

Summary
CRISPR-Cas systems defend bacteria and archaea against infection by bacteriophage and other threats. The central component of these systems are surveillance complexes that use guide RNAs to bind specific regions of foreign nucleic acids, marking them for destruction. Surveillance complexes must locate targets rapidly to ensure timely immune response, but the mechanism of this search process remains unclear. Here, we used single-molecule FRET to visualize how the Type I–E surveillance complex Cascade searches DNA in real time. Cascade rapidly and randomly samples DNA through nonspecific electrostatic contacts, pausing at short PAM recognition sites that may be adjacent to the target. We identify Cascade motifs that are essential for either nonspecific sampling or positioning and readout of the PAM. Our findings provide a comprehensive structural and kinetic model for the Cascade target-search mechanism, revealing how CRISPR surveillance complexes can rapidly search large amounts of genetic material en route to target recognition.
In Brief (eTOC blurb)
Xue et al. show that the Cascade surveillance complex rapidly and randomly samples DNA through nonspecific electrostatic contacts, pausing at PAM sites that may be adjacent to the target. They identify three Cascade motifs that are essential for this process and together enable efficient target recognition and immune response.
Introduction
CRISPR-Cas (clustered regularly interspaced short palindromic repeats–CRISPR associated) adaptive immune systems protect bacteria and archaea by detecting and destroying foreign nucleic acids (Marraffini, 2015; van der Oost et al., 2014; Sorek et al., 2013). CRISPR–Cas systems generate immunological memories by inserting short DNA segments from the invader DNA as spacers in the host CRISPR array (Barrangou et al., 2007; Jackson et al., 2017). The CRISPR array is transcribed and processed into mature CRISPR RNAs (crRNAs), which assemble with one or more effector Cas proteins (Hochstrasser and Doudna, 2015; Jackson and Wiedenheft, 2015). This crRNA-guided complex surveys the cell, searching for the complementary “protospacer” target in the invader nucleic acid. Following target binding through complementary base pairing with the crRNA, the invader is cleaved and degraded (Marraffini, 2015; van der Oost et al., 2014).
CRISPR-Cas systems are clustered into six types (Types I–VI), each having distinct Cas proteins and immune mechanisms (Koonin et al., 2017; van der Oost et al., 2014). In the Type I–E CRISPR-Cas system of Escherichia coli, five stoichiometrically unequal proteins (Cse11, Cse22, Cas76, Cas5e1, and Cas6e1) assemble with a 61-nt crRNA to form the Cascade surveillance complex (Fig. 1A) (Brouns et al., 2008; Hayes et al., 2016; Jackson et al., 2014; Jore et al., 2011; Mulepati et al., 2014; Zhao et al., 2014). Similar to the Type II surveillance complex Cas9, Cascade searches for targets by first recognizing a short sequence called the protospacer-adjacent motif (PAM) (Mojica et al., 2009; Redding et al., 2015; Rollins et al., 2015; Sashital et al., 2012; Semenova et al., 2011; Sternberg et al., 2014). PAM recognition destabilizes the DNA duplex by inserting a glutamine wedge located in the large Cse1 (Cas8e) subunit into the dsDNA adjacent to the PAM, enabling strand invasion and formation of an RNA-DNA heteroduplex in the seed region (positions 1–5 and 7–8 of the crRNA spacer) (Hayes et al., 2016; Xiao et al., 2017). Complementary base pairing in the seed region efficiently promotes further R-loop formation (Semenova et al., 2011; Szczelkun et al., 2014). Cascade bound to the dsDNA triggers the recruitment of Cas3, which degrades the non-target strand in a 3′–5′ direction (Hochstrasser et al., 2014; Huo et al., 2014; Mulepati and Bailey, 2013; Redding et al., 2015; Sinkunas et al., 2011; Westra et al., 2012)
Figure 1. Real-time observation of Cascade target searching and binding by smFRET.

(A) Schematic of Cascade labeled with Cy5 in the Cas5e subunit. (B) Schematic of smFRET experiment using dsDNAtarget and targeting Cascade. (C–D) A representative smFRET time trajectory of (C) long-lived or (D) short-lived FRET events for targeting Cascade and dsDNAtarget. (E) Schematic of smFRET experiment using dsDNAtrunc and targeting Cascade. (F) A representative smFRET time trajectory of long-lived FRET for targeting Cascade and dsDNAtrunc. (G) Transition density plot of long-lived FRET events for targeting Cascade and dsDNAtarget with a FRET peak centered at 0.51. (H) Transition density plot of short-lived FRET events for targeting Cascade and dsDNAtarget with a FRET peak centered at 0.46. (I) Transition density plot of all FRET events for targeting Cascade and dsDNAtrunc with a FRET peak centered at 0.52. See also Figure S1–S4 and Table S3.
Single-molecule studies have provided remarkable insight into how Cascade binds dsDNA targets (Blosser et al., 2015; Jung et al., 2017; Redding et al., 2015; Rutkauskas et al., 2015; Szczelkun et al., 2014). Magnetic tweezers experiments have revealed directional R-loop formation by the Cascade complex during dsDNA binding (Rutkauskas et al., 2015; Szczelkun et al., 2014). Single-molecule fluorescence resonance energy transfer (smFRET) studies using FRET pairs on each strand of the dsDNA target revealed two distinct Cascade-DNA binding modes for bona fide and mutant targets (Blosser et al., 2015). These studies provided mechanistic details of Cascade R-loop formation following the initial target encounter, but did not probe the mechanism by which Cascade locates its target. Single-molecule imaging experiments using DNA curtains have been used to visualize Cascade target searching and binding with intermediate spatial resolution (~30 nm) (Redding et al., 2015). This study suggested that Cascade dwells at potential PAM sites located throughout the DNA during target searching, providing Cascade with time to interrogate the adjacent DNA sequence for complementarity with the crRNA. However, it is still unclear how Cascade locates PAM sites and whether it employs other target searching mechanisms to accelerate the surveillance process. It has been proposed that target searching begins by Cascade randomly sampling dsDNA through three-dimensional diffusion and rapidly dissociating from non-PAM sites (Redding et al., 2015), although this process has not been observed by existing single-molecule systems.
Detecting putative transient searching events requires a highly sensitive experimental system that enables observation of both the location and duration of Cascade-dsDNA binding. Here, we developed a smFRET assay that meets these requirements, enabling the direct visualization of the Cascade target-searching process in real time. We observed that Cascade randomly samples dsDNA through short-lived nonspecific electrostatic contacts between two lysine-rich loops in two of the Cas7 subunits and the phosphate backbone of the dsDNA. In the absence of a PAM, Cascade quickly dissociates from dsDNA, but this interaction is significantly stabilized through interactions between PAM sequences and two motifs within the Cse1 subunit. Together, these results provide a kinetic and structural model that explains how Cascade is able to rapidly locate DNA targets.
Results
Single-molecule FRET assay for Cascade target binding
Using a previously developed fluorescently-labeled Cascade (Xue et al., 2016), we established a system to directly observe individual Cascade complexes binding to a complementary target dsDNA in real time using smFRET (Fig. 1A). We initially sought to observe Cascade binding to a bona fide target (dsDNAtarget) containing a perfectly complementary protospacer and canonical PAM (5′-AAG-3′ on the non-target strand). We immobilized biotinylated dsDNAtarget labeled with donor (Cy3) fluorophore at the 5′ end of the non-target strand on a passivated quartz surface (Fig. 1B, S1A). Cascade bearing a complementary crRNA was labeled with an acceptor (Cy5) fluorophore in the Cas5e (E102C) subunit (Fig. 1A, S1B). Bulk Cascade binding experiments showed that the fluorescent label on Cas5e did not affect Cascade DNA binding activity (Fig. S1C–D).
Injection of Cascade into a flow cell with immobilized dsDNAtarget resulted in long-lived FRET between Cy3-dsDNAtarget and Cy5-Cascade for ~20% of single-molecule trajectories (n = 512), reflecting binding of Cascade to the target (Fig. 1C). This long-lived FRET state persisted until fluorophore photobleaching after ~190 s (Fig. S2), consistent with the formation of a stable R-loop. The FRET efficiency value of 0.5 was consistent with the distance between Cy5 and Cy3 fluorophores as measured in the Cascade-dsDNA crystal structure (Fig. S1B). For the remaining trajectories, we observed short-lived FRET events, suggesting that Cascade quickly dissociates from the DNA (Fig. 1D and Fig. S3). We interpreted these events as random target sampling or incomplete R-loop formation. Previous bulk biochemical studies have shown that Cascade binds to full length dsDNA oligonucleotides with low affinity at room temperature, which may account for the low percentage of full R-loop formation events (Xue et al., 2016). These results suggest that Cascade requires additional energy to efficiently unwind dsDNA for full R-loop formation, contributed either by thermal energy or negative supercoiling (Westra et al., 2012). Indeed, single-molecule tweezer experiments have shown that DNA supercoiling is necessary for full R-loop formation by Cascade binding to linear dsDNA at room temperature (Rutkauskas et al., 2015; Szczelkun et al., 2014).
We hypothesized that Cascade may more efficiently form a full R-loop in our smFRET assay if the double-stranded region of the target were truncated, as has previously been shown in bulk biochemical assays (Xue et al., 2016). To test this, we designed a dsDNA target with a truncated non-target strand (dsDNAtrunc) forming only 10 base pairs (bp) with the PAM-proximal region of the target (Fig. 1E). As expected, we observed long-lived Cascade-target binding for most of the trajectories (~90%, n = 445) for dsDNAtrunc. For a small number of trajectories (less than 5%), we observed transient FRET events prior to long-lived Cascade-target binding, similar to short-lived events observed for the fully double-stranded target (Fig. 1F). Control experiments performed for either Cse2-Cas6e or Cse1 labeled with Cy5 had no detectable FRET events (Fig. S4), indicating that all DNA-binding events require the entire Cascade complex.
Analysis of single-molecule trajectories with dsDNAtarget and dsDNAtrunc revealed the relative frequencies of transitions between an unbound state (EFRET ~ 0) and bound states, which we visualized using transition density plots (TDPs) (Fig. 1G–I). For dsDNAtarget, we separately analyzed trajectories with long-lived (Fig. 1G) or short-lived (Fig. 1H) FRET events. The TDPs for the dsDNAtarget long-lived FRET trajectories and for all dsDNAtrunc trajectories are non-symmetrical (Fig. 1G and I), showing that Cascade binds to the target tightly and does not dissociate from the DNA once it finds the target, consistent with the long-lived and highly stable formation of an R-loop, as has been previously observed (Rutkauskas et al., 2015; Szczelkun et al., 2014). Almost all binding events initiated from EFRET ~ 0 and ended at EFRET ~ 0.51, indicating that Cascade finds and binds the target simultaneously (Fig. 1G and I). For dsDNAtarget trajectories containing only short-lived events, the TDP is symmetrical with peaks indicating transitions between EFRET ~ 0 and EFRET ~ 0.46 (Fig. 1H).
Real-time observation of Cascade PAM searching process
The transient FRET events for dsDNAtarget suggests that intermediate states adopted during Cascade target search can be observed by our smFRET assay (Fig. 1D, H and Fig. S3). If the transient binding events are intermediates along the path to stable target binding, we hypothesized that a similar transient Cascade searching process would also be observed with a non-matching crRNA-DNA pair. To test this, we prepared Cy5-Cascade bearing a non-matching crRNA (hereafter non-targeting Cascade) and observed binding to various dsDNA substrates using our smFRET assay (Fig. 2 and Fig. S1A). We first measured binding of non-targeting Cascade to dsDNAtarget, enabling a direct comparison of binding to the same substrate with targeting Cascade. As expected, long-lived stable binding events were not observed for non-targeting Cascade binding to dsDNAtarget. Instead, we observed abundant but highly transient events with variable FRET efficiencies, consistent with a rapid search process across the DNA target (Fig. 2A).
Figure 2. Real-time observation of target sampling by non-targeting Cascade on four substrates.

(A) Representative smFRET time trajectory of non-targeting Cascade binding to dsDNAtarget. (B) Survival probabilities of dwell time distribution of FRET events for non-targeting Cascade and dsDNAtarget (green curve), dsDNA0PAM (gray curve), dsDNA1PAM (red curve), and dsDNA3PAM (dark red curve) fit to double-exponential decays (black curve). The number of molecules for each decay ranged from 1050 to 4,775. (C) The time and amplitude (longer bound-state lifetime) parameters of the double-exponential decays for dsDNAtarget (green), dsDNA0PAM (gray), dsDNA1PAM (red), and dsDNA3PAM (dark red). All dwell times are at least two orders of magnitude shorter than fluorophore photobleaching under the same measurement conditions (Fig. S2), indicating that the sudden disappearance of FRET was caused by the dissociation of Cascade from the dsDNA instead of photobleaching. (D–E) Representative smFRET time trajectories of non-targeting Cascade for (D) dsDNA0PAM, (E) dsDNA1PAM, and (F) dsDNA3PAM. See also Figure S5–S6 and Table S3.
To determine the binding kinetics of the observed search process, we analyzed the bound-state lifetimes and association rates for all short-lived FRET events for targeting and non-targeting Cascade binding to dsDNAtarget (Fig. 2B–C). The bound-state lifetimes of FRET events were best described by double-exponential decay (Fig. 2B). For both targeting and non-targeting Cascade, the shorter bound-state lifetime (τ1) was similar to the 0.1 s frame rate used during data collection (Fig. 2C), suggesting that the detection of short events may be limited by the time resolution of the experiment. The longer bound-state lifetime (τ2) and the amplitude-weighted lifetime (τavg) were both significantly higher (p < 0.05 using unpaired t-test for all significance tests) for targeting Cascade, which may be due to partial R-loop formation prior to dissociation (Fig. 2C). Consistently, the FRET efficiency distribution for targeting Cascade is relatively narrow in comparison to non-targeting Cascade (Fig. 1H, S5A), indicating that Cascade mainly binds at the target site when loaded with a matching crRNA but binds at random sites along the DNA in the absence of a matching target.
Previous studies have shown that Cascade binds promiscuously to targets with a variety of non-canonical “functional” PAM sequences (Jung et al., 2017; Leenay et al., 2016; Xue et al., 2015), and dsDNAtarget contains functional PAMs throughout the sequence (Table S3). Therefore, it is possible that the broad FRET distribution observed for non-targeting Cascade may be due to Cascade binding promiscuously at functional PAM sequences, rather than at random non-specific sites. To address this possibility, we designed three dsDNA substrates containing 0, 1 or 3 canonical 5′-AAG-3′ PAM sequences (dsDNA0PAM, dsDNA1PAM and dsDNA3PAM, respectively), and otherwise consisting mainly of 5′-CCG-3′ triplets, a well-characterized non-functional PAM motif (Fineran et al., 2014; Jung et al., 2017; Leenay et al., 2016; Westra et al., 2012; Xue et al., 2015) (Table S3). Similar to non-targeting Cascade binding to dsDNAtarget, individual FRET trajectories for these three substrates revealed transient FRET events (Fig. 2D–F), although the frequency and duration of the events varied for each substrate.
As with dsDNAtarget, the bound-state lifetimes for FRET events measured for the three variable PAM substrates were best described by double-exponential decay (Fig. 2B). We again observed a short ~0.1 s bound-state lifetime (τ1) for all three substrates. Strikingly, τ2 increases significantly as the number of PAMs increases, although no significant difference was observed for τ1 values (Fig. 2C). We observe differences in τavg that reflect not only the variations in τ2, but also the low amplitude of the τ2 component for targets with fewer PAMs. In particular, the amplitude of the longer dwell events is less than 2% for dsDNA0PAM, and increases to ~8% for dsDNA1PAM and ~25% for dsDNA3PAM (Fig. 2C). These data suggest that the longer-lived binding events are PAM-dependent, while the short events are PAM-independent. We also observed a small but significant increase in the association rate (kon) in the presence of a large number of canonical (dsDNA3PAM) or non-canonical (dsDNAtarget) PAMs, suggesting that the rate of binding for Cascade may be dependent on the presence of favorable PAMs (Fig. 2C). Importantly, association rates were concentration dependent while concentration had no effect on dissociation rates, indicating that FRET transitions were due to Cascade association and dissociation (Fig. S6).
As for dsDNAtarget, non-targeting Cascade binding to the variable PAM substrates exhibited broad FRET distributions based on TDP analysis (Fig. S5). The broad FRET distributions are not crRNA specific, as the same distribution was also observed for targeting Cascade binding to dsDNA0PAM (Fig. S5B–C). A mid-FRET state around 0.4 (0.35–0.45) was observed for all substrates, and is likely attributable to the short-lived sampling that comprised the vast majority of FRET events for dsDNA0PAM. This FRET state may represent the time-averaged FRET value arising from local diffusion, as has been previously proposed for the Type II Cas9 surveillance complex (Singh et al., 2016). Besides this ~0.4 FRET state, an additional FRET state at ~0.61 was observed for dsDNA3PAM (Fig. S5E), consistent with localization of Cascade at the first PAM closest to the Cy3-labeled DNA terminus. PAM-dependent localization is not observable for dsDNA1PAM because of overlap between the expected peak (~0.4) with the main mid-FRET state that arises from the abundant short dwell events. When the PAM site was moved to the same position as the first PAM in dsDNA3PAM (Table S3), we observed a new FRET state around ~0.62, consistent with the FRET peak observed for dsDNA3PAM (Fig. S5F). These data suggest that Cascade specifically localizes at PAM sites during the target search process.
Our kinetic analysis of Cascade-DNA binding suggests that dsDNA targets containing more PAM sites may increase the time needed for Cascade to find the target (Fig. 2C, S6). Similarly, previous studies have suggested that target recognition by the Type II Cas9 surveillance complex is slowed in the presence of competitor DNA containing several PAM sites, although these measurements were based on the rate of endonucleolytic cleavage by Cas9 rather than the rate of binding (Sternberg et al., 2014). Because Cascade does not have a catalytic readout, we developed a competition assay to directly monitor the rate of Cascade-target binding in the presence of competitor DNA containing multiple PAM sequences (Fig. 3A, see Experimental Procedures). To determine the rate of Cascade-target binding, we measured the fraction of Cascade-bound target DNA with or without competitor DNA at different incubation times (Fig. 3B). In control experiments without any competitor, Cascade could bind more than 50% of the target within 5 min. To test the effects of competitor DNA containing PAM sequences, we used three competitors with 0, 4 or 8 5′-AAG-3′ PAMs. Target binding kinetics were slightly reduced in the presence of competitor DNA with 0 PAMs (Fig. 3B–C). However, the target was fully bound at 30 min with the 0 PAM competitor, indicating Cascade can quickly sample non-PAM regions of DNA before binding to the target (Fig. 3B–C). Increasing PAM density in the competitor correlated with decreased Cascade-target binding, with no binding observed in the presence of these competitors until the 30 min time point and full binding only at substantially longer incubation times (240 min) (Fig. 3B–C). Altogether, our smFRET and bulk biochemical results are consistent with a model in which Cascade quickly leaves dsDNA in the absence of PAMs, allowing for rapid discovery of the target, but is slowed by the presence of PAMs, substantially increasing the amount of time required to locate the target.
Figure 3. PAM sampling slows Cascade target binding kinetics.

(A) Schematic of competition assay with different competitors (PAMs labelled red). (B) Electrophoretic mobility shift assay of Cascade with different competitors. (C) Quantification of competition data. Average fraction bound from three replicates is plotted, with error bars representing standard deviation. See also Table S2.
Three Cascade motifs are required for target search
We next sought to identify protein motifs that are involved in the Cascade target search process. Crystal structures and molecular dynamics simulations of Cascade bound to a dsDNA target have revealed several regions of Cascade that may be important for target searching (Van Erp et al., 2015; Hayes et al., 2016). We first investigated motifs in the Cse1 subunit, which is required for PAM recognition (Van Erp et al., 2015; Hayes et al., 2016; Sashital et al., 2012) (Fig. 4, Fig. 5A). In the Cascade R-loop crystal structure, the minor groove of the −1 PAM base pair is recognized by the backbone amide of Cse1 G160, an interaction that may also be important for PAM recognition during target searching (Hayes et al., 2016). We additionally identified a lysine-rich β hairpin in Cse1 (K289, K290, and K296) located close to the PAM recognition motif that we hypothesized may be important for dsDNA positioning during PAM binding. To test the importance of these motifs on Cascade-PAM searching, we introduced alanine substitutions at Cse1 G160 or the lysine residues in the Cse1 β hairpin and measured binding to dsDNA1PAM using real-time smFRET. Similar to WT Cascade, the bound-state lifetimes for both mutants were best described by a double-exponential decay (Fig. 4A), although the amplitude of the longer dwell events was less than 2% for both mutants (Fig. 4B). These results and TDP analysis of FRET transitions are strikingly similar to WT Cascade binding to dsDNA0PAM (Fig. 2C, S4B, S7), and indicate an overall reduction in PAM sampling and dwelling when each motif is mutated. The longer bound-state lifetime (τ2) for the mutants were both significantly lower than that of WT Cascade (Fig. 4B). Importantly, there is no significant difference in τ2 between the two Cse1 mutants, suggesting that the β hairpin may facilitate PAM recognition by positioning the DNA in the proper orientation through non-specific contacts.
Figure 4. Three protein motifs in Cascade are important for target sampling and PAM recognition.

(A) Survival probabilities of dwell time distribution of FRET events for non-targeting Cascade with WT Cse1 (red curve), Cascade with Cse1 G160A mutation (yellow curve), and Cascade with Cse1 Lys-rich β hairpin mutation (K289A, K290A, and K296A) (blue curve) on dsDNA1PAM fit to double-exponential decays (black curve). The number of events for each decay ranged from 1089 to 4,775. (B) The time and amplitude (longer bound-state lifetime) parameters of the double-exponential decays for WT Cascade (red), G160A mutant (yellow), and Lys-rich β hairpin mutant (blue). Parameters could not be measured for mutants for which FRET events were not detected (Cse1 double mutant, Cas7 quadruple mutant) or were very rare (Cas7 K137A, 11 events in >4000 trajectories). (C) Electrophoretic mobility shift assay for Cascade mutants binding at different time point (2, 5, and 30 min) and at various Cascade concentrations (1, 5, 10, 50, and 500 nM). Red dashed boxes indicate the concentration at which all DNA is bound. See also Figure S7 and Table S3.
Figure 5. Cascade sampling structural model of Cse1 (green), Cas5e (purple) and Cas7.5 (light blue), and Cas7.6 (blue).

(A) The apo structure. (B) A simulated model of Cascade PAM sampling with Cse1 in the closed conformation (model was fit in PDB 4TVX). The dsDNA helix is bent by ~20°. (C) A simulated model of Cascade PAM sampling a linear dsDNA helix with Cse1 in a partially open conformation. Residues for three protein motifs are shown as spheres: Lys-rich vise (hot pink) in Cas7, PAM recognition motif (G160 in yellow) and Lys-rich β hairpin (blue) in Cse1 subunit. The PAM is colored red.
The association rates for the two Cse1 mutants were significantly lower (~2.5x) than for WT Cascade (Fig. 4B). These results indicate that the PAM recognition motifs in Cse1 are important for DNA sampling, and are not simply involved in reading out the PAM. When both G160 and the lysine-rich β hairpin were mutated together, no binding events were observed (Fig. S7A–B), further implicating PAM recognition in dsDNA sampling.
We next investigated two lysine-rich loops (K137, K138, K141 and K144) in adjacent Cas7.5 and Cas7.6 subunits of the Cascade backbone, which have previously been implicated in target binding (Van Erp et al., 2015) (Fig. 5A). These lysine-rich loops form a vise that positions the dsDNA helix upstream of the PAM through electrostatic contacts with the DNA backbone (Hayes et al., 2016), an interaction that we hypothesized may also be important for random dsDNA sampling. To test the role of these motifs during the target search process, we performed smFRET experiments to observe Cascade binding to dsDNA1PAM when mutations were introduced in the Cas7 lysine-rich loops. A single lysine mutation (K137A) significantly reduced the observed binding rate (11 FRET events in over 4000 trajectories), ~100x lower than for WT Cascade (Fig. 4B). No FRET events were observed in over 4000 trajectories when we mutated all four residues within the lysine-rich vise (Fig. S7C). The loss of binding was not due to defects in complex formation, as Cas7 mutations did not affect stoichiometry of Cse2-Cas6e (Fig. S7D). These results indicate that Cascade-dsDNA sampling relies on non-specific interactions between the lysine-rich vise in Cas7 and the DNA phosphate backbone. Combined, we propose that the Cas7 lysine-rich vise along with the Cse1 lysine-rich β hairpin positions dsDNA in an orientation that enables PAM readout by the PAM-recognition motif.
Based on our smFRET data, we hypothesized that target search kinetics would be slowed for the Cse1 and Cas7 mutants. We tested this hypothesis by performing bulk binding experiments at different Cascade-target incubation times to capture binding affinities for the Cascade mutants (Fig. 4C, S7G). At the indicated incubation time, binding reactions were quenched by loading onto a gel and immediately initiating electrophoresis at 4° C to detect binding based on electrophoretic mobility shift. For long incubation times (30 min), mutation of the whole Cas7 Lys-rich vise or the double mutation of both Cse1 motifs completely abolished binding, consistent with the lack of observable FRET events for these constructs (Fig. S7G). However, individual mutations within each of the motifs cause only modest decreases in binding affinity after 30 min incubation (Fig. 4C). WT Cascade shows no binding affinity difference between 2 min and 30 min incubation, suggesting that Cascade can find its target rapidly. Interestingly, for the Cse1 and Cas7 mutants, binding affinity defects are far more pronounced after 2 min incubation and affinity increases over time, indicating that the Cascade mutants require longer incubation periods to recognize and bind the target. Consistent with our smFRET results, these bulk binding experiments strongly indicate that the search kinetics are slowed and mutant Cascade requires substantially longer time to find and bind the target.
Structural model for Cascade-DNA sampling
Our results indicate that the Cas7 lysine vise and the Cse1 PAM recognition motif and lysine-rich β hairpin interact with dsDNA during target searching (Fig. 5A). Based on these data, we developed a structural model to simulate how Cascade may bind to dsDNA during the target-search process. Using HADDOCK, we created two models to simulate interactions between the Cas7 lysine vise and dsDNA or between the Cse1 motifs and dsDNA (Van Zundert et al., 2016). The models were then fit to a crystal structures of apo Cascade (Fig. 5B) (Jackson et al., 2014). For the apo-Cascade structure, Cse1 adopts a “closed” conformation, and the dsDNA helix must be bent by ~20° to accommodate all interactions enforced in our model (Fig. 5B). This model suggests that PAM recognition during target searching may be accompanied by a DNA bending event that enables local melting of the dsDNA helix. A recent Cascade/seed-bubble structure also suggests that PAM recognition is coupled with DNA bending, which drives the melting of dsDNA at the seed region (Xiao et al., 2017). However, this model does not account for how Cascade may interact with DNA prior to helical bending. Interestingly, when the two models are docked to ensure linearity of the DNA, Cse1 adopts a partially “open” conformation, as has been observed in the crystal structure of ssDNA-bound Cascade and through FRET studies of Cse1 conformation (Mulepati et al., 2014; Xue et al., 2016) (Fig. 5C). Together, these models suggest that the conformational flexibility of Cse1 may be required to initiate target searching prior to DNA bending.
Discussion
A universal question for all CRISPR–Cas immune systems is how subtype-specific surveillance complexes are able to locate targets rapidly, enabling timely immune response against infection. Here, we developed a smFRET assay to visualize the Type I–E Cascade target searching process in real time. We observed that Cascade rapidly and randomly samples DNA in the absence of PAMs, and dwells longer on DNA when PAMs are present. Our study reveals the rapidity of Cascade binding kinetics during PAM searching, with the majority of binding events exhibiting extremely short dwell times in the absence of PAM sites (~0.1 s). Similar transient binding states were also observed in a previous smFRET study of the Type II Cas9 surveillance complex (Singh et al., 2016). For crRNAs with non-complementary dsDNA target, Singh et al. observed short lifetime (~0.1 s) FRET events, which may represent non-specific DNA sampling during PAM surveillance similar to the sampling observed in our study. Together, these studies indicate distinct CRISPR surveillance complexes employ similar strategies for non-specific target searching.
Our single-molecule and bulk biochemical data indicate that the Cascade search kinetics are slowed in the presence of increasing numbers of PAM sites. The Cascade dwell time increases with the number of PAMs, although all dwell times measured in our study are shorter than the transient PAM dwelling (~3 s) observed for Cascade in a previous study using DNA curtains (Redding et al., 2015). These differences may be due to different buffer conditions used in each study. It is also possible that higher PAM density of the bacteriophage λ genome used in the DNA curtain experiment could result in the slightly longer dwell time. A recent Cas9 in vivo tracking study revealed that a single Cas9 molecule takes 6 hours to find its target in an E. coli cell. Notably, the estimated residence time for Cas9 at nonspecific sites (<30 ms) was substantially shorter than the time resolution (100 ms) of our experimental setup; therefore, it is possible that nonspecific Cascade binding occurs more rapidly than we could measure. It is also possible that the more rapid searching kinetics observed in vivo may be caused by the crowded DNA environment in the cell. If this is the case, it is likely that Cascade may also exhibit more rapid search kinetics when exposed to crowded or tightly packaged DNA.
Upon PAM recognition, the dwell time can be further increased by partial crRNA-target formation. A previous smFRET study of Cascade-target binding revealed a longer-lived bound state (~25 s) for mutant targets containing mutated PAM sequences or partial complementarity to the crRNA (Blosser et al., 2015). This study relied on target unwinding to detect Cascade binding, and likely reflects binding states following partial formation of the crRNA-target duplex. Overall, these studies indicate that target searching is likely slowed when Cascade encounters random sites of higher PAM density or partial complementarity to the crRNA. Previous studies have suggested that partial Cascade R-loops are sufficient to enable primed spacer acquisition (Blosser et al., 2015; Datsenko et al., 2012). Thus, while random complementary regions may slow target discovery, Cascade may mitigate these detrimental effects by eliciting an alternative immune response.
Recent structures of Cascade bound to dsDNA have revealed how Cascade engages the PAM and target upon R-loop formation (Hayes et al., 2016; Xiao et al., 2017). However, it remains unclear how Cascade recognizes the PAM during target searching. Our smFRET data indicate that the Cascade sampling process relies on motifs that maintain contacts with the DNA following R-loop formation. The Cas7 lysine-rich loops are hypothesized to be important for target positioning during duplex unwinding (Van Erp et al., 2015). Consistent with this hypothesis, our data implicate this motif in target sampling, revealing that interactions between the lysine vise and dsDNA are initiated prior to duplex unwinding and maintained following R-loop formation. T. fusca Cascade (type I–E) contains similar Lys-rich loops in its Cas7 subunits (K-vise and K-Rim) indicating that type I–E Cascade uses a common mechanism to engage dsDNA (Xiao et al., 2017). Although no corresponding domain exists in Cas7 subunits of the Csy complex (type I–F), it has been proposed that the Cas8f “hook” domain clamps onto dsDNA and holds it in a secure vise (Chowdhury et al., 2017; Guo et al., 2017).
The Cse1 PAM recognition motif identifies PAMs during target searching and maintains contact following target binding, an interaction that is thought to be important for stabilizing Cse1 in a conformation required for Cas3 recruitment and target degradation (Hayes et al., 2016; Xiao et al., 2017; Xue et al., 2016). In addition, our data also implicate the Cse1 lysine-rich β hairpin in the PAM searching process, although this motif does not contact DNA following target binding. We hypothesize that the β hairpin is critical for guiding the dsDNA to position the PAM for recognition by Cse1 and this is mainly achieved through non-specific DNA binding. Structural models that account for interactions between the target DNA and these three Cascade motifs suggest that DNA can be bound linearly when Cse1 adopts a partially open conformation observed in Cascade-ssDNA bound structures (Mulepati et al., 2014). However, in order to accommodate the DNA in the closed Cse1 conformation observed in the apo Cascade structure, the DNA must be bent by ~20°. Bending of dsDNA may favor unwinding, allowing for crRNA strand invasion and interrogation of the adjacent seed region. Future studies will be required to determine which conformation Cse1 adopts during the target search process.
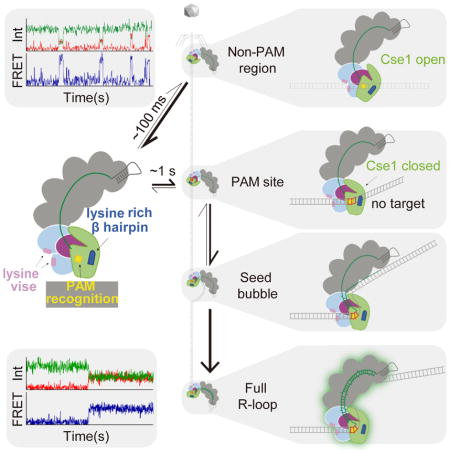
Together with previous studies, our smFRET data suggest a model for the Cascade target searching process (Fig. 6). Cascade searches the target through three-dimensional diffusion by randomly sampling dsDNA. The complex dissociates rapidly in the absence of PAM sites, but is slowed in the presence of increasing number of PAM sequences. Cascade pauses at PAMs due to favorable interactions between Cse1 and the PAM, which may enable dsDNA bending based on conformational flexibility of Cse1. Helical bending destabilizes the dsDNA and drives local melting at the PAM-proximal region. DNA unwinding is facilitated by crRNA strand invasion and the formation of an RNA-DNA heteroduplex within the seed region. Favorable energetic conditions, including perfect target complementarity and negative supercoiling, efficiently promote further R-loop formation to the PAM-distal end, resulting a stable R-loop structure. In contrast, unfavorable energetic conditions, including target mismatches, may inhibit complete R-loop formation and allow Cascade to dissociate. These findings reveal how Cascade quickly searches PAMs to locate targets and uncover several motifs in Cascade that are essential for this process.
Figure 6. Model of Cascade target search process.

Cascade randomly samples dsDNA through nonspecific electrostatic contacts between phosphate groups and the Lys-rich vise in Cas7. The dsDNA is positioned close to the PAM recognition motif in Cse1 with the guidance of a lysine-rich β hairpin in Cse1. Cascade quickly dissociates from dsDNA (within 0.1 s) when no PAM is present, but this interaction is significantly stabilized as PAM density increases (dwell time ~1 s or greater). PAM recognition causes a ~20° bend in the DNA, enabling local DNA unwinding. In the absence of a target, Cascade rapidly dissociates to continue searching. In the presence of a target and under favorable energetic conditions, Cascade forms a seed bubble. Seed sequence complementary promotes stable full R-loop formation.
Experimental Procedures
Plasmid Construction
Primers and plasmids are reported in Table S1 and S2. Point mutations were introduced using “round-the-horn” (RTH) (see Supplemental Experimental Procedures).
Protein purification and Dye labeling
All proteins were expressed in BL21(DE3) cells using plasmids reported in Table S1. The proteins were purified as described previously (Xue et al., 2016) (see Supplemental Experimental Procedures).
Preparation of dsDNA used for smFRET and EMSA
DNA oligos were purchased from Integrated DNA Technologies (IDT). Oligos used for smFRET were labeled at the 5′ end with Cy3 (performed by IDT) (Table S3). Oligos were purified as described in Supplemental Experimental Procedures.
Electrophoretic mobility shift assay (EMSA)
For Cascade oligonucleotide competition binding assays (Fig. 3B), XCY1185 and XCY1186 were 5′ end labeled with γ-32P-ATP using T4 polynucleotide kinase (NEB) for 30 min at 37 °C. PNK was heat denatured at 65 °C for 20 min, and excess γ-32P-ATP was removed using a G-25 spin column (GE Healthcare). All binding experiments were performed in binding buffer: 20 mM Tris, pH 7.5, 100 mM NaCl, and 5% glycerol. Cascade (10 nM) was incubated with 0.5 nM 32P labeled dsDNA with 1 μM indicated competitor for different times at 37 °C. Samples were quenched by adding 50 μM of a competitor with 16 AAG and stored on ice before loading (Table S1). In control experiments, Cascade does not bind the target DNA with the same amount of DNA quencher when incubated on ice for 30 min. Samples were run on 6% native polyacrylamide gel for 2 h at 250 volts at 4 °C. Gels were dried and DNA was visualized by phosphorimaging. Bound and unbound DNA fractions were quantified using ImageJ. For Cascade plasmid binding assays (Fig. 4F), Cascade at indicated concentrations and 100 ng target plasmid (~6 nM) were incubated at 37 °C for indicated time. Samples were immediately loaded on a 0.8% agarose gel stained with SYBR Safe and run at 15 volts at 4°C overnight (~18 hr).
Single-molecule FRET set-up
Single-molecule FRET experiments were performed with a prism-type total internal reflection fluorescence microscope at room temperature (23 ± 2°C). Quartz slides and microfluidic chambers were prepared as described (Chandradoss et al., 2014). Cy3 and Cy5 molecules were excited with a solid-state laser at 532 nm and a helium-neon (HeNe) laser at 635 nm, respectively. Fluorescence signals of Cy3 and Cy5 were collected through a 60× Water immersion objective with an inverted microscope and split by a dichroic mirror (T660lpxr, Chroma). Cy3 and Cy5 signals were recorded using an EM-CCD camera (iXon DU897E, Andor Technology).
For each chamber, 50 μL streptavidin (0.1 mg/mL) was incubated for 1 minute followed by a washing step with 500–600 μL of T50 buffer (10 mM Tris-HCl pH 8.0, 50 mM NaCl). Then, 50 μL of 40 pM Cy3-labeled dsDNA targets were immobilized on the microscope slides using biotin-streptavidin conjugation for 5 minutes. After incubation, unbound Cy3-labeled dsDNA targets was removed by washing with 500–600 μL of T50 buffer. Before imaging, 200 μL of Cascade imaging buffer (10 mM Tris-HCl pH 8.0, 50 mM NaCl, 0.1 mg/mL glucose oxidase, 4 μg/mL Catalase, 1 mM Trolox ((±)-6-Hydroxy-2,5,7,8, -tetramethylchromane-2-carboxylic acid) and 0.8% (v/v) glucose) was flowed into the chamber. For real-time experiments, Cy5-labeled Cascade (5 nM or as labelled) in the imaging buffer was flowed into the chamber 10–30 s after initiating imaging.
smFRET data acquisition and analysis
A series of CCD images were acquired with the smCamera software at a time resolution of 0.1 s/frame. For all smFRET experiments, fluorescence time traces resulting from the excitation of Cy3 were recorded and analyzed using smCamera software. FRET efficiencies (E) were calculated by the following equation:
Where IA and ID represent the fluorescence intensities of acceptor and donor. The background signal of Cy3 and Cy5 fluorescence intensities in each trajectory was determined by averaging the background noise intensity after fluorephore photobleaching, and then subtracting this value for all trajectories. All smFRET trajectories were idealized based on hidden Markov modeling (HMM) analysis using the HaMMy software (McKinney et al., 2006). The minimum number of frames for a FRET event is two, composed of a single frame of signal appearance and a single frame of signal disappearance. The dwell times of FRET events (τon) and dwell times between FRET events (τoff) of the idealized time traces were extracted using custom written RStudio codes. To avoid extracting any background signal, the FRET value cut-off was set to 0.2. Dwell time histograms were made in Origin (Origin lab). The dwell time histograms were fit by either a single-exponential decay curve or a double-exponential decay curve. The transition data formats generated from HaMMy were used to generate transition density plots using the TDP software (McKinney et al., 2006). For each experiment, three data sets were collected using three different slides. For each data set, 4 movies were collected for WT Cascade and 15 movies were collected for mutant Cascade. For each target, TDPs were generated by combining the three data sets. For dwell time histograms, the average of 3 or 4 datasets was shown. Dissociation (koff) and association rate (kon) were determined by by the following equations (Singh et al., 2016):
Reported dwell times, amplitudes, koff and kon constants are the average of the values calculated for the three data sets, and errors represent the standard deviation of the three values. The photobleaching rate of Cy3 was determined as previously described (Roy et al., 2008).
Structural modelling
Structural models of Cascade target searching were created using HADDOCK (Van Zundert et al., 2016). An idealized 21-bp dsDNA containing an AAG PAM was used to simulate the target. To simplify calculations, a minimal Cascade model was used for docking, based on PDB 4TVX containing Cse1, Cas7.5, Cas7.6, Cas5e subunits and the crRNA. Two separate models were simulated using restraints to enforce interactions between the dsDNA and either (1) the PAM recognition motif and β hairpin in Cse1 or (2) the lysine-rich vise in the Cas7 subunits. The top 3 best scored models for each simulation were selected and docked into apo (PDB 4TVX) and dsDNA-bound (PDB 5H9F) Cascade crystal structures, and the one with no obvious steric conflicts was selected. The best fitting model was docked into the apo-Cascade structure (PDB 4TVX) to create overall structural models for Cascade-PAM binding using UCSF Chimera (Goddard et al., 2007). To simulate linear DNA binding, the Cse1-dsDNA model was aligned with the Cas7-dsDNA model based on the PAM sequences in the dsDNA. All structure figures were made in UCSF Chimera (Goddard et al., 2007).
Supplementary Material
Figure S1. Development of smFRET assay. Related to Figure 1. (A) Schematic of two crRNAs correspond to targeting Cascade and non-targeting Cascade and a surface-immobilized, Cy3 labelled dsDNA that contains the target sequence (green) and the PAM (red). A 33-nt biotinylated adaptor is used as an anchor for surface immobilization. (B) Cy3 (green dot) and Cy5 (red dot) labelling locations shown in the Cascade-dsDNA bound crystal structure (PDB ID: 5H9F). (C) Electrophoretic mobility shift assay for WT Cascade and Cy5 labelled Cascade (labelling efficiency > 90%) (1, 2, 5, 10, 20, 50, and 500 nM) binding to plasmid containing target sequence. (D) Fraction of DNA bound versus Cascade concentration were fit with a binding isotherm (solid lines).
Figure S2. Photobleaching rate for Cy3-labeled dsDNA. Related to Figures 1 and 2. Mean donor photobleaching time was obtained from fitting donor survival probability to a single-exponential decay equation.
Figure S3. Representative smFRET trajectories of targeting Cascade with dsDNAtarget. Related to Figure 1.
Figure S4. FRET distribution for whole Cascade, Cse1, and Cse2-Cas6e with dsDNAtrunc. Related to Figure 1. (A–C) FRET histograms for FRET events existed in all extracted traces of Cascade with Cy5 labelled Cse2-Cas6e (A), Cy5 labelled Cse1 (B), and Cy5 labelled Cse2-Cas6e (C). The number of molecules used for FRET histograms was 392 (Cascade), 378 (Cse1), and 400 (Cse2).
Figure S5. Transition density plots for Cascade binding to different dsDNA substrates. Related to Figure 2. Transition density plots for (A) dsDNAtarget with non-targeting Cascade, (B) dsDNA0PAM with non-targeting Cascade, (C) dsDNA0PAM with targeting Cascade, (D) dsDNA1PAM with non-targeting Cascade, (E) dsDNA3PAM with non-targeting Cascade, and (F) dsDNA1PAM-12bp with non-targeting Cascade. The number of molecules for each TDP ranged from 1050 to 4,775.
Figure S6. FRET appearance and disappearance at different Cascade concentrations. Related to Figure 2. (A) kbinding for dsDNA0PAM, dsDNA1PAM and dsDNA3PAM at three different Cascade concentrations (2.5 nM, 5 nM and 10 nM). (B) koff for dsDNA0PAM, dsDNA1PAM and dsDNA3PAM at three different Cascade concentrations (2.5 nM, 5 nM and 10 nM).
Figure S7. Cse1 and Cas7 motif mutations abolish target binding. Related to Figure 4. (A–C) FRET distribution for (A) WT Cascade, (B) G160A and lysine-rich β hairpin (K289A, K290A, and K296A) double mutation in Cse1, (C) Cas7 quadruple Lys-rich vise mutation (K137A, K138A, K141A and K144A) binding to dsDNA1PAM. The number of molecules used for FRET histograms was 1517 (WT Cascade), 850 (Cse1 double mutant), and 744 (Cas7 Lys-rich vise mutant). (D) Coomassie blue-stained SDS-PAGE gel analysis of Cse2-Cas6e. Gel lanes: (1) WT Cse2-Cas6e; (2) WT Cse2-Cas6e with K137A Cas7; (3) WT Cse2-Cas6e with K137A, K138A, K141A and K144A Cas7. (E–F) Transition density plots for Cascade with (E) Cse1 G160A mutation and (F) Cascade with Cse1 Lys-rich β hairpin mutation (K289A, K290A, and K296A) binding to dsDNA1PAM. (G) Electrophoretic mobility shift assay for G160A and lysine-rich β hairpin (K289A, K290A, and K296A) double mutation in Cse1 and quadruple Lys-rich vise mutation (K137A, K138A, K141A and K144A) in Cas7 at different time point (2, 5, and 30 min) and various Cascade concentrations (1, 5, 10, 50, and 500 nM).
Highlights.
Cascade rapidly and randomly samples DNA through nonspecific electrostatic contacts
Cascade slows its search in the presence of PAM sites
Three Cascade motifs that are essential for the target searching process
Conformational flexibility of Cse1 may facilitate DNA searching and unwinding
Acknowledgments
We thank Jaewook Kim, Linxiang Yin and Brenden J. Hawk for helpful discussion; Samuel H. Sternberg for advice on data analysis and critical reading of the manuscript; Drena Dobbs for help with structural modelling; and Maria Spies and Fletcher E. Bain for advice on data analysis. This project was funded by NIH R01 GM115874 (to D.G.S.), start-up funds from Iowa State University and the Roy J. Carver Charitable Trust (to D.G.S.), and NIH R01 GM051290 (to Y.K.S.).
Footnotes
Author Contributions
X.C.Y. and D.G.S. conceived the project and designed the experiments. X.C.Y. and Z.Y.C. developed the smFRET system. X.C.Y. performed all experiments and smFRET data analysis. Z.X.M. wrote R codes used for data analysis. Y.K.S. assisted with smFRET data interpretation. X.C.Y. and D.G.S. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Blosser TR, Loeff L, Westra ER, Vlot M, Künne T, Sobota M, Dekker C, Brouns SJJ, Joo C. Two distinct DNA binding modes guide dual roles of a CRISPR-cas protein complex. Mol Cell. 2015;58:60–70. doi: 10.1016/j.molcel.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, Van Der Oost J. Small Crispr Rnas Guide Antiviral Defense in Prokaryotes. Science (80) 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandradoss SD, Haagsma AC, Lee YK, Hwang J-H, Nam J-M, Joo C. Surface passivation for single-molecule protein studies. J Vis Exp. 2014:4–11. doi: 10.3791/50549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Carter J, Rollins MCF, Golden SM, Jackson RN, Hoffmann C, Nosaka L, Bondy-Denomy J, Maxwell KL, Davidson AR, et al. Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell. 2017;169:47–57. e11. doi: 10.1016/j.cell.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun. 2012;3:945. doi: 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- Van Erp PBG, Jackson RN, Carter J, Golden SM, Bailey S, Wiedenheft B. Mechanism of CRISPR-RNA guided recognition of DNA targets in Escherichia coli. Nucleic Acids Res. 2015;43:8381–8391. doi: 10.1093/nar/gkv793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, Gerritzen MJH, Suarez-Diez M, Kunne T, Boekhorst J, van Hijum SAFT, Staals RHJ, Brouns SJJ. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc Natl Acad Sci. 2014;111:E1629–E1638. doi: 10.1073/pnas.1400071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Ferrin TE. Visualizing density maps with UCSF Chimera. J Struct Biol. 2007;157:281–287. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Guo TW, Bartesaghi A, Yang H, Earl LA, Patel DJ, Subramaniam S, Guo TW, Bartesaghi A, Yang H, Falconieri V, et al. Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell. 2017;171:414–419. e12. doi: 10.1016/j.cell.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes RP, Xiao Y, Ding F, van Erp PBG, Rajashankar K, Bailey S, Wiedenheft B, Ke A. Structural basis for promiscuous PAM recognition in type I–E Cascade from E. coli. Nature. 2016;530:499–503. doi: 10.1038/nature16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser ML, Doudna JA. Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem Sci. 2015;40:58–66. doi: 10.1016/j.tibs.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Hochstrasser ML, Taylor DW, Bhat P, Guegler CK, Sternberg SH, Nogales E, Doudna Ja. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc Natl Acad Sci. 2014;111:6618–6623. doi: 10.1073/pnas.1405079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Nam KH, Ding F, Lee H, Wu L, Xiao Y, Farchione MD, Zhou S, Rajashankar K, Kurinov I, et al. Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nat Struct Mol Biol. 2014;21:771–777. doi: 10.1038/nsmb.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RN, Wiedenheft B. A Conserved Structural Chassis for Mounting Versatile CRISPR RNA-Guided Immune Responses. Mol Cell. 2015;58:722–728. doi: 10.1016/j.molcel.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RN, Golden SM, van Erp PBG, Carter J, Westra ER, Brouns SJJ, van der Oost J, Terwilliger TC, Read RJ, Wiedenheft B. Structural biology. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science. 2014;345:1473–1479. doi: 10.1126/science.1256328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SA, McKenzie RE, Fagerlund RD, Kieper SN, Fineran PC, Brouns SJJ. CRISPR-Cas: Adapting to change. Science (80) 2017;356:eaal5056. doi: 10.1126/science.aal5056. [DOI] [PubMed] [Google Scholar]
- Jore MM, Lundgren M, van Duijn E, Bultema JB, Westra ER, Waghmare SP, Wiedenheft B, Pul U, Wurm R, Wagner R, et al. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat Struct Mol Biol. 2011;18:529–536. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
- Jung C, Hawkins JA, Jones SK, Xiao Y, Rybarski JR, Dillard KE, Hussmann J, Saifuddin FA, Savran CA, Ellington AD, et al. Massively Parallel Biophysical Analysis of CRISPR-Cas Complexes on Next Generation Sequencing Chips. Cell. 2017;170:35–47. e13. doi: 10.1016/j.cell.2017.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78. doi: 10.1016/j.mib.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, Maksimchuk KR, Slotkowski RA, Agrawal RN, Gomaa AA, Briner AE, Barrangou R, Beisel CL. Identifying and Visualizing Functional PAM Diversity across CRISPR-Cas Systems. Mol Cell. 2016;62:137–147. doi: 10.1016/j.molcel.2016.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature. 2015;526:55–61. doi: 10.1038/nature15386. [DOI] [PubMed] [Google Scholar]
- McKinney SA, Joo C, Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- Mulepati S, Bailey S. In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA Target. J Biol Chem. 2013;288:22184–22192. doi: 10.1074/jbc.M113.472233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulepati S, Héroux A, Bailey S. Structural biology. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science. 2014;345:1479–1484. doi: 10.1126/science.1256996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Oost J, Westra ER, Jackson RN, Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol. 2014;12:479–492. doi: 10.1038/nrmicro3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding S, Sternberg SH, Marshall M, Gibb B, Bhat P, Guegler CK, Wiedenheft B, Doudna JA, Greene EC. Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell. 2015;163:854–865. doi: 10.1016/j.cell.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins MF, Schuman JT, Paulus K, Bukhari HST, Wiedenheft B. Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res. 2015;43:2216–2222. doi: 10.1093/nar/gkv094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkauskas M, Sinkunas T, Songailiene I, Tikhomirova M, Siksnys V, Seidel R. Directional R-loop formation by the CRISPR-cas surveillance complex cascade provides efficient off-target site rejection. Cell Rep. 2015;10:1534–1543. doi: 10.1016/j.celrep.2015.01.067. [DOI] [PubMed] [Google Scholar]
- Sashital DG, Wiedenheft B, Doudna JA. Mechanism of Foreign DNA Selection in a Bacterial Adaptive Immune System. Mol Cell. 2012;46:606–615. doi: 10.1016/j.molcel.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Jore MM, Datsenko Ka, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJJ, Severinov K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Sternberg SH, Fei J, Ha T, Doudna JA. Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease Cas9. Nat Commun. 2016;7:1–8. doi: 10.1038/ncomms12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkunas T, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 2011;30:1335–1342. doi: 10.1038/emboj.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R, Lawrence CM, Wiedenheft B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem. 2013;82:237–266. doi: 10.1146/annurev-biochem-072911-172315. [DOI] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Natl Acad Sci. 2014;111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Erp PBG, Künne T, Wong SP, Staals RHJ, Seegers CLC, Bollen S, Jore MM, Semenova E, Severinov K, et al. CRISPR Immunity Relies on the Consecutive Binding and Degradation of Negatively Supercoiled Invader DNA by Cascade and Cas3. Mol Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Luo M, Hayes RP, Kim J, Ng S, Ding F, Liao M, Ke A. Structure Basis for Directional R-loop Formation and Substrate Handover Mechanisms in Type I CRISPR- Cas System Cryo-EM structures of type I CRISPR-Cas system resolve the mechanisms governing the PAM-dependent R-loop formation, Cas3 recruitment, and subst. Cell. 2017;170:48–60. e11. doi: 10.1016/j.cell.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C, Seetharam AS, Musharova O, Severinov K, Brouns SJJ, Severin AJ, Sashital DG. CRISPR interference and priming varies with individual spacer sequences. Nucleic Acids Res. 2015;43:10831–10847. doi: 10.1093/nar/gkv1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C, Whitis NR, Sashital DG. Conformational Control of Cascade Interference and Priming Activities in CRISPR Immunity. Mol Cell. 2016;64:826–834. doi: 10.1016/j.molcel.2016.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sheng G, Wang J, Wang M, Bunkoczi G, Gong W, Wei Z, Wang Y. Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature. 2014;515:147–150. doi: 10.1038/nature13733. [DOI] [PubMed] [Google Scholar]
- Van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, Van Dijk M, De Vries SJ, Bonvin AMJJ. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J Mol Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Development of smFRET assay. Related to Figure 1. (A) Schematic of two crRNAs correspond to targeting Cascade and non-targeting Cascade and a surface-immobilized, Cy3 labelled dsDNA that contains the target sequence (green) and the PAM (red). A 33-nt biotinylated adaptor is used as an anchor for surface immobilization. (B) Cy3 (green dot) and Cy5 (red dot) labelling locations shown in the Cascade-dsDNA bound crystal structure (PDB ID: 5H9F). (C) Electrophoretic mobility shift assay for WT Cascade and Cy5 labelled Cascade (labelling efficiency > 90%) (1, 2, 5, 10, 20, 50, and 500 nM) binding to plasmid containing target sequence. (D) Fraction of DNA bound versus Cascade concentration were fit with a binding isotherm (solid lines).
Figure S2. Photobleaching rate for Cy3-labeled dsDNA. Related to Figures 1 and 2. Mean donor photobleaching time was obtained from fitting donor survival probability to a single-exponential decay equation.
Figure S3. Representative smFRET trajectories of targeting Cascade with dsDNAtarget. Related to Figure 1.
Figure S4. FRET distribution for whole Cascade, Cse1, and Cse2-Cas6e with dsDNAtrunc. Related to Figure 1. (A–C) FRET histograms for FRET events existed in all extracted traces of Cascade with Cy5 labelled Cse2-Cas6e (A), Cy5 labelled Cse1 (B), and Cy5 labelled Cse2-Cas6e (C). The number of molecules used for FRET histograms was 392 (Cascade), 378 (Cse1), and 400 (Cse2).
Figure S5. Transition density plots for Cascade binding to different dsDNA substrates. Related to Figure 2. Transition density plots for (A) dsDNAtarget with non-targeting Cascade, (B) dsDNA0PAM with non-targeting Cascade, (C) dsDNA0PAM with targeting Cascade, (D) dsDNA1PAM with non-targeting Cascade, (E) dsDNA3PAM with non-targeting Cascade, and (F) dsDNA1PAM-12bp with non-targeting Cascade. The number of molecules for each TDP ranged from 1050 to 4,775.
Figure S6. FRET appearance and disappearance at different Cascade concentrations. Related to Figure 2. (A) kbinding for dsDNA0PAM, dsDNA1PAM and dsDNA3PAM at three different Cascade concentrations (2.5 nM, 5 nM and 10 nM). (B) koff for dsDNA0PAM, dsDNA1PAM and dsDNA3PAM at three different Cascade concentrations (2.5 nM, 5 nM and 10 nM).
Figure S7. Cse1 and Cas7 motif mutations abolish target binding. Related to Figure 4. (A–C) FRET distribution for (A) WT Cascade, (B) G160A and lysine-rich β hairpin (K289A, K290A, and K296A) double mutation in Cse1, (C) Cas7 quadruple Lys-rich vise mutation (K137A, K138A, K141A and K144A) binding to dsDNA1PAM. The number of molecules used for FRET histograms was 1517 (WT Cascade), 850 (Cse1 double mutant), and 744 (Cas7 Lys-rich vise mutant). (D) Coomassie blue-stained SDS-PAGE gel analysis of Cse2-Cas6e. Gel lanes: (1) WT Cse2-Cas6e; (2) WT Cse2-Cas6e with K137A Cas7; (3) WT Cse2-Cas6e with K137A, K138A, K141A and K144A Cas7. (E–F) Transition density plots for Cascade with (E) Cse1 G160A mutation and (F) Cascade with Cse1 Lys-rich β hairpin mutation (K289A, K290A, and K296A) binding to dsDNA1PAM. (G) Electrophoretic mobility shift assay for G160A and lysine-rich β hairpin (K289A, K290A, and K296A) double mutation in Cse1 and quadruple Lys-rich vise mutation (K137A, K138A, K141A and K144A) in Cas7 at different time point (2, 5, and 30 min) and various Cascade concentrations (1, 5, 10, 50, and 500 nM).