

Abstract
The Hedgehog (Hh) signaling pathway plays a central role during embryonic development and its aberrant activation has been implicated in the development and progression of several human cancers. Major efforts toward the identification of chemical modulators of the Hedgehog pathway has yielded several antagonists of the GPCR-like Smoothened receptor. In contrast, potent inhibitors of the Sonic Hedgehog/Patched interaction, the most upstream event in ligand-induced activation of this signaling pathway, have been elusive. To address this gap, a genetically encoded cyclic peptide was designed based on the Shh-binding loop of Hedgehog-Interacting Protein (HHIP) and subjected to multiple rounds of affinity maturation through the screening of macrocyclic peptide libraries produced in E. coli cells. Using this approach, an optimized macrocyclic peptide inhibitor (HL2-m5) was obtained that binds Shh with a KD of 170 nM, which corresponds to a 120-fold affinity improvement compared to the parent molecule. Importantly, HL2-m5 is able to effectively suppress Shh-mediated Hedgehog signaling and Gli-controlled gene transcription in living cells (IC50 = 250 nM), providing the most potent inhibitor of the Sonic Hedgehog/Patched interaction reported to date. This first-in-class macrocyclic peptide modulator of the Hedgehog pathway is expected to provide a valuable probe for investigating and targeting ligand-dependent Hedgehog pathway activation in cancer and other pathologies. This work also introduces a general strategy for the development of cyclopeptide inhibitors of protein-protein interactions.
Keywords: Hedgehog pathway, peptide macrocycles, Sonic Hedgehog/Patched protein-protein interaction, amber stop codon suppression, unnatural amino acids
The Hedgehog (Hh) signaling pathway plays a central role during embryonic development controlling cell growth and differentiation, tissue patterning, and organogenesis.1 Stimulation of the Hedgehog pathway is mediated by a complex sequence of molecular events at the level of the membrane and primary cilia of vertebrate cells, resulting in an intracellular signaling cascade and transcriptional response (Figure 1).2 Canonical activation of this pathway is initiated by binding of the Hedgehog signaling proteins (i.e., Sonic (Shh), Indian (Ihh), and/or Desert (Dhh) Hedgehog), to the extracellular domain of the transmembrane receptor Patched (PTCH1).3 This event relieves Patched-mediated inhibition on the Smoothened (Smo) receptor, allowing Smo to translocate from the plasma membrane and endoplasmic vesicles to the primary cilium.4 Smo activation results in the accumulation of the active forms of Gli2 and Gli3 transcription factors,5 which stimulate the transcription of Gli-controlled genes, including Gli1 and PTCH1 (Figure 1).6
Figure 1.
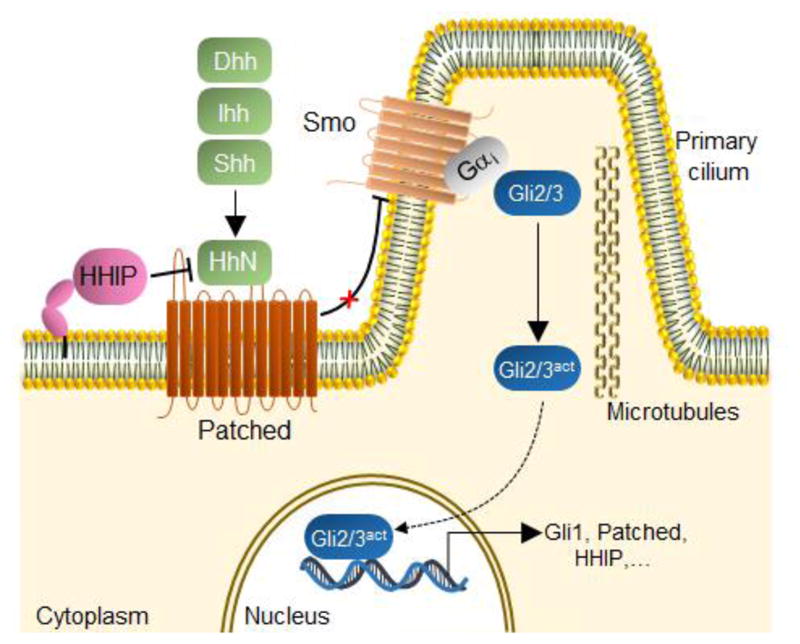
Hedgehog signaling pathway. Binding of the Hedgehog ligand(s) (HhN, corresponding to Shh, Dhh, or Ihh) to the Patched receptor relieves its inhibitory effect on Smoothened (Smo), resulting in the activation of Gli transcription factors and induction of Gli-controlled genes. Hedgehog-Interacting Protein (HHIP) inhibits the signaling pathway by competing with Patched for binding to the Hedgehog ligands. Adapted from www.phosphosite.org.20
Aberrant activation of the Hedgehog pathway has been associated with tumorigenesis in several human tissues.7 In particular, an increasing number of tumors, including leukemia,8 small-cell lung9, pancreas10, and colon11 cancer, have been found to rely on ligand-dependent Hedgehog signaling for sustained growth and proliferation. Hh signaling is also implicated in the maintenance and propagation of cancer stem cells,8a,8c,12 which are believed to play a crucial role in tumor self-renewal, survival against chemotherapy, and metastasis.13
Because of the therapeutic potential of the Hedgehog pathway, major efforts have been devoted to the development of chemical modulators of this signaling pathway.14 These efforts have led to the identification of several potent inhibitors of GPCR-like Smoothened (Smo) receptor.14 These include cyclopamine15 and vismodegib16, which correspond to the archetypal member and the first FDA-approved drug, respectively, belonging to this class of Hh pathway antagonists. Compounds that target downstream components of this pathway14 or processes involved in Shh maturation17 have also been reported. In stark contrast, potent inhibitors of the Shh/Patched protein-protein interaction have remained elusive. While a neutralizing anti-Hedgehog antibody (5E1) is available,18 small-molecular-weight agents capable of disrupting this interaction would be desirable. To date, the only compound of this type is robotnikinin, a small-molecule Shh antagonist developed by Schreiber and coworkers.19 Despite this progress, this compound has only moderate Shh inhibitory activity in vitro and in cells (IC50 ~ 15 μM)19, highlighting the need for more potent inhibitors directed against this component of the Hedgehog pathway.
Macrocyclic peptides are promising molecular scaffolds for targeting biomolecular interfaces, including those mediating protein-protein interactions.21 In view of their attractive features as chemical probes and potential therapeutics, we previously developed methodologies to access macrocyclic peptides through the cyclization of ribosomally derived polypeptides by means of a genetically encoded noncanonical amino acid (ncAA).22 NcAA-mediated peptide cyclization offers the opportunity to rapidly generate genetically encoded cyclic peptide libraries directly in bacterial cells, which can facilitate molecular discovery efforts.23 Here, we successfully applied this strategy to develop and evolve a macrocyclic peptide that targets Shh with high binding affinity and effectively inhibits Shh-mediated Hedgehog pathway signaling in living mammalian cells. This work makes available a valuable probe molecule for investigating the functional role and therapeutic potential of the Shh/Patched interaction. In addition, it introduces and validates an integrated platform for the development of bioactive macrocyclic peptides.
RESULTS AND DISCUSSION
Design of Shh-binding macrocyclic peptide HL2-m1
Recent crystallographic studies have provided insights into the structure of Shh in complex with Hedgehog-Interacting Protein (HHIP), a membrane protein that acts as a negative regulator of the Hh pathway (Figure 1).24 In this complex, HHIP is found to interact with Shh primarily via an extended loop (L2) located in the extracellular domain of HHIP (Figure 2a).25 These previous studies also indicated that the Shh binding site involved in the interaction with the HHIP L2 loop is shared by Patched, as evinced by (a) the sequence similarity between HHIP L2 and a L2-like sequence within Patched extracellular domain, and (b) the ability of a linear HHIP L2-derived peptide (HHIP370–390) to inhibit the Shh/Patched interaction in vitro, albeit with only very weak activity (IC50: 150 μM).25a Based on this information, we envisioned that a macrocyclic peptide encompassing the HHIP L2 loop sequence would provide a viable starting point for the development of an agent capable of disrupting the Shh/Patched protein-protein interaction. In particular, we recognized that the distance between the alpha carbon atoms of residue Met379 and Leu385 within the L2 loop of HHIP (Figure S1) is compatible with the inter-side-chain thioether bridge provided by a peptide macrocyclization method previously reported by our group.22a The latter involves a crosslinking reaction between a cysteine residue and a genetically encodable O-2-bromoethyl-tyrosine (O2beY), which bears a cysteine-reactive alkyl bromide group.22a The side chains of the Met379 and Leu385 residues point away from the HHIP L2-binding cleft in Shh, suggesting that a bridge connecting these positions would not directly interfere with Shh binding. At the same time, the conformational restriction imposed by the inter-side-chain linkage was expected to be beneficial toward improving Shh affinity compared to a linear L2-derived peptide, as a result of reduced entropic costs upon binding to the protein. Based on these considerations, a molecular model of the resulting O2beY/Cys-bridged peptide, called HL2-m1, was generated and docked into the structure of Shh using Rosetta simulations. Briefly, viable conformations of HL2-m1 that accommodate the thioether crosslink were generated based on the crystal structure of the HHIP-Shh complex, followed by energy minimization using the Rosetta FastRe-lax protocol26 in the modeled Shh-bound state. These analyses provided support to the design by showing a good overlap between the backbone of the modeled cyclic peptide and that of HHIP L2 loop in the Shh-bound structure as well as the absence of steric clashes between the thioether bridge and the L2-binding cleft in the Shh protein (Figure 2b).
Figure 2.
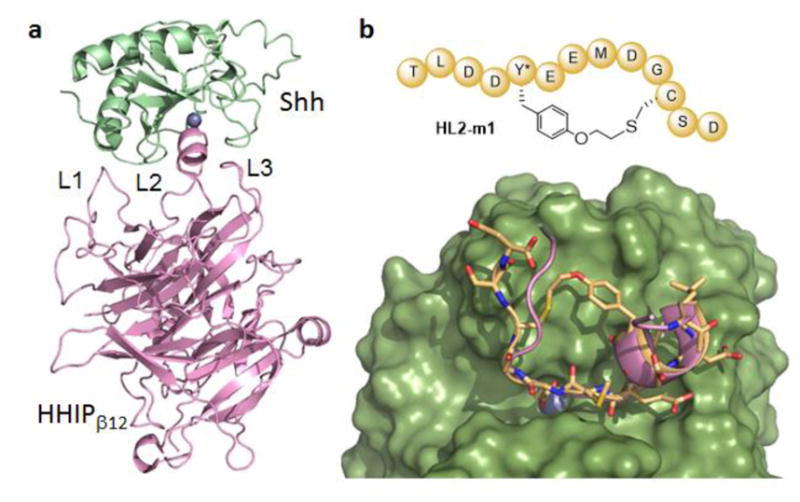
Macrocyclic HHIP L2 loop mimic. (a) Crystal structure of Shh (green) in complex with the extracellular domain of HHIP (pink) (pdb 3HO525a). The three loop regions of HHIP involved in Shh binding are labeled and the zinc ion in the L2 binding cleft of Shh is shown as sphere model (blue). (b) Top: schematic structure of the macrocyclic peptide HL2-m1. Bottom: model of HL2-m1 (yellow, stick model) bound to Shh (green, surface model). The L2 loop of HHIP (pink, ribbon model) is superimposed to the modeled complex.
Characterization of HL2-m1
With the goal of assessing both the biosynthetic accessibility and Shh binding properties of HL2-m1, the designer cyclic peptide was targeted for production by recombinant means. To this end, a gene encoding for a 13mer peptide sequence spanning the HHIP L2 loop (HHIP375–387) was cloned into a pET-based expression vector. The codon corresponding to Met5 residue in the HL2-m1 sequence (Met375 in HHIP) was mutated to an amber stop codon (TAG) to allow for the site-selective incorporation of O2beY via amber stop codon suppression.27 Residue Leu11 (Leu385 in HHIP) was mutated to cysteine to mediate the formation of the desired thioether bridge. The distance between these residues (i/i+6) was expected to be compatible with O2beY/Cys cyclization based on our previous studies with model peptide sequences.22a The HL2-m1 encoding sequence was then fused to an N-terminal FLAG tag for detection purposes (vide infra) and to a C-terminal GyrA intein28 containing a poly-histidine tag to facilitate purification and isolation. The resulting polypeptide construct (FLAG-HL2m1-T-GyrA-H6) was expressed in E. coli cells in the presence of O2beY and a O2beY-specific orthogonal aminoacyl-tRNA synthetase/tRNA pair.22a After protein purification via Ni-affinity chromatography, the FLAG-tagged HL2-m1 peptide was cleaved from the GyrA intein with thiophenol, followed by HPLC purification. MALDI-TOF MS analysis of the thiol-induced cleavage reaction mixture showed the release of the desired cyclic peptide and no detectable amounts of the acyclic peptide (Figure S6a), indicating that O2beY/Cys cyclization had occurred efficiently and quantitatively upon expression in E. coli cells.
To measure the Shh binding affinity of HL2-m1, an in vitro assay was developed in which recombinant GST-fused Shh was immobilized on microtiter plates and then exposed to the FLAG-tagged peptide. The Shh-bound peptide is then quantified colorimetrically (λ450) using a horseradish peroxidase (HRP)-conjugated anti-FLAG antibody. Using this assay, the FLAG-HL2-m1 peptide was determined to bind Shh with a KD of 3.6 μM (Figure 3). In comparison, a linear peptide encompassing the L2 sequence (FLAG-L2-pep) exhibited significantly lower binding affinity for Shh (KD = 20 μM). These results demonstrated the functionality of the designer macrocyclic L2 loop mimic as a Shh targeting agent. In addition, the 5.5-fold higher Shh binding affinity of HL2-m1 compared to its linear counterpart highlighted the anticipated beneficial effect of macrocyclization toward stabilizing the bioactive conformation of the peptide.
Figure 3.
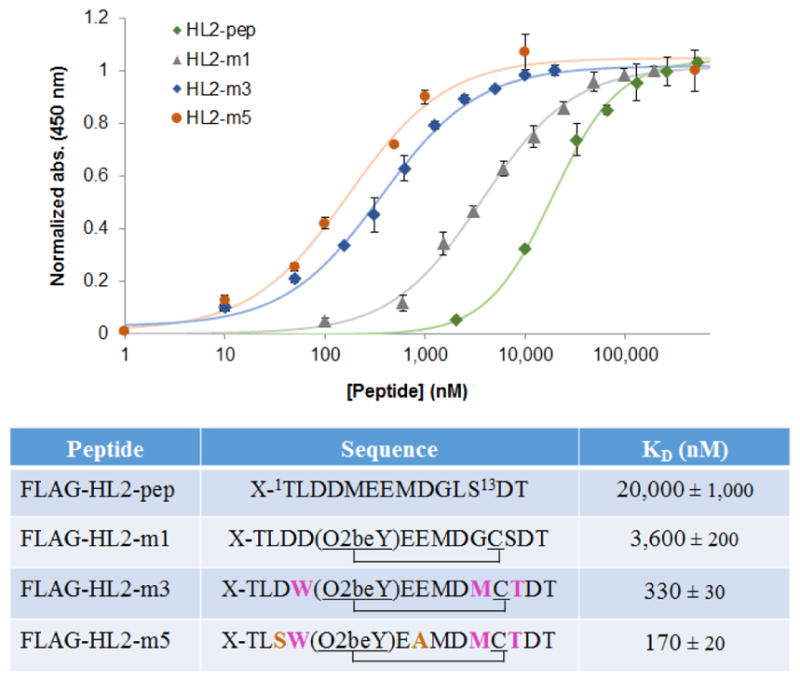
Shh binding affinity of linear and macrocyclic L2 mimics. (a) Dose-response curves for direct binding of the recombinantly produced FLAG tag-fused peptides to plate-immobilized GST-Shh as determined using HRP-conjugated anti-FLAG antibody. (b) Sequences and KD values corresponding to macrocyclic L2 mimics and linear L2-based peptide. X = MDYKDDDDKGSGS-. The mutated positions in the evolved macrocyclic peptides compared to the initial cyclic peptide HL2-m1 are highlighted.
Affinity maturation of macrocyclic HHIP L2 loop mimics
Next, we sought to improve the Shh binding affinity of HL2-m1 by leveraging the ability to genetically encode and produce the macrocyclic peptide in bacterial cells. To this end, the strategy outlined in Figure 4 was applied, which entails the generation of HL2-m1 variant libraries in multiwell plates followed by screening of the recombinantly produced macrocyclic peptides directly in cell lysates. In this system, the amino acid residue (Thr) at the junction between the macrocycle-encoding sequence and the GyrA intein was removed to leave an Asp residue at the ‘intein-1’ position. This residue was previously determined to promote the spontaneous, post-translational cleavage of the C-terminal intein moiety directly in cells, thereby releasing the peptide macrocycle.22a Upon cell lysis, the FLAG-tagged macrocycles are screened for improved Shh binding activity using the colorimetric assay described above.
Figure 4.

Overview of strategy for evolution of macrocyclic peptides. A plasmid library encoding for partially randomized peptide sequences fused to a FLAG tag and a C-terminal GyrA intein are transformed into E. coli cells and arrayed on multiwell plates. The corresponding pre-cursor polypeptides are produced via ribosomal translation and O2beY incorporation via amber stop codon (TAG) suppression. The macro-cyclic peptides are produced inside cells through ‘self-processing’ of the biosynthetic precursors via O2beY/Cys cyclization and aspartate-induced intein cleavage. After cell lysis, peptide binding to immobilized Shh is quantified colorimetrically. The variants showing improved Shh binding activity are deconvoluted via DNA sequencing. The best variant and acquired SAR data are used for the next round of affinity maturation.
For the first round of affinity maturation, five positions within the HL2-m1 macrocycle were selected for mutagenesis based on the modeled HL2-m1/Shh complex (Figure 2b). These positions include three interfacial residues located within the α-helical (Glu6, Glu7) and turn region (Gly10) of the molecule and two solvent-exposed residues neighboring the O2beY/Cys linkage (Asp4, Ser12). In this and subsequent steps (vide infra), residue Asp9 was left unaltered since the corresponding residue in HHIP (Asp383) mediates an energetically important interaction by coordinating a Zn(II) ion in the L2 binding cleft of Shh (Figure S1).25 Accordingly, five macrocycle libraries were prepared via site-saturation mutagenesis (NNK degenerate codon) of the aforementioned positions within the HL2-m1 encoding sequence. For each library, ~90 recombinant clones were arrayed on 96-well plates, followed by in-cell production of the corresponding macrocycles. Upon screening of library with the immunoassay, several library members were found to exhibit improved Shh binding activity compared to the parent compound HL2-m1 (Figure S3). Interestingly, each of the single-site libraries yielded two or more improved variants. Among them, the variant containing a Ser12Met mutation, renamed HL2-m2, emerged as the most promising hit and it was thus selected as the reference compound during the second round of affinity maturation.
Importantly, detailed structure-activity information for each mutated site were gathered at this point by sequencing the multiple hits identified from the initial macrocycle libraries (Figure S3). Based on this information, three second-generation libraries were prepared by recombining beneficial mutations at position 4 (A/D/G/W), 6 (L/S/V/W/E), 7 (K/Y/A/E), 10 (G/M/T), and 12 (L/M/T/S). Upon screening of the resulting libraries (~500 recombinants) according to the strategy of Figure 4, a macrocycle variant (HL2-m3) showing improved Shh binding activity compared to HL2-m2 was identified (Figure S4). By sequencing, HL2-m3 was determined to contain a total of three mutations, namely Asp4Trp, Gly10Met, and Ser12Thr (Figure 3). The cyclic structure of HL2-m3 was further confirmed by expressing this sequence as stable GyrA intein fusion (i.e., by introducing Thr at ‘intein-1’ position), followed by thiol-induced intein cleavage and MS analysis. These tests showed the occurrence of the macro-cyclic peptide as the only detectable species (Figure S6b). After purification, this compound was determined to bind Shh with a KD of 330 nM (Figure 3), corresponding to a 11-fold improvement compared to HL2-m1.
As the next step, all of the yet unmodified positions within the HL2-m3 sequence (relative to HL2-m1) were randomized by site-saturation mutagenesis. From the resulting libraries, an improved HL2-m3-derived variant was obtained, which carries a Glu→Ala mutation at the level of residue 7 (HL2-m4). A second hit carrying a Asp3Ser mutation was also identified at this stage. Upon combining these mutations, a further improved Shh-binding macrocycle was obtained, which was named HL2-m5. HL2-m5 contains a total of five amino acid substitutions compared to HL1-m1 and it undergoes quantitative cyclization in vivo (Figure S6c), further demonstrating the robustness and reliability of the O2beY-mediated peptide cyclization chemistry. Upon purification, FLAG-HL2-m5 was found to bind Shh with a KD of 170 nM (Figure 3), which corresponds to a more than 20-fold in-crease in affinity compared to the initially designed macro-cycle (HL2-m1) and to a nearly 120-fold improvement compared to the linear L2-derived peptide. Altogether, these results supported the effectiveness of the strategy outlined in Figure 4 toward enabling the affinity maturation of the initially designed macrocyclic L2 mimic.
Molecular Modeling and Circular Dichroism Experiments
To gain further insights into the role of the beneficial mutations accumulated in HL2-m5, a model of the cyclic peptide in complex with Shh was generated using Rosetta simulations (Figure S2). Inspection of the complex suggested the occurrence of potential interactions between the Trp4 and Met10 residues of HL2-m5 with regions of the Shh surface that are not contacted by the corresponding residues in the HL2-m1 peptide (Figure 2) or within the L2 loop of HHIP (Figure S1). Specifically, the side chain of Met10 is inserts into the L2 binding cleft of Shh (Figure S2), establishing new contacts between the protein and the HL2-m5 peptide that are not present in the Shh/HHIP complex due to the presence of a Gly residue at this position. Particularly interesting is also the case of Trp4, whose aryl ring inserts into a nearby cleft on the Shh surface according to the energy-minimized model of the complex. Experimentally, the energetic importance of this interaction is corroborated by the identification of an identical mutation, i.e. Asp4→Trp, among the most active compounds isolated from the single site mutagenesis libraries derived from HL2-m1 (Figure S3). On the other hand, the positive effect of the Asp7Ala substitution accumulated in HL2-m5 is supported by the approximately 2-fold higher Shh binding affinity of HL2-m4 compared to HL2-m3. The same substitution was also identified as beneficial during screening of the HL2-m1 derived libraries (Figure S3). Residue 7 is located at the C-terminal end of the two-turn alpha-helix and the beneficial effect of the alanine substitution at this position can be rationalized based on stabilization of an α-helical conformation in this region of the molecule.
To better examine the conformational properties of HL2-m5, a FLAG tag-free version of this peptide along with that of the linear L2-derived peptide (Ac-TLDDMEEMDGLSD-NH2) were prepared by solid-phase peptide synthesis (vide infra) and analyzed by circular dichroism (CD). As shown in Figure 5a, the near-UV CD spectrum of the linear L2-based peptide is consistent with that of a random coil polypeptide, indicating that it lacks a well-defined structure in solution. In contrast, the HL2-m5 macrocycle exhibits more pronounced negative bands in 215–222 nm range along with a positive band in the 190–195 nm region of the CD spectrum (Figure 5b). These features are consistent with the presence of a more structured peptide containing a α-helical motif. In addition, unlike for the linear peptide, intensification of the spectral features of the HL2-m5 macrocycle was observed upon addition of the helix-inducing solvent trifluoroethanol.29 Thus, in addition to more favorable contacts with Shh surface as suggested by molecular modeling, the improved Shh binding affinity of HL2-m5 compared to the linear L2 peptide likely arises from a stabilization of the bioactive conformation as a result of the cyclic backbone and other sequence alterations (e.g., Asp7Ala mutation).
Figure 5.
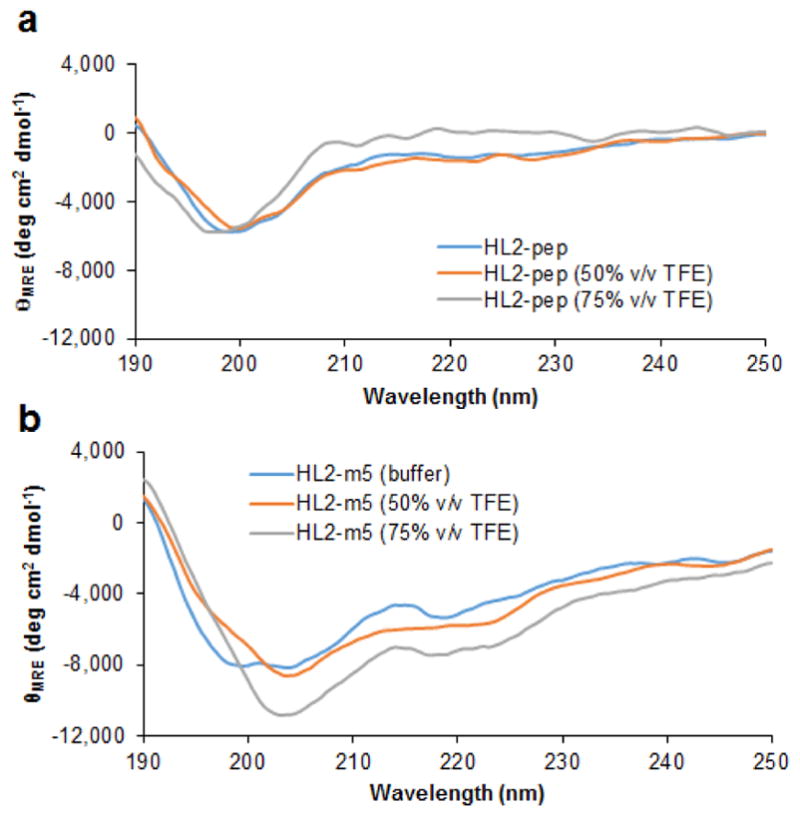
Circular dichroism (CD) spectra corresponding to the linear HL2-pep (a) and the macrocyclic HL2-m5 peptide (b) in buffer and in the presence of trifluoroethanol (TFE) at varying concentration. The signals are reported as mean residue molar ellipticity (θMRE).
Synthesis of Macrocyclic Peptides via SPPS
To provide further access to the macrocyclic peptides, a synthetic strategy was devised to afford these compounds. Inspired by approaches previously adopted for the synthesis of lantibiotics,30 this strategy involves the incorporation of a dipeptide building block encompassing the O2beY/Cys thioether crosslink during solid-phase peptide synthesis (SPPS), followed by on-resin cyclization and cleavage/deprotection of the peptide from the resin (Scheme 1). As shown in Scheme 1a, the dipeptide building block 7 was prepared via alkylation of N-Alloc-(O-2-bromoethyl)-tyrosine allyl ester (3) with N-Fmoc-(L)-cysteine tert-butyl ester (6), followed by removal of the tert-butyl group under acidic conditions. For the synthesis of HL2-m5, the first two C-terminal amino acids were loaded on a Rink amide MBHA resin, followed by incorporation of the dipeptide building block via amide coupling with COMU, yielding 8. The peptide chain was then further extended to include amino acid residues Met10 to Glu7, affording 9. The side-chain Alloc and allyl ester protecting groups were then removed using Pd(PPh3)4 catalyst in the presence of PhSiH3, whereas the N-terminal amino group was exposed via Fmoc deprotection. On-resin cyclization was then realized under amide coupling conditions with PyBOP and HOBt in the presence of DIPEA, to afford 11. The peptide was then further extended via SPPS to include the N-terminal tail of the peptide, followed by Fmoc deprotection and N-acetylation. The synthesis of HL2-m5 was completed by cleavage of the peptide from the resin using a 95:2.5:2.5 trifluoroacetic acid/TIS/water mixture. After purification by reverse-phase HPLC, the desired macrocyclic peptide was obtained with an overall yield of 1.5% (Figure S8). The same protocol could be then applied to afford HL2-m1 (Figure S9) in comparable yields.
Scheme 1.

Synthesis of macrocyclic peptides. (a) Synthetic route for the preparation of the diamino acid building block encompassing the Cys/O2beY thioether linkage. (b) Solid-phase syn-thesis of macrocyclic peptide HL2-m5.
Suppression of Hedgehog Pathway Activation in Living Cells
Having demonstrated the ability of HL2-m5 to target Shh in vitro, we next examined its activity toward disrupting Shh-mediated Hedgehog pathway signaling in cells. To this end, we utilized a cell-based luciferase reporter assay,31 in which mouse embryo fibroblasts (NIH3T3) are transfected with vectors encoding for a Firefly luciferase (FF) gene under a Gli-controlled promoter and a Renilla luciferase (Ren) gene under a constitutive promoter. Hh pathway suppression is measured based on the decrease in Firefly/Renilla luminescence ratio in the presence of the inhibitor. In preliminary experiments, this assay was validated using the Smo inhibitor cyclopamine, which caused full inhibition of Shh-induced luminescence in the cells at a concentration of 10 μM, in accordance with previous reports.31
After transfection with the luciferase reporter plasmids, NIH3T3 cells showed strong luminescence in the presence of recombinant N-palmitoylated Sonic Hedgehog (Shh-N) and low luminescence in the absence of Shh-N, thereby confirming Shh-dependent activation of the Hedgehog pathway in the cells. Upon incubation of Shh-N-stimulated cells with HL-m5, a dose-dependent suppression of the luminescence signal was observed (Figure 6a), from which a half-maximal inhibitory concentration (IC50) of 250 nM was determined. A residual pathway activity was observed at the highest dose tested, which could be attributed to the limited solubility of the peptide at a concentration ≥ 5–10 μM in the medium used for the cell-based assay. In contrast to the cyclic peptide, the linear L2-pep peptide showed no inhibitory activity at concentrations up to 30 μM under identical conditions. Incubation of HL-m5-treated cells with purmorphamine, a Smo agonist,32 restored activation of the signaling pathway (Figure 6b), demonstrating that HL2-m5-dependent inhibition occurs the level of Shh/Patched interaction. Notably, the inhibitory activity of HL2-m5 toward blocking Hedgehog pathway activation in cells is nearly two orders of magnitude higher than that of robotnikinin (IC50 ~ 15 μM),19 as determined using a similar cell-based assay. Noteworthy is also the fact that the IC50 value exhibited by HL2-m5 in the cell-based assay (250 nM) is very similar to the KD value measured for FLAG-HL2-m5 in the in vitro Shh binding assay (170 nM, Figure 3) and to the IC50 value of HL2-m5 determined using this assay in a competition format (280 nM, Figure S10). These results indicate that the macrocyclic peptide targets Shh with high affinity and specificity even in the presence of cells and a complex growth medium.
Figure 6.
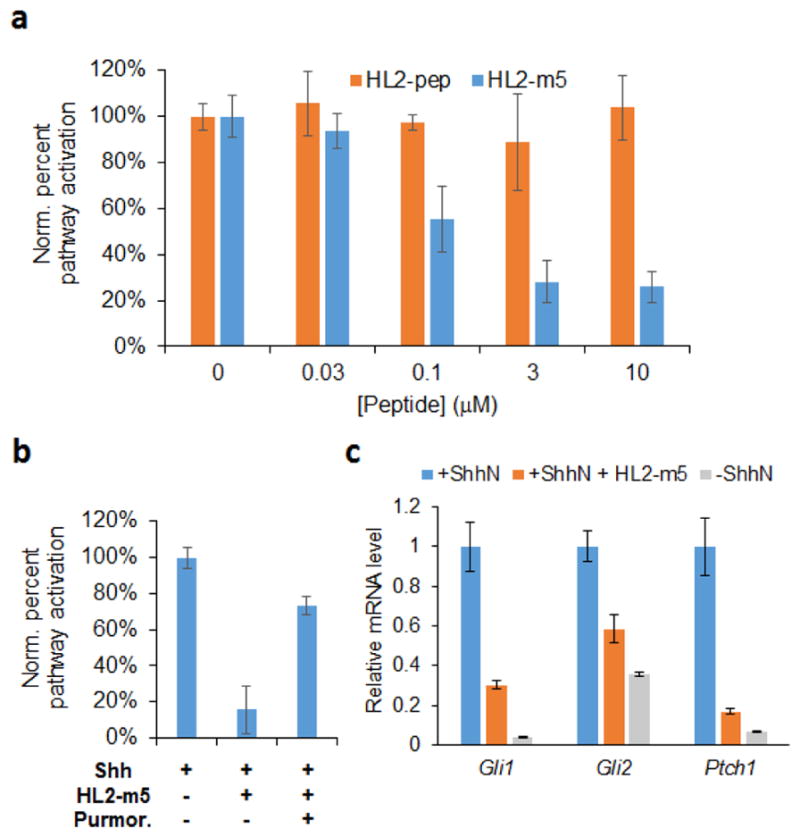
HL2-m5-induced suppression of Hh pathway signaling. (a) Dose dependent inhibition of luciferase expression (FF/Ren ratio) in Shh-stimulated NIH3T3 cells containing a dual luciferase reporter system. (b) Restoration of Hedgehog pathway signaling upon addition of purmorphamine (5 μM) to cells treated with HL2-m5 (10 μM). (c) Relative transcriptional levels of Gli1, Gli2, and Ptch1 genes in Shh-stimulated NIH3T3 cells in the presence and in the absence of HL2-m5 (10 μM) as determined by real-time PCR. mRNA levels in unstimulated cells are included for comparison.
To further validate HL2-m5 as a Hedgehog pathway antagonist, the effect of this compound on the transcriptional activity of two canonical target genes of the pathway, Gli1 and Ptch1 (Figure 1) was examined via real-time PCR. As shown in Figure 6c, a significant reduction (75–85%) of the mRNA levels corresponding to these genes was observed in ShhN-stimulated cells upon incubation with HL2-m5 at 10 μM, relative to compound-untreated cells. Treatment with the macrocyclic peptide also suppresses the mRNA level for the transcription factor Gli2. For both Ptch1 and Gli2, the corresponding transcriptional levels in HL2-m5-treated cells approach those observed in unstimulated cells grown in the absence of Shh-N ligand (Figure 6c). No changes in cell morphology, growth behavior and titer were noted in the presence of HL2-m5, indicating a lack of cytotoxicity at the highest concentration range applied in these experiments. Taken together, these results demonstrate that the macrocyclic peptide is able to potently inhibit Shh-dependent Hedgehog pathway activation in living cells and suppress signature transcriptional responses resulting from ligand-induced stimulation of the pathway.
Hedgehog Analog Selectivity
While Shh is the most abundant analog among Hedgehog proteins, paracrine/autocrine Hedgehog signaling in normal and cancer cells is also mediated by the Indian (Ihh) and Desert (Dhh) analogs.33 Hh-targeted inhibitors capable of targeting multiple analogs of this protein are thus expected to be particularly useful toward suppressing ligand-induced activation of this pathway. Since the Hh analog selectivity of robotnikinin had not previously investigated, this property was examined by means of a competition assay, whereby inhibition of FLAG-HL2-m5 binding to plate-immobilized GST-Shh, GST-Ihh, or GST-Dhh is measured via the HRP-conjugated anti-FLAG antibody. These experiments showed that robotnikinin has significantly lower affinity toward Ihh and Dhh relative to Shh (Figure 7). By comparison, HL2-m5 was found to interact with all three analogs of Hh proteins, showing nearly identical activity toward Shh and Ihh (Figure 7). Consistent with this trend, direct binding experiments showed that HL2-m5 interacts with Ihh and Dhh with a KD of 160 and 330 nM, respectively (Figure S11). Thus, the affinity of HL2-m5 for Ihh and Dhh is nearly identical and only two-fold lower, respectively, than that for Shh (170 nM) as determined using the same assay. These results indicate that the macrocyclic peptide can act as an effective inhibitor for all analogs of the Hedgehog protein.
Figure 7.
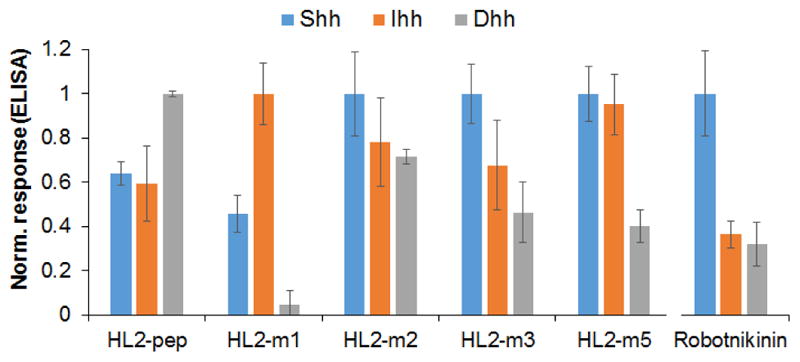
Hh analog selectivity of linear and cyclic L2 mimics. Data relative to the peptides are derived from direct binding experiments to immobilized Hh proteins. Data relative to robotnikinin are derived from competition experiments (10 μM robonikinin + 400 nM FLAG-HL2-m5). For each compound, values are normalized to the highest binding response measured across the three Hedgehog analogs.
In the interest of determining how the affinity maturation process affected the Hh analog selectivity of the peptides, these experiments were extended to the other linear and cyclic L2 mimics. These analyses showed that the linear L2-based peptide (HL2-pep) binds preferentially to Dhh over Shh and Ihh (Dhh > Shh ≈ Ihh; Figure 7). This selectivity profile mirrors that of full-length HHIP.34 Interestingly, macrocyclization of the L2 peptide sequence via the O2beY/Cys linkage (= HL2-m1) results in a complete shift in Hh analog selectivity (Ihh > Shh ≫ Dhh), leading to a preference for Ihh over Shh and nearly abolishing its affinity for Dhh. In the HL2-m1 → HL2-m2 transition, the selectivity is then shifted toward Shh. This result is reasonable given that improved Shh binding was the selection criteria applied during library screening process. At the same time, it is interesting to see how a single mutation accumulated during this step (Ser12Met) restores binding to Dhh and leads to Hh analog cross-reactivity (Figure 7). With HL-m3, the preference for Shh over the other two Hh analogs becomes more pronounced (Shh > Ihh ≈ Dhh). As noted above, HL2-m5 shows comparable affinity toward Shh and Ihh and higher preference toward these analogs over Dhh (Shh ≈ Ihh > Dhh). Altogether, these results illustrate the potential of tuning the Hh analog selectivity of these macrocyclic peptide scaffolds.
CONCLUSION
In summary, we have reported the development of a potent macrocyclic peptide inhibitor of the Shh/Patched interaction, a key protein-protein interaction implicated in the activation of the Hedgehog pathway. HL2-m5 binds Shh with high affinity in vitro and is able to effectively suppress Shh-mediated stimulation of Hedge-hog pathway signaling in living mammalian cells. The in-hibitory activity of HL2-m5 is about two orders of magnitude higher than that of robonitnikinin, the only compound previously reported to target the Shh/Patched interaction. Furthermore, unlike robonitnikinin, HL2-m5 exhibits high affinity toward all three analogs of the Hedgehog protein. HL2-m5 shows also promising stability against proteolytic degradation (t1/2 > 6–8 hours in blood serum; Figure S12). Collectively, these features should make HL2-m5 a valuable probe for investigating the biological role and therapeutic potential of the Hh/Patched interaction in the context of pathologies that are associated with aberrant ligand-dependent activation of the Hedgehog pathway.
From a methodological standpoint, this work demonstrates the value of the strategy outlined in Figure 4 toward the development of potent and selective macrocyclic peptide disruptors of protein-protein interactions. Using this approach, a low-affinity linear peptide encompassing a Shh recognition motif from HHIP could be rapidly evolved into a high-affinity Shh-targeting agent (120-fold lower KD) through the generation and screening of macrocyclic peptide libraries generated in bacteria. This process was further facilitated by the ability to produce and isolate the macrocyclic peptides by recombinant means, which expedites hit evaluation in secondary functional assays. At the same time, an efficient methodology was implemented to afford these compounds by synthetic means, which will facilitate further optimization of these compounds using non-proteinogenic amino acids. We expect that the overall strategy presented here will prove valuable for the development of bioactive cyclopeptides against a variety of other challenging protein-protein interactions.
EXPERIMENTAL PROCEDURES
Cloning, expression, and purification of GST-Hh proteins
Vectors containing human Shh, Ihh and Dhh genes were kindly provided by the Riobo-Del Galdo laboratory.34 Genes encoding for Shh, Ihh, and Dhh were amplified by PCR (primers #1–6; Table S1) and cloned into the Nco I/Xho I cassette of the expression vector pET42b (Novagen), resulting in the C-terminal fusion of the Hh protein sequence to that of glutathione-S-transferase (GST) protein containing a poly-histidine tag. The GST-Hh fusion proteins were expressed in BL21(DE3) cells by growing recombinant cells in LB medium with kanamycin (30 μg/mL). At an OD600 of 0.6, cells were induced with IPTG (1 mM) and grown for 20 hours at 27°C. The proteins were purified by Ni-NTA chromatography (Invitrogen) according to the manufacturer’s instructions. After elution, the proteins were buffer exchanged with PBS buffer (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4) and stored at −80°C. The identity and purity of the purified proteins were confirmed by MS spectrometry and SDS-PAGE (Figure S5).
Cloning of HL2-m1 constructs
A DNA sequence encoding for the HL2-m1 sequence fused to a N-terminal FLAG tag and a C-terminal GyrA intein from M. xenopi28 (MDYKDDDDK-(GS)2-TLDD(stop) EEMDGCSD-T-(GyrA)) was assembled by PCR. The resulting gene was cloned into the BamH I/Xho I cassette of the expression vector pET22b (Novagen), resulting in the fusion of poly-histidine (H6) tag to the C-terminus of the intein. Using a similar procedure, a ‘self-cleaving’ vari-ant22a of the FLAG-HL2-m1-GyrA construct was prepared by removing the Thr residue preceding the intein sequence (= ‘intein-1’ position), thereby leaving an Asp residue at the intein-1 position. In a similar manner, a stable and a self-cleaving variant of the FLAG tag-fused L2-derived peptide (= MDYKDDDDK-(GS)2- TLDDMEEMDGLSD-(T)-(GyrA))) were prepared. The sequences of the recombinant vectors were confirmed by DNA sequencing.
Library construction and screening
The single-site site-saturation libraries were constructed via overlap extension PCR using pET22_FLAG-(HL2-m1)-D-GyrA as the template and the appropriate mutagenizing primers (NNK codon at target position; forward primers #12–16, #23, #24, reverse primer #8; Table S1). The PCR product was cloned into the BamH I/Xho I cassette of pET22_FLAG-(HL2-m1)-D-GyrA. The recombinant plasmids were transformed in DH5α cells and selected on LB plates containing ampicillin (100 μg/mL). The re-combination libraries were prepared in a similar manner but using primers with partially randomized codons (codons: KGB, WAM, KGG, WYG, AYG; forward primers #17–22; reverse primer #08) to encode for the desired subset of amino acids at each target position. The resulting plasmid libraries were pooled and transformed into cells containing a pEVOL_O2beY-RS vector22a en-coding for of the orthogonal O2beY-RS/tRNACUA pair. Recombinant cells were selected on LB plates containing ampicillin (100 μg/mL) and chloramphenicol (34 ug/mL) and individual colonies from these plates were used to inoculate 1.0 mL LB media containing the two antibiotics in 96-deep well plates. After overnight growth at 37°C, 50 μL from each well was used to inoculate a replica plate containing 1 mL M9 medium containing ampicillin (100 μg/mL) and chloramphenicol (34 μg/mL). Cells were grown to an OD600 of 0.6 in a plate shaker at 37°C, and then induced with arabinose (0.06% m/v) and O2beY (2 mM). After one hour, cells were induced with IPTG (1 mM) and grown at 27°C for 18–20 hours. For library screening, the 96-well plate cell cultures were pelleted by centrifugation and then washed once with PBS buffer. Cell pellets were then resuspended in lysis buffer (50 mM potassium phosphate, 150 mM NaCl, 10 mM MgCl2, 0.8 μg/ml DNase, 0.8 mg/mL lysozyme, pH 7.5) and incubated for 1 hour and 15 minutes at 37°C. After centrifugation, 200 uL of the clarified lysate was used for measuring Shh binding activity using the immunoassay described further below (see Hedgehog binding assay). Positive hits were identified upon comparison with the reference macrocycle and then validated through rescreening in triplicate using the same overall procedure and assay. The validated hits were then deconvoluted via DNA sequencing of the plasmids extracted from the master plate.
Recombinant synthesis and purification of macrocyclic peptides
Plasmids for the expression of stable GyrA intein fusions of the macrocyclic peptides were prepared by substituting the Asp residue at the ‘intein-1’ position with Thr in the corresponding pET22-based plasmids via site-directed mutagenesis. The plasmids were co-transformed along with the pEVOL_O2beY-RS plasmid into E. coli BL21(DE3) cells. The recombinant cells were grown in LB media containing ampicillin (100 μg/mL) and chloramphenicol (34 μg/mL) overnight at 37°C. Overnight cultures were then used to inoculate 1.0 L M9 media ampicillin (100 μg/mL) and chloramphenicol (34 μg/mL). After growth at 37°C to an OD600 of 0.6, the cells were induced with arabinose (0.06% m/v) and O2beY (2 mM). After one hour, cells were induced with IPTG (1 mM) and grown at 27°C for 18–20 hours. The GyrA-fused peptides were purified by Ni-NTA chromatography (Invitrogen) and the eluted proteins were buffer exchanged with potassium phosphate buffer (10 mM potassium phosphate, 150 mM NaCl, pH 7.5). Cleavage of the intein was carried out using a 200 μM solution of purified proteins in potassium phosphate buffer containing 20 mM TCEP and 10 mM thiophenol at pH 8.5. The reaction mixtures were incubated over-night at room temperature with gentle shaking and then dialyzed against water. The cleaved peptide was purified using solid phase extraction with a step gradient of acetonitrile in water (+ 0.1% TFA). The peptides generally eluted between 10% and 25% acetonitrile. After lyophilization, the peptides were further purified by reverse-phase HPLC using a Grace C18 column (monomeric; 120Å; 250 × 10 mm) and a 5→95% gradient of acetonitrile in water (+ 0.1% TFA). The peptide identity was confirmed by MALDI-TOF MS (Figure S7) and the concentration was determined by HPLC (OD220) using a calibration curve generated with a reference peptide of identical length. Typical yields for the recombinantly produced cyclic peptides obtained using this procedure was between 0.5 and 1.5 mg/L culture.
Molecular modeling
The peptide variants were mapped onto the backbone scaffold derived from an HHIP L2 loop structure in the Shh-bound state (PDB code: 3HO525a; residues 375–387 chain B). Based on visual examination of the L2 structure, the backbone psi dihedrals of residue 383 was perturbed by up to 37.5° and RosettaMatch35 was used to select backbone conformations that accommodate the O2beY-Cys thioether crosslink between positions 5 and 11. Geometric constraints to model the preferred geometry of the O2beY-Cys thioether crosslink were derived from the Cambridge Structural Database. The resulting conformations were optimized using Rosetta FastRelax protocol.26 Total and per-residue energies of the macrocycle residues were used for scoring, and visual examination of models was used to identify favorable interactions. During the energy minimization, geometric constraints were placed on both the metal-chelating Asp10 residue and the O2beY/Cys crosslink. Atom coordinate constraints were placed for backbone atoms of Shh residues outside of the L2 binding cleft to maintain them in their crystallographic conformations. All Rosetta files required to perform simulations are provided as Supporting Information.
Synthesis of O2beY and dipeptide building block
O2beY was synthesized as described previously.36 Detailed procedures for the synthesis of 7 are provided as Supporting Information.
Solid-phase peptide synthesis
The macrocyclic peptides were manually synthesized via standard solid-phase Fmoc chemistry using MBHA rink amide resin (loading: 0.25 mmol/g) in a polypropylene reaction vessel. Standard Fmoc-protected amino acids were used as building blocks, with the exception of Asp10, for which N-Fmoc-Asp(OEpe)CO2H was used to avoid aspartimide formation. Loading of the first amino acid and subsequent elongation steps were carried out using 5 equiv Fmoc-protected amino acid preactivated with COMU (4.95 equiv) and DIPEA (10 equiv) in DMF for 1 hour at room temperature. The Fmoc protecting group was removed with 30% piperidine in DMF (2 × 10 min). To introduce the dipeptide building block, compound 7 (75 mg 0.11 mmol) was pre-activated with COMU/DIPEA in DMF and added to the resin for 3 hours at room temperature. Prior to the cyclization reaction, deprotection of the Al-loc/allyl groups was carried out using Pd(PPh3)4 (1 equiv)/PhSiH3 (20 equiv) in dry DCM (2 × 45 minutes). Peptide cyclization was carried out at millimolar pseudo-dilution using a mixture of PyBop (2 equiv), HOBt (2equiv), and DIPEA (4 equiv) in DMF, for two cycles of 12 hours. After addition of the last amino acid in the sequence, the resin-bound peptide was acetylated by two treatments with a mixture of acetic anhydride (0.5 M), DIPEA (0.015 M, and HOBt (0.125 M) in DMF for 10 minutes. The peptides were cleaved from the solid support using a solution of TFA:H2O:TIS (95:2.5:2.5 v/v/v) for 3 hours at room temperature. After removal of the resin by filtration, the crude peptide was precipitated with cold MTBE, re-dissolved in 1:1 water/acetonitrile solution, and lyophilized. The crude peptide was purified by reverse-phase HPLC using an Agilent 1200 system equipped with a Grace C18 column (10 μm; 90 Ǻ; 250 × 10 mm) at a flow rate of 2.5 mL/min and a linear gradient starting from 20% to 80% acetonitrile in water (+ 0.1% TFA) over 25 min. The purity and identity of all peptide was confirmed by analytical HPLC and LC-MS (Figure S8–S9). The overall yield of the macrocyclic peptides obtained using this procedure was around 1.5%.
Hedgehog binding assay
Shh binding activity/affinity of the linear and cyclic peptides was measured using the immunoassay outlined in Figure 4. For these experiments, GST-Shh was immobilized on microtiter plates by incubating 100 μL of a 4 μM GST-Shh solution in PBS buffer overnight at 4°C, followed by washing (3 × 150 μL PBS with 0.5% Tween-20) and blocking with 0.5% BSA in PBS for 1.5 hours at room temperature. After washing, each well was incubated with 200 μL cell lysate for 1 hour at room temperature (for library screening). For the KD determination experiments (Figure 3), each well was incubated under the same conditions with 100 μL of purified FLAG-fused peptide at varying peptide concentrations. The FLAG-tagged peptides were prepared by recombinant means as described above (see Recombinant synthesis and purification of macrocyclic peptides). After washing, each well was incubated with 100 μL of 1:2,500 dilution of HRP-conjugate mouse anti-FLAG polyclonal antibody (Sigma-Aldrich) for 1 hour at room temperature. After washing, 100 μL of 2.2 mM o-phenylenediamine dihydrochloride, 4.2 mM urea hydrogen peroxide, 100 mM dibasic sodium phosphate and 50 mM sodium citrate, pH 5.0 was added to each well, followed by measurement of the absorbance at 450 nm after 10–20 minutes using a Tecan Infinite 1000 plate reader. Equilibrium dissociation constants (KD) were determined by fitting the dose-response curves (Figure 3) to a 1:1 binding isotherm equation via non-linear regression using SigmaPlot. KD values for HL2-m5 binding to Ihh and Dhh were determined in a similar manner using GST-Ihh and GST-Dhh-coated plates, respectively. The peptide relative binding affinity for the three analogs of Hedgehog (Figure 7) was determined using the same assay and peptide solutions at a fixed concentration of 0.5–1 μM. In this case, binding responses were subtracted against the blank (no peptide sample) and normalized to the highest value measured across the three Hh analogs. Mean values and standard deviations were calculated from experiments performed at least in triplicate.
Competition assay
A PBS solution (100 uL) containing 10 μM robotnikinin and 400 nM FLAG-HL2-m5 was added to GST-Shh-, GST-Ihh-, and GST-Dhh-coated wells in a microtiter plate. The plates were then treated and developed as described above. The relative affinity of robotnikinin for the three Hh analogs was expressed as follows: (1 − % inhibition)GST-Hh/(1 − % inhibition)GST-Shh, where % inhibition in the presence of GST-Shh was 34%. Mean values and standard deviations were calculated from experiments performed in triplicate. The same assay was applied to determine the IC50 value for the inhibition of FLAG-HL2-m5 binding to immobilized GST-Shh induced by the synthetic peptide HL2-m5 (Figure S10).
Circular dichroism analyses
CD analyses were performed using solutions of the purified, synthetic peptides at a concentration of 0.4 μM in 20 mM potassium phosphate buffer (pH 7) in the absence and in the presence of trifluoroethanol at 50% or 75% (v/v). CD spectra were recorded at 26°C at a scan rate of 50 nm/min with a bandwidth of 1 nm and an averaging time of 10 seconds per measurement using a JASCO J-1100 CD spectrophotometer. The raw signal (θd, mDeg) was background subtracted against buffer and converted to molar residue ellipticity (θMRE) using θMRE = θd/(c l nR), where c is the peptide concentration (M), l is the path length (1 mm), and nR is the number of residues in the peptide.
Serum Stability Assay
The serum stability assay was carried out by dissolving the peptide at a final concentration of 25 μM in 300 uL of 50% human male serum (Sigma Aldrich) in 20 mM potassium phosphate buffer (pH 7.0). Prior to the assay, the serum was clarified by centrifugation at 14,000 rpm for 15 minutes and pre-activated at 37°C for 10 minutes. Each peptide was incubated at 37°C and aliquots (45 μL) were removed over the course of 26 hours and quenched with 45 μL of 20% of trichloroacetic acid solution, followed by incubation at 4°C for 15 minutes and centrifugation at 14,000 rpm for 5 minutes. The supernatants were analyzed by analytical RP-HPLC (Grace Vision HT C18 HL column; 21.2 × 250 mm; 5μ) using a gradient from 10 to 75% of acetonitrile (0.1% TFA) in water (0.1% TFA) at a flow rate of 1 mL/min. The residual peptide was quantified based on the corresponding peak area (210 nm) as relative to the sample at time zero. Mean values and standard deviations were calculated from experiments performed in triplicate.
Gli-reporter Assay
NIH3T3 cells (AATC CRL-1658) were passaged twice and then plated in 24-well culture dishes at 5 × 105 cells/well in DMEM containing 10% FBS and 1% penicillin/streptomycin. After 24 hours, the cells were transfected (TransIT-2020) with a mixture of a firefly luciferase reporter construct under the control of a Gli1 inducible promoter and a Renilla luciferase reporter construct under a constitutive promoter (40:1) (Cignal GLI Reporter Luciferase Kit, Qiagen). Cells were allowed to reach con-fluency, at which point the media was changed to Opti-MEM containing 1% FBS and added with 4 nM Shh-N (R&D Systems, Minneapolis, MN) in sterile PBS buffer. Synthetic HL2-pep and HL2-m5 were added at the same time at varying concentration (0.01–30 μM), and control cells were prepared by adding vehicle only (1% DMSO). Purmorphamine-treated cells were prepared by adding 5 μM purmorphamine to wells containing 10 μM HL2-m5. After growth for 24 hours at 37°C in a humidified chamber, the cells were harvested and analyzed for Firefly and Renilla luciferase activity using a Tecan Spark-20 plate reader and a DLR kit (Promega) according to the manufacturer’s instructions. Luminescence values were normalized to those of the Shh pathway activated control cells. Mean values and standard deviations were calculated from experiments performed at least in duplicate.
Gene transcription analyses
NIH3T3 cells we passaged twice and plated at a density of 1:3 in DMEM containing 10% FBS and 1% penicillin/streptomycin in 6-well cell culture dishes. Cells were allowed to reach confluency, at which point the media was changed to Opti-MEM containing 1% FBS and added with Shh-N (4 nM). At the same time, the cells were incuated with HL2-m5 (25 uM) or vehicle-only (1% DMSO). After growth for 24 hours at 37°C in a humidified chamber, the cells were harvested, and total mRNA was collected using TRIazol reagent (ThermoFisher) according to the manufacturer’s instructions. cDNA was generated using 1 ug of mRNA using First-Strand RT-PCR with random hexamers (Super-Script First-Strand RT-PCR, ThermoFisher). The relative amounts of Gli1, Gli2, and Ptch1 mRNA transcripts were determined by real-time PCR (Bio-Rad CFX thermocycler) using the primers listed in Table S1 and SYBR green TAQ reagent (Bio-Rad) according to the manufacture’s protocol. The mRNA levels for the biomarker genes were normalized to that of the reference house-keeping gene cyclophilin. Mean values and standard deviations were calculated from measurements performed in quadruplicate.
Supplementary Material
Acknowledgments
We are grateful to Prof. Natalia Riobo-Del Galdo (University of Leeds) for providing plasmids containing genes of human Hedgehog proteins and to Prof. Vera Gorbunova (U. Rochester) for providing access to the Tecan Spark-20 plate reader and Bio-Rad CFX thermocycler. This research was supported by the U.S. National Institute of Health grant CA187502 awarded to R.F. and U.S. National Science Foundation grant MCB-1330760 awarded to S.K. MS instrumentation at the University of Rochester was supported by the National Science Foundation grants CHE-0840410 and CHE-0946653.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. Supporting figures and graphs, procedures for synthesis of compound 7, oligonucleotide sequences, compound characterization data (HPLC chromatograms, NMR spectra, MS spectra, protein gels), Rosetta files. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Chiang C, Ying LTT, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Nature. 1996;383:407. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]; (b) Ingham PW, McMahon AP. Gene Dev. 2001;15:3059. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]; (c) Ingham PW, Placzek M. Nat Rev Genet. 2006;7:841. doi: 10.1038/nrg1969. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ingham PW, McMahon AP. Genes Dev. 2001;15:3059. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]; (b) Jiang J, Hui CC. Dev Biol. 2008;15:801. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wong SY, Reiter JF. Curr Top Dev Biol. 2008;85:225. doi: 10.1016/S0070-2153(08)00809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carpenter D, Stone DM, Brush J, Ryan A, Armanini M, Frantz G, Rosenthal A, de Sauvage FJ. Proc Natl Acad Sci USA. 1998;95:13630. doi: 10.1073/pnas.95.23.13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denef N, Neubuser D, Perez L, Cohen SM. Cell. 2000;102:521. doi: 10.1016/s0092-8674(00)00056-8. [DOI] [PubMed] [Google Scholar]
- 5.(a) Wen X, Lai CK, Evangelista M, Hongo JA, de Sauvage FJ, Scales SJ. Mol Cell Biol. 2010;30:1910. doi: 10.1128/MCB.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim J, Kato M, Beachy PA. Proc Natl Acad Sci USA. 2009;106:21666. doi: 10.1073/pnas.0912180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P. J Biol Chem. 2002;277:5548. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]; (b) Kasper M, Regl G, Frischauf AM, Aberger F. Eur J Cancer. 2006;42:437. doi: 10.1016/j.ejca.2005.08.039. [DOI] [PubMed] [Google Scholar]
- 7.(a) Rubin LL, de Sauvage FJ. Nature Rev Drug Discov. 2006;5:1026. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]; (b) Theunissen JW, de Sauvage FJ. Cancer Res. 2009;69:6007. doi: 10.1158/0008-5472.CAN-09-0756. [DOI] [PubMed] [Google Scholar]
- 8.(a) Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, Warmuth M. Cancer Cell. 2008;14:238. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]; (b) Hegde GV, Peterson KJ, Emanuel K, Mittal AK, Joshi AD, Dickinson JD, Kollessery GJ, Bociek RG, Bierman P, Vose JM, Weisenburger DD, Joshi SS. Mol Cancer Res. 2008;6:1928. doi: 10.1158/1541-7786.MCR-08-0142. [DOI] [PubMed] [Google Scholar]; (c) Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, Munchhof M, VanArsdale T, Beachy PA, Reya T. Nature. 2009;460:652. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watkins DN, Berman DM, Burkholder SG, Wang BL, Beachy PA, Baylin SB. Nature. 2003;422:313. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 10.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo CF, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. Nature. 2003;425:851. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berman DM, Karhadkar SS, Maitra A, de Oca RM, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. Nature. 2003;425:846. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 12.Liu SL, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Cancer Res. 2006;66:6063. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Reya T, Morrison SJ, Clarke MF, Weissman IL. Nature. 2001;414:105. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]; (b) Beachy PA, Karhadkar SS, Berman DM. Nature. 2004;432:324. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 14.(a) Stanton BZ, Peng LF. Mol Biosyst. 2010;6:44. doi: 10.1039/b910196a. [DOI] [PubMed] [Google Scholar]; (b) Peukert S, Miller-Moslin K. ChemMedChem. 2010;5:500. doi: 10.1002/cmdc.201000011. [DOI] [PubMed] [Google Scholar]; (c) Sharpe HJ, Wang W, Hannoush RN, de Sauvage FJ. Nat Chem Biol. 2015;11:246. doi: 10.1038/nchembio.1776. [DOI] [PubMed] [Google Scholar]
- 15.Cooper MK, Porter JA, Young KE, Beachy PA. Science. 1998;280:1603. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 16.Robarge KD, Brunton SA, Castanedo GM, Cui Y, Dina MS, Goldsmith R, Gould SE, Guichert O, Gunzner JL, Halladay J, Jia W, Khojasteh C, Koehler MFT, Kotkow K, La H, LaLonde RL, Lau K, Lee L, Marshall D, Marsters JC, Murray LJ, Qian CG, Rubin LL, Salphati L, Stanley MS, Stibbard JHA, Sutherlin DP, Ubhayaker S, Wang SM, Wong S, Xie ML. Bioorg Med Chem Lett. 2009;19:5576. doi: 10.1016/j.bmcl.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 17.Owen TS, Xie XJ, Laraway B, Ngoje G, Wang CY, Callahan BP. Chembiochem. 2015;16:55. doi: 10.1002/cbic.201402421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Ericson J, Morton S, Kawakami A, Roelink H, Jessell TM. Cell. 1996;87:661. doi: 10.1016/s0092-8674(00)81386-0. [DOI] [PubMed] [Google Scholar]; (b) Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Genes Dev. 2010;24:2001. doi: 10.1101/gad.1951710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, Taveras KM, Hyman JM, Lee SW, Koehler AN, Chen JK, Fox JL, Mandinova A, Schreiber SL. Nat Chem Biol. 2009;5:154. doi: 10.1038/nchembio.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. Nucleic Acids Res. 2015;43:D512. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Driggers EM, Hale SP, Lee J, Terrett NK. Nat Rev Drug Discov. 2008;7:608. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]; (b) Robinson JA, Demarco S, Gombert F, Moehle K, Obrecht D. Drug Discov Today. 2008;13:944. doi: 10.1016/j.drudis.2008.07.008. [DOI] [PubMed] [Google Scholar]; (c) Marsault E, Peterson ML. J Med Chem. 2011;54:1961. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]; (d) Hill TA, Shepherd NE, Diness F, Fairlie DP. Angew Chem Int Ed Engl. 2014;53:13020. doi: 10.1002/anie.201401058. [DOI] [PubMed] [Google Scholar]; (e) Cardote TA, Ciulli A. ChemMedChem. 2016;11:787. doi: 10.1002/cmdc.201500450. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Villar EA, Beglov D, Chennamadhavuni S, Porco JA, Jr, Kozakov D, Vajda S, Whitty A. Nat Chem Biol. 2014;10:723. doi: 10.1038/nchembio.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Bionda N, Cryan AL, Fasan R. ACS Chem Biol. 2014;9:2008. doi: 10.1021/cb500311k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Frost JR, Jacob NT, Papa LJ, Owens AE, Fasan R. ACS Chem Biol. 2015;10:1805. doi: 10.1021/acschembio.5b00119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bionda N, Fasan R. Chembiochem. 2015;16:2011. doi: 10.1002/cbic.201500179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tavassoli A. Curr Opin Chem Biol. 2017;38:30. doi: 10.1016/j.cbpa.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 24.Chuang PT, McMahon AP. Nature. 1999;397:617. doi: 10.1038/17611. [DOI] [PubMed] [Google Scholar]
- 25.(a) Bosanac I, Maun HR, Scales SJ, Wen X, Lingel A, Bazan JF, de Sauvage FJ, Hymowitz SG, Lazarus RA. Nat Struct Mol Biol. 2009;16:691. doi: 10.1038/nsmb.1632. [DOI] [PubMed] [Google Scholar]; (b) Bishop B, Aricescu AR, Harlos K, O’Callaghan CA, Jones EY, Siebold C. Nat Struct Mol Biol. 2009;16:698. doi: 10.1038/nsmb.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D. PLoS One. 2011;6 doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tyka MD, Keedy DA, Andre I, DiMaio F, Song YF, Richardson DC, Richardson JS, Baker D. J Mol Biol. 2011;405:607. doi: 10.1016/j.jmb.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu CC, Schultz PG. Annu Rev Biochem. 2010;79:413. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 28.Smith JM, Vitali F, Archer SA, Fasan R. Angew Chem Int Ed Engl. 2011;50:5075. doi: 10.1002/anie.201101331. [DOI] [PubMed] [Google Scholar]
- 29.Nelson JW, Kallenbach NR. Proteins. 1986;1:211. doi: 10.1002/prot.340010303. [DOI] [PubMed] [Google Scholar]
- 30.(a) Pattabiraman VR, McKinnie SM, Vederas JC. Angew Chem Int Ed Engl. 2008;47:9472. doi: 10.1002/anie.200802919. [DOI] [PubMed] [Google Scholar]; (b) Knerr PJ, Tzekou A, Ricklin D, Qu H, Chen H, van der Donk WA, Lambris JD. ACS Chem Biol. 2011;6:753. doi: 10.1021/cb2000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Proc Natl Acad Sci USA. 2002;99:14071. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha S, Chen JK. Nat Chem Biol. 2006;2:29. doi: 10.1038/nchembio753. [DOI] [PubMed] [Google Scholar]
- 33.(a) Azoulay S, Terry S, Chimingqi M, Sirab N, Faucon H, Gil Diez de Medina S, Moutereau S, Maille P, Soyeux P, Abbou C, Salomon L, Vacherot F, de La Taille A, Loric S, Allory Y. J Pathol. 2008;216:460. doi: 10.1002/path.2427. [DOI] [PubMed] [Google Scholar]; (b) Ibuki N, Ghaffari M, Pandey M, Iu I, Fazli L, Kashiwagi M, Tojo H, Nakanishi O, Gleave ME, Cox ME. Int J Cancer. 2013;133:1955. doi: 10.1002/ijc.28193. [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Chinchilla P, Riobo NA. Method Enzymol. 2008;446:189. doi: 10.1016/S0076-6879(08)01611-X. [DOI] [PubMed] [Google Scholar]
- 35.(a) Zanghellini A, Jiang L, Wollacott AM, Cheng G, Meiler J, Althoff EA, Rothlisberger D, Baker D. Protein Sci. 2006;15:2785. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Richter F, Leaver-Fay A, Khare SD, Bjelic S, Baker D. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bionda N, Fasan R. Methods Mol Biol. 2017;1495:57. doi: 10.1007/978-1-4939-6451-2_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.