→TATGGG; Catr | This study |
Mutations are indicated with bold letters. Kanr, the plasmid confers resistance to the antibiotic kanamycin; Catr, the plasmid confers resistance to the antibiotic chloramphenicol.
A series of ect promoter mutants was constructed by site-directed mutagenesis using the ectB-lacZ reporter plasmid pGJK4 as the DNA template, custom-synthesized DNA primers (Table 5), and the Q5 site-directed mutagenesis (Q5 Site) kit (New England BioLabs GmbH, Frankfurt, Germany). This resulted in the construction of plasmids pPH1 to pPH17 and plasmids pASTI16 and pASTI17 (Table 6).
For the heterologous production of ectoine and 5-hydroxyectoine in E. coli, a 5,065-bp DNA fragment containing the natural ect promoter and the complete ectoine gene cluster (ectABCD-ask_ect) from P. stutzeri A1501 (17, 67) was cloned into the low-copy-number plasmid pHSG575 (Cmr) (89). The corresponding fragment was amplified from genomic DNA of P. stutzeri A1501 using the primers Pstutz_EctClu_Blunt_for and Pstutz_EctClu_Blunt_rev (Table 5). The resulting DNA fragment was phosphorylated with the T4 polynucleotide kinase (Thermo Fisher Scientific Inc., Waltham, MA, USA) and was blunt-end-cloned into the linearized (by cutting with SmaI) and dephosphorylated (with FastAP; Thermo Fisher Scientific Inc., Waltham, MA, USA) DNA of pHSG575 (89). The resulting plasmid was pLC68 (Fig. 1B; Table 6). After verification of the DNA sequence of the entire cloned ectABCD-ask_ect operon (17), selected point mutations were introduced via site-directed mutagenesis (by using the Q5 Site kit) into the −10 and −35 regions of the ect promoter and into the spacer region that separates these promoter elements; the resulting plasmids were pASTI1 (with a point mutation in the −35 region) and pASTI9 (with a perfect Sig70-type promoter with a 17-bp spacing) (Table 6). Plasmid pASTI1 was used to delete the ectD gene from the ect operon by using the primers EctD_KO_for and EctD_KO_rev (Table 5); the resulting plasmid was pLC75 (Table 6). The backbone of the pHSG575 cloning vector carries a lac promoter (89) that will generate an antisense transcript in plasmid pLC68 (Fig. 1B) and its derivatives. To inactivate the lac promoter on plasmid pASTI1, a consecutive change of two base pairs (see Fig. 9C) was introduced into the −10 region of the lac promoter via site-directed mutagenesis (by using the Q5 Site kit) using the primers plac_kaputt_for and plac_kaputt_rev (Table 5). The resulting plasmid was pASTI14 (Table 6). The correct DNA sequences of the chromosomal inserts in all plasmids constructed in this study (Table 6) were verified by DNA sequence analysis (Eurofins MWG Operon, Ebersberg, Germany); all DNA primers (Table 5) used in this study were custom synthesized by Microsynth AG (Balgach, Switzerland).
Cultivation of recombinant E. coli strains for the production of ectoines.
For the heterologous production of ectoine and hydroxyectoine in E. coli, various strains were transformed with different plasmids carrying either the wild-type ectABCD-ask_ect gene cluster or mutant derivatives of it that either carried alterations in the ect promoter sequence or lacked the ectD gene (Table 6). Twenty-milliliter cultures (in 100-ml baffled Erlenmeyer flasks) of these plasmid-carrying E. coli strains were cultivated in MMA with the addition of different concentrations of NaCl and sodium aspartate and with chloramphenicol (30 μg ml−1) to select for the recombinant plasmids. The cultures were inoculated with a preculture grown in MMA overnight at 37°C to an OD587 of 0.15 and incubated for 24 or 48 h (as indicated in the individual experiments) in a shaking water bath (set to 220 rpm and 37°C). Two-milliliter samples were collected from these cultures, and the cells were harvested by centrifugation (16,000 × g for 10 min) in an Eppendorf tabletop centrifuge. The supernatant was separated from the pellet, and both samples were stored at −20°C until further analysis.
Detection and quantification of ectoines.
Low-molecular-weight compounds of the E. coli cell pellets were extracted with 20% ethanol. For this purpose, the cell pellets were resuspended in 1 ml of 20% ethanol and were shaken for 1 h. After centrifugation at 16,000 × g (4°C for 30 min) to remove cell debris, the ethanolic extracts were transferred into fresh Eppendorf tubes, and the ethanol was removed by evaporation (at 55°C for 20 h). The resulting dried material was resuspended in 100 μl of distilled water, and insoluble material was removed by centrifugation (16,000 × g at 4°C for 30 min). The extracted samples and the cell-free supernatant derived from the corresponding cell cultures were diluted 10-fold with distilled water and acetonitrile (the final concentration of acetonitrile was 50%) and analyzed for their ectoine/5-hydroxyectoine content by isocratic high-performance liquid chromatography (HPLC) (24). For these measurements, we employed an Agilent 1260 Infinity LC system (Agilent, Waldbronn, Germany) and a Grom-Sil Amino 1PR column (Grom, Rottenburg-Hailfingen, Germany) essentially as described previously (24) with the exception that a 1260 Infinity diode array detector (DAD) (Agilent) was used instead of the previously used UV/visible detector system (24, 85). The ectoine and 5-hydroxyectoine contents of HPLC-analyzed samples were quantified using the OpenLAB software suite (Agilent). Standard curves for the calculation of the ectoine and 5-hydroxyectoine concentrations were determined with commercially available samples.
Quantification of intracellular trehalose.
Cells of E. coli strains MC4100, FF4169, SK51, LC6, and LC7 (Table 4) were grown in MMA without and with the addition of 0.4 M NaCl until they reached an OD578 of 1. One-milliliter samples of the cultures were harvested by centrifugation, and trehalose was extracted from the cells using the ethanolic extraction protocol used for the recovery of ectoines. The dried pellet was resuspended in 100 μl distilled water (dH2O). The trehalose content of the samples was determined with a commercially available kit from Megazyme (Wicklow, Ireland) according to the manufacturer's protocol. The otsBA+ strain MC4100 accumulated 0.65 μM trehalose per OD578 unit when it was grown in MMA without NaCl and 25.5 μM trehalose per OD578 unit when it was grown in MMA in the presence of 0.4 M NaCl. Strains FF4169, SK51, LC6, and LC7, all of which carry the (otsA::Tn10)1 mutation (Table 4), contained between 0.56 μM and 1.5 μM trehalose per OD578 unit regardless of the absence or presence of 0.4 M NaCl in the growth medium.
Detection and quantification of aspartate.
Two-milliliter samples of 200-ml cultures of strain E. coli SK51(pASTI1) (grown for 24 h in 2-liter baffled Erlenmeyer flasks) were collected by centrifugation (16,000 × g for 10 min) in an Eppendorf tabletop centrifuge. The supernatant was separated from the pellet and stored at −20°C until further analysis. For the analysis of the aspartate content, the samples were diluted to an estimated aspartate concentration of 0.5 to 1 mM with distilled water, and 1-μl aliquots were derivatized using 2 μl of 10 mM 9-fluorenylmethoxy carbonyl (FMOC). Excessive FMOC was removed by adding 3 μl of 40 mM 1-adamantylamine hydrochloride (ADAM) to the samples. Distilled water (494 μl) was then added to this solution and analyzed for its aspartate content by isocratic high-performance liquid chromatography (HPLC). For these measurements, we employed an Agilent 1260 Infinity LC system (Agilent, Waldbronn, Germany) and a Gemini 5-μm C18 110-Å column (Phenomenex, Torrance, CA, USA). Two different solvents (solvent A, 40 mM NaH2PO4, pH 7.8; solvent B, 55% HPLC-grade distilled water plus 45% acetonitrile plus 0.1% trifluoroacetic acid [TFA]) were used to create the following gradient: 0 min, 0% solvent B; 2 min, 50% solvent B; 11 min, 70% solvent B; 16 min, 100% solvent B; 24 min, 100% solvent B; 24.5 min, 0% solvent B; and 27 min, 0% solvent B. The flow rate was set to 1 ml min−1, and the temperature for the measurements of the samples was 40°C. The aspartate content of the analyzed samples was quantified using the OpenLAB software suite (Agilent). Standard curves for the calculation of the aspartate concentrations were determined with commercially available samples.
Determination of extracellular glucose concentrations.
Two-milliliter samples of various cell cultures were collected by centrifugation (16,000 × g for 10 min) in an Eppendorf tabletop centrifuge. The supernatants were stored at −20°C until further analysis. The glucose content of these samples was determined with a commercially available kit from Megazyme (Wicklow, Ireland) according to the manufacturer's protocol. Standard curves for the calculation of the glucose concentrations were determined with commercially available reference material.
β-Galactosidase reporter enzyme assays.
The ect-lacZ reporter gene plasmids (Table 6) were introduced into various E. coli strains via routine DNA transformation procedures. Twenty-milliliter cultures (in 100-ml Erlenmeyer flasks) of these plasmid-bearing strains were grown in a shaking water bath at 37°C in MMA with or without the addition of NaCl and in the presence of various types of compatible solutes until the cultures reached an OD587 of between 1.8 and 2. Two-milliliter samples of the cultures were harvested by centrifugation (16,000 × g for 10 min) and assayed for β-galactosidase activity as described previously (108) using the chromogenic compound ONPG as the substrate. The β-galactosidase enzyme activity is expressed in Miller units (108). Plasmid pBBR1MCS-2-lacZ (114), which was used to construct the ect-lacZ reporter fusions, was used as a control, and its presence in strains with the entire lacIZYA operon deleted (Table 4) yielded a background activity of 0 to 2 Miller units.
SDS gels and Western blot analysis.
The production of the β-galactosidase protein in E. coli strains harboring pBBR1MCS-2 ect-lacZ reporter fusions was analyzed by SDS-polyacrylamide gel electrophoresis as described previously (81). For this purpose, the cultures were harvested when they reached late log phase (OD587 between 1.8 and 2.2); the volume of the harvested cell culture (2 ml) was adjusted such that the harvested cells of the various cultures possessed the same OD587. For Western blot analysis of these samples, cell pellets were resuspended in 350 μl of 2× loading buffer (0.5 ml β-mercaptoethanol, 0.25 ml 0.1% bromophenol blue, 4 ml 10% SDS, 5.3 ml 2× sample buffer [consisting of 21 ml 0.5 M Tris-HCl [pH 6.8], 0.4% SDS, and 34 ml glycerol per liter]) and incubated for 5 min at 95°C. After centrifugation of the disrupted cells in an Eppendorf tabletop centrifuge (5 min, 16,000 × g), 30-μl portions of the supernatants of these samples were applied to a 7% SDS-polyacrylamide gel. The electrophoretically separated proteins from the E. coli cell extracts were transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-FL; purchased from Merck Millipore Ltd., Darmstadt, Germany) via semidry blotting. Western blotting of the transferred proteins was performed with a primary rabbit antiserum directed against a hybrid ProW-LacZ fusion protein. This antiserum recognizes the inner membrane component (ProW) of the E. coli ProU ABC transport system (80) and the β-galactosidase portion of the hybrid protein (115). The immune complex formed on the blotting membrane was detected with a secondary IRDye 800CW fluorescently labeled donkey anti-rabbit IgG antibody (purchased from Li-Cor, Lincoln, NE, USA). Incubation with the ProW-LacZ antibody was performed in Odyssey blocking buffer (purchased from Li-Cor) with 0.2% Tween. For the incubation of the blotting membrane with the second antibody, 0.01% SDS was additionally added to the bathing solution. Signals were detected by fluorescence imaging at 800 nm using an imager system (Odyssey Fc; Li-Cor, Lincoln, NE, USA) by exposing the membrane for 2 min.
Supplementary Material
ACKNOWLEDGMENTS
We thank Oliver Dähn and Florian Kindinger for their participation during the exploratory phase of the project, Susanne Kneip for help with strain constructions, and Alexandra Richter for help with the measurement of the aspartate contents of supernatants of various bacterial cultures by HPLC analysis. We are very grateful to Jutta Gade and Jochen Sohn for their dedicated and expert technical assistance during part of this project. We thank bitop AG (Witten, Germany) for the generous gifts of ectoine and hydroxyectoine reference samples. We appreciate the kind donation of E. coli strains by Arne Strom (University of Trondheim, Norway), Regine Hengge (Humboldt University, Berlin, Germany), and Tom Silhavy (Princeton University, USA), of the P. stutzeri A1501 strain by Claudine Elmerich (Institute Pasteur, Paris, France), and of the reporter plasmid pBBR1MCS2-lacZ by Jürgen Lassak (Ludwig Maximilian University, Munich, Germany). We thank Katrin Ritter for a sample of the ProW-LacZ antiserum. We are grateful to Vickie Koogle for her expert help in the language editing of our manuscript, and we thank our colleague Tamara Hoffmann for insightful discussions.
This work was financially supported by grants from the Deutsche Forschungsgemeinschaft (DFG) through the SFB 987 and by the LOEWE excellence program of the state of Hessen (via the Center for Synthetic Microbiology). L.C. was supported by a Ph.D. fellowship from the International Max Planck Research School for Environmental, Cellular and Molecular Microbiology (IMPRS-Mic, Marburg).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01772-17.
REFERENCES
- 1.Pastor JM, Salvador M, Argandona M, Bernal V, Reina-Bueno M, Csonka LN, Iborra JL, Vargas C, Nieto JJ, Canovas M. 2010. Ectoines in cell stress protection: uses and biotechnological production. Biotechnol Adv 28:782–801. doi: 10.1016/j.biotechadv.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Kempf B, Bremer E. 1998. Uptake and synthesis of compatible solutes as microbial stress responses to high osmolality environments. Arch Microbiol 170:319–330. doi: 10.1007/s002030050649. [DOI] [PubMed] [Google Scholar]
- 3.Widderich N, Höppner A, Pittelkow M, Heider J, Smits SH, Bremer E. 2014. Biochemical properties of ectoine hydroxylases from extremophiles and their wider taxonomic distribution among microorganisms. PLoS One 9:e93809. doi: 10.1371/journal.pone.0093809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Widderich N, Czech L, Elling FJ, Könneke M, Stöveken N, Pittelkow M, Riclea R, Dickschat JS, Heider J, Bremer E. 2016. Strangers in the archaeal world: osmostress-responsive biosynthesis of ectoine and hydroxyectoine by the marine thaumarchaeon Nitrosopumilus maritimus. Environ Microbiol 18:1227–1248. doi: 10.1111/1462-2920.13156. [DOI] [PubMed] [Google Scholar]
- 5.Harding T, Brown MW, Simpson AG, Roger AJ. 2016. Osmoadaptative strategy and its molecular signature in obligately halophilic heterotrophic protists. Genome Biol Evol 8:2241–2258. doi: 10.1093/gbe/evw152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Street TO, Bolen DW, Rose GD. 2006. A molecular mechanism for osmolyte-induced protein stability. Proc Natl Acad Sci U S A 103:13997–14002. doi: 10.1073/pnas.0606236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Street TO, Krukenberg KA, Rosgen J, Bolen DW, Agard DA. 2010. Osmolyte-induced conformational changes in the Hsp90 molecular chaperone. Protein Sci 19:57–65. doi: 10.1002/pro.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borwankar T, Rothlein C, Zhang G, Techen A, Dosche C, Ignatova Z. 2011. Natural osmolytes remodel the aggregation pathway of mutant huntingtin exon 1. Biochemistry 50:2048–2060. doi: 10.1021/bi1018368. [DOI] [PubMed] [Google Scholar]
- 9.Bourot S, Sire O, Trautwetter A, Touze T, Wu LF, Blanco C, Bernard T. 2000. Glycine betaine-assisted protein folding in a lysA mutant of Escherichia coli. J Biol Chem 275:1050–1056. doi: 10.1074/jbc.275.2.1050. [DOI] [PubMed] [Google Scholar]
- 10.Cayley S, Lewis BA, Record MT Jr. 1992. Origins of the osmoprotective properties of betaine and proline in Escherichia coli K-12. J Bacteriol 174:1586–1595. doi: 10.1128/jb.174.5.1586-1595.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stadmiller SS, Gorensek-Benitez AH, Guseman AJ, Pielak GJ. 2017. Osmotic shock induced protein destabilization in living cells and its reversal by glycine betaine. J Mol Biol 429:1155–1161. doi: 10.1016/j.jmb.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bremer E, Krämer R. 2000. Coping with osmotic challenges: osmoregulation through accumulation and release of compatible solutes, p 79–97. In Storz G, Hengge-Aronis R (ed), Bacterial stress responses. ASM Press, Washington DC, USA. [Google Scholar]
- 13.Wood JM. 2011. Bacterial osmoregulation: a paradigm for the study of cellular homeostasis. Annu Rev Microbiol 65:215–238. doi: 10.1146/annurev-micro-090110-102815. [DOI] [PubMed] [Google Scholar]
- 14.Wood JM, Bremer E, Csonka LN, Krämer R, Poolman B, van der Heide T, Smith LT. 2001. Osmosensing and osmoregulatory compatible solute accumulation by bacteria. Comp Biochem Physiol A Mol Integr Physiol 130:437–460. doi: 10.1016/S1095-6433(01)00442-1. [DOI] [PubMed] [Google Scholar]
- 15.Csonka LN. 1989. Physiological and genetic responses of bacteria to osmotic stress. Microbiol Rev 53:121–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo CC, Bonner CA, Xie G, D'Souza M, Jensen RA. 2009. Cohesion group approach for evolutionary analysis of aspartokinase, an enzyme that feeds a branched network of many biochemical pathways. Microbiol Mol Biol Rev 73:594–651. doi: 10.1128/MMBR.00024-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stöveken N, Pittelkow M, Sinner T, Jensen RA, Heider J, Bremer E. 2011. A specialized aspartokinase enhances the biosynthesis of the osmoprotectants ectoine and hydroxyectoine in Pseudomonas stutzeri A1501. J Bacteriol 193:4456–4468. doi: 10.1128/JB.00345-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bestvater T, Louis P, Galinski EA. 2008. Heterologous ectoine production in Escherichia coli: by-passing the metabolic bottle-neck. Saline Sys 4:12. doi: 10.1186/1746-1448-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reshetnikov AS, Khmelenina VN, Trotsenko YA. 2006. Characterization of the ectoine biosynthesis genes of haloalkalotolerant obligate methanotroph “Methylomicrobium alcaliphilum 20Z.” Arch Microbiol 184:286–297. [DOI] [PubMed] [Google Scholar]
- 20.Ono H, Sawada K, Khunajakr N, Tao T, Yamamoto M, Hiramoto M, Shinmyo A, Takano M, Murooka Y. 1999. Characterization of biosynthetic enzymes for ectoine as a compatible solute in a moderately halophilic eubacterium, Halomonas elongata. J Bacteriol 181:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bursy J, Pierik AJ, Pica N, Bremer E. 2007. Osmotically induced synthesis of the compatible solute hydroxyectoine is mediated by an evolutionarily conserved ectoine hydroxylase. J Biol Chem 282:31147–31155. doi: 10.1074/jbc.M704023200. [DOI] [PubMed] [Google Scholar]
- 22.Höppner A, Widderich N, Lenders M, Bremer E, Smits SHJ. 2014. Crystal structure of the ectoine hydroxylase, a snapshot of the active site. J Biol Chem 289:29570–29583. doi: 10.1074/jbc.M114.576769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis P, Galinski EA. 1997. Characterization of genes for the biosynthesis of the compatible solute ectoine from Marinococcus halophilus and osmoregulated expression in Escherichia coli. Microbiology 143:1141–1149. doi: 10.1099/00221287-143-4-1141. [DOI] [PubMed] [Google Scholar]
- 24.Kuhlmann AU, Bremer E. 2002. Osmotically regulated synthesis of the compatible solute ectoine in Bacillus pasteurii and related Bacillus spp. Appl Environ Microbiol 68:772–783. doi: 10.1128/AEM.68.2.772-783.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vargas C, Argandona M, Reina-Bueno M, Rodriguez-Moya J, Fernandez-Aunion C, Nieto JJ. 2008. Unravelling the adaptation responses to osmotic and temperature stress in Chromohalobacter salexigens, a bacterium with broad salinity tolerance. Saline Sys 4:14. doi: 10.1186/1746-1448-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwibbert K, Marin-Sanguino A, Bagyan I, Heidrich G, Lentzen G, Seitz H, Rampp M, Schuster SC, Klenk HP, Pfeiffer F, Oesterhelt D, Kunte HJ. 2011. A blueprint of ectoine metabolism from the genome of the industrial producer Halomonas elongata DSM 2581T. Environ Microbiol 13:1973–1994. doi: 10.1111/j.1462-2920.2010.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calderon MI, Vargas C, Rojo F, Iglesias-Guerra F, Csonka LN, Ventosa A, Nieto JJ. 2004. Complex regulation of the synthesis of the compatible solute ectoine in the halophilic bacterium Chromohalobacter salexigens DSM 3043T. Microbiology 150:3051–3063. doi: 10.1099/mic.0.27122-0. [DOI] [PubMed] [Google Scholar]
- 28.Mustakhimov II, Reshetnikov AS, Glukhov AS, Khmelenina VN, Kalyuzhnaya MG, Trotsenko YA. 2010. Identification and characterization of EctR1, a new transcriptional regulator of the ectoine biosynthesis genes in the halotolerant methanotroph Methylomicrobium alcaliphilum 20Z. J Bacteriol 192:410–417. doi: 10.1128/JB.00553-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shikuma NJ, Davis KR, Fong JNC, Yildiz FH. 2013. The transcriptional regulator, CosR, controls compatible solute biosynthesis and transport, motility and biofilm formation in Vibrio cholerae. Environ Microbiol 15:1387–1399. doi: 10.1111/j.1462-2920.2012.02805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao Z, Deng W, Li S, He J, Ren S, Huang W, Lu Y, Zhao G, Cai Z, Wang J. 2015. GlnR-mediated regulation of ectABCD transcription expands the role of the GlnR regulon to osmotic stress management. J Bacteriol 197:3041–3307. doi: 10.1128/JB.00185-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bursy J, Kuhlmann AU, Pittelkow M, Hartmann H, Jebbar M, Pierik AJ, Bremer E. 2008. Synthesis and uptake of the compatible solutes ectoine and 5-hydroxyectoine by Streptomyces coelicolor A3(2) in response to salt and heat stresses. Appl Environ Microbiol 74:7286–7296. doi: 10.1128/AEM.00768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kunte HJ, Lentzen G, Galinski E. 2014. Industrial production of the cell protectant ectoine: protection, mechanisms, processes, and products. Curr Biotechnol 3:10–25. doi: 10.2174/22115501113026660037. [DOI] [Google Scholar]
- 33.Kuhlmann AU, Hoffmann T, Bursy J, Jebbar M, Bremer E. 2011. Ectoine and hydroxyectoine as protectants against osmotic and cold stress: uptake through the SigB-controlled betaine-choline-carnitine transporter-type carrier EctT from Virgibacillus pantothenticus. J Bacteriol 193:4699–4708. doi: 10.1128/JB.05270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Estepa R, Argandona M, Reina-Bueno M, Capote N, Iglesias-Guerra F, Nieto JJ, Vargas C. 2006. The ectD gene, which is involved in the synthesis of the compatible solute hydroxyectoine, is essential for thermoprotection of the halophilic bacterium Chromohalobacter salexigens. J Bacteriol 188:3774–3784. doi: 10.1128/JB.00136-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma Y, Wang Q, Xu W, Liu X, Gao X, Zhang Y. 2017. Stationary phase-dependent accumulation of ectoine is an efficient adaptation strategy in Vibrio anguillarum against cold stress. Microbiol Res 205:8–18. doi: 10.1016/j.micres.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 36.Zaccai G, Bagyan I, Combet J, Cuello GJ, Deme B, Fichou Y, Gallat FX, Galvan Josa VM, von Gronau S, Haertlein M, Martel A, Moulin M, Neumann M, Weik M, Oesterhelt D. 2016. Neutrons describe ectoine effects on water H-bonding and hydration around a soluble protein and a cell membrane. Sci Rep 6:31434. doi: 10.1038/srep31434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hahn MB, Solomun T, Wellhausen R, Hermann S, Seitz H, Meyer S, Kunte HJ, Zeman J, Uhlig F, Smiatek J, Sturm H. 2015. Influence of the compatible solute ectoine on the local water structure: implications for the binding of the protein G5P to DNA. J Phys Chem B 119:15212–15220. doi: 10.1021/acs.jpcb.5b09506. [DOI] [PubMed] [Google Scholar]
- 38.Knapp S, Ladenstein R, Galinski EA. 1999. Extrinsic protein stabilization by the naturally occurring osmolytes beta-hydroxyectoine and betaine. Extremophiles 3:191–198. doi: 10.1007/s007920050116. [DOI] [PubMed] [Google Scholar]
- 39.Tanne C, Golovina EA, Hoekstra FA, Meffert A, Galinski EA. 2014. Glass-forming property of hydroxyectoine is the cause of its superior function as a dessication protectant. Front Microbiol 5:150. doi: 10.3389/fmicb.2014.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schnoor M, Voss P, Cullen P, Boking T, Galla HJ, Galinski EA, Lorkowski S. 2004. Characterization of the synthetic compatible solute homoectoine as a potent PCR enhancer. Biochem Biophys Reses Commu 322:867–872. doi: 10.1016/j.bbrc.2004.07.200. [DOI] [PubMed] [Google Scholar]
- 41.Lippert K, Galinski EA. 1992. Enzyme stabilization by ectoine-type compatible solutes: protection against heating, freezing and drying. Applied Microbial Biotechnology 37:61–65. doi: 10.1007/BF00174204. [DOI] [Google Scholar]
- 42.Manzanera M, Garcia de Castro A, Tondervik A, Rayner-Brandes M, Strom AR, Tunnacliffe A. 2002. Hydroxyectoine is superior to trehalose for anhydrobiotic engineering of Pseudomonas putida KT2440. Appl Environ Microbiol 68:4328–4333. doi: 10.1128/AEM.68.9.4328-4333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manzanera M, Vilchez S, Tunnacliffe A. 2004. High survival and stability rates of Escherichia coli dried in hydroxyectoine. FEMS Microbiol Lett 233:347–352. doi: 10.1111/j.1574-6968.2004.tb09502.x. [DOI] [PubMed] [Google Scholar]
- 44.Kolp S, Pietsch M, Galinski EA, Gutschow M. 2006. Compatible solutes as protectants for zymogens against proteolysis. Biochim Biophys Acta 1764:1234–1242. doi: 10.1016/j.bbapap.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 45.Harishchandra RK, Wulff S, Lentzen G, Neuhaus T, Galla HJ. 2010. The effect of compatible solute ectoines on the structural organization of lipid monolayer and bilayer membranes. Biophys Chem 150:37–46. doi: 10.1016/j.bpc.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 46.Roychoudhury A, Bieker A, Haussinger D, Oesterhelt F. 2013. Membrane protein stability depends on the concentration of compatible solutes—a single molecule force spectroscopic study. Biol Chem 394:1465–1474. doi: 10.1515/hsz-2013-0173. [DOI] [PubMed] [Google Scholar]
- 47.Barth S, Huhn M, Matthey B, Klimka A, Galinski EA, Engert A. 2000. Compatible-solute-supported periplasmic expression of functional recombinant proteins under stress conditions. Appl Environ Microbiol 66:1572–1579. doi: 10.1128/AEM.66.4.1572-1579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lentzen G, Schwarz T. 2006. Extremolytes: natural compounds from extremophiles for versatile applications. Appl Microbiol Biotechnol 72:623–634. doi: 10.1007/s00253-006-0553-9. [DOI] [PubMed] [Google Scholar]
- 49.Graf R, Anzali S, Buenger J, Pfluecker F, Driller H. 2008. The multifunctional role of ectoine as a natural cell protectant. Clin Dermatol 26:326–333. doi: 10.1016/j.clindermatol.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Booth IR. 2014. Bacterial mechanosensitive channels: progress towards an understanding of their roles in cell physiology. Curr Opin Microbiol 18:16–22. doi: 10.1016/j.mib.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sauer T, Galinski EA. 1998. Bacterial milking: a novel bioprocess for production of compatible solutes. Biotechnol Bioeng 57:306–313. doi:. [DOI] [PubMed] [Google Scholar]
- 52.Schulz A, Stöveken N, Binzen IM, Hoffmann T, Heider J, Bremer E. 2017. Feeding on compatible solutes: a substrate-induced pathway for uptake and catabolism of ectoines and its genetic control by EnuR. Environ Microbiol 19:926–946. doi: 10.1111/1462-2920.13414. [DOI] [PubMed] [Google Scholar]
- 53.Grammann K, Volke A, Kunte HJ. 2002. New type of osmoregulated solute transporter identified in halophilic members of the bacteria domain: TRAP transporter TeaABC mediates uptake of ectoine and hydroxyectoine in Halomonas elongata DSM 2581T. J Bacteriol 184:3078–3085. doi: 10.1128/JB.184.11.3078-3085.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fallet C, Rohe P, Franco-Lara E. 2010. Process optimization of the integrated synthesis and secretion of ectoine and hydroxyectoine under hyper/hypo-osmotic stress. Biotechnol Bioeng 107:124–133. doi: 10.1002/bit.22750. [DOI] [PubMed] [Google Scholar]
- 55.Van-Thuoc D, Hashim SO, Hatti-Kaul R, Mamo G. 2013. Ectoine-mediated protection of enzyme from the effect of pH and temperature stress: a study using Bacillus halodurans xylanase as a model. Appl Microbiol Biotechnol 97:6271–6278. doi: 10.1007/s00253-012-4528-8. [DOI] [PubMed] [Google Scholar]
- 56.Schiraldi C, Maresca C, Catapano A, Galinski EA, De Rosa M. 2006. High-yield cultivation of Marinococcus M52 for production and recovery of hydroxyectoine. Res Microbiol 157:693–699. doi: 10.1016/j.resmic.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 57.Onraedt AE, Walcarius BA, Soetaert WK, Vandamme EJ. 2005. Optimization of ectoine synthesis through fed-batch fermentation of Brevibacterium epidermis. Biotechnol Prog 21:1206–1212. doi: 10.1021/bp0500967. [DOI] [PubMed] [Google Scholar]
- 58.Zhang LH, Lang YJ, Nagata S. 2009. Efficient production of ectoine using ectoine-excreting strain. Extremophiles 13:717–724. doi: 10.1007/s00792-009-0262-2. [DOI] [PubMed] [Google Scholar]
- 59.Seip B, Galinski EA, Kurz M. 2011. Natural and engineered hydroxyectoine production based on the Pseudomonas stutzeri ectABCD-ask gene cluster. Appl Environ Microbiol 77:1368–1374. doi: 10.1128/AEM.02124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen W, Zhang S, Jiang PX, Yao J, He YZ, Chen LC, Gui XW, Dong ZY, Tang SY. 2015. Design of an ectoine-responsive AraC mutant and its application in metabolic engineering of ectoine biosynthesis. Metab Eng 30:149–155. doi: 10.1016/j.ymben.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 61.Schubert T, Maskow T, Benndorf D, Harms H, Breuer U. 2007. Continuous synthesis and excretion of the compatible solute ectoine by a transgenic, nonhalophilic bacterium. Appl Environ Microbiol 73:3343–3347. doi: 10.1128/AEM.02482-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez-Garcia F, Ziert C, Risse JM, Wendisch VF. 2017. Improved fermentative production of the compatible solute ectoine by Corynebacterium glutamicum from glucose and alternative carbon sources. J Biotechnol 258:59–69. doi: 10.1016/j.jbiotec.2017.04.039. [DOI] [PubMed] [Google Scholar]
- 63.Becker J, Schäfer R, Kohlstedt M, Harder BJ, Borchert NS, Stöveken N, Bremer E, Wittmann C. 2013. Systems metabolic engineering of Corynebacterium glutamicum for production of the chemical chaperone ectoine. Microb Cell Fact 12:110. doi: 10.1186/1475-2859-12-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eilert E, Kranz A, Hollenberg CP, Piontek M, Suckow M. 2013. Synthesis and release of the bacterial compatible solute 5-hydroxyectoine in Hansenula polymorpha. J Biotechnol 167:85–93. doi: 10.1016/j.jbiotec.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 65.Ning Y, Wu X, Zhang C, Xu Q, Chen N, Xie X. 2016. Pathway construction and metabolic engineering for fermentative production of ectoine in Escherichia coli. Metab Eng 36:10–18. doi: 10.1016/j.ymben.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez-Moya J, Argandona M, Iglesias-Guerra F, Nieto JJ, Vargas C. 2013. Temperature- and salinity-decoupled overproduction of hydroxyectoine by Chromohalobacter salexigens. Appl Environ Microbiol 79:1018–1023. doi: 10.1128/AEM.02774-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yan Y, Yang J, Dou Y, Chen M, Ping S, Peng J, Lu W, Zhang W, Yao Z, Li H, Liu W, He S, Geng L, Zhang X, Yang F, Yu H, Zhan Y, Li D, Lin Z, Wang Y, Elmerich C, Lin M, Jin Q. 2008. Nitrogen fixation island and rhizosphere competence traits in the genome of root-associated Pseudomonas stutzeri A1501. Proc Natl Acad Sci U S A 105:7564–7569. doi: 10.1073/pnas.0801093105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jebbar M, Talibart R, Gloux K, Bernard T, Blanco C. 1992. Osmoprotection of Escherichia coli by ectoine: uptake and accumulation characteristics. J Bacteriol 174:5027–5035. doi: 10.1128/jb.174.15.5027-5035.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoffmann T, Wensing A, Brosius M, Steil L, Völker U, Bremer E. 2013. Osmotic control of opuA expression in Bacillus subtilis and its modulation in response to intracellular glycine betaine and proline pools. J Bacteriol 195:510–522. doi: 10.1128/JB.01505-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brill J, Hoffmann T, Bleisteiner M, Bremer E. 2011. Osmotically controlled synthesis of the compatible solute proline is critical for cellular defense of Bacillus subtilis against high osmolarity. J Bacteriol 193:5335–5346. doi: 10.1128/JB.05490-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weber H, Polen T, Heuveling J, Wendisch VF, Hengge R. 2005. Genome-wide analysis of the general stress response network in Escherichia coli: sigmaS-dependent genes, promoters, and sigma factor selectivity. J Bacteriol 187:1591–1603. doi: 10.1128/JB.187.5.1591-1603.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weber A, Jung K. 2002. Profiling early osmostress-dependent gene expression in Escherichia coli using DNA macroarrays. J Bacteriol 184:5502–5507. doi: 10.1128/JB.184.19.5502-5507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lucht JM, Dersch P, Kempf B, Bremer E. 1994. Interactions of the nucleoid-associated DNA-binding protein H-NS with the regulatory region of the osmotically controlled proU operon of Escherichia coli. J Biol Chem 269:6578–6578. [PubMed] [Google Scholar]
- 74.Pratt LA, Hsing W, Gibson KE, Silhavy TJ. 1996. From acids to osmZ: multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli. Mol Microbiol 20:911–917. doi: 10.1111/j.1365-2958.1996.tb02532.x. [DOI] [PubMed] [Google Scholar]
- 75.Balsalobre C, Johansson J, Uhlin BE. 2006. Cyclic AMP-dependent osmoregulation of crp gene expression in Escherichia coli. J Bacteriol 188:5935–5944. doi: 10.1128/JB.00235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang H, Chong H, Ching CB, Jiang R. 2012. Random mutagenesis of global transcription factor cAMP receptor protein for improved osmotolerance. Biotechnol Bioeng 109:1165–1172. doi: 10.1002/bit.24411. [DOI] [PubMed] [Google Scholar]
- 77.Landis L, Xu J, Johnson RC. 1999. The cAMP receptor protein CRP can function as an osmoregulator of transcription in Escherichia coli. Genes Dev 13:3081–3091. doi: 10.1101/gad.13.23.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feklistov A, Sharon BD, Darst SA, Gross CA. 2014. Bacterial sigma factors: a historical, structural, and genomic perspective. Annu Rev Microbiol 68:357–376. doi: 10.1146/annurev-micro-092412-155737. [DOI] [PubMed] [Google Scholar]
- 79.Voskuil MI, Chambliss GH. 1998. The −16 region of Bacillus subtilis and other Gram-positive bacterial promoters. Nucleic Acids Res 26:3584–3590. doi: 10.1093/nar/26.15.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lucht JM, Bremer E. 1994. Adaptation of Escherichia coli to high osmolarity environments: osmoregulation of the high-affinity glycine betaine transport system ProU. FEMS Microbiol Rev 14:3–20. doi: 10.1111/j.1574-6976.1994.tb00067.x. [DOI] [PubMed] [Google Scholar]
- 81.May G, Faatz E, Lucht JM, Haardt M, Bolliger M, Bremer E. 1989. Characterization of the osmoregulated Escherichia coli proU promoter and identification of ProV as a membrane-associated protein. Mol Microbiol 3:1521–1531. doi: 10.1111/j.1365-2958.1989.tb00138.x. [DOI] [PubMed] [Google Scholar]
- 82.Hengge-Aronis R, Klein W, Lange R, Rimmele M, Boos W. 1991. Trehalose synthesis genes are controlled by the putative sigma factor encoded by rpoS and are involved in stationary-phase thermotolerance in Escherichia coli. J Bacteriol 173:7918–7924. doi: 10.1128/jb.173.24.7918-7924.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Giaever HM, Styrvold OB, Kaasen I, Strom AR. 1988. Biochemical and genetic characterization of osmoregulatory trehalose synthesis in Escherichia coli. J Bacteriol 170:2841–2849. doi: 10.1128/jb.170.6.2841-2849.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boos W, Ehmann U, Bremer E, Middendorf A, Postma P. 1987. Trehalase of Escherichia coli. Mapping and cloning of its structural gene and identification of the enzyme as a periplasmic protein induced under high osmolarity growth conditions. J Biol Chem 262:13212–13218. [PubMed] [Google Scholar]
- 85.Czech L, Stöveken N, Bremer E. 2016. EctD-mediated biotransformation of the chemical chaperone ectoine into hydroxyectoine and its mechanosensitive channel-independent excretion. Microb Cell Fact 15:126. doi: 10.1186/s12934-016-0525-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peters P, Galinski EA, Trüper HG. 1990. The biosyntheis of ectoine. FEMS Microbiol Lett 71:157–162. doi: 10.1111/j.1574-6968.1990.tb03815.x. [DOI] [Google Scholar]
- 87.He YZ, Gong J, Yu HY, Tao Y, Zhang S, Dong ZY. 2015. High production of ectoine from aspartate and glycerol by use of whole-cell biocatalysis in recombinant Escherichia coli. Microb Cell Fact 14:55. doi: 10.1186/s12934-015-0238-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.MacMillan SV, Alexander DA, Culham DE, Kunte HJ, Marshall EV, Rochon D, Wood JM. 1999. The ion coupling and organic substrate specificities of osmoregulatory transporter ProP in Escherichia coli. Biochim Biophys Acta 1420:30–44. doi: 10.1016/S0005-2736(99)00085-1. [DOI] [PubMed] [Google Scholar]
- 89.Takeshita S, Sato M, Toba M, Masahashi W, Hashimoto-Gotoh T. 1987. High-copy-number and low-copy-number plasmid vectors for lacZ alpha-complementation and chloramphenicol- or kanamycin-resistance selection. Gene 61:63–74. doi: 10.1016/0378-1119(87)90365-9. [DOI] [PubMed] [Google Scholar]
- 90.Gralla JD, Huo YX. 2008. Remodeling and activation of Escherichia coli RNA polymerase by osmolytes. Biochemistry 47:13189–13196. doi: 10.1021/bi801075x. [DOI] [PubMed] [Google Scholar]
- 91.Cayley S, Record MT Jr. 2003. Roles of cytoplasmic osmolytes, water, and crowding in the response of Escherichia coli to osmotic stress: biophysical basis of osmoprotection by glycine betaine. Biochemistry 42:12596–12609. doi: 10.1021/bi0347297. [DOI] [PubMed] [Google Scholar]
- 92.Higgins CF, Dorman CJ, Stirling DA, Waddell L, Booth IR, May G, Bremer E. 1988. A physiological role for DNA supercoiling in the osmotic regulation of gene expression in S. typhimurium and E. coli. Cell 52:569–584. doi: 10.1016/0092-8674(88)90470-9. [DOI] [PubMed] [Google Scholar]
- 93.Lilley DM, Higgins CF. 1991. Local DNA topology and gene expression: the case of the leu-500 promoter. Mol Microbiol 5:779–783. doi: 10.1111/j.1365-2958.1991.tb00749.x. [DOI] [PubMed] [Google Scholar]
- 94.Hoffmann T, Bremer E. 2016. Management of osmotic stress by Bacillus subtilis: genetics and physiology, p 657–676. In de Bruijn FJ. (ed), Stress and environmental regulation of gene expression and adaptation in bacteria, vol 1 Wiley-Blackwell Publishers, Hoboken, NJ. [Google Scholar]
- 95.Borowiec JA, Gralla JD. 1987. All three elements of the lac ps promoter mediate its transcriptional response to DNA supercoiling. J Mol Biol 195:89–97. doi: 10.1016/0022-2836(87)90329-9. [DOI] [PubMed] [Google Scholar]
- 96.Gemmill RM, Tripp M, Friedman SB, Calvo JM. 1984. Promoter mutation causing catabolite repression of the Salmonella typhimurium leucine operon. J Bacteriol 158:948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhi X, Dages S, Dages K, Liu Y, Hua ZC, Makemson J, Leng F. 2017. Transient and dynamic DNA supercoiling potently stimulates the leu-500 promoter in Escherichia coli. J Biol Chem 292:14566–14575. doi: 10.1074/jbc.M117.794628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dorman CJ, Dorman MJ. 2016. DNA supercoiling is a fundamental regulatory principle in the control of bacterial gene expression. Biophys Rev 8:209–220. doi: 10.1007/s12551-016-0205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Travers A, Muskhelishvili G. 2005. DNA supercoiling—a global transcriptional regulator for enterobacterial growth? Nat Rev Microbiol 3:157–169. doi: 10.1038/nrmicro1088. [DOI] [PubMed] [Google Scholar]
- 100.Edwards MD, Black S, Rasmussen T, Rasmussen A, Stokes NR, Stephen TL, Miller S, Booth IR. 2012. Characterization of three novel mechanosensitive channel activities in Escherichia coli. Channels (Austin) 6:272–281. doi: 10.4161/chan.20998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lamark T, Styrvold OB, Strom AR. 1992. Efflux of choline and glycine betaine from osmoregulating cells of Escherichia coli. FEMS Microbiol Lett 75:149–154. doi: 10.1111/j.1574-6968.1992.tb05408.x. [DOI] [PubMed] [Google Scholar]
- 102.Mikkat S, Hagemann M. 2000. Molecular analysis of the ggtBCD gene cluster of Synechocystis sp. strain PCC6803 encoding subunits of an ABC transporter for osmoprotective compounds. Arch Microbiol 174:273–282. doi: 10.1007/s002030000201. [DOI] [PubMed] [Google Scholar]
- 103.Hoffmann T, von Blohn C, Stanek A, Moses S, Barzantny S, Bremer E. 2012. Synthesis, release, and recapture of the compatible solute proline by osmotically stressed Bacillus subtilis cells. Appl Environ Microbiol 78:5753–5762. doi: 10.1128/AEM.01040-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Börngen K, Battle AR, Möker N, Morbach S, Marin K, Martinac B, Krämer R. 2010. The properties and contribution of the Corynebacterium glutamicum MscS variant to fine-tuning of osmotic adaptation. Biochim Biophys Acta 1798:2141–2149. doi: 10.1016/j.bbamem.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 105.Bay DC, Turner RJ. 2012. Small multidrug resistance protein EmrE reduces host pH and osmotic tolerance to metabolic quaternary cation osmoprotectants. J Bacteriol 194:5941–5948. doi: 10.1128/JB.00666-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jones CM, Lozada NJH, Pfleger BF. 2015. Efflux systems in bacteria and their metabolic engineering applications. Appl Microbiol Biotechnol 99:9381–9393. doi: 10.1007/s00253-015-6963-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hoffmann T, Bremer E. 2011. Protection of Bacillus subtilis against cold stress via compatible-solute acquisition. J Bacteriol 193:1552–1562. doi: 10.1128/JB.01319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 109.Ferenci T, Zhou Z, Betteridge T, Ren Y, Liu Y, Feng L, Reeves PR, Wang L. 2009. Genomic sequencing reveals regulatory mutations and recombinational events in the widely used MC4100 lineage of Escherichia coli K-12. J Bacteriol 191:4025–4029. doi: 10.1128/JB.00118-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Haardt M, Kempf B, Faatz E, Bremer E. 1995. The osmoprotectant proline betaine is a major substrate for the binding-protein-dependent transport system ProU of Escherichia coli K-12. Mol Gen Genet 246:783–786. doi: 10.1007/BF00290728. [DOI] [PubMed] [Google Scholar]
- 111.Hengge-Aronis R, Fischer D. 1992. Identification and molecular analysis of glgS, a novel growth-phase-regulated and rpoS-dependent gene involved in glycogen synthesis in Escherichia coli. Mol Microbiol 6:1877–1886. doi: 10.1111/j.1365-2958.1992.tb01360.x. [DOI] [PubMed] [Google Scholar]
- 112.Lange R, Hengge-Aronis R. 1991. Identification of a central regulator of stationary-phase gene expression in Escherichia coli. Mol Microbiol 5:49–59. doi: 10.1111/j.1365-2958.1991.tb01825.x. [DOI] [PubMed] [Google Scholar]
- 113.Dersch P, Schmidt K, Bremer E. 1993. Synthesis of the Escherichia coli K-12 nucleoid-associated DNA-binding protein H-NS is subjected to growth-phase control and autoregulation. Mol Microbiol 8:875–889. doi: 10.1111/j.1365-2958.1993.tb01634.x. [DOI] [PubMed] [Google Scholar]
- 114.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. 4 new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 115.Ritter K. 1996. Struktur und Funktion der Cytoplasmamembran-Komponente (ProW) des Bindeprotein-abhängigen Glycin-Betain Transportsystems ProU aus Eschericia coli. Philipps-Universität Marburg, Marburg, Germany. [Google Scholar]
- 116.Casadaban MJ. 1976. Transposition and fusion of lac genes to selected promoters in Escherichia coli using bacteriophage lambda and bacteriophage-Mu. J Mol Biol 104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.