

ABSTRACT
Over 90% of the world's population is persistently infected with Epstein-Barr virus. While EBV does not cause disease in most individuals, it is the common cause of acute infectious mononucleosis (AIM) and has been associated with several cancers and autoimmune diseases, highlighting a need for a preventive vaccine. At present, very few primary, circulating EBV genomes have been sequenced directly from infected individuals. While low levels of diversity and low viral evolution rates have been predicted for double-stranded DNA (dsDNA) viruses, recent studies have demonstrated appreciable diversity in common dsDNA pathogens (e.g., cytomegalovirus). Here, we report 40 full-length EBV genome sequences obtained from matched oral wash and B cell fractions from a cohort of 10 AIM patients. Both intra- and interpatient diversity were observed across the length of the entire viral genome. Diversity was most pronounced in viral genes required for establishing latent infection and persistence, with appreciable levels of diversity also detected in structural genes, including envelope glycoproteins. Interestingly, intrapatient diversity declined significantly over time (P < 0.01), and this was particularly evident on comparison of viral genomes sequenced from B cell fractions in early primary infection and convalescence (P < 0.001). B cell-associated viral genomes were observed to converge, becoming nearly identical to the B95.8 reference genome over time (Spearman rank-order correlation test; r = −0.5589, P = 0.0264). The reduction in diversity was most marked in the EBV latency genes. In summary, our data suggest independent convergence of diverse viral genome sequences toward a reference-like strain within a relatively short period following primary EBV infection.
IMPORTANCE Identification of viral proteins with low variability and high immunogenicity is important for the development of a protective vaccine. Knowledge of genome diversity within circulating viral populations is a key step in this process, as is the expansion of intrahost genomic variation during infection. We report full-length EBV genomes sequenced from the blood and oral wash of 10 individuals early in primary infection and during convalescence. Our data demonstrate considerable diversity within the pool of circulating EBV strains, as well as within individual patients. Overall viral diversity decreased from early to persistent infection, particularly in latently infected B cells, which serve as the viral reservoir. Reduction in B cell-associated viral genome diversity coincided with a convergence toward a reference-like EBV genotype. Greater convergence positively correlated with time after infection, suggesting that the reference-like genome is the result of selection.
KEYWORDS: EBV, DNA sequencing, viral diversity, phylogenetic analysis, genome analysis, Epstein-Barr virus
INTRODUCTION
With a worldwide infection rate greater than 90%, Epstein-Barr Virus (EBV) ranks as one of the most successful human pathogens. The majority of primary EBV infections occur early in life and are asymptomatic; primary infection in older children or young adults frequently results in acute infectious mononucleosis (AIM) (1, 2). EBV establishes a persistent infection in human hosts, characterized by nearly continuous lytic infection in the oropharynx and latent infection of memory B cells (3). While EBV does not cause disease in the majority of persistently infected individuals, it has been associated with several cancers (Hodgkin's and Burkitt's lymphomas and nasopharyngeal and gastric carcinomas), as well as autoimmune diseases (systemic lupus erythematosus and multiple sclerosis) (4–6). Although a vaccine is clearly needed, efforts to date have failed to provide the level of sterilizing protection required to prevent EBV infection and persistence (7–12).
Effective vaccine development benefits from a detailed understanding of circulating primary viral genome sequences and proteins. It has been generally assumed that double-stranded DNA (dsDNA) viral genomes such as EBV are relatively stable due to the proofreading capacity of eukaryotic DNA polymerases which restrict mutation rates to approximately 10−9 substitutions/site/year (13). However, several recent reports from our lab and others have demonstrated that even dsDNA viral genes and genomes (e.g., cytomegalovirus [CMV] and EBV) display a measurable amount of variability within an infected host at any time after infection (14–16). The origin of this variability is unknown; it could be present in the initial viral inoculum, arise early during initial rounds of infection and replication in epithelial cells or seeding of the B cell compartment, or evolve as a result of immune escape over the course of chronic infection. In addition, different levels of variation were detected among EBV genes within similar cohorts (15, 16). Sequencing full-length genomes directly from patient samples over the course of primary through persistent EBV infection is necessary to resolve these questions.
Following primary infection, EBV establishes lifelong persistence characterized by a latent reservoir of memory B cells and nearly continuous lytic replication in the nasopharynx and tonsils. To date, direct amplification and sequencing of EBV genomes from peripheral blood cells (BC) and saliva or oral wash (OW) samples has been hampered by the small proportion of viral genomes present compared to those of human and other genomic DNA. The recent development of strategies to selectively enrich EBV genomes from human samples and to remove contaminating human genome reads have facilitated EBV sequencing directly from patient samples (17, 18).
Recently, several groups have demonstrated considerable global genomic diversity over the full-length sequences of greater than 100 EBV genomes (17–25). However, the bulk of these studies were conducted using diseased tissue or transformed primary cells (18, 19, 23–26). Either of these two conditions (disease or tissue culture passage) may have imposed selective pressures on the viral genomes, raising the question of how representative they are of circulating and transmitted viral genomes (27, 28).
In this study, we have used methods to enrich and amplify EBV genomes from peripheral blood B cell and oral wash samples obtained from 10 young adults presenting with acute infectious mononucleosis (AIM) and in convalescence (5 to 11 months post-primary infection [CONV]). Genetic variability was detected in primary infection across the length of the EBV genome and was most pronounced in latency genes, consistent with previous reports. Most importantly, we observed a significant reduction in diversity in circulating B cell-derived viral sequences in convalescence, with convergence toward a reference-like EBV genome (B95.8). To our knowledge, this is the largest body of EBV sequencing data obtained directly ex vivo that clearly demonstrates early genomic diversity and convergence of EBV genome sequences over the course of primary EBV infection.
RESULTS
Enrichment of EBV genomes facilitates sequencing directly from patient samples.
As noted in previous reports, the small proportion of viral genomes present in even purified B cell fractions presents a challenge for generating complete full-length EBV next-generation sequencing libraries from infected patient samples (29). In the absence of any purification or enrichment strategy, the majority of the sequencing reads align to the human genome. The recent development of biochemical strategies to remove contaminating human genome reads has facilitated EBV sequencing directly from patient samples (17, 18, 26).
Using an approach similar to one successfully employed to generate overlapping RNA probes against the larger and more complex genome of Plasmodium falciparum (30), EBV genomes were enriched from patient B cell or oral wash samples using biotinylated RNA probes based on templates from type I reference EBV strains, B95.8 and Akata. Probe-genome hybrids were immobilized on NeutrAvidin-coated magnetic beads, and stringent washes were employed to remove nonhybridized, contaminating sequences. This markedly reduced the human genomic material in each sample, thus increasing EBV-specific reads in each library, with commensurate increases in both depth and breadth of coverage (31).
As proof of principle, the above enrichment protocol was used to capture and resequence the EBV Akata bacterial artificial chromosome (BAC) mixed with 1.0 × 105 copies of human genome isolated from EBV-negative cultured cells. Following successful genome enrichment and sequencing, paired reads with 99 to 100% presumed base call accuracy, as indicated by FastQC statistics, were mapped to the B95.8 reference genome (NCBI accession number NC_007605.1) for alignment and genome assembly. The B95.8 reference genome was selected as a scaffold for our data set based on recent reports suggesting that it is a representative type I EBV genome (18). Additionally, several genomes were assembled by scaffolding to the Mutu reference genome (NCBI accession number KC207814), and the full-length sequences were compared to those scaffolded against B95.8; no important differences were noted, and, using a phylogenetic approach, it was shown that these genomes consistently branched identically to their paired B95.8 assemblies. The resequenced EBV genome aligned to the Akata reference genome deposited in NCBI (KC207813.1) to 96.7% identity, with the loss of alignment coinciding with previously described areas of low sequencing depth and high sequence repetition (18).
Utilizing these techniques, 40 EBV genomes were successfully enriched and sequenced from all 10 patients (two time points and two tissue types per patient) at an average depth of 5,420× across the full-length genome. A consensus sequence for each of the 40 sequenced samples was compiled and designated by patient code, location of sample collection (BC or OW), and sample collection time (AIM or CONV). Alignment of all 40 patient sequences to the B95.8 reference genome indicated completeness of coverage (Fig. 1). Many single nucleotide polymorphisms (SNPs) and identical bases were located throughout the alignment, as demonstrated by the height and color (green to light green) in the identity plot. Several regions in the alignment, denoted by blue boxes, were representative of EBV repeat regions, which typically cannot be assembled when Illumina sequencing is used; previously reported sequencing information obtained by either Sanger sequencing or sequencing of high-copy-number viral genomes isolated from transformed cell lines was used (32). These regions were masked for all subsequent analysis. Comparison of fragment per kilobase million (FPKM) values, taken as a measure of coverage per EBV open reading frame (ORF), demonstrated comparable levels of sequencing depth, regardless of tissue of origin or time of collection (see Table S2 in the supplemental material). Also highlighted is a region containing the LF3 ORF; this ORF, located between bases 142000 and 145000 of the reference genome, was not present in the original sequencing of B95.8 and has been added to the reference using EBV sequences from Raji cells (33, 34). The function of this region has yet to be determined, with some studies suggesting a coding function and others suggesting a noncoding function (25, 35). In addition, although this region is present in EBV associated with tumors, it is conspicuously absent in the only other published full-length, primary EBV genome taken directly from saliva of an otherwise healthy individual, as well as in our samples (18).
FIG 1.

Overview of the features of the EBV genome and sequence diversity: alignment of 40 full-length genome sequences. The identity plot derived from this alignment is shown at the top and is colored as follows: dark green, 100% identity; light green, 30 to <100% identity; red, <30% identity. The black bar below represents a consensus sequence drawn from the alignment of the genomes of 40 EBV strains. Below the identity plot, EBV coding regions are shown in dark red, with arrows indicating the direction of the reading frame. The blue boxes indicate regions of the multigenome alignment that failed to align properly. The bold red box indicates the duplication in the LF3 ORF of the B95.8 reference genome that is missing in our patient cohort. These boxed regions were masked for phylogenetic and genetic distance calculations.
Primary EBV genomes derived from cohort donors were exclusively type I.
EBV is segregated into two subtypes based largely on the sequences of four EBV genes, specifically, EBNA2, EBNA3A, EBNA3B, and EBNA3C (36). The type I strain, predominantly found in the Western Hemisphere as well parts of Southeast Asia, is represented by reference genomes B95.8, Akata, Mutu, C666-1, M81, GD1, and GD2 (19, 24, 25, 32, 37, 38). Type II, which is codominant with type I and until recently was believed to be restricted in distribution largely to sub-Saharan Africa, is represented by the reference genomes of AG876, Jijoye, and Wewak (18, 39, 40). A midpoint-rooted maximum-likelihood phylogenetic tree that includes these type I and II reference genomes demonstrated that all EBV genomes sequenced from our patient cohort segregated with type I strains (Fig. 2). None of the patient sequences branched with type II reference genomes, indicating that our cohort was infected exclusively with type I EBV.
FIG 2.

Forty full-length EBV viral genomes sequenced from 10 patients align almost exclusively with type I reference strains. EBV sequences are labeled by source (peripheral blood B cells, BC; oral wash, OW) and timing of sample (AIM, within 2 weeks of diagnosis of acute infectious mononucleosis; CONV, at least 6 months postinfection [highlighted with black dots]) during primary EBV infection. A maximum-likelihood, midpoint-rooted phylogeny is shown, with each patient represented independently by color. EBV type II sequences are boxed. All other sequences are derived from EBV type I. Sequences from the cohort are most closely related to B95.8 and Mutu isolates (circled) and least related to Asian EBV type I isolates (GD1, Akata, GD2, M81, C666-1, and saliva sample from a healthy Asian donor), shown at the top of the tree.
In addition to subtypes, strong geographic segregation has been observed for type I EBV strains (41). The Western reference strain, B95.8, was isolated from marmoset B cells transformed by EBV from an elderly patient diagnosed with AIM following multiple blood transfusions, while an African type I reference, Mutu, was taken from the tissue of a Kenyan Burkitt lymphoma patient (25, 42). Several sequences of Asian origin have been described: Akata, which was derived from a Japanese Burkitt lymphoma patient; GD-1, GD-2, C666-1, and M81, which were sequenced from nasopharyngeal carcinoma tissue and cell lines originating from Chinese patients (19, 24, 25, 37, 38). Also, a recently reported full-length primary EBV sequence isolated from saliva of a healthy individual believed by the authors to be of Asian descent clustered with this group (Fig. 2, healthy donor saliva) (18). All patient sequences clustered closer to the prototypical type I reference, B95.8, or to Mutu (blue ovals), the type I sequence presumed to originate from Kenya, with the E1578 AIM sequences displaying the most diversity, as is evident by the long branch lengths in comparison to those of other subject viruses.
EBV genomes sequenced from AIM patients display nucleotide variation across the full genome.
A recent report by Palser et al. provided sequence information for 83 different global EBV genomes and showed the highest levels of genetic variation, including an increase in nonsynonymous substitutions, in open reading frames (ORFs) associated with latent EBV genes (18). Only one primary saliva specimen was analyzed; the remainder of EBV sequences were derived primarily from lymphoblastoid cell lines (LCL) or tumors.
Access to a cohort of young adults experiencing primary EBV infection provided the opportunity to sequence circulating EBV genomes directly from peripheral blood B cells and oral wash samples over the course of primary infection and transition to persistence. High levels of variation were detected in latent EBV genes, including EBNA1 (BKRF1), EBNA2, EBNA3A, EBNA3B, EBNA3C, LMP1, and LMP2, in agreement with previously published reports (Fig. 3A) (18). Synonymous and nonsynonymous substitutions were evaluated across the EBV genome ORFs for all 40 sequences. For the analyses, we segregated the EBV genome into latent, early lytic (early), and late lytic (late) genes and compared the mean levels of synonymous and nonsynonymous nucleotide changes within these gene groups (Fig. 3B). Nonsynonymous variation was significantly higher in the latency genes than in either early or late lytic genes (Mann-Whitney test; nonsynonymous latent versus nonsynonymous early lytic, P = 0.0004; nonsynonymous latent versus late lytic, P = 0.0002). At the gene level, only BCRF2 (tegument), EBNA-LP (transcription cofactor), EBNA1/BKRF1 (DNA-binding, genome replication), and BNLF2a (interleukin-10 [IL-10] homologue) demonstrated higher ratios of nonsynonymous/synonymous (dN/dS) changes. All remaining EBV ORFs displayed higher levels of synonymous than nonsynonymous variation, suggesting increased conservation at the protein level, a finding that was supported by dN/dS analysis (Fig. 3C). Latent genes had a statistically higher dN/dS ratio as a group than early or late genes (Mann-Whitney test; latent versus early, P = 0.0011; latent versus late, P = 0.002); the mean ratio was 0.69, suggesting some positive selection pressure, most likely due to the increased number of nonsynonymous changes in EBNA-LP and EBNA1.
FIG 3.

The majority of variations in EBV ORFs are synonymous amino acid changes to viral genes required for latent infection. (A) Sites of synonymous and nonsynonymous nucleotide variations present in patient samples organized by gene. The height of the bar represents nucleotide changes per 1,000 base pairs. (B) Both synonymous and nonsynonymous sites are increased in the latent gene subset compared to these sites in the early and late gene groups. (C) Ratio of nonsynonymous to synonymous nucleotide changes (dN/dS) suggests some positive selective pressure on genes in the latent subset. The bars represent the mean accumulated variation determined in the 40 sequenced patient samples. Genes were annotated based on B95.8 (NCBI accession number NC_007605.1) and allotted to latent, late, and early groups based on a previously established convention. (Mann-Whitney test; *, P < 0.0001).
Intrahost and intracompartment EBV whole-genome diversity varies among patients and sample time points.
Having detected variation across the EBV genome in our patient cohort, we next investigated intrahost genetic diversity for each patient sequence population (Fig. 4A). Substantial differences in variation were identified across our patient pool. One patient (E1503) displayed very little intrahost variation across compartments (blood and oral wash) or time. In contrast, consensus sequences from six patients (E1563, E1577, E1578, E1583, E1587, and E1590) showed higher intrapatient variability. Next, pairwise genetic distances were calculated for consensus sequences grouped by time point and compartment (BC AIM, OW AIM, BC CONV, and OW CONV) (Fig. 4B). Mean levels of genetic variation were similar in viral sequences from the B cell fraction and oral wash samples during AIM. However, the overall genetic variation was significantly reduced (P < 0.01) in both compartments during CONV. We then calculated the Tamura-Nei (TN93) genetic distance (see Materials and Methods) between each consensus sequence and the B98.5 reference genome (Fig. 4C). Again we observed significant differences between the mean pairwise distance during AIM and CONV (Wilcoxon matched-pairs test; BC AIM versus BC CONV, P < 0.001; OW AIM versus OW CONV, P < 0.01). This analysis suggested that genetic diversity was decreasing over time and, in addition, converging to a genotype that was more closely related to the B95.8 reference genome.
FIG 4.
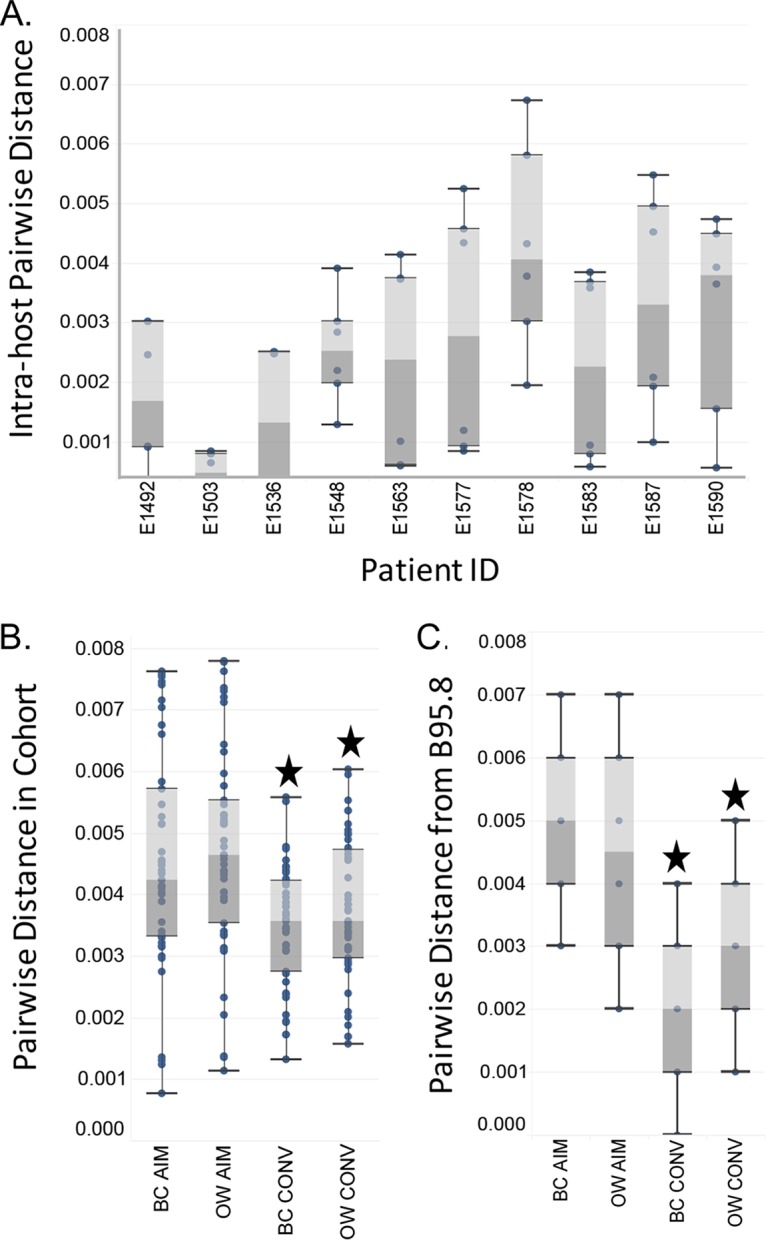
Diversity in EBV genomes is detectable intrahost and varies by compartment and time. (A) Intrahost pairwise diversity for all four sequences generated from each patient is plotted as a box-and-whisker plot showing the median (between light and dark gray), upper and lower bins, and outliers. Whiskers extend to 1.5 times the interquartile range. Each of the four sequences results in six paired-distance values. (B) Pairwise distances for sequences from each compartment and time point grouped together and plotted as described for panel A, with 10 patient samples per compartment, or a total of 45 paired values. (C) Pairwise distances for each sequence in panel B from the reference genome B95.8 (10 values). Stars indicate significant differences in distances from AIM to CONV (Wilcoxon test; P < 0.01).
Whole EBV genomes become more similar to the B95.8 reference strain over time.
Several independent observations suggested that EBV genomes sequenced from circulating B cells and oral wash samples tend to become more similar to the reference strain, B95.8, over time. For example, phylogenetic analysis (Fig. 2) displayed clear clustering of B cell CONV sequences (Fig. 2, marked by black dots) with the B95.8 reference genome, regardless of the branching pattern from related sequences from the same patient (e.g., E1578 sequences). To get a clearer sense of the relationship between length of infection and level of convergence with the reference strain, the genetic distance of each full-length patient consensus genome from the reference genome of B95.8 was determined and plotted as a function of time (number of days between AIM and CONV visits [Table 1]). As Fig. 5A indicates, several EBV genomes sequenced from patient oral washes during CONV were more similar (shorter genetic distance) to the B95.8 reference sequence than those sequenced from the same patients during AIM. In a similar analysis of the full-length EBV genomes sequenced from B cells, 7 of the 10 matched sequences showed convergence toward the reference strain of B95.8 during CONV (Fig. 5B). A Spearman rank-order correlation test indicated a negative correlation (r = −0.5589; P = 0.0264) between genetic distance to B95.8 and length of infection, which is to say that patient samples sequenced after a greater length of infection were more likely to be closer in sequence identity to B95.8. This convergence phenomenon was specific for the B95.8 genome and was not observed for any other type I or type II EBV reference genome (Fig. 5C).
TABLE 1.
Participant characteristics
| Participant no. | Gender | Age (yr) | Time of visit | Serologya |
Viral load by sample type |
|||
|---|---|---|---|---|---|---|---|---|
| IgM VCA | IgG VCA | EBV nuclear Ag | B cell (copies/106 B cells) | Oral wash (copies/ng of DNA) | ||||
| E1492 | M | 20.4 | AIM | Pos | Neg | Neg | 55,094 | 234.3 |
| 21.0 | CONV | 1,983 | 0.7 | |||||
| E1503 | M | 19.3 | AIM | Pos | Neg | Neg | 111,092 | 2.9 |
| 19.7 | CONV | 271 | 2.9 | |||||
| E1536 | F | 19.3 | AIM | Pos | Neg | Neg | 28,247 | 23.7 |
| 19.8 | CONV | 4,581 | 0.8 | |||||
| E1548 | F | 18.7 | AIM | Pos | Neg | Neg | 23,263 | 0.4 |
| 19.2 | CONV | 1,081 | 0.2 | |||||
| E1563 | M | 21.8 | AIM | Pos | Neg | Neg | 2,391 | 3.1 |
| 22.3 | CONV | 388 | 1.0 | |||||
| E1577 | M | 20.3 | AIM | Pos | Pos | Neg | 64,825 | 1.9 |
| 21.1 | CONV | 375 | 0.6 | |||||
| E1578 | F | 20.0 | AIM | Pos | Pos | Neg | 4,523 | 10.8 |
| 20.8 | CONV | 3,283 | 0.1 | |||||
| E1583 | F | 18.9 | AIM | Pos | Pos | Neg | 410,435 | 13.1 |
| 19.6 | CONV | 839 | 0.5 | |||||
| E1587 | F | 19.9 | AIM | Pos | Neg | Neg | 54,060 | 1.8 |
| 20.6 | CONV | 596 | 0.1 | |||||
| E1590 | M | 18.4 | AIM | Pos | Pos | Neg | 8,127 | 1.4 |
| 19.0 | CONV | 271 | 0.3 | |||||
VCA, viral capsid antigen; Ag, antigen.
FIG 5.

Full-length EBV genomes sequenced from circulating B cells, but not oral wash samples, of patients during primary EBV infection converge toward a common B95.8 reference-like genome over time. EBV genomes were sequenced from either oral wash (A) or B cell compartment (B) samples during AIM and CONV, and their genetic distances from the prototypical reference genome B95.8 were determined. Repeat regions and incompletely sequenced stretches greater than 10 nucleotides were masked in all samples as well as in the reference genome prior to alignment and subsequent phylogenetic tree formation. (C) The observed convergence toward a common reference genome appears to specifically favor a B95.8-like sequence rather than alternate type I EBV strains such as Akata, Mutu, GD, etc., or type II strains.
EBV genomic diversity varies by gene, compartment, and over time.
We then evaluated diversity within individual EBV genes at the nucleotide level. Pairwise genetic distances for each time point and compartment were calculated for six glycoproteins (Fig. 6) and six latency genes (Fig. 7). We examined within-cohort distance for each gene (Fig. 6 and 7, top panels) and distance from B95.8 (Fig. 6 and 7, bottom panels). Overall, glycoproteins were much less diverse than latent genes (note scale on the y axis). However, despite their low diversity, significant differences were observed in the intrahost distances of gp350, gp42, gH, gB, and gL between time point and compartments (Fig. 6, top panel). For example, gp350 demonstrated significantly reduced diversity of B cell- and oral wash-derived EBV sequences from AIM to CONV although a much greater reduction was observed in the BC population. A similar pattern of reduction in diversity was also observed for gB and gH sequence populations. In contrast, sequence diversity in gp42, gp85, and gL increased significantly in BC-derived virus sequenced during CONV (Wilcoxon matched-pairs test, P < 0.01). When pairwise distance was compared to that of the reference genome (Fig. 6, lower panel), gp350 BC sequence populations showed significantly reduced diversity from AIM to CONV, indicating a convergence toward the reference genome (BC AIM versus BC CONV gp350, P < 0.05).
FIG 6.

Overall genetic diversity in EBV glycoproteins is low but varies between time and compartment. The top panel shows the box-and-whisker plot of the genetic distances between all sequences in our cohort for each indicated gene (total of 45 values per time point/compartment). The bottom panel shows the box-and-whisker plot genetic distances from each sequence to the reference genome B95.8 for the same gene (10 values). Several data points for each gene overlapped and caused the apparent reduction in sample number. For each gene, the plots are ordered BC AIM (blue), OW AIM (red), BC CONV (orange), and OW CONV (green). Stars indicate significant changes from AIM to CONV for BC and OW sequence populations (Wilcoxon matched-pairs signed-rank test; P < 0.05).
FIG 7.

Latent genes from circulating EBV genomes are diverse during AIM but become less diverse over time and more similar to B95.8 latent genes. The top panel shows the box-and-whisker plot of the genetic distances between all sequences in our cohort for each indicated latent gene (total of 45 values per time point/compartment). The bottom panel shows the box-and-whisker plot of genetic distances from each sequence to the reference genome, B95.8, for the same gene (10 values). For each gene, the plots are ordered BC AIM (blue), OW AIM (red), BC CONV (orange), and OW CONV (green). Stars indicate significant changes from AIM to CONV for BC and OW sequence populations (Wilcoxon matched-pairs signed-rank test; P < 0.05).
Similarly, each of the six latency genes demonstrated different patterns of variation, indicating that the genes were not strictly linked. For example, EBNA1 mean distances were significantly lower during AIM than during CONV, in both BC and OW (Wilcoxon test; BC AIM versus BC CONV, P = 0.0013; OW AIM versus OW CONV, P = 0.0012); this trend was reversed for EBNA2, EBNA3C, and LMP1 (Fig. 7, top panel) where there was significantly lower diversity during CONV than during AIM (Wilcoxon test; P < 0.05 for all pairings). In EBNA3B the mean diversity for BC-associated viral sequences increased significantly from AIM to CONV in B cells although a significant reduction was noted in OW-associated sequences (Wilcoxon test; P < 0.05). LMP2 distances remained about the same for collection time and compartment.
The pairwise distance for each gene to annotated ORF of the full-length B95.8 reference genome was also calculated. In five of the six genes studied (EBNA1, EBNA3B, EBNA3C, LMP1, and LMP2), the BC CONV sequences were more similar to those of the reference genome than BC AIM sequences; measurements of mean genetic distance indicated significant differences between the AIM and CONV time points (Fig. 7, lower panel) (Wilcoxon ranked-pairs test, significance values from P < 0.05 to P < 0.01). In contrast to the observations in B cell-associated virus, only one gene, LMP2, was found to converge toward B95.8 in virus sequenced from OW during the same time frame (Wilcoxon ranked-pairs test; LMP2 OW AIM versus OW CONV, P < 0.01).
DISCUSSION
Next-generation sequencing, coupled with methods used to enrich viral genomes from contaminating human genomes, has proven to be a powerful tool for sequencing and characterization of full-length viral genomes, including EBV. In the past 5 years alone, the number of publicly available EBV genome sequences has increased from under 10 to greater than 100 (26, 29). While these studies have provided novel sequence data that increase our understanding of EBV biology, the overwhelming majority of genome sequences originated in either diseased tissue or from immortalized cell lines. Either of these two conditions may have imposed selective pressures on the viral genome, and the extent to which these sequences represent circulating EBV strains has not been clear. Although our cohort is limited in size, this study is the first to directly sequence whole EBV genomes from otherwise healthy patients over the course of primary infection. These sequences originated from circulating B cells and oral wash samples and were collected during both the acute phase of infection and convalescence.
Phylogenetic analyses of our sequences suggested that all 10 patients were infected with a type I strain of EBV, which was expected, given the geographic location of the study. As noted, almost all of the sequences aligned with the B95.8 and Mutu reference genomes, two type I EBV strains isolated from North America and Kenya, respectively. Sequences collected from our patient cohort clustered away from type I Asian strains (e.g., Akata, GD1, and GD2). While this could possibly be explained by the choice of using two type I genomes (B95.8 and Akata) as templates for our baits, similar probe sets have been used by other groups to successfully capture both type I and type II EBV genomes (17, 18), suggesting that our sequences accurately represent the strains circulating in the sampled population.
The levels of individual gene and protein variation detected in the viral genomes of our patient cohort are consistent with those previously published (18). High levels of variability (both synonymous and nonsynonymous) were detected in latent genes (EBNA1, EBNA2, EBNA3C, and LMP1), particularly compared to the variation observed in early and late lytic genes. We did not detect higher levels of nonsynonymous variation reported in a recent analysis of 83 full-length genomes primarily from tumor tissues and cell lines. This may reflect different selective pressures operating across tissue type and disease process; variability may also be higher in EBV genomes sequenced from different cancerous tissues where DNA proofreading and repair may be impaired. In addition, the 40 genomes reported here originated from only 10 patients, making it much more likely that they would share the same variants. The technique employed in our study, enriching for viral genomes from infected human cells with low viral genome copy number, could be effectively utilized to directly compare circulating viral genomes to tissue-associated viral genomes in the same patient to better understand these apparent discrepancies.
Similar to our previously reported observations in a comparable cohort, we found significant intrahost EBV genomic diversity between samples taken from B cells and those from oral washes over time (15). We previously reported an increase in LMP1 diversity from AIM to CONV in OW-derived virus; in this study, diversity at both the full-length genome level and in LMP1 were determined to decline from AIM to CONV in both BC- and OW-derived virus (Fig. 4 and 7). This discrepancy can be attributed to the nature of the comparison in our prior work as well as to the increased sensitivity of the current library preparation and sequencing. Our previous study focused solely on LMP1 sequences directly amplified from patients with acute or chronic infection; because we were not able to study all patients at both time points, we were unable to directly compare diversity at the two time points for each compartment, and comparisons were made between group values (15). In addition, in the patient sets that were complete (contained all four samples), we failed to detect any LMP1 variants in three of the five sets, which thus disproportionately skewed the effect of variants in the convalescent oral wash samples. The increased sensitivity of our new enrichment and sequencing approach allowed us to accurately detect and determine LMP1 variants in all patient samples.
This finding highlights one of the particular strengths of our current study with this cohort, i.e., the ability to quantify changes in viral diversity over time within the same patient, beginning with primary infection. The few full-length EBV genomes that have been sequenced directly from patients have described samples from a single collection time point and from only one sample type. While these data reflect the overall level of EBV variation potential present in circulating virus, they do not reflect the identity of the infecting virus or the rate of viral evolution following infection. Our data suggest that all 10 patients may have been infected with different strains of type I EBV. The diversity detected in both the B cell- and oral wash-associated virus during AIM could be the result of initial infection with a diverse pool of EBV, or the diversity could have arisen over the course of several rounds of early virus replication in the newly infected host (43, 44). Regardless, changes in EBV genomic variation within each patient (as measured by genetic distance) indicated continuing evolution of the viral genome in both the peripheral blood B cells and the oral compartment (Fig. 4).
A significant reduction in overall EBV genomic diversity was detected in the sequences of B cell-associated virus from AIM to CONV (Fig. 4B). Changes in latent genes accounted for most of the loss of viral genomic diversity in B cell EBV sequences over time (Fig. 7, top panel). Three of six latent genes (EBNA2, EBNA3C, and LMP1) demonstrated significantly lower mean genetic distances in B cells during CONV than during AIM, a trend that was also observed in OW samples. Significantly lower mean genetic distances were also detected in CONV blood gp350, gH, and gB sequences and oral wash gp350, gH, and gB sequences (Fig. 6, top panel). Additional studies will be helpful in discerning whether this indicates separate and discrete selection pressures operant on these genes. Certainly, envelope glycoprotein variation is subject to selection pressure by antibody activity (16), whereas latent gene variability may be driven more by CD8 T cell selective pressures.
Perhaps the most striking finding was the apparent convergence of B cell-associated EBV genomes toward a B95.8-like reference genotype over time (Fig. 5B); this was apparent at both the whole-genome level as well at the level of selected, individual genes (Fig. 6 and 7, bottom panels). With our access to longitudinal samples, we showed a strong negative correlation between length of infection and genetic distance from B95.8 (Spearman correlation; r = −0.5589; P = 0.0264); that is, the longer an individual was infected, the more likely the B cell EBV sequence was to resemble B95.8. Though B95.8 was isolated from a patient during AIM, the suspected origin of the virus was latently infected B cells delivered via transfusion; the reference genome was subsequently selected by its ability to transform marmoset B cells, perhaps explaining how this strain is representative of longer-term infection seen in our samples (42). We also demonstrated that the convergence toward a reference genome is specific for B95.8, the prototypic type I virus geographically linked to North America, and not another type I reference genome (e.g., Akata or Mutu) (Fig. 5C). This observation was even more pronounced at the protein level for gp350 and LMP1 (data not shown). We anticipate that a similar cohort assembled from a different geographic locale might demonstrate the same trend toward the nearest reference strain for that region.
It is unlikely that utilization of a B95.8 BAC for the generation of our hybridization probes reduced the diversity of our libraries, skewing our results toward the reference genome. Prior studies utilizing hybridization to enrich EBV genomes have used probes designed from the B95.8 reference genome and were able to assemble type I and type II EBV genomes (17, 18). Probes were a 50:50 mix of B95.8 and Akata BAC genomes (see Materials and Methods), yet we observed convergence toward B95.8 only, and not Akata or a B95.8-Akata hybrid (Fig. 5C). Finally, identity with B95.8 was independent of total sequencing reads or depth and thus independent of viral load (Table 1, viral load; see also read information in Table S1 in the supplemental material). In fact, low depth would be predicted to result in a greater amount of variation as there are insufficient reads to determine true SNPs from sequencing errors; in contrast, we find that regardless of overall sequencing depth (particularly in CONV B cells where copy numbers are expected to be lower) similarity to B95.8 is striking. Similarly, even when samples had comparable viral loads (e.g., E1578 BC AIM and BC CONV) (Table 1), detectable differences in the EBV genomes were observed.
Convergence of viral genomes toward a consensus sequence over time following infection has previously been described in RNA viruses. A recent study reported duplication in a region of the respiratory syncytial virus (RSV) G glycoprotein; this duplication was noted to occur independently in two separate and otherwise unrelated strains, likely as an adaptation for immune evasion (45). Interestingly, the hepatitis C virus (HCV) G proteins from infected patients sequenced 20 years after a common-source outbreak indicated convergent evolution in HCV in the absence of specific HLA alleles (46). The latter data suggest that the consensus sequence provides a selective advantage, perhaps favoring persistence within a reservoir or for transmission to a new host. Indeed, convergence of HIV env sequences toward an ancestral version better adapted for transmission has been reported in HIV-positive patients (47, 48).
We note the obvious differences in convergence levels across the genes discussed here; not all genes appear to converge, and those that do likely converge at different rates. Also, some of the most significant convergence detected is within the most variable genes. Both of these observations can be explained by the complex replication cycle and gene expression pattern of EBV. For example, although genes such as EBNA1 and LMP1 are indeed the most variable, they are also the most expressed during the establishment of latent infection, exposing them to both functional and immunological pressure, particularly cytotoxic T lymphocyte (CTL) recognition. However, all study participants were HLA-A2 positive, and analysis of common HLA-A2 epitopes did not reveal evolution of any potential CTL escape mutations in either BC or OW samples from AIM to CONV (Table S3 and data not shown) (49).
Interestingly, we did not find any relationship between peripheral blood viral load and intrahost diversity; EBV copy number did not correlate with total variation in B cell-derived virus at either time point (data not shown). As noted above, this was not likely due to any artifactual effect of sequencing depth as samples with similar viral loads generated measurably different genomic sequences. Likewise, the reciprocal interaction was also observed; despite a greater than 400-fold difference in BC viral loads from AIM to CONV (Table 1), sequences obtained from patient E1503 demonstrated remarkable conservation, clustering together with matched OW sequences from both time points to form a distinctive group (Fig. 2). Furthermore, no correlation was detected between depth of coverage and detection of viral variants; Spearman analysis of FPKM scores versus variation at the individual ORF failed to indicate any correlation (data not shown). Last, no correlation was detected between viral load values and genome diversity (data not shown), suggesting that the reduction in viral diversity measured during CONV was not due to reduction in genome sample size. This evidence indicates that reduced viral copy number during convalescence, particularly in B cells, did not negatively impact the findings in this study.
The current model posits transfer of EBV in saliva from an infected donor and subsequent infection of naive B cells in the new host (50). These naive cells undergo a germinal cell-like reaction and transition into memory B cells persistently infected with a latent EBV genome expressing a severely limited number of gene products (51–53). Activation of these memory B cells results in lytic replication of EBV and produces virus that greatly favors infection of epithelial cells, further amplifying the virus (54). During early infection, in the absence of an adaptive immune response, this cycle can result in greater than 50% of circulating memory B cells being positive for EBV (55). Alternatively, it has also been suggested that incoming virus released from donor B cells is first amplified by direct infection of epithelial tissue to generate a lymphotropic virus prior to naive B cell infection (56, 57). Both of these paths of infection may affect the levels of viral diversity detected in either compartment during AIM as any replication event has the potential to increase genomic variation (13). Our compartment sequencing data and those from similar studies moving forward may provide further information on which proposed infection mechanism may be operant.
Although the data described here provide valuable information regarding overall viral diversity as well as viral genome evolution during EBV infection, additional questions will need to be addressed. Our data indicate that certain EBV glycoprotein and latent genes required for the establishment and persistence of B cell infection, despite being some of the most variable, demonstrated the greatest degree of convergence over time. This suggests that conservation of genomic segments is important for persistent infection. However, additional analyses at the individual gene level are required to determine what specific functions may be selected for over the course of persistent infection. This quasi-species-level analysis is important to answering these questions and to better appreciate the various push-pull mechanics of immune evasion versus gene functionality. Additionally, the origin of the B95.8-like genome sequenced in our patients will need to be resolved. Does it arise through spontaneous mutation of diverse transmitted EBV genomes, or is it present during transmission and out-competes other EBV variants? Answering these questions will require additional studies that leverage new technologies, including single-cell sequencing to track individual, complete viral genomes through time.
MATERIALS AND METHODS
Study cohort.
Peripheral blood mononuclear cell (PBMC) and saliva samples were obtained from 10 young adults (5 male and 5 female) presenting with symptoms compatible with AIM; AIM diagnosis was confirmed by a positive monospot assay (University of Massachusetts Amherst Health Services). Primary infection was confirmed by the detection of serum IgM specific for the EBV viral capsid antigen (Table 1) (58). Additional blood and oral wash samples were collected from the same 10 patients at least 5 months postinfection (CONV).
Quantification of viral genomes in peripheral blood.
EBV genomes present in the peripheral blood and saliva were quantified using a previously described quantitative PCR (qPCR)-based viral load assay (16). Briefly, circulating B cells were isolated from PBMCs using a RosetteSep human B cell kit (StemCell Technologies), and total DNA was extracted using a Qiagen DNeasy blood and tissue kit (Qiagen). DNA from oral wash samples was isolated using a High Pure Viral Nucleic Acid kit (Roche Diagnostics) following preclearing of samples by low-speed centrifugation. The EBV copy number per human genome was determined using duplexed qPCRs to simultaneously quantify the number of EBV BALF5 copies (forward primer, 5′-CGGAAGCCCTCTGGACTTC-3′; reverse primer, 5′-CCCTGTTTATCCGATGAATG-3′; and probe 5′-FAM-TGTACACGCACGAGAAATGCGCCT-BHQ-1-3′, where FAM is 6-carboxyfluorescein and BHQ is Black Hole quencher); human genome copies were determined by quantifying the copy number of the CCR5 gene (forward primer, 5′-GCTGTGTTTGCGTCTCTCCCAGGA-3′; reverse primer, 5′-CTCACAGCCCTGTGCCTCTTCTTC-3′; and probe 5′-Cy5-AGCAGCGGCAGGACCAGCCCCAAG-BHQ-1-3′). DNA extracted from the Namalwa cell line, containing two integrated copies of the EBV genome, was used to establish standards and controls (59).
EBV bait preparation.
Two sets of EBV whole-genome baits (WGB) from BACs containing the full-length EBV genomes of either B95.8 or Akata were prepared as described previously (31). Briefly, 3 μg of DNA was sheared to an average size of 250 bp using a Covaris S220 instrument. End repair, addition of a 3′ A, adapter ligation, and reaction cleanup followed the Illumina's genomic DNA sample preparation protocol except that the adapter consisted of oligonucleotides 5′-TGTAACATCACAGCATCACCGCCATCAGTCxT-3′ (where x refers to an exonuclease I-resistant phosphorothioate linkage) and 5′-[PHOS]GACTGATGGCGCACTACGACACTACAATGT-3′. The T7 RNA polymerase promoter sequence was added by PCR using the forward primer 5′-GGATTCTAATACGACTCACTATAGGCGCTCAGCGGCCGCAGCATCACCGCCATCAGT-3′, and the purified PCR product was used as the template to generate biotinylated RNA baits using a MEGAshortscript T7 kit (Thermo Fisher). WGB from EBV B95.8 and Akata genomes were mixed at a 1:1 ratio and used for hybrid selection.
Patient sample pond library prep.
Three micrograms of DNA extracted from either patient oral washes or peripheral blood B cells was sheared using a Covaris S220 Sonicator (Covaris) and concentrated using a MinElute kit (Qiagen). Sheared DNA was enzymatically blunted and end repaired with T4 DNA polymerase and polynucleotide kinase (PNK), and 3′ A tails were added using Klenow DNA polymerase prior to ligation of sequencing adapters. Forty-one unique nucleotide-barcoded sequencing adapters were used for each of the patient samples, as well as the resequenced EBV Akata-containing BAC. Following ligation, libraries were amplified by PCR for either 8, 10, or 12 cycles using the forward primer 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGA-3′ and reverse primer 5′-CAAGCAGAAGACGGCATACGAGAT-3′ and 2× Phusion PCR Mastermix (New England BioLabs). The final amplified patient pond libraries were purified using AMPure XP beads, and DNA concentration was determined using a Qubit 2.0 Fluorometer (Life Technologies).
Enrichment of EBV genomes from patient samples.
We applied hybrid selection with WGB to enrich EBV genomes in patient pond libraries prepared from oral washes or peripheral blood B cells. Forty-one unique barcoded sequencing adapters were used for each of the patient samples as well as for the resequenced EBV Akata-containing BAC control. For enrichment, we hybridized DNA of the pond library (1 to 2 μg) with EBV B95.8-Akata WGB (0.5 μg) as described previously (30, 31).
Sequencing.
Each sample was sequenced by Beckman Coulter Genomics; all four patient libraries constructed from AIM and CONV B cell fraction (BC)- and oral wash (OW)-extracted viral genomes were run on the same lane of an Illumina HiSeq 2500, across two flow cells, as 2- by 125-bp runs. Sequences were parsed according to barcodes prior to sample analysis and genome assembly.
EBV sequence assembly methods.
Fastq files were quality checked using FastQC (version 0.11.4) and imported into Geneious Software (version 9.1.3). Any remaining adapter sequences were trimmed using the BBduk plug-in in Geneious. Guided by FastQC statistics, reads for each file were further trimmed to a level of 99 to 100% inferred base call accuracy. Paired reads were mapped to the NCBI EBV reference genome (NC_007605.1) using the Geneious Read Mapper set to a medium to high sensitivity. A consensus sequence was called for each set of contigs using the highest quality threshold based on the chromatograms, with an N called for any position where coverage was less than five contigs. A summary of the paired reads, assembled reads, percent mapped reads, and reference coverage is provided (see Table S1 in the supplemental material). Mean coverage denotes coverage across the entire length of the EBV genome, including regions of high-GC content and repetitive DNA sequences; in keeping with the current standards of analysis, these regions were masked for all alignments and subsequent analyses. To ensure that coverage levels of all EBV ORFs were comparable, fragment per kilobase million (FPKM) reads for each reported gene for each patient were calculated (Table S2). Consensus sequences for each patient (BC AIM, BC CONV, OW AIM, and OW CONV) were aligned to each other using MAFFT (http://mafft.cbrc.jp/alignment/server/) and manually proofed against their assemblies to determine if additional bases could be called.
All subsequent analyses utilized consensus sequences generated from Illumina assemblies. MAFFT (version 7) was used to align the 40 subjects' consensus sequences to the NCBI reference genome. The alignment was optimized by hand to correct for poorly sequenced and/or repeat regions. Repeat regions with assembly errors were masked for subsequent distance and phylogenetic calculations. A maximum-likelihood phylogenetic tree was generated using PhyML (http://www.atgc-montpellier.fr/phyml/) and the Akaike information criterion (AIC) (60). The alignment was annotated according to the reference genome in Geneious (Fig. 1). Coding regions were extracted from the alignment and checked for proper translation prior to distance-based calculations. Within each coding region, overall nonsynonymous (dN) and synonymous (dS) genetic distances were calculated using SNAP (www.lanl.gov). Estimates of genomic diversity within and between sequence populations were calculated in MEGA using the Tamura-Nei (TN93) molecular model (identified as the best-fitting model using the hierarchical test based on the Bayesian information criterion), and standard errors were calculated using a bootstrap procedure (1,000 replicates) (61, 62). Pairwise distances were calculated for each subject's consensus sequences (4 sequences each; 6 pairwise comparisons), for each compartment during either AIM or CONV (10 patient sequences; 45 pairwise comparisons), and relative to the NCBI reference genome (the genetic distance of each sequence to the reference genome; 10 pairwise comparisons) (Fig. 4). Similarly, pairwise distances were calculated for specific genes (Fig. 6).
Statistical analysis.
Statistical analyses were performed using GraphPad Prism, version 7.03, for Windows (GraphPad Software, San Diego, CA). Correlations for non-normally distributed data were calculated using Spearman's rank correlation coefficient (rho). Comparison of pairwise distance measurements for AIM patients during AIM and CONV were calculated using Wilcoxon matched-pairs signed-rank test; comparisons between EBV gene groups were calculated using a Mann-Whitney test. All statistical tests were two-sided, and a P value of <0.05 was considered statistically significant.
Ethics statement.
All study participants provided written informed consent, and the University of Massachusetts Medical School IRB approved these studies.
Accession number(s).
Consensus sequence data from this study have been deposited in the NCBI database under accession numbers MF547453 to MF547492 (Table 2). NCBI accession numbers of previously determined full-length EBV sequences used in analysis are as follows: Akata, KC207813; B95.8, NC_007605.1; C666-1, LN827525; GD1, AY961628; GD2, HQ020558; healthy donor saliva, LN824142; Mutu, KC207814; M81, KF373730; AG876, NC_009334; Jijoye, LN827800; and Wewak, LN827544.
TABLE 2.
Sample accession numbers by each participant, compartment, and time point
Supplementary Material
ACKNOWLEDGMENTS
We thank the study participants for their participation in the research. We thank Alan Calhoun, Jessica Conrad, George Corey, and Gail Gnatek at the University of Massachusetts Amherst Student Health Service for providing clinical care and research samples. We also thank Fred Wang and Teru Kanda for their generous donations of the B95.8 and Akata BACs, respectively.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01466-17.
REFERENCES
- 1.Luzuriaga K, Sullivan JL. 2010. Infectious mononucleosis. N Engl J Med 362:1993–2000. doi: 10.1056/NEJMcp1001116. [DOI] [PubMed] [Google Scholar]
- 2.Balfour HH Jr, Odumade OA, Schmeling DO, Mullan BD, Ed JA, Knight JA, Vezina HE, Thomas W, Hogquist KA. 2013. Behavioral, virologic, and immunologic factors associated with acquisition and severity of primary Epstein-Barr virus infection in university students. J Infect Dis 207:80–88. doi: 10.1093/infdis/jis646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. 2013. The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol 3:227–232. doi: 10.1016/j.coviro.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nielsen TR, Rostgaard K, Nielsen NM, Koch-Henriksen N, Haahr S, Sorensen PS, Hjalgrim H. 2007. Multiple sclerosis after infectious mononucleosis. Arch Neurol 64:72–75. doi: 10.1001/archneur.64.1.72. [DOI] [PubMed] [Google Scholar]
- 5.Hjalgrim H, Engels EA. 2008. Infectious aetiology of Hodgkin and non-Hodgkin lymphomas: a review of the epidemiological evidence. J Intern Med 264:537–548. doi: 10.1111/j.1365-2796.2008.02031.x. [DOI] [PubMed] [Google Scholar]
- 6.Thacker EL, Mirzaei F, Ascherio A. 2006. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann Neurol 59:499–503. doi: 10.1002/ana.20820. [DOI] [PubMed] [Google Scholar]
- 7.Cohen JI, Fauci AS, Varmus H, Nabel GJ. 2011. Epstein-Barr virus: an important vaccine target for cancer prevention. Sci Transl Med 3:107fs7. doi: 10.1126/scitranslmed.3002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen JI, Mocarski ES, Raab-Traub N, Corey L, Nabel GJ. 2013. The need and challenges for development of an Epstein-Barr virus vaccine. Vaccine 31(Suppl 2):B194–B196. doi: 10.1016/j.vaccine.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moutschen M, Leonard P, Sokal EM, Smets F, Haumont M, Mazzu P, Bollen A, Denamur F, Peeters P, Dubin G, Denis M. 2007. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein-Barr virus vaccine in healthy adults. Vaccine 25:4697–4705. doi: 10.1016/j.vaccine.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 10.Sokal EM, Hoppenbrouwers K, Vandermeulen C, Moutschen M, Leonard P, Moreels A, Haumont M, Bollen A, Smets F, Denis M. 2007. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis 196:1749–1753. doi: 10.1086/523813. [DOI] [PubMed] [Google Scholar]
- 11.Rees L, Tizard EJ, Morgan AJ, Cubitt WD, Finerty S, Oyewole-Eletu TA, Owen K, Royed C, Stevens SJ, Shroff RC, Tanday MK, Wilson AD, Middeldorp JM, Amlot PL, Steven NM. 2009. A phase I trial of Epstein-Barr virus gp350 vaccine for children with chronic kidney disease awaiting transplantation. Transplantation 88:1025–1029. doi: 10.1097/TP.0b013e3181b9d918. [DOI] [PubMed] [Google Scholar]
- 12.Cui X, Cao Z, Sen G, Chattopadhyay G, Fuller DH, Fuller JT, Snapper DM, Snow AL, Mond JJ, Snapper CM. 2013. A novel tetrameric gp350 1-470 as a potential Epstein-Barr virus vaccine. Vaccine 31:3039–3045. doi: 10.1016/j.vaccine.2013.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duffy S, Shackelton LA, Holmes EC. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet 9:267–276. doi: 10.1038/nrg2323. [DOI] [PubMed] [Google Scholar]
- 14.Renzette N, Bhattacharjee B, Jensen JD, Gibson L, Kowalik TF. 2011. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog 7:e1001344. doi: 10.1371/journal.ppat.1001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Renzette N, Somasundaran M, Brewster F, Coderre J, Weiss ER, McManus M, Greenough T, Tabak B, Garber M, Kowalik TF, Luzuriaga K. 2014. Epstein-Barr virus latent membrane protein 1 genetic variability in peripheral blood B cells and oropharyngeal fluids. J Virol 88:3744–3755. doi: 10.1128/JVI.03378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss ER, Alter G, Ogembo JG, Henderson JL, Tabak B, Bakis Y, Somasundaran M, Garber M, Selin L, Luzuriaga K. 2017. High Epstein-Barr virus load and genomic diversity are associated with generation of gp350-specific neutralizing antibodies following acute infectious mononucleosis. J Virol 91:e01562-16. doi: 10.1128/JVI.01562-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Depledge DP, Palser AL, Watson SJ, Lai IY, Gray ER, Grant P, Kanda RK, Leproust E, Kellam P, Breuer J. 2011. Specific capture and whole-genome sequencing of viruses from clinical samples. PLoS One 6:e27805. doi: 10.1371/journal.pone.0027805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palser AL, Grayson NE, White RE, Corton C, Correia S, Ba Abdullah MM, Watson SJ, Cotten M, Arrand JR, Murray PG, Allday MJ, Rickinson AB, Young LS, Farrell PJ, Kellam P. 2015. Genome diversity of Epstein-Barr virus from multiple tumor types and normal infection. J Virol 89:5222–5237. doi: 10.1128/JVI.03614-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu P, Fang X, Feng Z, Guo YM, Peng RJ, Liu T, Huang Z, Feng Y, Sun X, Xiong Z, Guo X, Pang SS, Wang B, Lv X, Feng FT, Li DJ, Chen LZ, Feng QS, Huang WL, Zeng MS, Bei JX, Zhang Y, Zeng YX. 2011. Direct sequencing and characterization of a clinical isolate of Epstein-Barr virus from nasopharyngeal carcinoma tissue by using next-generation sequencing technology. J Virol 85:11291–11299. doi: 10.1128/JVI.00823-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song KA, Yang SD, Hwang J, Kim JI, Kang MS. 2015. The full-length DNA sequence of Epstein Barr virus from a human gastric carcinoma cell line, SNU-719. Virus Genes 51:329–337. doi: 10.1007/s11262-015-1248-z. [DOI] [PubMed] [Google Scholar]
- 21.Zhou L, Chen JN, Qiu XM, Pan YH, Zhang ZG, Shao CK. 2017. Comparative analysis of 22 Epstein-Barr virus genomes from diseased and healthy individuals. J Gen Virol 98:96–107. doi: 10.1099/jgv.0.000699. [DOI] [PubMed] [Google Scholar]
- 22.Santpere G, Darre F, Blanco S, Alcami A, Villoslada P, Mar Alba M, Navarro A. 2014. Genome-wide analysis of wild-type Epstein-Barr virus genomes derived from healthy individuals of the 1,000 Genomes Project. Genome Biol Evol 6:846–860. doi: 10.1093/gbe/evu054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwok H, Wu CW, Palser AL, Kellam P, Sham PC, Kwong DL, Chiang AK. 2014. Genomic diversity of Epstein-Barr virus genomes isolated from primary nasopharyngeal carcinoma biopsy samples. J Virol 88:10662–10672. doi: 10.1128/JVI.01665-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng MS, Li DJ, Liu QL, Song LB, Li MZ, Zhang RH, Yu XJ, Wang HM, Ernberg I, Zeng YX. 2005. Genomic sequence analysis of Epstein-Barr virus strain GD1 from a nasopharyngeal carcinoma patient. J Virol 79:15323–15330. doi: 10.1128/JVI.79.24.15323-15330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin Z, Wang X, Strong MJ, Concha M, Baddoo M, Xu G, Baribault C, Fewell C, Hulme W, Hedges D, Taylor CM, Flemington EK. 2013. Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains. J Virol 87:1172–1182. doi: 10.1128/JVI.02517-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiara M, Manzari C, Lionetti C, Mechelli R, Anastasiadou E, Chiara Buscarinu M, Ristori G, Salvetti M, Picardi E, D'Erchia AM, Pesole G, Horner DS. 2016. Geographic population structure in Epstein-Barr virus revealed by comparative genomics. Genome Biol Evol 8:3284–3291. doi: 10.1093/gbe/evw226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamers SL, Salemi M, Galligan DC, de Oliveira T, Fogel GB, Granier SC, Zhao L, Brown JN, Morris A, Masliah E, McGrath MS. 2009. Extensive HIV-1 intra-host recombination is common in tissues with abnormal histopathology. PLoS One 4:e5065. doi: 10.1371/journal.pone.0005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wahl SM, Greenwell-Wild T, Peng G, Hale-Donze H, Doherty TM, Mizel D, Orenstein JM. 1998. Mycobacterium avium complex augments macrophage HIV-1 production and increases CCR5 expression. Proc Natl Acad Sci U S A 95:12574–12579. doi: 10.1073/pnas.95.21.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwok H, Chiang AK. 2016. From conventional to next generation sequencing of Epstein-Barr virus genomes. Viruses 8:60. doi: 10.3390/v8030060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. 2009. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melnikov A, Galinsky K, Rogov P, Fennell T, Van Tyne D, Russ C, Daniels R, Barnes KG, Bochicchio J, Ndiaye D, Sene PD, Wirth DF, Nusbaum C, Volkman SK, Birren BW, Gnirke A, Neafsey DE. 2011. Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol 12:R73. doi: 10.1186/gb-2011-12-8-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, Tuffnell PS, Barrell BG. 1984. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310:207–211. doi: 10.1038/310207a0. [DOI] [PubMed] [Google Scholar]
- 33.Skare J, Edson C, Farley J, Strominger JL. 1982. The B95-8 isolate of Epstein-Barr virus arose from an isolate with a standard genome. J Virol 44:1088–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parker BD, Bankier A, Satchwell S, Barrell B, Farrell PJ. 1990. Sequence and transcription of Raji Epstein-Barr virus DNA spanning the B95-8 deletion region. Virology 179:339–346. doi: 10.1016/0042-6822(90)90302-8. [DOI] [PubMed] [Google Scholar]
- 35.Xue SA, Jones MD, Lu QL, Middeldorp JM, Griffin BE. 2003. Genetic diversity: frameshift mechanisms alter coding of a gene (Epstein-Barr virus LF3 gene) that contains multiple 102-base-pair direct sequence repeats. Mol Cell Biol 23:2192–2201. doi: 10.1128/MCB.23.6.2192-2201.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tzellos S, Farrell PJ. 2012. Epstein-Barr virus sequence variation—biology and disease. Pathogens 1:156–174. doi: 10.3390/pathogens1020156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tso KK, Yip KY, Mak CK, Chung GT, Lee SD, Cheung ST, To KF, Lo KW. 2013. Complete genomic sequence of Epstein-Barr virus in nasopharyngeal carcinoma cell line C666-1. Infect Agents Cancer 8:29. doi: 10.1186/1750-9378-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai MH, Raykova A, Klinke O, Bernhardt K, Gartner K, Leung CS, Geletneky K, Sertel S, Munz C, Feederle R, Delecluse HJ. 2013. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep 5:458–470. doi: 10.1016/j.celrep.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Dolan A, Addison C, Gatherer D, Davison AJ, McGeoch DJ. 2006. The genome of Epstein-Barr virus type 2 strain AG876. Virology 350:164–170. doi: 10.1016/j.virol.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 40.Correia S, Palser A, Elgueta Karstegl C, Middeldorp JM, Ramayanti O, Cohen JI, Hildesheim A, Fellner MD, Wiels J, White RE, Kellam P, Farrell PJ. 2017. Natural variation of Epstein-Barr virus genes, proteins, and primary microRNA. J Virol 91:e00375-17. doi: 10.1128/JVI.00375-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neves M, Marinho-Dias J, Ribeiro J, Sousa H. 2017. Epstein-Barr virus strains and variations: geographic or disease-specific variants? J Med Virol 89:373–387. doi: 10.1002/jmv.24633. [DOI] [PubMed] [Google Scholar]
- 42.Blacklow NR, Watson BK, Miller G, Jacobson BM. 1971. Mononucleosis with heterophil antibodies and EB virus infection. Acquisition by an elderly patient in hospital. Am J Med 51:549–552. [DOI] [PubMed] [Google Scholar]
- 43.Sitki-Green D, Covington M, Raab-Traub N. 2003. Compartmentalization and transmission of multiple Epstein-Barr virus strains in asymptomatic carriers. J Virol 77:1840–1847. doi: 10.1128/JVI.77.3.1840-1847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sitki-Green DL, Edwards RH, Covington MM, Raab-Traub N. 2004. Biology of Epstein-Barr virus during infectious mononucleosis. J Infect Dis 189:483–492. doi: 10.1086/380800. [DOI] [PubMed] [Google Scholar]
- 45.Schobel SA, Stucker KM, Moore ML, Anderson LJ, Larkin EK, Shankar J, Bera J, Puri V, Shilts MH, Rosas-Salazar C, Halpin RA, Fedorova N, Shrivastava S, Stockwell TB, Peebles RS, Hartert TV, Das SR. 2016. Respiratory syncytial virus whole-genome sequencing identifies convergent evolution of sequence duplication in the C terminus of the G gene. Sci Rep 6:26311. doi: 10.1038/srep26311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. 2005. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med 201:1753–1759. doi: 10.1084/jem.20050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herbeck JT, Nickle DC, Learn GH, Gottlieb GS, Curlin ME, Heath L, Mullins JI. 2006. Human immunodeficiency virus type 1 env evolves toward ancestral states upon transmission to a new host. J Virol 80:1637–1644. doi: 10.1128/JVI.80.4.1637-1644.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lythgoe KA, Gardner A, Pybus OG, Grove J. 2017. Short-sighted virus evolution and a germline hypothesis for chronic viral infections. Trends Microbiol 25:336–348. doi: 10.1016/j.tim.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hislop AD, Taylor GS, Sauce D, Rickinson AB. 2007. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol 25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553. [DOI] [PubMed] [Google Scholar]
- 50.Thorley-Lawson DA. 2001. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol 1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 51.Tracy SI, Kakalacheva K, Lunemann JD, Luzuriaga K, Middeldorp J, Thorley-Lawson DA. 2012. Persistence of Epstein-Barr virus in self-reactive memory B cells. J Virol 86:12330–12340. doi: 10.1128/JVI.01699-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hadinoto V, Shapiro M, Greenough TC, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. 2008. On the dynamics of acute EBV infection and the pathogenesis of infectious mononucleosis. Blood 111:1420–1427. doi: 10.1182/blood-2007-06-093278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roughan JE, Thorley-Lawson DA. 2009. The intersection of Epstein-Barr virus with the germinal center. J Virol 83:3968–3976. doi: 10.1128/JVI.02609-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. 2009. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog 5:e1000496. doi: 10.1371/journal.ppat.1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hochberg D, Souza T, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. 2004. Acute infection with Epstein-Barr virus targets and overwhelms the peripheral memory B-cell compartment with resting, latently infected cells. J Virol 78:5194–5204. doi: 10.1128/JVI.78.10.5194-5204.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borza CM, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med 8:594–599. doi: 10.1038/nm0602-594. [DOI] [PubMed] [Google Scholar]
- 57.Hutt-Fletcher LM. 2017. The long and complicated relationship between Epstein-Barr virus and epithelial cells. J Virol 91:e01677-16. doi: 10.1128/JVI.01677-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hess RD. 2004. Routine Epstein-Barr virus diagnostics from the laboratory perspective: still challenging after 35 years. J Clin Microbiol 42:3381–3387. doi: 10.1128/JCM.42.8.3381-3387.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lawrence JB, Villnave CA, Singer RH. 1988. Sensitive, high-resolution chromatin and chromosome mapping in situ: presence and orientation of two closely integrated copies of EBV in a lymphoma line. Cell 52:51–61. doi: 10.1016/0092-8674(88)90530-2. [DOI] [PubMed] [Google Scholar]
- 60.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 61.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tamura K. 1992. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol Biol Evol 9:678–687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.