

Abstract
Red and green are two of the most‐preferred colors from the entire chromatic spectrum, and red and green dyes are widely used in biochemistry, immunohistochemistry, immune‐staining, and nanochemistry applications. Selective dyes with green and red excitable chromophores can be used in biological environments, such as tissues and cells, and can be irradiated with visible light without cell damage. This critical review, covering a period of five years, provides an overview of the most‐relevant results on the use of red and green fluorescent dyes in the fields of bio‐, chemo‐ and nanoscience. The review focuses on fluorescent dyes containing chromophores such as fluorescein, rhodamine, cyanine, boron–dipyrromethene (BODIPY), 7‐nitobenz‐2‐oxa‐1,3‐diazole‐4‐yl, naphthalimide, acridine orange, perylene diimides, coumarins, rosamine, Nile red, naphthalene diimide, distyrylpyridinium, benzophosphole P‐oxide, benzoresorufins, and tetrapyrrolic macrocycles. Metal complexes and nanomaterials with these dyes are also discussed.
1. Introduction
Due to their sensitivity, technical simplicity, and fast response time, fluorescent probes, also known as fluorescent chemosensors, have emerged as very useful tools in analytical sensing and optical imaging.1 The main parts of a fluorescent probe are the signaling unit (chromophore), the spacer (chemical bridge), and the binding unit (receptor); manipulating these three key components allows the design of probes specifically tailored to particular targets. Organic dyes that absorb light in the visible region of the spectrum (λ=400–700 nm) can be used as chromophores. These dyes can contain different auxochromes and functional groups such as amino, carboxylic acid, carbonyl, hydroxy, sulfonic acid, and nitro groups, which modify the ability of the chromophore to absorb light.2 These auxochromes can increase the intensity of the color and/or shift the emitted color in the spectra as well as increase the solubility of the dye.3, 4 Selective probes with red and green emission, two of the most‐desired colors from the entire electromagnetic spectrum, are used in biochemistry, immuno‐ histochemistry, immunostaining, and nanochemistry. For example, translational applications have arisen in the field of imaging of biological tissues to minimize cellular autofluorescence and colocalization in confocal microscopy in multicolored experiences.5, 6
To achieve a better understanding of how biological systems work, researchers need to be able to visualize and quantify events happening at the cellular level with high levels of spatial and temporal resolution.7 Despite great advances in the field, creating selective and sensitive fluorescent probes remains a challenge and generally requires a long process of trial and error. There are many requirements that a fluorescent probe must meet to be used in biological systems, such as nontoxicity, specificity, and solubility in aqueous solutions. Moreover, probes to be used in intracellular labeling need to be able to cross plasma membranes.8 Theoretical models specific to particular chromophores have been created to facilitate the design of better probes, for example, to design boron–dipyrromethene (BODIPY)9 and benzothiazole derivatives.10 The majority of the current fluorescent probes have been designed by using a limited number of core chromophores, with coumarin, BODIPYs, cyanines, fluoresceins, rhodamines, and phenoxazines among the most‐popular ones.11, 12 The most‐studied green dyes are fluoresceins,13 Oregon green 488 and 514,14 perylene diimide,15, 16, 17, 18, 19 the rhodamine green family,20 chlorophyll,21 and eosin.22 Red dyes typically come from rhodamines,11, 20 porphyrins,23 and corroles.24, 25
Red and green pigments have long attracted the interest of researchers. For example, the ability of primates to discriminate between red and green has been linked to foraging advantages, allowing animals to detect more easily ripe fruit and young leaves against mature foliage. Research also suggests it may help intraspecies sociosexual communication in primates by aiding them in the selection of their reproductive partner.26 The primary function of pigments in plants is the process of photosynthesis, in which chlorophylls play a key role.27 Chlorophylls are a group of natural pigments based on a chlorin magnesium macrocycle ring that absorbs yellow and blue wavelengths and reflects green color.28, 29 Chlorophyll is present in photosynthetic organisms (e.g. plants, algae, and cyanobacteria).30 Other red and yellow pigments can help chlorophyll capture light and convert it in energy. There are many colored natural plant pigments, such as porphyrins, anthocyanins, carotenoids, and betalains.31 In the food industry, emphasis has been placed on replacing synthetic colorants with natural pigments.32 In other fields such as environmental monitoring and biomedical diagnosis, smart materials have been synthesized to be used as colorimetric biosensors: color changes can be seen by the naked eye, which makes them easier to visualize and reduces the need for expensive or sophisticated instrumentation.33
Some of the most useful red, orange, and yellow pigments are carotenoids, also called tetraterpenoids.2, 34 The most‐common carotenoids are carotene, the orange pigment present in carrots; lutein, the yellow pigment present in fruits and vegetables; and lycopene, the red pigment found in tomatoes.35 Anthocyanins, a type of flavonoid pigment, are mostly responsible for the purple and blue color of flowers and are soluble in water. Covalent bonding of anthocyanins to organic acids, other flavonoids, or aromatic acyl groups can result in changes to the color intensity and hue.36, 37, 38 Betalains (which can have red or yellow color) are indole derivatives derived from the amino acid tyrosine and are also soluble in water. They are present in plants belonging to the caryophyllales families, such as cactus plants, ice plants, amaranth, and carnivorous plants. Betalains from red beets are often used in the food industry as a natural colorant.39
It is well known that if white light goes through a substance, the substance will absorb particular wavelengths. The residual light being reflected will then result in the color complementary to the wavelength that was absorbed. The color wheel shown in Figure 1 demonstrates this relationship. Here, complementary colors are diametrically opposite each other. Consequently, absorption at λ=420–430 nm makes a substance yellow, and absorption at λ=500–520 nm makes it red. Interestingly, green is a unique color, as it can be created by absorption at about λ=400 nm as well as near λ=800 nm.
Figure 1.
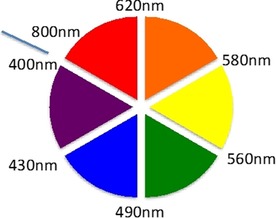
Color wheel.
Several reviews have described the performance of fluorescent probes with specific applications, for example, used as intracellular pH indicators; sensors for reactive oxygen and nitrogen species, for metal ions, and for anions; and as diagnostic imaging tools.40, 41, 42 However, most of the reviews published so far tend to be either exclusively focused on sensors for a specific target or centered around a specific chromophore.43, 44, 45, 46 The aim of this review is to provide a comprehensive, critical, and readable overall overview of the latest research on green and red fluorescent probes and their application in the fields of bio‐, chemo‐, and nanoscience. This review focuses on research published over a five‐year period and looks at both the structure of the different probes and their applications. Section 2 provides a general overview of key advances in the design and applications of green dyes derived from fluorescein, rhodamine, cyanine, boron–dipyrromethene (BODIPY‐FL), 7‐nitobenz‐2‐oxa‐1,3‐diazole‐4‐yl, naphthalimide, acridine, and perylene diimide. Section 3 presents some recent examples of red probes based on cyanine, BODIPY, coumarin, xanthene, Nile red, naphthalene diimide, distyrylpyridinium dyes, benzophosphole P‐oxide scaffold derivatives, and benzoresorufins. Metal complexes with lanthanides, iridium, and ruthenium are discussed in Section 4. Section 5 discusses briefly different nanomaterials such as quantum dots, fluorescent metallic nanoclusters, and semiconductor nanocrystals.
2. Green Fluorescent Dyes
Considerable effort has been focused on the design of new fluorescent probes with the aim to synthesize increasingly more sophisticated structures to enhance further their properties and applications.47 High sensitivity and specificity and the ability to fine‐tune the optical properties (e.g. lifetime, emission and excitation spectra, intensity, and anisotropy) are some of the advantages of fluorescent probes. Probes bearing visible excitable chromophores are particularly appealing, because they can be applied in biological models, such as in cells and tissues, and can be irradiated with light without cell damage.48
Fluorescein and rhodamine dyes are the ones most commonly used to develop biological sensing probes.49 These probes possess excellent optical properties such as a long excitation wavelength (λ≈500 nm) and high fluorescence quantum yields and extinction coefficients, and thus, they are widely used in biochemistry, cell imaging, cell biology, clinical diagnosis, and drug delivery.50, 51, 52 Green fluorescent dyes show an emission wavelength in the λ=500–550 nm range. Fluorophores with green fluorescence include fluorescein, rhodamine, cyanine, BODIPY‐FL, 7‐nitrobenz‐2‐oxa‐1, 3‐diazole‐4‐yl, naphthalimide (lucifer yellow), and acridine orange. This section provides a general overview of the key advances in the design and applications of these types of probes.
2.1. Fluorescein
Probes containing fluorescein have been well studied: they produce a bright signal, are nontoxic, can be obtained on a gram scale, and have many possible reactive sites in their skeletons.53, 54, 55, 56, 57 Fluorescein has several sites at which modifications can be introduced, such as the xanthene unit (at positions 4 and/or 5, 3, and/or 6), the hydroxy groups, and the phenyl ring (positions 4′ and/or 5′). Derivatization of the carboxylic acid is very common, as it leads to spirolactam‐based chemosensors, which in their open ring form result in highly emissive probes. Most fluorescein derivatives are synthesized by substitution on the benzene unit, which leaves the 3′‐ and 6′‐positions available for conjugation and the ability to form the strongly emissive fluorescent dianion.58 Modification of both hydroxy groups to form the methyl ester of fluorescein also yields a highly fluorescent compound.59
The excitation wavelength of fluorescein is about λ=494 nm, which is close to the λ=488 nm spectral line of an argon laser. Thus, fluorescein can be a good fluorophore for probes used in confocal laser‐scanning microscopy and flow cytometry applications. However, these probes are not without problems. Fluorescein can be susceptible to photobleaching (photodegradation, which can eventually lead to the destruction of the probe), and it has a broad fluorescence emission spectrum. Moreover, it has the tendency to self‐quench if conjugated with polymers and high degrees of substitution, and consequently, fluorescein derivatives have limitations in multicolor applications.60 Fluorescein is also highly pH dependent: basic pH results in ring opening of fluorescein, which becomes strongly emissive and shows a very intense greenish‐yellow color.61 Depending on the pH, fluorescein can exist in four ionization forms: cationic (pH 2), neutral (pH<5), monoanionic (pH 6–7), and dianionic (pH>8).62
Oliveira and co‐workers58 report on fluorescent alanine–fluorescein probes 1 and 2 with green emission (see Figure 2). Probe 1 shows colorimetric and “turn‐off” fluorescence behavior for both the HgII ion and for the neurotransmitter dopamine in HEPES [4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid] buffer at pH 8; a change in color is observed from yellow to pink for HgII and to orange for dopamine. Probes 1 and 2 are highly dependent on pH. The absorption spectra of 1 show an increase in the absorbance at λ=490 nm, as well as a color change from colorless to yellow at high pH values. Different species in equilibrium are present depending on the pH: a neutral species (N) is detected below pH 5, a monoanionic species (M) is observed at pH 6–7, and a dianionic species (D) is seen above pH 8 (see Figure 2). A cationic form at pH 2 is not observed. The emission intensity for 1 reaches a maximum at pH 8 (dianionic form) and then decreases. For comparative purposes, the researchers have performed a similar pH study with free fluorescein: the free chromophore does not present a decline in the emission signal above pH 8. The authors conclude that quenching of the emission above pH 8 can be attributed to the presence of the protected amino‐acid unit.63
Figure 2.
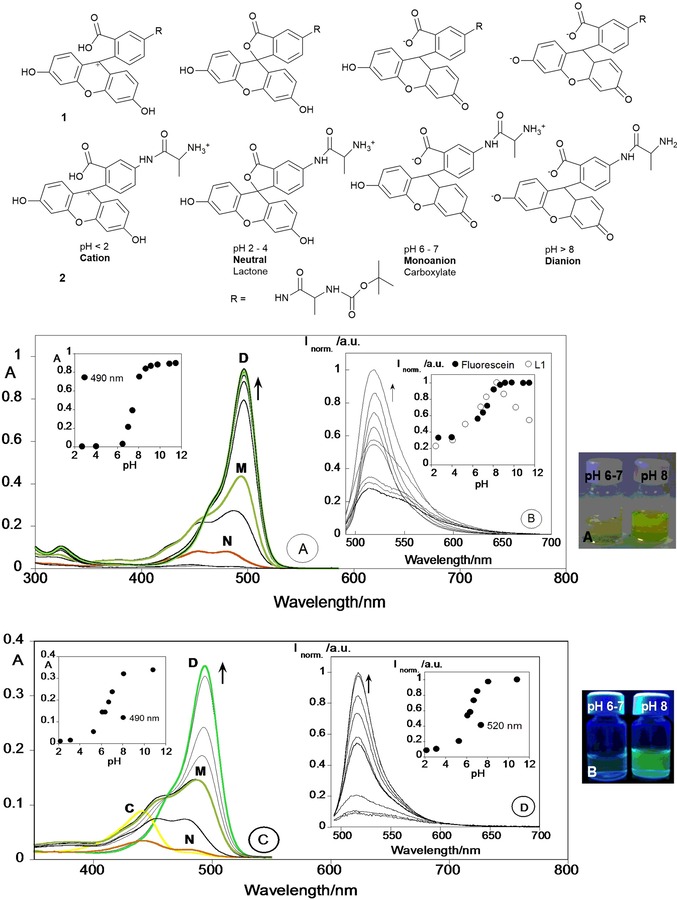
Chemical structures of fluorescein in compounds 1 and 2. Pictures of compounds 1 or 2 at pH 6–7 and pH 8 a) under the naked eye and b) under a UV lamp at λ=365 nm. a) Spectrophotometric and b) spectrofluorimetric titrations of compound 1 and c) spectrophotometric and d) spectrofluorimetric titrations of compound 2 as a function of pH in ethanol/Milli‐Q water (40:60) solution. The insets in panels b, d show the emission fluorescence intensity at λ=520 nm for 1, free fluorescein (panel b), and 2 (panel d). ([Fluorescein]=[1]=1.0×10−5 m, [2]=4.0×10−6 m, λ ex=490 nm, T=298 K). Adapted with permission from Ref. 58. Copyright 2013 Elsevier B. V.
Extensive work has been performed to design ion‐selective fluorescence sensors for alkali, alkali‐earth, and transition‐metal ions. “Turn‐on” sensors are the most appealing, as they can be used as fluorescent sensors in cells and tissues. Zinc(II) is the second most‐abundant d‐block metal ion in the human brain,64 and its closed‐shell nature produces a chelation enhancement of fluorescence (CHEF). Zinc(II) has been widely studied by Lippard and co‐workers, who have synthesized several CHEF families of fluorescein derivatives that are able to detect ZnII in biological systems (see Figure 3): Zinpyr sensors 3–5,65, 66 Zinspy sensors 6–9,67, 68 and QZ sensors 10 and 11.69 Zinpyr sensors ZP1 (3) and ZP2 (4) are able to detect ZnII in sub‐nanomolar concentrations: chelation with the ZnII ion at the di(2‐picolyl)amine (DPA) ligand causes an enhancement in the fluorescence. X‐ray crystallography reveals a 1:2 (ligand/metal) stoichiometry for the 3/ZnII complex, with a bipyramidal trigonal geometry for both ZnII centers.65 ZP3 (5) is asymmetrical, and coordination by the ZnII ion gives a sixfold enhancement in the fluorescence; the 5/ZnII complex has a 1:1 stoichiometry, which results in a distorted octahedral geometry.66 Zinspy (ZS) sensors ZS1 (6), ZS2 (7), ZS3 (8), and ZS4 (9) have sulfur units. Due to the fact that sulfur has lower affinity for ZnII than nitrogen and oxygen, only a 1.4–2.0‐fold enhancement in the fluorescence is observed; interestingly, these compounds also show a “turn‐on” effect in the presence of CdII ions. Chemosensors QZ1 (10) and QZ2 (11) contain one and two 8‐aminoquinoline units, respectively. QZ1 is asymmetrical and shows a 42‐fold enhancement in the fluorescence upon the addition of ZnII. QZ2 (11) binds two metal ions with a 150‐fold “turn‐on” effect for ZnII. Chemosensors QZ are ZnII selective and can detect this ion in the presence of other biologically relevant metal ions such as NaI, KI, MgII, and CaII.
Figure 3.
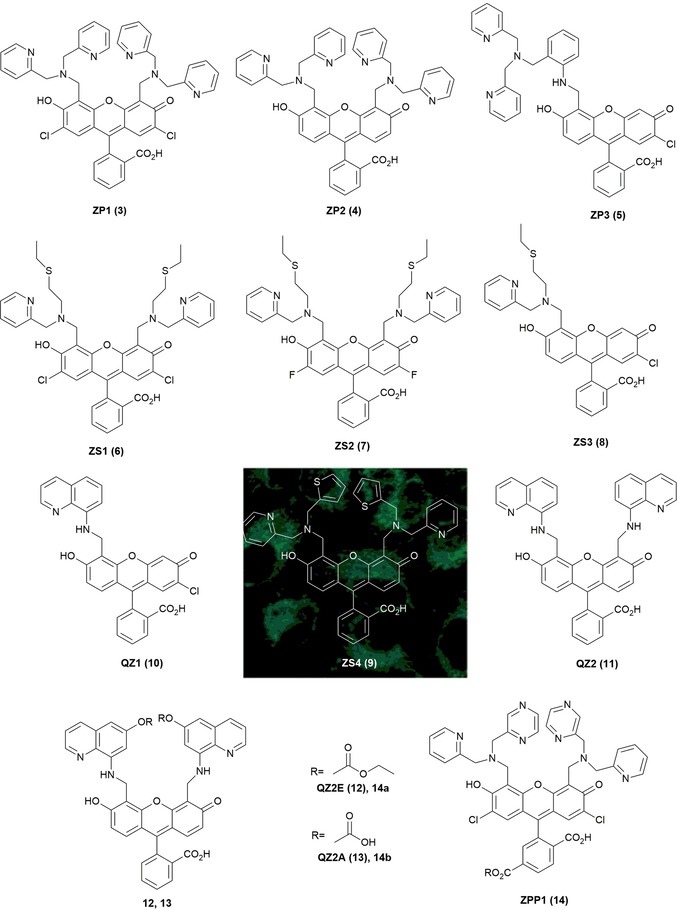
Chemical structures of ZnII probes: Zinpyr sensors 3–5, Zinspy sensors 6–9, QZ sensors 10 and 11, QZ2E (12), QZ2A (13), ZPP1 (14), 14 a, and 14 b; subcellular compartmentalization of ZS4 (9) (mitochondrial) in HeLa cells. Adapted with permission from Ref. 68 Copyright 2006 American Chemical Society.
Studies by Nolan et al. on these ZnII sensors, performed in living cells and brain tissue, show that tertiary amine probes 9 are more cell permeable than aniline probes 5 and 11. Imaging studies in HeLa cells show that ZP probe 3 accumulates primarily within the Golgi apparatus, and ZS probe 9 accumulates in the mitochondria (see Figure 3).68 Although QZ2 (11) can detect higher concentrations of free ZnII reversibly and without probe saturation, it has the limitation of not being cell trappable; to address this limitation, McQuade and Lippard70 have modified the structure to develop new “turn‐on” probes QZ2E (12) and QZ2A (13). Two esters are added to the quinolone rings of QZ2 to produce QZ2E (12). The esters allow cell permeability until the probe is inside the cells. After that, intracellular esterases hydrolyze the esters to carboxylates and QZ2A (13) is formed. QZ2A (13) is negatively charged, which prevents the probe from diffusing out of the cells. Following a similar approach, Buccella, Horowitz, and Lippard71 have designed the new probe ZPP1 (14) by incorporating carboxylate/ester groups in the 6‐position of fluorescein; these structural changes allow better control of the intra/extracellular distribution of the probe while maintaining the ZnII binding properties of ZP1 (3).
A shortcoming of these probes is that they suffer from unpredictable cellular localization. In an attempt to design a more accurate delivery system, Radford and co‐workers72 have published a peptide‐based targeting strategy, in which a series of targeting peptides are attached to the diacetylated Zinpyr sensor 15 for intracellular detection of ZnII (see Figure 4). Modification of probe 15 with a mitochondrial‐targeting peptide allows the use of the sensor in concentrations four times lower than previously reported for the non‐acetylated probe.
Figure 4.
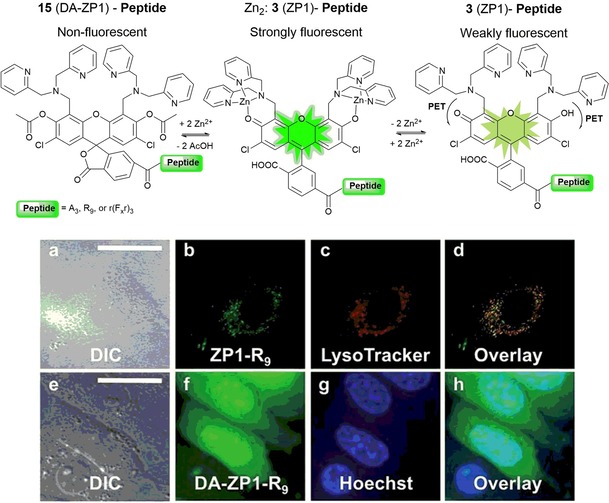
Illustrative scheme of the two zinc‐sensing mechanisms operating with peptide constructs. Deconvoluted fluorescence microscopy images of live HeLa cells pretreated with ZP1 (3)‐R9 (5 mm) or DA‐ZP1 (15)‐R9 (2.5 mm) and the indicated organelle stain. Top, ZP1‐R9) a) differential interference contrast (DIC) image, b) signal from ZP1‐R9, c) signal from Lyso‐Tracker Red, d) overlay of the images in panels b and c. Pearson's r=0.42±0.16 (n=8). Bottom, DA‐ZP1‐R9) e) DIC, f) signal from DA‐ZP1‐R9 after treatment with 25 mm zinc pyrithione (ZnPT), g) signal from Hoechst 33258, h) overlay of the images in panels f and g. Scale bar=25 μm. Reproduced from Ref. 72 with permission from The Royal Society of Chemistry.
Most fluorescent probes exhibit a quenching response upon chelation to paramagnetic transition‐metal ions; however, some probes with a “turn‐on” effect have been reported for those metal ions. Abebe and Sinn73 report on “turn‐on” and colorimetric fluorescein derivative probes 16 and 17 for CoII and NiII, two metal ions that are usually fluorescence quenchers. The probes are capable of detecting both ions in aqueous solutions quickly and selectively. Coordination occurs through the carbonyl O atom and the two N atoms and is reversible (see Figure 5).
Figure 5.
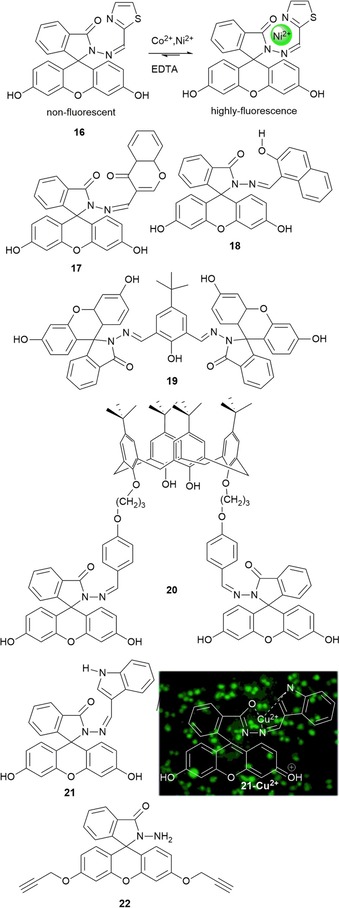
Chemical structures of probes 16–22. Fluorescence imaging of 21 in RAW 264.7 cells in the presence of CuII RAW 264.7 loaded with 21 and treated with copper for 1 h at 37 °C. Adapted with permission from Ref. 74. Copyright 2014 American Chemical Society.
CuII, a metal ion with an unfilled d shell, is also well known for quenching fluorescence upon binding to fluorescent probes. Like CoII and NiII, it usually produces colored metal complexes due to d–d electronic transitions. Probes 18, 19, and 20 are fluorescein derivatives with a highly selective “off–on” behavior towards CuII in aqueous solution (for 18 and 19) and acetonitrile (for 20).75, 76 For 18 and 19, a yellow color appears upon coordination with CuII due to the opening of the ring in the fluorescein unit, which results in the formation of complexes with a 1:1 (ligand/metal) stoichiometry. Probe 20 is formed by a calix[4]arene derivative with fluorescein substituents. The addition of CuII to an acetonitrile solution of the probe results in a color change from colorless to yellow; if the reaction is performed in aqueous solution, the color changes from colorless to purple. In acetonitrile, the probe also produces an enhancement in the fluorescence intensity and is able to detect nanomolar concentrations of CuII; this “turn‐on” effect is not observed in aqueous solution. In 2014, rapid “turn‐on” probe 21 for CuII detection in water was reported by Muthuraj et al.74 (see Figure 5). Probe 21, with an indole‐3‐carboxaldehyde‐functionalized fluorescein hydrazine, can selectively bind CuII both in vivo and in vitro. CuII coordination is the result of induced Forster resonance energy transfer (FRET): in the presence of CuII, interaction between the donor (indole‐3‐carboxaldehyde) and the acceptor fluorophore (xanthene) leads to intramolecular FRET between the two. The in vivo results performed in RAW 264.7 cells show bright fluorescence in the presence of CuII, with no interference from other metal ions; research has revealed that both probe 21 and its CuII complex are nontoxic in the cellular system and show considerable potential in biomedical applications (see Figure 5).
Another metal that often produces a quenching effect in the emission of the probe is PdII. However, fluorescein derivative 22 published by Wei and co‐workers77 can selectively sense PdII in the presence of CuII, with CuII acting as a synergic trigger. The addition of PdII and CuII to a solution of 22 in acetonitrile turns the solution from colorless to green, a color change visible to the naked eye; the reaction conditions are 2 h at room temperature with an excitation wavelength of 492 nm. The two metal ions hydrolyze the alkyne groups of 22, which leads to a significant fluorescent enhancement at λ=514 nm.
Swamy and co‐workers78 have designed fluorescein probes 23, 24, and 25 by Mannich reaction of 2′,7′‐dichlorofluorescein with morpholine, thiomorpholine, and 1‐methylpiperazine, respectively. pH studies conducted with the three probes show that, unlike the free chromophore, the three probes exhibit intense fluorescence at acidic pH values and weak fluorescence at basic pH values. Probes 23 and 24 show selectivity towards AgI ions in aqueous solution at pH 7.4; an enhancement in the emission intensity is only observed for probe 23, whereas quenching is seen for 24, followed by a color change from yellow to pink. These different spectral behaviors are related to the different binding modes of the AgI ion in both systems. Two AgI ions chelate to probe 24 through the sulfur atoms of the thiomorphine moieties and the phenyl oxygen atoms of fluorescein; this leads to photoinduced electron transfer (PET) from the released tertiary amine groups, which before were blocked by hydrogen bonding of the phenolic hydrogen atoms. For probe 23, only one AgI ion coordinates to the probe through the nitrogen donor atoms in the morpholine and benzylic amines, which blocks the PET phenomena; thus, “turn on” of fluorescence occurs (Figure 6). Recently, fluorescein spirolactam derivative 26 was design by Lin and co‐workers;79 these authors have used this probe as a “turn‐on” fluorescence probe for the detection of AgI ions in aqueous solutions (detection limit 0.08 μm), including tap, river, and lake waters; the results obtained are in excellent agreement with those obtained by studying the samples by flame atomic absorption spectrometry (Figure 6).
Figure 6.
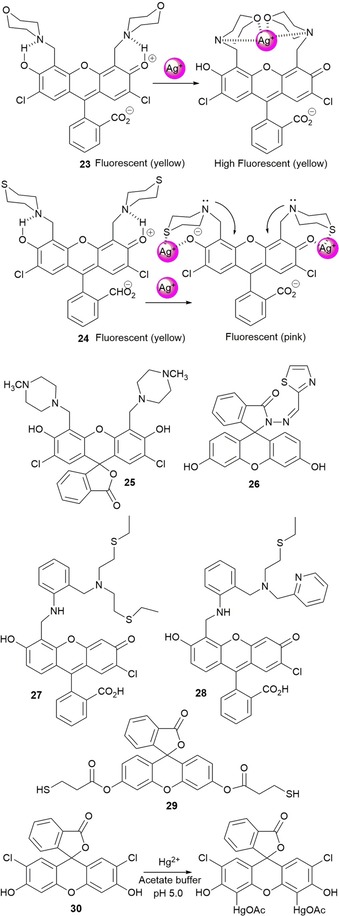
Chemical structures and mechanisms of probes 23–30. Adapted from Refs. 78, 79, 80, 81, 82, 83.
Water‐soluble “turn‐on” fluorescein derivative 27 has been reported by Nolan and Lippard.80 Probe 27 is highly sensitive and selective to HgII ions over alkali (LiI, NaI, and RbI), alkali‐earth (MgII, CaII, SrII, and BaII), and transition‐metal ions (CrIII, MnII, FeII, CoII, NiII, CuII, ZnII, CdII, and PbII). Coordination at the aniline nitrogen atoms by HgII blocks the PET mechanism and a fivefold increase in the emission intensity is observed. The same authors have published new asymmetrical fluorescein derivative 28, which selectively coordinates to HgII ions (see Figure 6); this probe shows a “turn on” of the emission intensity and is able to detect parts per billion levels of this metal ion in aqueous solutions.81
Probe 29, a fluorescein thiol ester derivative, has been synthesized by Fernández‐Lodeiro and co‐workers82 and has been grafted onto the surface of silver nanoparticles (AgNPs). This new hybrid system (i.e. AgNPs@29) shows an intense enhancement in fluorescence due to the presence of silver. The interaction of AgNPs@29 with HgII (16 μm) leads to a blueshift and an increase in the surface plasmon resonance (SPR) band from around λ=410 to 398 nm as well as an enhancement in the emission band at about λ=534 nm in toluene. Choi and co‐workers83 report on dichlorofluorescein probe 30, which shows an intense emission band at λ=528 nm; this band is quenched upon the addition of HgII with a color change from yellowish green to orange, visible by the naked eye. These changes are due to selective mercuration of the xanthene ring at positions 4 and 5.
Wang et al.84 report on fluorescein‐based sensor 31 bearing a nitroolefin that is able to detect biological thiols such as cysteine and glutathione. An increase in the intensity of the absorption band at λ=497 nm as well as an enhancement in the fluorescence intensity at λ=520 nm are noticed upon the addition of both thiols. The spectral changes are due to Michael addition of the thiols to the double bond of the nitroolefin moiety, which leads to the formation of 32 (see Figure 7). This mechanism has also been demonstrated in the imaging of thiols in PC‐12 cells. The amino acid l‐histidine in its free form acts as a tridentate ligand towards transition‐metal ions such as CuII. Wang and co‐workers85 report on water‐soluble fluorescent probe 33, which selectively binds to CuII ions; this binding causes quenching of the emission intensity through photoinduced electron transfer (limit of detection, LOD=1.6 μm). The 33–CuII complex can selectively detect histidine (LOD=1.6 μm) in the presence of the other naturally occurring amino acids and shows a “turn on” of the fluorescence signal. This behavior has also been verified in biological environments such as living HepG2 cells (see Figure 7).
Figure 7.
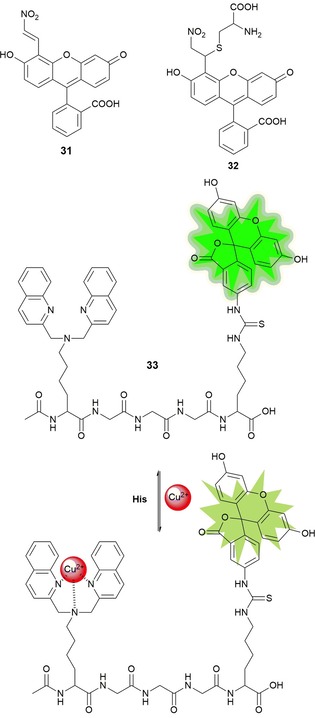
Chemical structures and reaction mechanism of probes 31–33. Adapted from Refs. 84,85
A series of CuII complexes with fluorescein probes 34 to 38 have been synthesized by Lim and co‐workers86 (see Figure 8). The structures suggest that CuII is coordinated with one oxygen and two nitrogen atoms to form nonfluorescent complexes. The addition of nitrogen monoxide (NO) to a buffered aqueous solution of the probes (pH 7.0) produces the “turn on” of the emission. The reduction of CuII to CuI produces the NO+ cation, which in turns reacts with the probes to form nitrosated probes (34–38)–NO. Probes 34–38 are selective to NO over other biologically important species, such as H2O2, NO2 −, NO3 −, HNO, ONOO−, and ClO−. The copper complex of probe 38 has been used to detect NO as a biological signaling agent in SK‐N‐SH neuroblastoma cells and in Raw 264.7 murine macrophages.87 The 38–CuII complex has significant advantages such as brightness, cell‐membrane permeability, minimal cytotoxicity, and selectivity. It also shows a rapid fluorescent enhancement in the presence of NO, with an immediate 11‐fold “turn on” in the emission intensity (see Figure 8). To be used successfully in biological tissues, the signaling agent must be retained inside cells; unfortunately, the 38–CuII complex shows a tendency to diffuse out of cells. To overcome this problem, McQuade and Lippard have prepared three new probes, 39, 39E, and 39A, by employing the ester/acid strategy to improve the ability of the probes to stay inside cells.70
Figure 8.
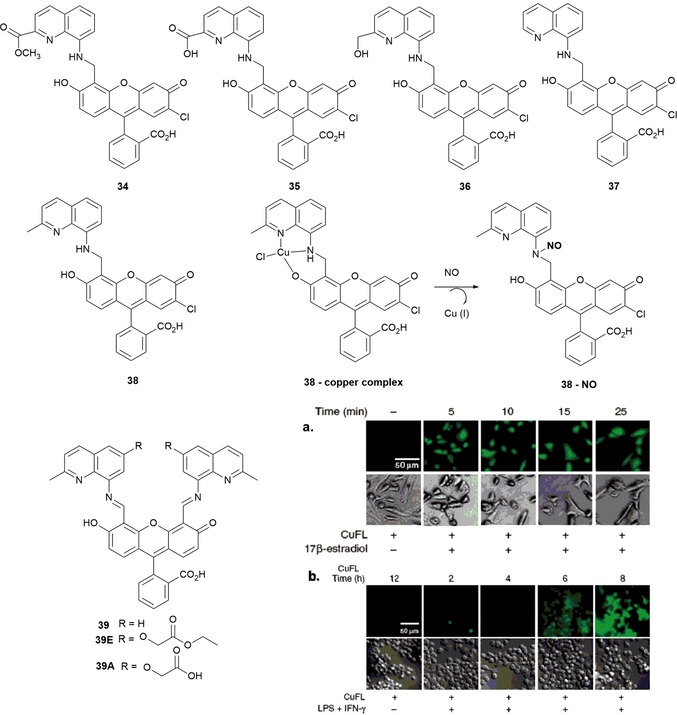
Chemical structure of probes 34–38. a) CuFL1 detection of NO in SK‐N‐SH neuroblastoma cells. [CuFL1]=1 μm, [17β‐estradiol]=100 nm. From left to right) 25 min exposure to CuFL1 without 17β‐estradiol and 5, 10, 15, and 25 min exposure to CuFL1 and 17β‐estradiol. Top) fluorescence images. Bottom) DIC images. Scale bars=50 μm. b) CuFL1 detection of NO in Raw 264.7 murine macrophages. [CuFL1]=1 μm, [lipopolysaccharide (LPS)]=500 ng mL−1, [interferon‐γ (IFN‐γ)]=250 U m−. From left to right) 12 h exposure to CuFL1 without LPS/IFN‐γ and 6, 8, 10, and 12 h exposure to CuFL1 and LPS/IFN‐γ. Top) fluorescence images. Bottom) DIC images. Scale bars=50 μm. Reprinted with permission from Ref. 87 Copyright 2010 American Chemical Society.
The development of methods that can quickly, sensitively, and selectively detect fluoride anions in aqueous samples is of great importance because of the impact of fluoride anions on human health and the environment. Zheng and co‐workers88 have synthesized probe 40, a biocompatible hydrophilic poly‐ (ethylene glycol) (PEG) polymer attached to a fluorescein derivative; the role of the PEG polymer is to guarantee water solubility and biocompatibility (see Figure 9). Quenching of fluorescence is observed upon linkage of tert‐butyldiphenylsilyl (TBDPS) groups. However, the addition of fluoride anions leads to selective fluoride‐mediated cleavage of the Si−O bond, which causes a “turn on” of the fluorescence emission; the detection limit for fluoride is 19 ppb. This effect is also observed in “real‐life” samples such as running water, urine, and serum, as well as in HeLa and L929 cells.
Figure 9.
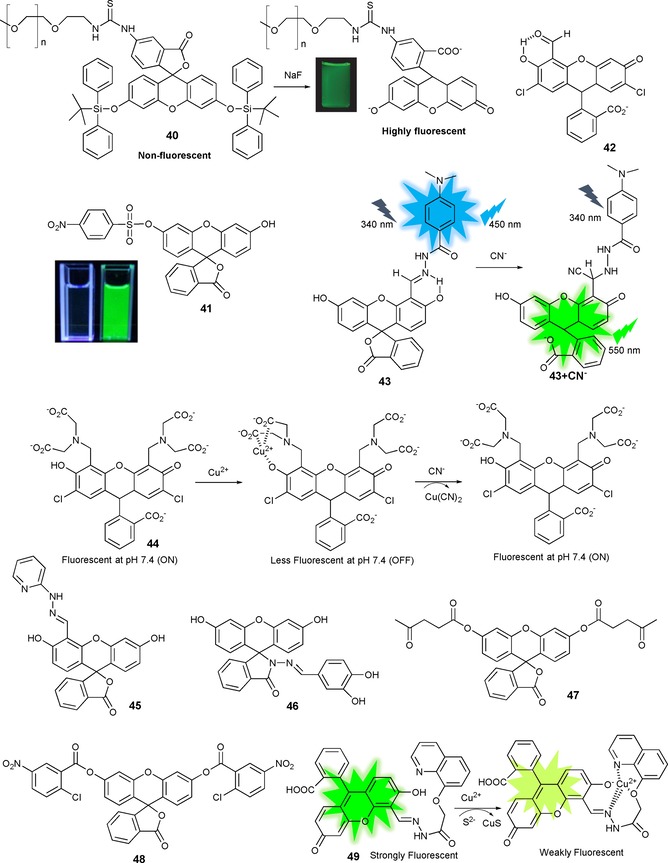
Chemical structures and/or mechanisms of compounds 40–49. Adapted from Refs. 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101.
Several fluorescein derivatives for fluoride detection have been synthesized;89, 90, 91, 92, 93 however, these probes do not selectively detect fluoride, as they suffer from interference from other anions. Asthana and co‐workers94 report the synthesis of fluoride‐selective “turn‐on” fluorescein probe 41 containing nitrobenzenesulfonyl chloride as a masking unit (see Figure 9). The addition of F− releases the NBS group from the fluorescein unit, which leaves the ring open. In the absorption spectrum, a new intense band appears at λ=517 nm; moreover, a change in color from colorless to yellow is observed, followed by a 60‐fold enhancement in emission at λ=530 nm (green emission). Probe 41 detects F− over sulfide anions and thiols (detection limit 27.5 nm); however, the probe works in an organic solvent, acetonitrile, not in aqueous solution.
Regarding the cyanide anion, “turn‐on” fluorescence probe 42 has been reported by Kwon and co‐workers.95 Probe 42 is able to detect selectively CN− ions over other anions in CH3CN/HEPES (9:1, v/v), at pH 7.4. The CN− anion attacks the aldehyde group in the salicylaldehyde unit, which results in intramolecular hydrogen transfer with the phenol proton (see Figure 9). This reaction mechanism implies that 42 behaves like a colorimetric (colorless to yellow) and off–on green fluorescent sensor for cyanide and can be used for live‐cell imaging in HaCaT cells. Lv et al.96 also report ratiometric fluorescein‐based probe 43 for CN− detection in water. The probe acts by a FRET mechanism, with 4‐(N,N‐dimethylamino)benzamide as the donor, fluorescein as the receptor, and the participation of the salicylaldehyde hydrazone unit. The CN− anion activates the hydrazone functionality, which is followed by fast proton transfer of the phenol to the developing nitrogen anion of the probe, generating the open form of fluorescein (see Figure 9). Unfortunately, the probe is not exclusively CN− selective and can also weakly sense F−, AcO−, and H2PO4 − anions.
Knowing the amazing ability of cyanide to react with copper ions to form very stable Cu(CN2) species, Chung and co‐workers97 describe selective off–on‐type CN− sensing probe 44, which is based on the CuII complex of a fluorescein derivative. The fluorescence of the system is quenched by the CuII ion: CN− added in aqueous solution (pH 7.4) coordinates with the metal, which enhances the fluorescence signal (Figure 9).
Regarding off–on anion sensing, fluorescein probes 45 84 and 46 98 show intense green emission upon the addition of the hypochlorite anion (ClO−) in aqueous solution with very low detection limits (7.3 and 40 nm, respectively); similar behavior is observed in studies performed in living cells. Probes 47 99 and 48 100 show high selectivity and sensitivity to the sulfide anion (S2−) with an enhancement in the emission signal in aqueous solutions, even in real samples such as white wine (47) and MGC‐803 cells (48). In the presence of CuII, sulfide forms very stable CuS species, and this has allowed Hou and co‐workers101 to design a nonemissive CuII complex with fluorescein probe 49 containing a 8‐hydroxyquinoline unit. This probe is able to detect S2− anions by showing an intense green fluorescence (see Figure 9).
2.2. Rhodamine
The spirolactam structure of rhodamine is nonfluorescent and colorless and exhibits high selectivity for metal ions; the rhodamine ring in its open form has strong fluorescence. Zhang and co‐workers102 have reported rhodamine derivative 50 containing an ethylenediamine‐N,N‐diacetic acid moiety (see Figure 10). Experiments performed in acetonitrile show that probe 50 is CuII selective. The addition of up to 6 equivalents of CuII to 50 produces a 49‐fold enhancement in the fluorescence, followed by a blueshift of Δλ=45 nm, which results in intense green fluorescence at λ=530 nm. However, the addition of HgII results in only a minimal enhancement in the emission. Recently, Park and co‐workers103 have described “turn‐on” fluorescence sensor 51 based on rhodamine that selectively detects CuII ions over others metal ions such as HgII, CoII, KI, CsI, AgI, PbII, ZnII, MgII, FeIII, NiII, LiI, and AlIII. Probe 51 shows unprecedented colorimetric (from colorless to pink) and fluorometric (from colorless to yellowish green) changes in the presence of CuII on the basis of the ring‐opening mechanism of the rhodamine spirolactam with a 1:1 (ligand/metal) binding ratio. The detection limit is 0.14 μm, and the solvent used is CH3CN/H2O (9:1, v/v). Taking advantage of the same reaction mechanism, Lee and co‐workers104 have developed “turn‐on” fluorescence rhodamine 6G phenylthiourea derivative 52 to detect HgII ions in acetonitrile. The addition of HgII opens the spirolactam ring of the rhodamine moiety, which leads to a 700‐fold increase in the fluorescence emission (detection limit 0.45 μm); the authors also prove the reversibility of this system with the addition of ethylenediaminetetraacetic acid (EDTA) (see Figure 10). Lozano and co‐workers105 have immobilized a rhodamine 6G spirocyclic phenylthiosemicarbazide derivative on a nylon membrane to form probe 53, which shows intense green emission in the presence of HgII (see Figure 10). This behavior can be attributed to mercury‐promoted ring opening of the spirolactam in the rhodamine moiety (detection limit 0.4 ng mL−1). This mechanism has been tested through a recovery study by using several spiked water samples from different locations.
Figure 10.
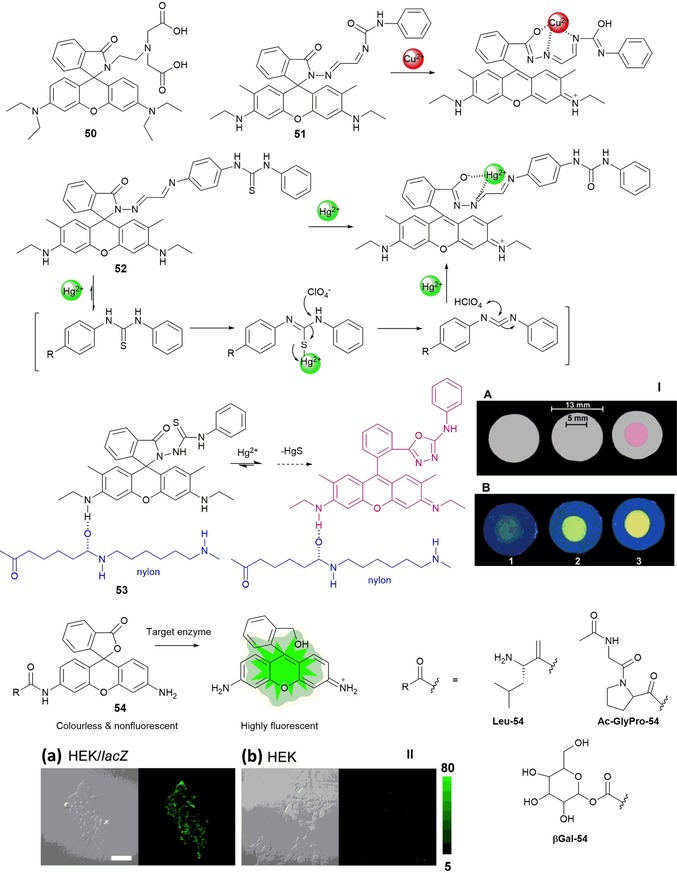
Chemical structures and reaction mechanisms of probes 50–54. Panel I) Photographs of 53‐doped nylon membranes irradiated with a) visible and b) UV lamps, after treatment with 10 mL of 1) HgII‐free water, 2) 5 ng mL−1 HgII solution, and 3) 12 ng mL−1 HgII solution. The bars indicate the diameters of the nylon membrane and the area exposed to the solution flow. Reproduced from Ref. 105 with permission from The Royal Society of Chemistry. Panel II) Fluorescence confocal imaging of a) HEK 293/lacZ cells and b) HEK 293 cells loaded with βGal‐HMRG. Scale bar represents 50 μm. Reprinted (adapted) with permission from Ref. 106. Copyright 2013 American Chemical Society.
Highly sensitive fluorescence probes for proteases and glycosidases have been developed by Sakabe and co‐workers106 by replacing the acetyl group of rhodamine derivative 54, which is colorless and nonfluorescent, with a target enzyme substrate moiety R (see Figure 10), leading to a hydroxymethyl rhodamine derivative highly fluorescent. The addition of the target enzyme to the probe generates an open nonspirocyclic structure that is strongly fluorescent. The authors have synthesized nonfluorescent probes for leucine aminopeptidase (Leu‐54), fibroblast activation protein (Ac‐GlyPro‐54), and β‐galactosidase (βGal‐54); in all cases, upon the addition of the enzymes, a green fluorescence emission is observed. They have also conducted imaging studies in living cells (see Figure 10). Oliveira and co‐workers107 report an unusual green‐luminescent IrIII complex based on a lissamine chromophore (see Figure 11). Probe 55, a lissamine rhodamine B sulfonyl derivative bearing alanine, produces orange emission in water; the addition of IrIII at room temperature results in a highly emissive green Ir@55 complex, with a fluorescence quantum yield of 0.47. NMR spectroscopy and theoretical studies suggest coordination of the IrIII ion at the alanine moiety through involvement of the amide and carbonyl groups. This complex is selective towards HgII, which results in quenching of the green emission; the addition of cysteine results in recovery of the emission signal. The Ir@55 complex can detect minimal amounts of HgII [(13.63±0.11) μm] and cysteine [(6.56±0.10) μm].
Figure 11.
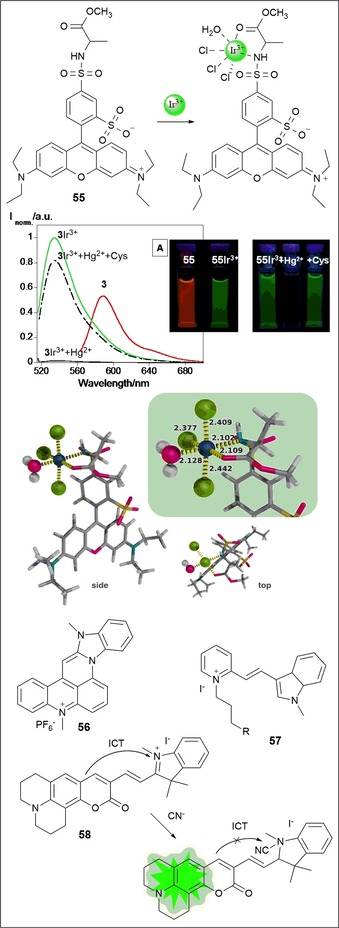
Chemical structures and/or mechanisms of probes 55–58. a) Spectrofluorometric spectra and image under a UV lamp (λ ex=365 nm) of 55, 55–IrIII, 55–IrIII+HgII, 55–IrIII+HgII+cysteine in aqueous solution, λ ex=510 nm. Side and top view of the lowest energy coordination mode of IrIII to the lissamine receptor, with the inset highlighting the interaction sites and coordination distances (color code: Cl, green; S, yellow; O, pink; C, gray; Ir, dark blue; H, white). Reproduced from Ref. 107 with permission from The Royal Society of Chemistry.
2.3. Cyanine
Cyanine dyes are fluorescent molecules commonly used as labels or probes in proteomics, imaging, and biomolecular labeling.108, 109, 110, 111 They have high biocompatibility, are bright, and have high molar absorptivity; they also have the advantage of covering a wide spectral range, from blue to near infrared.60 Cyanine dye families are mostly formed by a polymethine chain linked to two nitrogen‐containing heterocycles, such as indoles, benzothiazoles, benzoxazoles, or quinolones.112 These probes have excellent ability to bind DNA through noncovalent interactions by intercalation between base pairs.113 Probe 56, a hexacyclic acridine–monomethine cyanine dye, was designed by Mahmood and co‐workers114 (see Figure 11). The probe exhibits green emission with a quantum yield close to 1 for dimethyl sulfoxide, methanol, and glycerol and 0.5 for water. The ability of 56 to bind to DNA has led the researchers to study its potential as an intracellular DNA stain by using human breast carcinoma cells, MD231 and ECACC; the results show that the probe accumulates primarily in the nucleus. Taking advantage of the ability of cyanine to bind DNA, Bohhänder and Wagenknecht115 have synthesized green fluorescent cyanine styryl probe 57, which can be further incorporated into oligonucleotides through “click”‐type chemistry to be used as an energy donor in DNA. Energy transfer from 57 to a red emitter leads to a significant wavelength shift from green to red, with both emitters working as “DNA traffic lights” (see Figure 11). Colorimetric “turn‐on” fluorescent probe 58, with a coumarin–hemicyanine structure, is selective for the cyanide anion in HEPES buffer (pH 9.3).116 The addition of the CN− anion to 58 leads to a gradual decrease in the absorption band at λ=603 nm with the emergence of a new band at λ=440 nm; at the same time, a remarkable fluorescence enhancement at λ=502 nm occurs. The cyanide anion is highly reactive to indolium groups; thus, nucleophilic attack of CN− to the indolium group blocks π conjugation between this group and the coumarin, which leads to the observed spectral and color changes (see Figure 11).
2.4. BODIPY‐FL
Boron–dipyrromethene (BODIPY) dyes are very stable against light degradation and chemical attack; they have very high extinction coefficients, high fluorescence quantum yields, and are soluble in most organic solvents. By simply varying the substituents in the different positions, a wide range of emissive fluorophores can be synthesized, covering the blue to the near‐IR part the electromagnetic spectrum. Probe 59 (see Figure 12) shows bright‐green fluorescence with an emission band at λ=512 nm in acetonitrile/water solutions. This probe shows high selectivity towards CuII cations, which causes quenching of the fluorescence emission. The 59–Cu complex is selective to cysteine (Cys) and homocysteine (Hcy) over other amino acids such as Thr, Asp, Leu, Iso, Pro, Met, Glu, Try, Gly, Ser, Asn, Phe, Gln, Tyr, Arg, Lys, His, Ala, and Val; recovery of the emission with a sixfold enhancement in the intensity is observed.117 Probes 60 and 61, selective towards Cys, are reported by Guo and co‐workers.118 These probes are not emissive, but the presence of Cys leads to a 300‐fold increase in the emission intensity at λ=516 nm for probe 60 and a 54‐fold increase at λ=512 nm for 61, generating green‐emissive species 60 a and 61 a (Figure 12). Cysteine causes cleavage of the 2,4‐dinitrobenzenesulfonyl (DNBS) groups in the probes, which generates the new species. These probe have been used for fluorescence imaging of intracellular thiols in NCI‐H446 cells.
Figure 12.
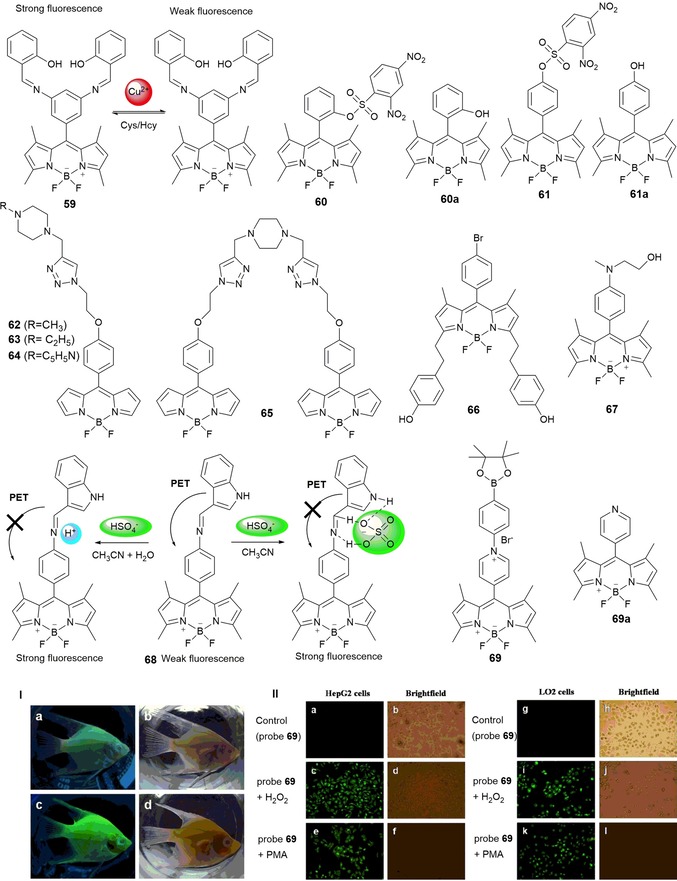
Chemical structures and mechanisms of compounds 59–69. Panel I) a) In vivo fluorescence images of 20‐day‐old angelfish treated with 10 μm probe 69 for 30 min at 25 °C and b) the bright‐field image. c) Probe‐stained angelfish treated with 100 μm H2O2 for 120 min at 25 °C and d) the bright‐field image. Panel II) a) Fluorescence images of HepG2 cells incubated with 10 μm probe 69 for 30 min at 37 °C and b) the bright‐field overlay to confirm viability. c) Probe‐stained HepG2 cells treated with 100 μm H2O2 for 90 min at 37 °C and d) the bright‐field overlay. e) Probe‐stained HepG2 cells stimulated with 1 μg mL−1 (PMA=phorbol myristate acetate) for 90 min at 37 °C and f) the bright‐field overlay. g) Fluorescence images of LO2 cells incubated with 10 μm probe 69 for 30 min at 37 °C and h) the bright‐field overlay. i) Probe‐stained LO2 cells treated with 100 μm H2O2 for 90 min at 37 °C and j) the bright‐field overlay. k) Probe‐stained LO2 cells stimulated with 1 μg mL−1 PMA for 90 min at 37 °C and l) the bright‐field overlay. Reproduced with permission from Ref. 123. Copyright 2014 Elsevier B. V.
A series of green‐fluorescent BODIPY–piperazine conjugates separated by alkyl spacers (probes 62–65) have been synthesized by Singh and co‐workers119 (see Figure 12). The sensing ability of these probes towards a variety of metal ions has been studied. Probes 62–64 are inactive towards NaI, KI, CaII, MgII, AgI, NiII, ZnII, CdII, CuII, PbII, HgII, AlIII, FeIII, and CrIII in acetonitrile/water solutions. However, probe 65 shows selectivity towards HgII, with a “turn off” in the fluorescence signal at λ=521 nm. This quenching of the fluorescence signal is due to HgII coordination with the donor atoms in the piperazine unit and the triazlyl nitrogen atom, which leads to the formation of a 65–HgII complex. Wang et al.120 have prepared BODIPY‐based chemosensors 66 and 67 (see Figure 12). In experiments performed in ethanol, probe 66 is selective to FeIII with little interference from AlIII and CrIII; in aqueous solution, probe 67 is selective to AlIII over CrIII and FeIII. Probe 66 shows the typical bands of BODIPY dyes, with absorption maximum at λ=337 nm and an emission band at λ=571 nm. Upon the addition of FeIII, the band at λ=571 nm decreases and a new intense band at λ=502 nm appears; a change in color from pink to yellow occurs, which can be observed by the naked eye.120 Probe 67 showed a “turn‐on” effect on the fluorescence signal at λ=515 nm upon metal coordination. Coordination takes place with the N‐methyl‐N‐(2‐hydroxyethyl) moiety at the lone pair of electrons on the nitrogen atom, which blocks the PET process and gives rise to an enhancement in the emission intensity.121 Also taking advantage of a PET mechanism, Li and co‐workers122 have designed probe 68, a “turn‐on” green fluorescence BODIPY derivative bearing an indole moiety; the probe detects hydrogen sulfate (HSO4 −) anions in acetonitrile and aqueous solutions. In acetonitrile, the PET effect is blocked due to coordination between the HSO4 − anion and the amine groups; in aqueous solutions, blocking is due to protonation. Li and co‐workers have also published interesting N‐alkylated BODIPY derivative 69.123 This probe shows a “turn on” of the fluorescence signal in the presence of H2O2 and is selective to H2O2 without interference from other reactive oxygen species (ROS) and reactive nitrogen species (RNS) under physiologic conditions. The “turn on” in the fluorescence signal can be attributed to the formation of probe 69 a, which is a product of oxidation by H2O2 (see Figure 12).
To design probes that are able to detect gas molecules, Gotor and co‐workers124 have developed probes 70 and 70 a, which are able to detect the nerve gases diethylcyanophosphonate (DCNP) and diisopropylfluorophosphate (DFP) in buffered water/acetonitrile solutions (see Figure 13). Initially, both probes have no emission due to intramolecular charge transfer (ICT) processes promoted by the nonbonding electrons on the nitrogen atom. The addition of the nerve agents leads to a large enhancement in the corresponding emission bands due to the formation of ammonium salts through a displacement reaction. To assess the potential use of these probes in handheld sensing kits, the authors have also immobilized the probes on a solid support; an enhancement in the fluorescence is also observed in this case. Pan and co‐workers125 report off–on fluorescence BODIPY derivative 71, which can detect carbon dioxide gas. Probe 71 is highly sensitive to pH with a 500‐fold enhancement in the fluorescence signal at acidic pH values (pH 1.42 to 4.12) on the basis of a PET mechanism. The authors postulate that the green fluorescence observed in the presence of CO2 gas can be attributed either to the reaction of the tethered phenylamino group of 71 with CO2 with the formation of carbamic acid or to the formation of the protonated phenylammonium species of probe 71, which induces the conversion of CO2 into carbonic acid (see Figure 13).
Figure 13.
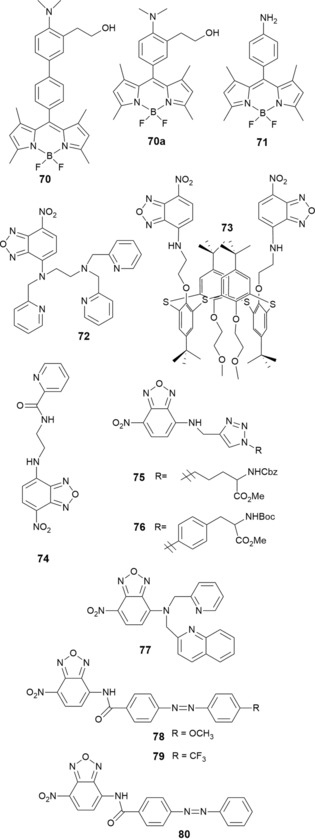
Chemical structures of compounds 70–80.
2.5. 7‐Nitrobenz‐2‐oxa‐1,3‐diazole‐4‐yl
7‐Nitrobenz‐2‐oxa‐1,3‐diazole‐4‐yl (NBD), a chromophore with high cell permeability and long wavelength absorbance, is often used in biological applications such as the fluorescence labeling of proteins and to study structural and conformational modifications in proteins. Xu and co‐workers126 have synthesized NBD‐based probe 72 for the selective detection of ZnII. In aqueous solution and upon the addition of ZnII, probe 72 develops a red‐to‐yellow color change and an enhancement in the fluorescence signal due to the combination of ICT and PET mechanisms caused by the formation of the 72–ZnII chelate (see Figure 13). Using the NDB chromophore as the signaling unit, probes 73 127 and 74 128 have been developed (see Figure 13). Probe 73, formed by addition of (1,3‐alternate‐25,27‐bis(2‐aminoethoxy)‐26,28‐bis(2‐methoxyethyl)thiacalix[4]arene) to NBD‐Cl, is a colorimetric and fluorescent sensor for AgI and AcO− in water/THF (1/4, v/v; pH 7.5); the addition of these ions causes quenching of the emission signal. Sensor 74 contains a N‐(2‐aminoethyl)picolinamide moiety. Studies performed in aqueous solutions show that upon binding to CuII, the green emission of the probe at λ=544 nm is completely quenched; CaII, ZnII, MgII, CdII, CoII, FeII, HgII, MnII, and NiII are not detected by this probe. The 74–CuII complex has 1:1 (ligand/metal) stoichiometry, with an association constant (K a) of 1.22×103 m −1. Regarding the detection of HgII by using NBD derivatives, NBD‐based probes 75 and 76 containing triazole units129 and probe 77 containing a 2‐pyridylmethyl‐(2‐quinolylmethyl)amine unit130 are shown in Figure 13. These three probes behave as on–off fluorescence sensors in the presence of HgII. For probe 77, besides a change in the fluorescence emission, a colorimetric effect is also observed, and the color changes from yellow to colorless. The stoichiometry of the 77–HgII complex is 1:2 (ligand/metal) with K a=8.6×109 m −1.
Tsui and co‐workers131 describe chemosensors 78–80 containing NBD and 4‐[(4‐methoxyphenyl)diazenyl]benzoic acid moieties for cyanide detection in aqueous solution. The recognition of CN− is due to the strong affinity of this anion to the carbonyl C atom of the amide group; nucleophilic attack occurs, which is followed by proton transfer of the amide hydrogen atom, and this leads to the formation of the alkoxide anions of 78–80. A colorimetric effect, with a color change from yellow to red, is produced by the enhancement in the charge‐transfer interactions between the electron‐rich and electron‐deficient units, and a “turn on” in the emission intensity is also observed.
Niu and co‐workers132 report the ability of NBD‐Cl probe 81 (see Figure 14) to detect thiols on the basis of an intramolecular displacement mechanism. Probe 81 can detect selectively intracellular cysteine and homocysteine in HeLa cells.133 Chen and co‐workers134 have designed NBD‐SCN fluorogenic agent 82 for accurate detection of cysteine and homocysteine. The thiocyanate group present in the structure of 82 increases its push–pull characteristics, which results in an enhancement in the emission intensity. At λ=550 nm, probe 82 shows a 470‐fold enhancement in the fluorescence signal for cysteine and a 745‐fold enhancement in the fluorescence signal for homocysteine; the detection limits are 2.99 and 1.43 nm, respectively. The efficiency and cell permeability of 82 have been studied in Raw 264.7 cells (see Figure 14).
Figure 14.
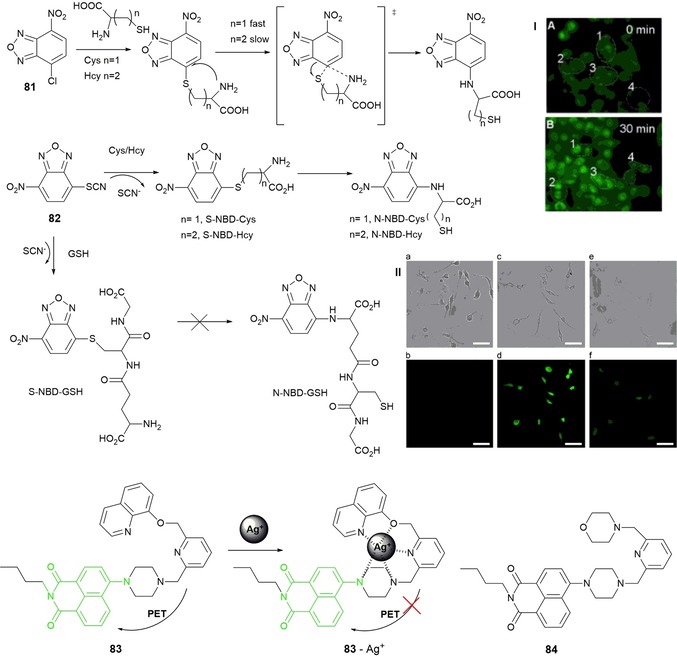
Chemical structures and/or mechanisms of probes 81–84. Panel I) a) Fluorescence images of HeLa cells incubated with only probe 4 and b) followed by incubation with Cys for 30 min. Reproduced with permission from Ref. 134. Copyright 2014 Elsevier B. V. Panel II) Bright‐field (top) and fluorescence (bottom) images of Raw 264.7 macrophages a, b) in the absence and c, d) in the presence of NBD‐SCN (5 μm). e, f) Bright‐field and fluorescence images of Raw 264.7 macrophages, which were pretreated with NEM (12.5 μm) for 30 min and then incubated with NBD‐SCN for 30 min, respectively. The scale bar represents 50 μm. GSH=glutathione; NEM=N‐ethylmaleimide. Reproduced with permission from Ref. 134. Copyright 2014 Elsevier B. V.
2.6. Naphthalimide Derivatives and Acridine Orange
1,8‐Naphthalimide (NP) derivatives have a pure‐blue fluorescence emission at λ=460 nm; the introduction of different electron‐donating substituents at the 4‐position allows fine‐tuning of the emission with colors ranging from blue to yellowish green. Lucifer yellow is an example of a substituted NP derivative, and it is widely used in cell biology to visualize fixed and living cells.135 Xu and co‐workers report fluorescent probes 83 136 and 84 137 based on naphthalimide that exhibit highly selective “turn‐on” fluorescence for AgII and HgII ions, respectively, in aqueous media. Probe 83 has a hydroxyquinoline receptor, and it is responsible for coordination to the AgI metal ion. PET from the electron‐rich receptor to the excited naphthalimide fluorophore makes the probe weakly fluorescent; coordination with AgI inhibits transfer, which leads to a significant enhancement in fluorescence at λ=533 nm (see Figure 14). The 83–AgI complex has a 1:1 (ligand/metal) ratio and an association constant of 9.0×105 m −1. Cell permeability and imaging of AgI in living cells by probe 83 have also been tested. Upon replacing the hydroxyquinoline moiety by a morpholine one, the same authors have designed “turn‐on” fluorescence probe 84, which is selective for HgII. The mechanism is similar to that of probe 83 and involves inhibition of the PET mechanism, which leads to an 11‐fold enhancement in the emission spectra with an association constant of 6.06×106 m −1. The authors conclude that, for both probes, coordination of the pyrazine nitrogen atom that links the naphthalimide ring is involved in chelation of the AgI and HgII ions and that this involvement is crucial for the “turn‐on” effect; reaction of probe 84 with CuII produces a “turn‐off” effect, which indicates that this nitrogen atom is not involved in coordination to this metal ion.
Acridine orange is another green‐emissive dye used for cellular staining due to its good cell permeability and its capability to bind nucleic acids through electrostatic interaction. It is a selective dye that emits green light if linked to double‐stranded DNA (dsDNA) with an emission wavelength of 520 nm and emits red if it is coordinated to single‐stranded DNA (ssDNA) or RNA (λ=650 nm). Due to its metachromatic properties, acridine orange is often used in flow cytometry analysis, fluorescence microscopy, and cellular physiology.138, 139
2.7. Perylene Diimides
Perylene diimide (PDI) is a very attractive chromophore and has been widely studied as an active component in solar cells, field‐effect transistors (OFETs), and organic light‐emitting diodes (OLEDs). The dye has great optical properties such as a high fluorescence quantum yield and a high extinction coefficient, and it has good chemical stability, photostability, and thermostability.18, 19 The absorption and emission maxima of PDI are above λ=500 nm, which avoids problems with autofluorescence background in experiments with living cells. However, because of its tendency to form aggregates, its water solubility is poor. In an attempt to address solubility problems in aqueous solutions, Sun and co‐workers18 have published a review on the synthesis and applications of PDI derivatives with improved aqueous solubility. These probes are obtained by the insertion of multiple polar groups into the bay region, in the imide or ortho positions of the PDI dyes; these substituents hinder aggregation and improve the solubility of the probes in water. Studies performed on the potential of these water‐soluble PDIs to be used as biosensors and bioimaging agents in living cells, tissues, and the body are promising, and the authors propose that PDIs can be used as carriers for gene/drug delivery in gene therapy or chemotherapy.18
Heek et al.15 have designed a PDI derivative that can be used as a membrane marker, and it is able to track polyglycerol‐bound bioactive compounds in both artificial and cellular membranes. Upon dissolving the probe in water, it forms micellar self‐aggregates and the fluorescence signal is quenched; if the probe is in a lipophilic environment, such as a biological membrane, the probe remains in monomeric form and produces strong green fluorescence. Montalti and co‐workers16 have taken advantage of the ability of PDI derivatives to change their fluorescent color and have synthesized nonfluorescent nanoparticles (NPs) of a new strongly fluorescent perylene derivative that can be dispersed in water. The NPs are used as multicolor fluorescent imaging agents in yeast cells; by controlling the dosage, the researchers are able to produce green or red fluorescence, and by photoirradiating the samples, the authors are able to achieve a multicolor experience.
3. Red Fluorescent Dyes
Fluorescent probes are very useful tools for the analysis of physiological events such as ion‐channel activity, localization of metal ions in biological samples, and enzyme activity; this can be achieved by following changes in their optical properties (fluorescence intensity and excitation/emission wavelength) as a result of specific interactions with the target molecules.140 This section presents some recent examples of novel probes based on red dyes. Fluorescent dyes that emit in the red and near‐infrared (NIR) regions of the electromagnetic spectrum can be particularly useful for imaging living cells and tissues, as the fluorescence emission in the longer wavelength region can reduce autofluorescence effects from the biological matrix, encourage deeper tissue penetration in vivo, and avoid visible‐light absorption; moreover, typically they cause less photodamage.141 The optical setup used for working in the red region of the electromagnetic spectrum is simpler than that used for working in the green region, as the scattering effect for the red region is minimal and sources such as dye lasers in the red region are readily available. Red emitters are of interest in numerous fields such as optical communications142 and energy conversion;143 they are also extremely useful tools in biomedical analysis, for example, genetic analysis, DNA sequencing, in vivo imaging, and proteomics.144, 145 Dyes with red emission are employed in the areas of OLEDs,146 protein tracking,147 multicolor imaging,148 and far‐field optical nanoscopy.149 In OLEDs, red‐emitting dyes can be used to complement blue150 and green151 dyes in the construction of displays. Red‐emitting dyes are particularly valued in the imaging of biological tissues, due to their ability to produce emission signals that are distinguishable from the autofluorescence background of the tissues152 and to produce images with high spatial and temporal resolution.153
Organic red‐emitting fluorophores belong to various dye families. As red emission is linked to extensive π‐electron conjugation, fluorophores are usually large polycyclic aromatic hydrocarbons, porphyrin‐type compounds, or very polar push–pull heteroaromatic compounds. Developing red fluorophores that can be used as pure organic materials is particularly challenging. Because of their structure, they all show a tendency towards aggregation due to intermolecular π stacking or attractive dipole–dipole interactions. Aggregation is extremely detrimental to fluorescence,154 and most red‐emitting fluorophores become very weakly emissive, or not emissive at all, at high concentration and/or in the solid state; thus, they are generally used in solution at low concentrations or after dispersion in organic or inorganic matrices. However, the search for red dyes that can be used in the solid phase is essential to be able to synthesize self‐assembling fluorescent organic micro‐ and nanoparticles. These materials present distinct advantages in optics155 and biomedical imaging156 because of their increased brightness, chemical and photochemical stability, low cost, high structural flexibility, low toxicity, and bioavailability.
3.1. Cyanines
Cyanine derivatives are a key type of NIR fluorescent probes for biological applications, because of their high molar extinction coefficients, moderate‐to‐high fluorescence quantum yields, and a broad wavelength tunability.157 Guo and co‐workers158 describe the five‐stage synthesis of cyanine‐based fluorescence probe 85 composed of a tricarbocyanine fluorophore and tris(2‐pyridylmethyl)amine (TMPA); this probe is selective towards ZnII ions (see Figure 15). Upon binding of the TMPA moiety to the ZnII ion, the amine attached to the center of the polymethine chain of the tricarbocyanine is deprotonated to form an imine, which is cross‐conjugated with the less‐delocalized diaminotetraene group. Reduced delocalization results in a large hypsochromic shift in the emission maxima of 85 (Figure 15) and leads to lowering of the background signal and an increase in the signal fidelity for ZnII. In addition, 85 has a very strong binding affinity for ZnII, and it exhibits high sensitivity and selectivity towards ZnII over other metal ions, with a detection limit in the nanomolar range. Probe 85 has also been successfully used in live mammalian cells as an imaging tool for ZnII ions released during apoptosis.
Figure 15.
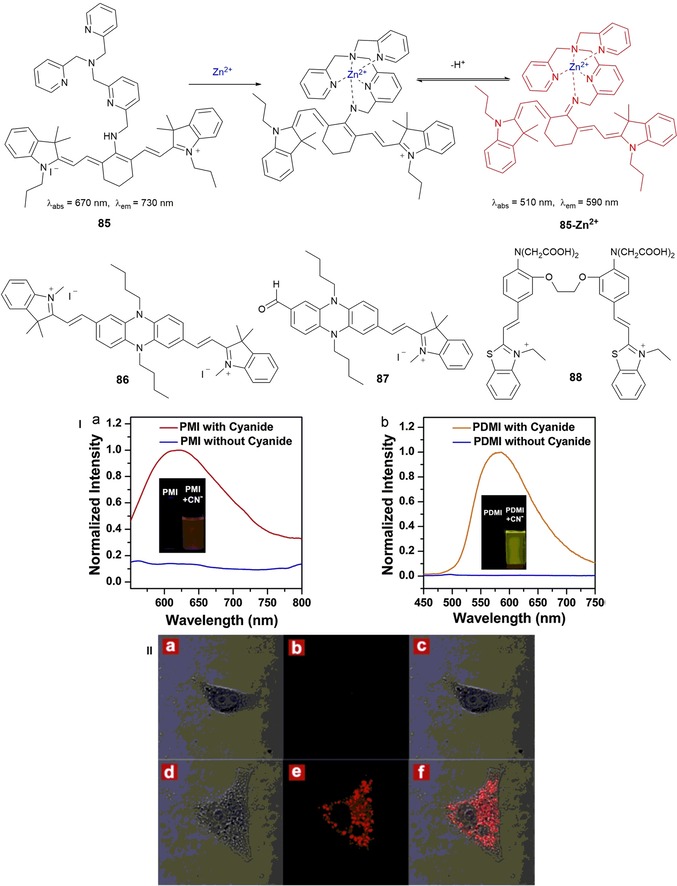
Chemical structures of compounds 85–88. Panel I) Normalized fluorescence emission spectra of a) PMI (87) (λ ex=530 nm) and b) PDMI (86) (λ ex=425 nm) before and after reaction with cyanide. Inset) Corresponding photographs of PMI (87) and PDMI (86) both in HEPES buffer/DMSO (7:3, v/v, pH 7.4) solution under a UV lamp at λ=365 nm. Panel II) Confocal laser scanning imaging of HeLa cells with PMI (87) incubated in RPMI‐1640, pH 7.4, 37 °C. First row) a) bright‐field, b) dark‐field, and c) merged images of HeLa cells incubated with PMI (10 μm) for 30 min. Second row) d) bright‐field, e) dark‐field, and f) merged images with following incubated with CN− (2.0 equiv.) for another 30 min. λ ex=543 nm. Emission was collected by the red channel from λ=650 to 750 nm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.). Reproduced with permission from Ref. 159. Copyright 2014 Elsevier B. V.
To improve the solubility of phenazine–cyanines in water, Yang and co‐workers159 have introduced a vinylindolium moiety into the phenazine skeleton to synthesize phenazine–cyanines derivatives PMDI (with two indolium moieties on both sides of phenazine) 86 and PMI (with one indolium group) 87 (see Figure 15). The quenching effect on the phenazine–cyanine fluorophore by strong intramolecular charge transfer (ICT) from the phenazine (donor) to the indolium (receptor) moieties makes both 86 and 87 nonemissive. Both compounds behave as chemodosimeters for CN− detection, with detection limits of 1.4 μm and 200 nm, respectively. Upon coordination to the CN− anion through the N‐methylindolium group, the ICT effect disappears, which leads to a dramatic “off–on” enhancement in the fluorescence signal with emission maxima at λ=580 nm for 86 and λ=630 nm for 87. Probe 87 seems to be a promising candidate for monitoring intracellular CN− in HeLa cells (see Figure 15, panel II).
The measurement of intracellular CaII has become an important topic in biological and medical research.160 Zhu and co‐workers161 have developed new visible‐light‐excited and red‐emitting probe 88, which is used as a ratiometric fluorescence probe for detecting intracellular CaII (see Figure 15). Probe 88 is made of 1,2‐bis(2‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid, which acts as the CaII‐chelating moiety, and two benzothiazolium hemicyanine dyes acting as fluorophores. An approximately 48‐fold enhancement in the intensity of the fluorescence signal and a blueshift of Δλ=20 nm (from λ=600 to 580 nm) in the emission spectrum are observed upon adding the CaII ion; probe 88 has successfully been used to distinguish simultaneously between CaII changes in the cytosol and nuclei of living cells.
3.2. BODIPY
Boron dipyrromethene (BODIPY) derivatives can be used as molecular probes,162 in photodynamic therapy,163 as laser dyes,164 in nonlinear optics,165 in dye‐sensitized solar cells,166 and as part of electrogenerated chemiluminescence (ECL) emitters for the study of organic and inorganic materials.167 The use of BODIPY derivatives in those applications is based on their narrow absorption and emission bands, high fluorescence quantum yields (even in aqueous media), large molar absorption coefficients, pH‐independent emissions, and excellent photostability; the characteristic emission of the original BODIPY fluorophore is centered at around λ=520 nm with a small Stokes shift (λ≈20 nm).168
Interest in BODIPYs has been mostly on red‐ or near‐infrared‐emitting probes constructed by the following: 1) intramolecular rigidification of the molecular structure by B−O ring formation;169 2) fusion of aryl or heteroaryl moieties into the BODIPY structure;170 3) using tetraaryl‐substituted azadipyrromethenes;171 and 4) with styryl linkers in the 3,5‐ or 1,3,5,7‐positions and, more recently, in the 8‐position.172 Following this last strategy, Shao and co‐workers173 have synthesized 89 for the specific detection of cysteine among other biological molecules, such as glutathione and amino acids (see Figure 16). Probe 89, a styryl BODIPY/DNBS dyad, shows intense absorption at λ=556 nm and is nonfluorescent; however, the DNBS moiety is cleaved by thiols, which results in a 46‐fold enhancement in the red emission band centered at around λ=590 nm. The probe has been used for the fluorescent imaging of cellular thiols, with the advantage that it is pH independent in the physiological pH range. Hiruta and co‐workers174 report on two distyryl‐BODIPY‐based NIR red‐shifting ratiometric fluorescent probes: KBHR‐1 (90) for pH and KBAHgR‐1 (91) for AgI and HgII (see Figure 16). Probe 90 shows a redshifting ratiometric response to pH in the NIR region. The use of a different dialkylamino moiety as the recognition unit for probe 91 results in a ratiometric response to AgI and HgII, also in the NIR region.
Figure 16.
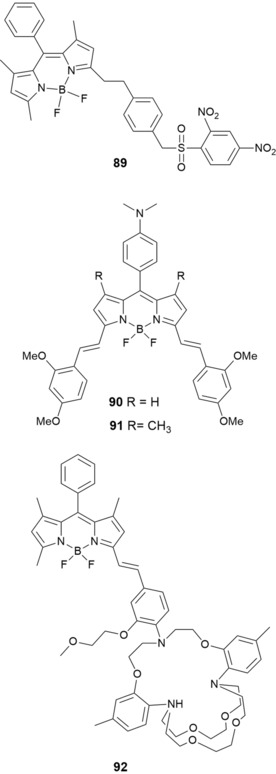
Chemical structures of compounds 89–92.
Hirata and co‐workers175 have developed probe B3TAC (92) (see Figure 16), which is synthesized by conjugating 2‐triazacryptand [2,2,3]‐1‐(2‐methoxyethoxy)benzene (TAC) to the 3‐position of the BODIPY fluorophore through a styryl linkage. Probe 92 is highly selective to KI in the physiological concentration range over NaI and other metal ions by using water/acetonitrile as the solvent. Although 92 shows good potential as a selective probe for the detection of intracellular or extracellular KI, further modifications of the structure are needed to improve its solubility in water to avoid the need for an organic solvent.
BODIPY derivatives 93–98 (see Figure 17), prepared by Fu and co‐workers, show similar solubility problems in water.176 The presence of trihexylsilyl (THS) and trimethylsilyl (TMS) groups makes them sensitive to fluoride ions, which induces a strong blueshift (Δλ=70–117 nm) and a significant increase (6‐ to 40‐fold) in the fluorescence intensity in the emission spectra. It is likely that the extended π system in all of these probes allows for the large shifts in the absorption and fluorescence bands in the presence of F−. In particular, probe 98 a turns from red to yellow upon adding F− ions (see Figure 17), but no color change is observed in the presence of Cl−, Br−, I−, SCN−, NO3 −, HCO3 −, CH3COO−, H2PO4 −, CO3 2−, and SO4 2−. The chemosensing abilities of 98 a have also been tested by monitoring the emission spectra of the probe in acetone. The probe exhibits the largest blueshift in the fluorescence band if in the presence of F− (Δλ=117 nm), along with a 40‐fold enhancement in the fluorescence intensity. Again, the promising sensing capability of these probes is somewhat hampered by the fact that they cannot be used in aqueous solution.
Figure 17.
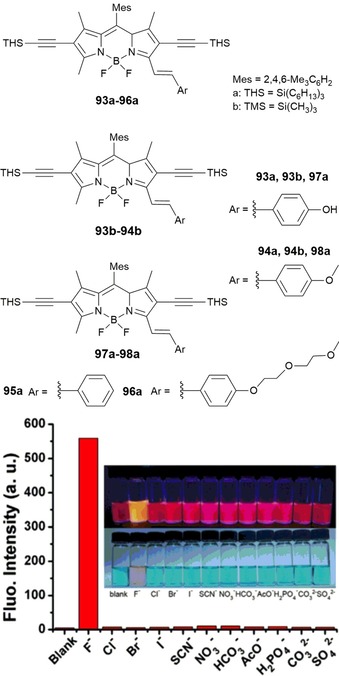
Chemical structures of compounds 93–98. Relative fluorescence spectral changes of probe 8a (98 a) (10 μm) after treatment with 50 equivalents of various anions in acetone at λ em=570 nm. Insets) corresponding solutions upon irradiation with a UV lamp at λ=365 nm (top) and under ambient light (bottom). Reproduced with permission from Ref. 176. Copyright 2015 Elsevier B. V.
Major effort has been devoted to improving the solubility of BODIPY dyes in water, so that they can be used in biological applications such as intracellular and tissue imaging. As an alternative to red‐emitting 3,5‐distyryl‐BODIPYs, 3,5‐dithienyl‐BODIPYs can provide a good compromise between solubility, stability, and hydrophobicity. Poirel and co‐workers177 report the synthesis of red‐emitting water‐soluble thienyl‐BODIPYs 99–102 (see Figure 18). The trimethyl(propargyl)ammonium group is chosen to improve water solubility. One or two cationic arms are introduced either in the 2‐position of the thienyl unit or in the 4‐position on the boron atom. Probes 99–102 have strong absorption (at λ≈600 nm) and intense emission (at λ≈650 nm) in water.
Figure 18.
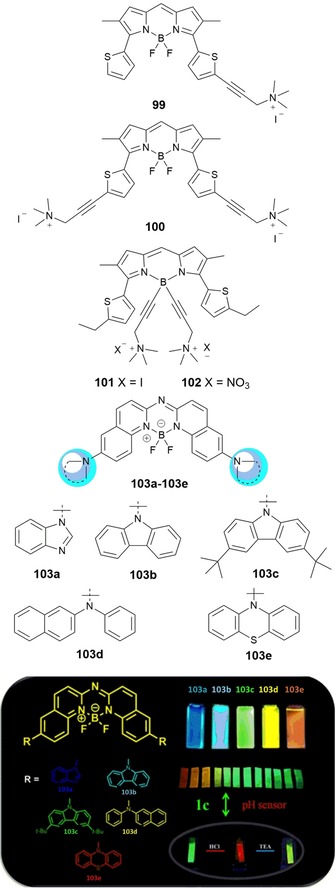
Chemical structures of compounds 99–103. Reproduced from Ref. 178 with permission from The Royal Society of Chemistry.
Zhu and co‐workers178 have designed new aza–boron–diquinomethene complexes 103 a–e containing different N‐arylamines (see Figure 18). Photoluminescence studies reveal that all of the probes show an intense and tunable luminescence signal from blue to red and good emission quantum yields. These probes show considerable potential as pH sensors, as bioimaging probes, and for the synthesis of organic light‐emitting materials. Probe 103 c shows a remarkable capacity to change color due to protonation effects and has been used as a pH sensor both in solution and in the solid state; the probe, supported on filter paper, is able to act as a pH sensor upon exposure to acid and base vapors.
Chen and co‐workers179 have synthesized two novel bis(methoxyphenyl)–BODIPY fluorescent probes, 104 and 105, for the detection of nitric oxide (NO) (see Figure 19). The probes show red fluorescence (λ=590 and 620 nm, respectively) and large Stokes shifts (Δλ=38 nm) in the far‐visible/NIR spectral region. Both probes are highly selective towards NO in the presence of other reactive oxygen species and reactive nitrogen species. Besides studying their fluorescent properties, the researchers have used both probes for NO imaging in living cells (RAW 264.7); the cytotoxicity studies are performed in a mixture of acetonitrile/water, as the probes are not soluble in water. Both dyes are promising candidates for fluorescence imaging of NO due to low background interference and high detection sensitivity.
Figure 19.
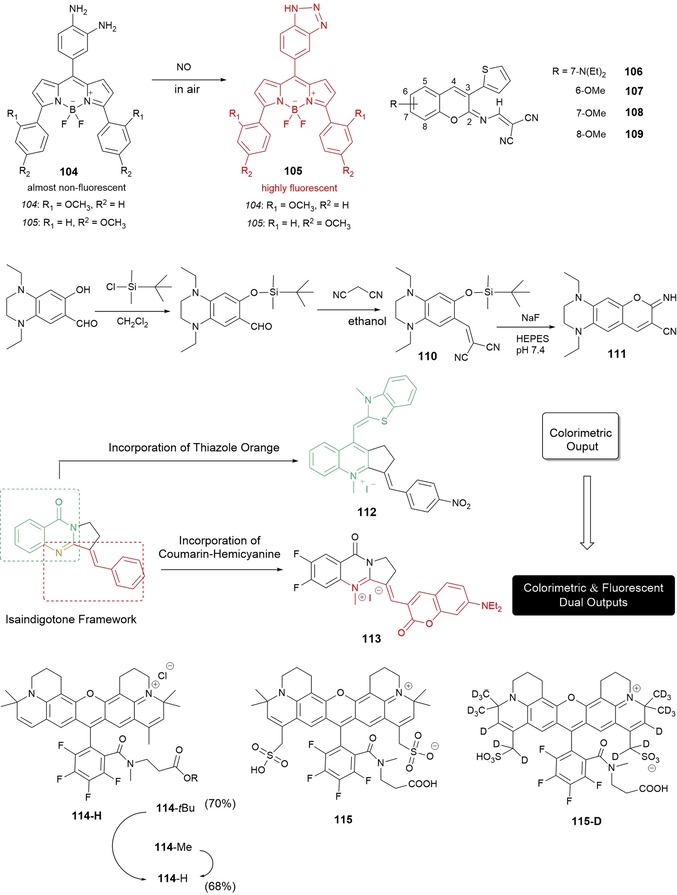
Chemical structures of probes 104–115. Synthesis route to probe 110 and its proposed reaction with NaF to give fluorophore 111. Structures of the isaindigotone framework, colorimetric probe 112, and upgraded colorimetric and fluorescent dual probe 113.
3.3. Coumarins
Probes containing coumarin, a fluorophore known to be highly fluorescent and with moderate to good quantum yields, have been used in a wide range of biological applications such as the fluorescent labeling of proteins, cellular imaging, and lasers.180, 181, 182, 183 Different strategies can be employed to ensure that coumarin derivatives have enough charge transfer in the coumarin molecule to push the absorption and emission in the red region: 1) introducing an electron donor in the 5‐ and 7‐positions; 2) introducing an electron‐withdrawing group at the 3‐ and 4‐positions; 3) increasing the rigidity of the donor and acceptor groups; and 4) extending the conjugation. Khemakhem and co‐workers184 show the value of using substituents with low molecular weights to prepare new orange‐ and red‐emitting fluorescent materials based on coumarin. In their work, the researchers explore the fluorescence properties of four derivatives of 3‐thienyl‐2‐(N‐dicyanovinyl)iminocoumarin, that is, 106–109, bearing a diethylamino group in the 7‐position (as in 106) or a methoxy group in the 6‐, 7‐, and 8‐positions (as in 107–109); studies have been performed both in solution, using a range of organic solvents as well as water, and in the solid state (see Figure 19). The fluorescence emission spectra of probe 106 ranges from yellow/orange to red; the compound emits strongly in dilute solutions and shows remarkable solvatochromic behavior. Stability studies have been conducted on probe 106, which shows the best solubility in aqueous solutions, and this probe resists pH changes without hydrolysis; 106 has successfully been used to stain the cytoplasm of HCT‐116 colon cancer cells. The fluorescence emission spectra of probes 107–109 range from yellow to green. The presence of the methoxy group in the structure makes the probes suitable for solid‐state emission; additionally, probes 107 and 109 exhibit crystallization‐enhanced emission. Moreover, the researchers also prepare nano‐ and microsized particles for all of the probes, including millimeter‐long microfibers, which exhibit clear wave‐guiding properties; this makes them promising candidates as optical guides for potential biomedical applications.
Hou and co‐workers185 have synthesized red‐fluorescence probe 110, with λ max(emission)=616 nm. The probe is able to detect F− in aqueous solution on the basis of F−‐triggered Si−O bond cleavage, which leads to a cyclization reaction in the probe and the formation of iminocoumarin dye 111. The reaction has a rapid response time (within 10 min), is highly selective towards F− in the presence of other anions, and can detect F− in concentrations ranging from 1.0 mm to a 10 μm. End‐product 111 emits red photons, which help to minimize interference from endogenous chromophores (Figure 19), and exhibits a relatively large Stokes shift (Δλ=143 nm), which is beneficial for signal detection in fluorescence microscopy. Fluorescence microscopy experiments have established the usefulness of probe 110 to detect F− in living cells, and the probe has been used to monitor and image F− in living HaCaT cells in aqueous media.
In biochemistry and clinical diagnosis, it is of key importance to develop highly sensitive and selective probes to detect nucleic acids,186 in particular G‐quadruplex structures.187 To detect G‐quadruplex structures, Yan and co‐workers188 have developed colorimetric probe 112 by merging thiazole orange into an isaindigotone framework (see Figure 19); the sensitivity of the probe and its further application are restricted due to weak fluorescence emission. Building on this work, Yan and co‐workers189 have synthesized a similar probe, 113, this time by merging a coumarin–hemicyanine fluorophore into the isaindigotone framework. Probe 113 is successful in detecting G‐quadruples structures, and a marked “turn‐on” effect in the fluorescence signal is observed. Remarkably, the probe also shows a color change from pink to blue, which is visible by the naked eye, so it could also act as a colorimetric probe. The researchers show the selectivity of 113 towards G‐quadruplexes in the presence of interference analytes such as ssDNA, dsDNA, and the bovine serum albumin (BSA) protein. Intracellular applications of 113 in live and fixed cells have been performed by using HeLa and A549 cells. The results reveal that 113 accumulates in the nucleus, mainly bound to the rDNA regions.
3.4. Rhodamine
Because of their high photostability, high fluorescent quantum yields, high extinction coefficients, and low degree of triplet formation, probes derived from rhodamine are widely used as laser dyes and fluorescent markers for the labeling of biomolecules.11,190 Kolmakov and co‐workers191 have developed novel red fluorescent dyes for imaging and labeling applications in the red optical region (see Figure 19). The probes have been used to image a range of biological substrates such as various animal antibodies and sphingomyelin lipids. Probe 114 and derivatives are lipophilic, whereas probes 115 and 115D are soluble in water and aqueous buffers. The probes are very photostable, have a low tendency to aggregate, and have relatively long excited‐state lifetimes (t=3.4 ns). High‐resolution GSDIM (ground‐state depletion with individual molecular return) images and live‐cell STED‐FCS (stimulated emission depletion‐fluorescence correlation spectroscopy) experiments performed on labeled microtubules and lipids show the capability of the probes to be used in fluorescence microscopy and nanoscopy.
3.5. Rosamine
Differences in pH can be used to differentiate cancer cells (pH 4.5–5.5) from normal cells (pH 5.0–6.0);192 thus, with the use of fluorescence probes sensitive to pH it is possible to visualize the acidic lysosomal lumen of cancer cells. Sun and co‐workers193 have developed rosamine‐based pH probe 116 (see Figure 20), which is capable of intracellular pH imaging in live cells. The presence of an electron‐donating side chain containing an amino group makes the probe nonfluorescent in neutral environments as a result of PET; however, in acidic pH environments, the probe behaves as an off–on probe with a 400‐fold enhancement in the fluorescence signal upon binding with H+; other physiologically important cations do not interfere with the recognition process. Research performed in live HeLa cells indicates that the probe has good cell permeability, and co‐staining with the commercial lysosome‐staining probe LysoTracker Green DND‐26 shows good agreement between both the commercial and the experimental dyes. The research thus shows that 116 can be successfully used as red‐emitting lysosome‐specific probe.
Figure 20.
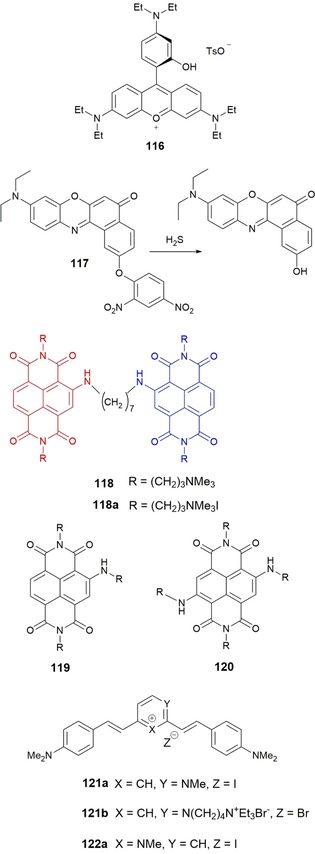
Chemical structures of probes 116–122. Mechanism of H2S sensing of compound 117. Structure of dimeric 118 (resulting from the merging of monomeric 119 and 120), and its quaternary ammonium salt 118 a, as the iodide.
3.6. Nile Red
Nile red is well established as an environment‐sensitive fluorescent dye for labeling and sensing biomolecules, because of its high fluorescence quantum yield, long‐wavelength emission, and good photostability.194 Tang and co‐workers195 have designed long‐wavelength‐emitting fluorescent “turn‐on” probe 117 for H2S detection (see Figure 20). The detection mechanism is based on the thiolysis of the dinitrophenyl ether, which is added to the 2‐position of Nile red to form 117. The fluorescence of the free probe is quenched by the PET process between the fluorophore and the dinitrophenyl ether group. In experiments performed in aqueous solution, the probe shows a 17‐fold increase in the fluorescence signal in the presence of H2S (detection limit of 2.7×10−7 m), with no interference from various biologically relevant species. The probe shows good membrane permeability and has successfully been used for the fluorescence imaging of H2S in MCF‐7 cells (human breast carcinoma).
3.7. Naphthalene Diimide Dyad
Naphthalene diimide (NDI) derivatives have been successfully used in a wide range of applications, including the construction of supramolecular materials, such as rotaxanes and catenanes, and as molecular sensors.196 Doria and co‐workers197 report the synthesis of 118, a water‐soluble dimeric NDI resulting from the conjugation of two monomeric NDIs, red trisubstituted dye 119 and blue tetrasubstituted 120, through a (CH2)7 flexible spacer. Probe 118 is nonfluorescent, but in the presence of G‐quadruplex DNA, a turn‐on effect in fluorescence is observed with the subsequent complex emitting in the red/NIR region. Although the probe exhibits good selectivity to G‐quadruplex DNA in the presence of dsDNA, it is not able to select between different G‐quadruplex structures.
3.8. Distyrylpyridinium
Styryl (or arylvinyl) dyes represent an important class of functional dyes that can be used in optical recording media, laser dyes, and optical sensitizers and as fluorescent probes in biomedical applications.198 Xie and co‐workers199 have synthesized three red‐emitting cationic distyryl probes (see Figure 20): two of them, 121 a and 121 b, are derived from the asymmetric 2,4‐bis(4‐dimethylaminostyryl)‐1‐methylpyridinium ion, and the third one,122 a, is derived from the symmetric 2,6‐bis(4‐dimethylaminostyryl)‐1‐methylpyridinium ion. The researchers have studied the ability of the probes to detect several quadruplex and duplex nucleic acids. Dyes 121 a and 121 b are able to distinguish between quadruplex DNA and dsDNA structures, both by a strong “light‐up” effect in the presence of quadruplex DNA (80–100‐fold “turn on” of the fluorescence signal), as well as by the position of the emission maxima in the fluorescence spectra. Symmetric analogue 122 a as not able to distinguish between quadruplex and duplex DNA, and the enhancement in the fluorescence signal is typically 20–40‐fold.
3.9. Benzophosphole P‐Oxide Scaffold
Ratiometric fluorescent probes are powerful diagnostic tools for the quantitative detection of metal ions in living systems. Using benzophosphole P‐oxide (123) as a fluorophore, Taki and co‐workers200 have developed ratiometric fluorescent probes 124 and 124‐AM for NaI detection (see Figure 21). These probes are examples of d‐π‐A systems (D=electron‐donating moiety; A=electron‐accepting moiety). Typically, d‐π‐A systems are fluorescent because of the existence of an ICT process. Once the probes interact with the target metal ion, the ICT character is reduced and hypsochromic shifts can be observed for both the absorption and emission maxima. This behavior is observed for probe 124; after excitation under visible light (λ ex=405 nm), and in the presence of NaI, the probe exhibits a hypsochromic shift in its emission spectrum upon complexation. Experiments consisting in blocking the Na+–K−+ pump in living mammalian cells show that probe 124‐AM (the membrane‐permeable form) can successfully be used for the ratiometric visualization of intracellular Na+ dynamics. The results suggest that this probe can potentially be used as an imaging tool to study NaI dynamics, for example, imaging of potential‐evoked NaI influx between axon and soma in neuronal cells.
Figure 21.
Structures of fluorescent probes based on a benzophosphole P‐oxide scaffold. Environment‐sensitive probe 123. Ratiometric NaI probe 124 and membrane‐permeable form 124‐AM. Chemical structures of probes 125–129.
3.10. Benzoresorufins
It is well known that ZnII and reactive nitrogen species (RNS) such as nitric oxide (NO) and peroxynitrite (ONOO−) are implicated in some neurological dysfunctions and play important physiological roles in the nervous system.201 Fluorescein‐based sensors can be used to study these species, but they are limited by their high‐energy absorptions and small Stokes shifts. The use of sensors emitting in the red or NIR region can avoid those shortcomings. With this aim, Lin and co‐workers202 report a new family of red‐emitting fluorescent sensors 127–129 that can be used to detect labile ZnII (see Figure 21). The probes are based on a benzoresorufin dye functionalized with dipicolylamine or picolylamine moieties as the metal‐binding groups. The researchers have also studied probes 125 and 126, which contain identical binding moieties (and thus the same [N3O] metal‐binding motif) but have fluorescein as the fluorophore. Benzoresorufin‐based dyes 127–129 perform well in terms of their binding capabilities towards ZnII and show binding affinities similar to those of their fluorescein‐based counterparts. A significant advantage of probes 127–129 is their ability to monitor organelle‐specific ZnII levels in live cells. This research shows that the benzoresorufin platform is a promising dye that can serve as the basis for the design of new red‐emitting sensors with improved performance.
3.11. Tetrapyrrolic Macrocycles
Metal‐ion probes based on porphyrins can present large Stokes shifts, high fluorescent quantum yields, and long lifetimes, and thus designing this type of probe constitutes an important area of research.203, 204 Our research group, in collaboration with the University of Aveiro, has explored the sensing ability of porphyrin‐based probes 130–145. Probes 130–134 are benzoporphyrins, and probes 135–139 are porphyrin‐2‐ylpyridines (see Figure 22).205 Probe 141 is a 3,5‐disubstituted pyridine with two porphyrin moieties (Figure 22).206 Porphyrin‐based probes 143 a–e contain an α,β‐unsaturated ketone unit in a β‐pyrrolic position,207 and probes 145 a–d are pyrazole–porphyrin conjugates (see Figure 23).208
Figure 22.
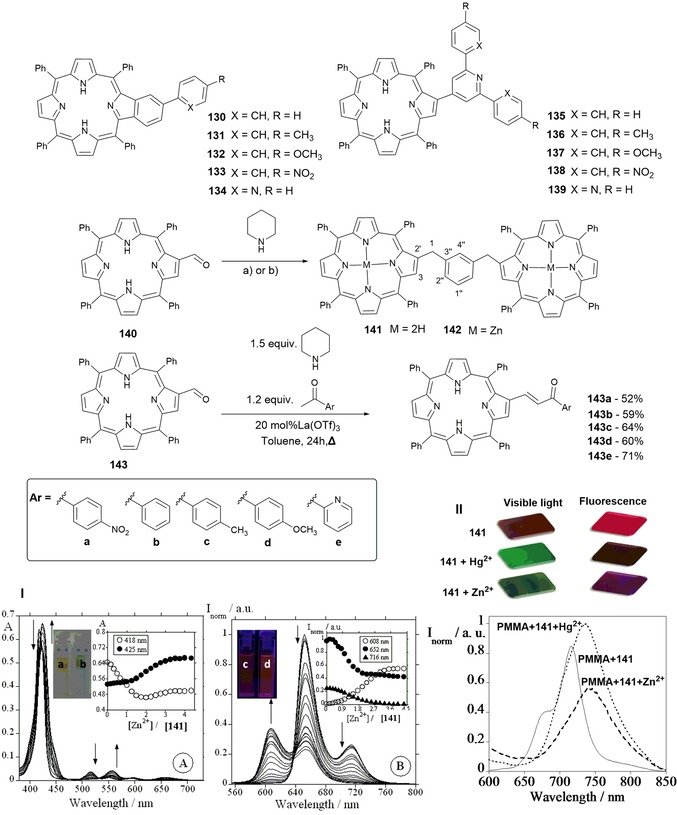
Chemical structures and mechanism of compounds 130–143. Panel I) a) Spectrophotometric and b) spectrofluorimetric titrations of 141 in chloroform as a function of added ZnII in acetonitrile at 298 K. The inset of panel a shows the absorption at λ=418 and 425 nm, and the inset of panel b shows the normalized fluorescence intensity at λ=608, 652, and 716 nm ([141]=1.00×10−6 m, λ ex=536 nm). The inset photograph in panel a shows a solution of 141 in CHCl3 before (left) and after (right) the addition of ZnII under visible light; the inset photograph in panel b shows a solution of 141 in CHCl3 before (left) and after (right) the addition of ZnII after excitation at λ=365 nm by a UV lamp. Panel II) Visual changes observed upon spraying a PMMA film with chemosensor 141 with an aqueous solution containing HgII or ZnII under visible light and after excitation at λ=365 nm by using a UV lamp (top). Emission spectra of the PMMA film doped with 141 and after spraying with ZnII or HgII at room temperature (bottom). Adapted with permission from Ref. 149. Copyright 2014 Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 23.
Chemical structures and mechanism for the synthesis of compounds 143–145.
Probes 135–139, which form complexes with a 2:1 (ligand/metal) stoichiometry, show higher stability constants than probes 130–134, with a 1:1 stoichiometry; it is suggested that a substituent in the 2‐position improves the host–guest interaction and thus allows the formation of a more stable complex. Importantly, benzoporphyrin‐based probes 130–134 show significant changes upon titration with HgII. First, there is a decrease in the two bands at λ=658 and 718 nm due to free‐base porphyrin emission Q (0–0) and Q (0–0)(0–1).209 Moreover, a new band appears at λ=687 nm: this new emission band, which increases with the addition of HgII, is attributed to metal‐to‐ligand charge transfer and indicates the generation of a new fluorophore arising from metal–porphyrin complexation. Upon titration with HgII, the color of the solution changes from yellowish‐brown to green, and this is accompanied by the appearance of a blueshifted emission and a color change in the emission from red to intense orange. The highest association constant is obtained for probe 130 [log K a=(8.71±4.81)×10−3]; this probe is able to quantify 32 ppb of HgII.
The results of the spectrophotometric and spectrofluorimetric studies for probe 141 are shown in Figure 22. Probes 141 and 143 b are able to quantify 79 and 80 ppb of ZnII, respectively. The ligand/metal stoichiometries of the complexes formed are 1:3 for the 141–ZnII complex and 1:1 for the (143 a–e)–ZnII complexes; probe 141 shows the highest association constant (log K a=15.50). The possibility of using the probes as metal‐ion chemosensors in the solid state has also been investigated. The probes, supported in polymethylmethacrylate (PMMA) and made into films, show promising results in the solid phase; in particular, compounds 130, 141, and 143 d in PMMA are able to distinguish between ZnII and HgII.
From the porphyrinoid family, corroles have merited special attention in recent years because of their high fluorescence quantum yields and high molar extinction coefficients; moreover, the corrole core can accommodate different metal ions that can act as active centers. In experiments performed in toluene, the sensing abilities of probes 146–151 towards a range of biologically and environmentally relevant anions have been studied (Figure 24).210 Spherical (F−, Br−, and Cl−) and linear (CN−) anions have been investigated; bulky anions such as CH3COO− and H2PO4 − have also been studied. Probe 146 with F− shows the highest association constant, and this probe is able to quantify 0.69 ppm of this anion. Probes 146 and 147 show high sensitivity towards CN−, and complexation with CN− produces a redshift in the absorption and emission spectra and an increase in the emission intensity; the probes are able to quantify 1.43 ppm of this anion.
Figure 24.
Reagents and conditions for Scheme I: a) i) GaCl3, pyridine, reflux, ii) 1. 36 % HCl, 2. aq NaHCO3; b) Vilsmeier–Haack reagent; c) CH3PPh3Br, NaH in THF, RT, N2. Reagents and conditions for Scheme II: a) reflux, toluene, N2; b) reflux, toluene, N2. Chemical structures of compounds 146–153. Panel III) a) Emission spectra of acrylamide gel doped with compound 146 in the presence of fluoride as a function of time (T=298 K, λ ex=570 nm). b) Polymethylmethacrylate film with 146 and c) polyacrylamide gel of 146 in the presence of fluoride (F−). Reproduced from Ref. 210 with permission from The Royal Society of Chemistry. Panel IV) a) Spectrophotometric and b) spectrofluorimetric titration of compound 152 with the addition of CN− in toluene. The inset represents a) the absorption at λ=605 and 630 nm and b) the emission intensity at λ=616 and 637 nm ([152]=1×10−5 m, λ ex=590 nm, T=298 K). Reproduced with permission from Ref. 211. Copyright 2015 Elsevier B. V.
Probes 146 and 150 have been used to prepare low‐costing solid polymers supported on PMMA and polyacrylamide. The PMMA films prepared with compounds 146 and 150 show very strong red emission and a purple (for 146) or blue color (for 150) to the naked eye. Upon supporting 146 on polyacrylamide, the resulting gel is not emissive; however, the probe is able to switch on the emission after being submerged in a solution containing F−, probably due to the large pores in the gel, which allow entry of the anion. An enhancement in the emission intensity with time is also observed (see Figure 24). Similar behavior is observed in the presence of CN−, and the acrylamide polymer doped with 146 is able to detect about 70.0 ppb of CN− in water. Santos and co‐workers report the sensing ability of probes 152 and 153 (Figure 24) towards different anions by using absorption and emission spectroscopy.211 Probe 152 is a colorimetric probe capable of detecting CN− with a change in color from green to colorless (Figure 24); the detection limit is around 1.00 μm.
Santos and co‐workers212 have synthesized probes 154–161 (see Figure 25). Probes 154–159 are porphyrin–coumarin derivatives; probes 160 and 161 are corrole–coumarin derivatives, for which the coumarin moiety is inserted to improve solubility in aqueous solution. This approach is somewhat successful, and both probes 160 and 161 are soluble in ethanol/water (50:50) mixtures. Studies on the ability of the probes to detect a range of anions and metal ions have been performed. Probe 158 shows high selectivity for HgII both in solution and in the solid state if supported on cellulose paper (i.e. filter paper).
Figure 25.
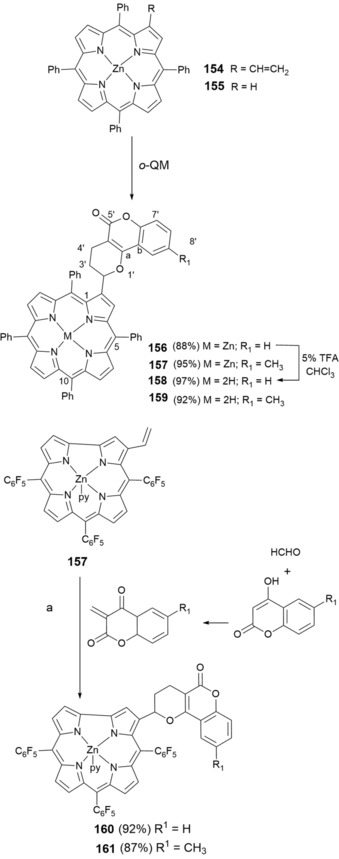
Chemical structures of compounds 154–161.
Probe 162, a ZnII complex of a coumarin–porphyrin unit, is sensitive to the alkaloids caffeine (163), nicotine (164), and cotinine (165) (see Figure 26).213 Spectrophotometric and fluorometric experiments, performed in ethanol, show a 1:1 162/alkaloid stoichiometry in all cases. The ability of probe 162 to detect these alkaloids in real environmental samples has been studied, and the probe is able to detect (2.5±0.3) μm of cotinine in samples of dam water.
Figure 26.
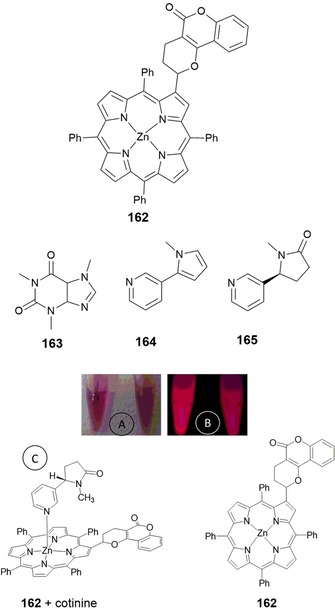
Structural formula of {2‐(5‐oxo‐2,3,4,5‐tetrahydro‐2H‐pyrano‐[3,2‐c]chromen‐2‐yl)‐5,10,15,20‐tetraphenylporphyrinato}zinc(II) (162), caffeine (163), nicotine (164), and cotinine (165). a, b) Naked‐eye picture and under a UV lamp (λ ex=365 nm) of 162 (left) and 162+cotinine (right). c) Hypothetical coordination structure of cotinine as an example of the alkaloids studied and probe 162.
3.12. Photostability
The optical properties, low toxicity, and high selectivity to accumulate in cancer cells make porphyrins very useful in photodynamic therapy (PDT).214, 215 In photodynamic therapy a good photosensitizer must have a high quantum yield for triplet formation, has to produce singlet oxygen very efficiently (predominant cytotoxic agent in PDT),216, 217 and must have an excitation wavelength to the first singlet state in the λ=700–800 nm region. Indeed, wavelengths longer than 800 nm are preferable to penetrate deeper into the tissue; however, they do not provide enough triplet energy to excite oxygen to its singlet state. Unfortunately, during this process, molecules tend to loss their fluorescence and are photobleached. Photobleaching occurs in green and red fluorophores, especially if they are irradiated with light. As an example, Benson and co‐workers218 report the different rates of photobleaching of acridine orange bound to DNA and RNA with green and red fluorescence. Photobleaching can be caused by photodynamic interactions between the excited fluorophores and molecular oxygen in the media. This generates singlet oxygen and other types of damaging oxygen free radicals and leads to photodamage. To avoid such an issue, it is very important to select a fluorophore with high photostability. Chemicals capable of quenching singlet oxygen can also be employed to reduce the effects of photobleaching. A good strategy is to design oxygen‐reactive protective molecules including vitamin E analogues, vitamin C, glutathione imidazole, cysteamine, and histidine.219
4. Metal Complexes
The usefulness of organic molecules as probes can sometimes be hampered by the need for complicated synthetic procedures to prepare them. Also, organic probes can suffer from short fluorescence lifetimes (sometimes in the range of nanoseconds), narrow energy gaps, and interference problems caused by autofluorescence from surrounding biological environments or light scattering.220 To address these shortcomings, considerable effort has been focused on synthesizing fluorescent sensors based on metal complexes.
4.1. Lanthanide Complexes
Fluorescent sensors based on lanthanide complexes can be used in a wide range of applications, including as temperature sensors, molecular sensors, and bioimaging agents. The sharp emission lines arising from the characteristic 4f electronic transitions from the lanthanide ions and the analyte‐induced hyperfine energy transfer or change in coordination environment mechanism imply that lanthanide‐based fluorescence sensors can offer considerable advantages over typical luminescent complexes. Lanthanide complexes have high luminous efficiency, large Stokes shifts, and long excited‐state lifetimes (up to milliseconds). Moreover, their high sensitivity to changes in the surrounding local environment allows their use in time‐resolved fluorescence (TRF) measurements.221, 222
Xu and co‐workers223 report the synthesis of lanthanide complexes, Ln2PQC6 (Ln=LaIII, PrIII, NdIII, SmIII, and EuIII), derived from the quinolinecarboxylate ligand 2‐phenyl‐4‐quinolinecarboxylic acid (PQC). Probe Eu2PQC6 (166) (see Figure 27) shows intense red emission both in solution and in the solid state. The probe displays high affinity towards HSO4 − and H2PO4 − anions, with very low detection limits (15.3 and 8.3 nm, respectively). The luminescence signal of 166 is quenched in the presence of these anions through hydrogen bonding for HSO4 − and through coordination to the metal ion for H2PO4 −. Probe 166 has been successfully used as a luminescent sensor for three nucleoside phosphates, adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine monophosphate (AMP), in mixed aqueous solution.
Figure 27.
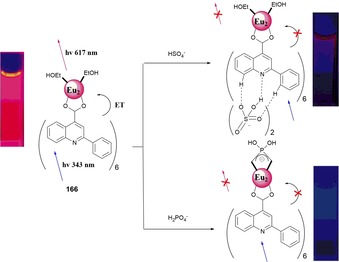
Proposed binding modes of HSO4 − and H2PO4 − with sensor 166. In the absence of the anion, excitation of PQC followed by energy transfer to the lanthanide enables bright‐red luminescence centered at λ=617 nm. Interaction of anions favors quenching of the antenna and, consequentially, also that of the EuIII luminescence. Reproduced from Ref. 223 with permission from The Royal Society of Chemistry.
Due to extremely weak absorption from f–f transitions, lanthanide ions possess low molar absorptivity. However, their luminescence can be increased by using organic antenna chromophores, which coordinate to the metal ion and act as sensitizers.224 If the organic antenna chromophore chosen as a sensitizing group is also sensitive to pH, it is possible to design pH probes that detect pH in two independent pH windows. Zhang and co‐workers225 have selected pH‐sensitive fluorophores such as hydroxyquinoline derivatives and rhodamine moieties as the binding sites to form EuIII complexes (Figure 28). The researchers have developed Eu(TTA)2‐DSQ (167) and Eu(TTA)3‐DR1 (168) {DSQ=5‐(dimethylamino)‐N‐(4‐{2‐[(8‐hydroxyquinolin‐2‐yl)methylene]hydrazinecarbonyl}phenyl)naphthalene‐1‐sulfonamide; DR1=N 1‐[4‐(dimethylamino)benzylidene]‐N 2‐(rhodamine 6G) lactamethylene‐diamine; TTA=thiophentrifluoroacetone} probes. Probe 167 shows high sensitivity to pH changes in neutral aqueous solution, and background fluorescence is negligible. For probe 168, the EuIII ion acts as a red emitter, and the rhodamine 6G fluorophore acts as a green emitter. Both components of the probe are pH sensitive, with pK a values of 7.2 (EuIII moiety) and 5.0 (rhodamine moiety). Luminescence titrations show the ability of the probe to detect pH changes in two different ranges, and this allows 168 to measure pH in both near‐neutral pH and acidic pH ranges (see Figure 28); the probe is also able to detect pH in both cultured cells and in vivo.
Figure 28.
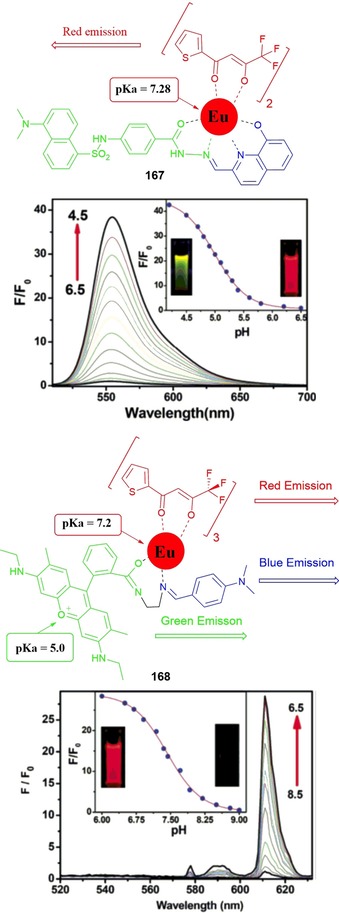
Chemical structures of dyes 167 and 168. Top) Fluorescence spectra of Eu(TTA)3‐168 (10 mm) in 0.02 m NaCl buffer solution with different pH values. The inset shows the fluorescence changes of Eu(TTA)3–168 with different pH values and the fluorescence photos of Eu(TTA)3–168 at pH 6.5 and 4.5; fluorescence intensity was recorded at λ=550 nm with excitation at λ=500 nm. Bottom) Fluorescence spectra of Eu(TTA)3–168 (10 mm) in MeCN/water (20:80, v/v) buffer (0.02 m NaCl buffer solution) with different pH values. The inset shows the fluorescence changes of Eu(TTA)3–168 with different pH values and the fluorescence photos of Eu(TTA)3–168 at pH 6.5 and 8.5; the intensities were recorded at λ=612 nm with excitation at λ=340 nm. Reproduced from Ref. 225 with permission from The Royal Society of Chemistry.
4.2. Iridium Complexes
Cyclometalated IrIII complexes have received considerable attention due to their high phosphorescent quantum yields, excellent color‐tuning capability, and large Stokes shifts. They also have long lifetimes (of the order of microseconds), especially if compared to the lifetimes of organic fluorescent probes (typically in the nanoseconds range).226, 227 These properties make them promising candidates to be used as phosphorescent emitters in OLEDs,228 in a range of biological applications as chemosensors,229 as cellular imaging probes,230 in vivo tumor imaging,231 and as photosensitizers for the production of singlet oxygen (1O2).232
Kando and co‐workers233 have published a series of pH‐activable IrIII complexes 169–178 (Figure 29) that can be used for tumor imaging. The emission intensity of these IrIII complexes is considerably enhanced upon protonation of their basic groups in aqueous solution. A strong orange‐red emission of 171 and 176 has also been reported. These probes have been successfully used for live‐cell imaging of HeLa‐S3 cells. Moreover, by photoirradiating probes 176–178 at λ=465 nm the researchers are able to generate singlet oxygen (1O2) from triplet oxygen (3O2). Photoirradiation of the IrII probes is also able to induce necrosis‐like cell death in HeLa‐S3 cells (Figure 30 a).
Figure 29.
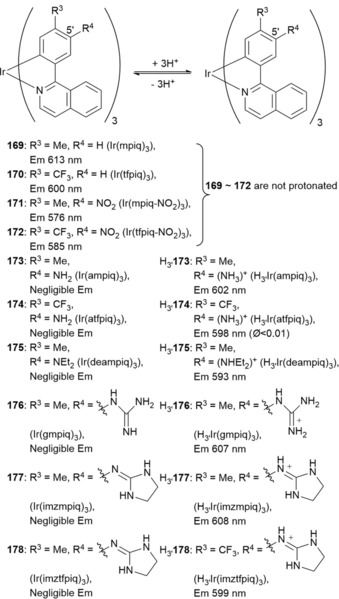
Chemical structures of probes 169—178, where mpiq = 1‐(4′‐methylphenyl)isoquinoline), tfpiq = 1‐(4′‐trifluoromethylphenyl)‐isoquinoline), ampiq = 1‐(5′‐amino‐4′‐methylphenyl)isoquinoline), atfpiq = 1‐(5′‐amino‐4′‐trifluoromethylphenyl)isoquinoline), deampiq = 1‐(5′‐diethylamino‐4′‐methylphenyl)isoquinoline), gmpiq = 1‐(5′‐guanidyl‐4′‐methylphenyl)isoquinoline), imzmpiq = 1‐(5′‐iminoimidazolidinyl‐4′‐methylphenyl)isoquinoline), and imztfpiq =1‐(5′‐iminoimidazolidinyl‐4′‐trifluoromethylphenyl)isoquinoline).
Figure 30.
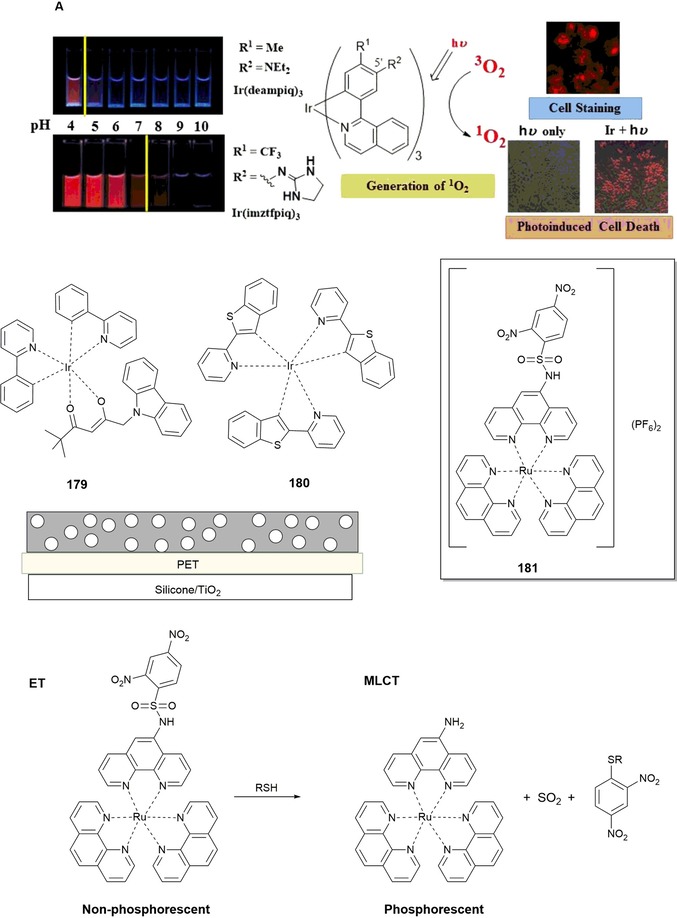
Photograph showing solutions of 175 (1 μm) and 178 (5 μm) in degassed DMSO/100 mm buffer (from pH 4 to 10) at 25 °C. Excitation at λ=365 nm (left). Luminescence microscopy images (Biorevo BZ‐9000, Keyence) of HeLa‐S3 cells irradiated at λ=465 nm (Twinlight 465, Relyon) for 30 min with 178 (right). deampiq = Ir(III) complexes that contain diethylamino groups on the 1‐(4′‐methylphenyl)isoquinoline) ligand; imztfpiq = Ir(III) complexes that contain iminoimidazolidinyl groups on the 1‐(4′‐methylphenyl)isoquinoline) ligand. Reprinted (adapted) with permission from Ref. 233. Copyright (2015) American Chemical Society. The chemical structures of compounds 179–181 are also shown.
Fischer and co‐workers234 report on the preparation and calibration of a dual sensor for barometric pressure and temperature. The sensor is made by combining two organometallic IrIII probes, green‐emitting complex 179 to measure temperature and red‐emitting complex 180 to function as a barometric (due to its oxygen‐sensing ability) probe (Figure 30). Probe 179 is then applied to poly(acrylonitrile) (PAN) microparticles; these 179/PAN microparticles are dispersed into a THF solution of cellulose acetate butyrate (CAB) also containing oxygen probe 180. The mixture is then spread onto solid poly(ethyleneterephthalate) (PET); once the solvent evaporates, a sensor film approximately 6 μm thick results. Due to a difference of about 75 nm in the emission maxima of both probes, both signals can be separated by using optical filters. The dual sensor can be successfully calibrated and has potential for luminescence lifetime imaging of temperature and barometric pressure.
4.3. Ruthenium Complexes
RuII polypyridine complexes235 have assumed a prominent status thanks to their multichannel sensing abilities.236 RuII polypyridine complexes can be used in colorimetric (UV/Vis),237 photoluminescence,238 electrochemiluminescence,239 and redox240 measurements. Ji and co‐workers241 have designed phosphorescent thiol probe 181 based on a RuII–poly(1,10‐phenanthroline) complex (Figure 30). This complex is considered a good luminophore candidate, as it shows strong metal‐to‐ligand charge‐transfer (MLCT) red emission (λ≈600 nm), a large Stokes shift (Δλ≈150 nm), and long luminescent lifetimes (of the order of microseconds).242 The luminescent properties of the RuII complex can be modified by introducing a 2,4‐dinitrobenzenesulfonyl (DNBS) moiety into the structure. Electron transfer (ET) from the RuII center (a strong electron donor) to the N^N coordination ligand is diverted to the DNBS moiety, a strong intramolecular electron acceptor; this results in quenching of the emission of the resulting probe, which becomes “switched off”. Cleavage of the DNBS moiety by thiols re‐establishes the MLCT of the RuII complex. The phosphorescence probe is “switched on” with a 90‐fold increase in the intensity of the signal at λ=598 nm, a Stokes shift of Δλ=143 nm, and a luminescent lifetime of 1.1 μs (Figure 30). This probe has been successfully used to image intracellular thiols in NCI‐H446 cells.
4.4. Iridium Complexes as Electrochemiluminescent Sensors
Despite their exciting photophysical and photochemical properties,243 only a few IrIII‐based chemosensors for cation sensing have been reported.244 Electrochemiluminescence (ECL) has emerged as a powerful alternative to photoluminescence (PL) in a wide range of fields such as clinical diagnostics, pharmaceutical analysis, environmental assays in food, and water testing.245, 246 A few examples of ECL‐based sensors for metal‐ion detection have been reported, such as those reported by High and co‐workers for the detection of Cu ions in water samples.247 The ECL properties of IrIII complexes appear to be superior to those of RuII complexes.248 Lin and co‐workers249 report ECL probes 182 and 183, two IrIII complexes with azacrown ethers, as probes for metal cations (Figure 31). The probes operate by an oxidation–reduction ECL process, with tri‐n‐propylamine as a co‐reactant and acetonitrile as the solvent. Complexes 182 and 183 behave as remarkable ECL sensors for BaII and AgI, respectively, with a ninefold enhancement in the intensity of the emission signal and a redshift in the emission λ max. In contrast, structurally analogous RuII complexes 184 and 185 do not display any significant ECL changes, and no wavelength shift is observed upon the addition of the same metal ions. This different behavior can be explained by the azacrown ether phenanthroline moiety, which is responsible for coordination to the target metal ions; for IrIII complexes 182 and 183, it represents the lowest unoccupied molecular orbital (LUMO) but is part of the highest occupied molecular orbital (HOMO) for RuII complexes 184* and 185*. If the azacrown ether moieties are part of the LUMO, the electronic situation produces a bathochromically shifted emission of the IrIII probe in the presence of the target metal ion. This work thus provides generally useful guidelines for improving the design of future ECL sensors based on metal complexes for metal‐ion recognition.
Figure 31.
Chemical structure of compounds 182–185.
5. Nanomaterials
5.1. Quantum Dots
Quantum dots (QDs) have unique optical properties, including high quantum yields, symmetric fluorescence emission spectra, wide excitation spectra, light resistance, and tunable spectra.250 QDs probes can be synthesized by linking QDs to peptides,251 antibodies,252 and organic molecules with specific ability to bind a particular metal.253 QDs can be used as molecular beacons to monitor enzymatic reactions,254 to track single vesicles following their endocytic uptake,255 and for membrane‐diffusion studies of individual QD‐tagged receptors;256 all these applications are based on the ability of QDs to report molecular position. Adding a sensing moiety to the QD adds a sensing functionality (e.g. by Förster resonance energy transfer, FRET) to the localization information. These functionalized QD probes become very powerful tools that can be used in a range of fields such as toxin detection, cell physiology, and pathology.257
Zamaleeva and co‐workers258 have designed cell‐penetrating FRET‐based CaII nanobiosensor 186 (Figure 32). The researchers use CANdot565QD as the donor and CaRuby, a red‐emitting CaII indicator derived from rhodamine, as the acceptor. To improve cell permeability and cytoplasmic delivery, the QDs are also functionalized with a small cell‐penetrating peptide (CPP) derived from hadrucalcin. The ability of 186 to act as a CaII sensor has been studied in the concentration range of 0 to 2 mm by using the relative increase in CaRuby fluorescence as a measure of sensitivity. In cell‐imaging experiments, the nanobiosensors penetrate inside the cells and distribute throughout the cytoplasm. Interestingly, the sensor shows a pointillistic distribution, that is, it is possible to determine localized CaII concentrations at discrete points. Imaging studies of intracellular CaII in HEK293 cells expressing N‐methyl‐d‐aspartate receptors have also been performed. By further functionalization with specific antibodies targeting high‐conductivity CaII channels, new CaII sensors can be developed to allow for optical single‐channel recording.
Figure 32.
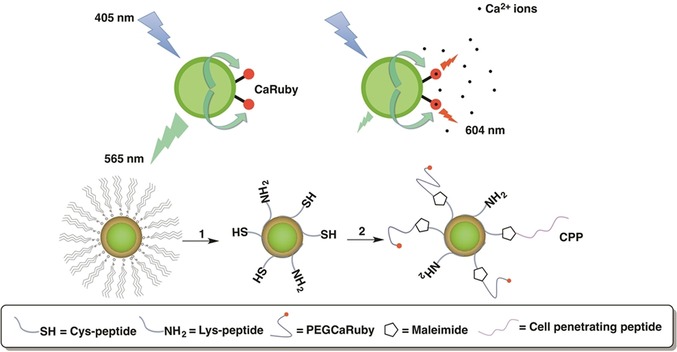
FRET‐based Ca2+ biosensor 186. Step 1) The QD TOP/TOPO passivating layer was replaced by a peptide coating made by mixing cysteine (SH function) and lysine (NH2 function) terminated peptides [pC, Ac‐CGSESGGSESG(FCC)3F‐amide; and pK, NH2‐KGSESGGSESG(FCC)3F‐amide, respectively]. Both components (hydrophobic QDs and peptides) were first dissolved in their respective solvents, pyridine and DMSO. After mixing, surfactant exchange and peptide binding were initiated by raising the pH. Step 2) Nanoparticles were further functionalized by adding CaRuby (red dots) and cell‐penetrating peptides (CPP, purple wiggles) onto peptide‐coated QDs by using a SH/maleimide linking reaction. Reprinted (adapted) with permission from Ref. 258. Copyright (2014) American Chemical Society.
Research is currently focused on labeling mammalian cells with QD probes. However, research on imaging plant cells and tissues is limited, because of concerns of the potential toxicity of QD probes to plant cells. Yu et al.259 have synthesized probe 187, the result of the conjugation of CdTe/ZnS QDs with 2‐amino‐3‐indolepropionic acid, to recognize indole–propionic acid (IPA) binding proteins in plant tissues (Figure 33). CdTe/ZnS‐IPA has the biological activity of the plant hormone IPA and is able to recognize IPA binding sites in plant tissues. The fluorescence emission wavelength of red‐emitting CdTe/ZnS‐IPA is λ=595 nm, which thus avoids interference from the intrinsic yellow‐green fluorescence background of plant tissues. CdTe/ZnS‐IPA has been used for the in situ imaging of IPA binding sites, and it has been revealed that the IPA binding sites in mung‐bean root tissues are concentrated in the membrane of endodermal cells.
Figure 33.
Structure diagram of CdTe/ZnS‐IPA (187).
Promising results have so far been obtained in research conducted on QD probes. Further research is necessary to develop QD probes with even lower toxicity and better fluorescence stability. The aim is to design probes that do not inhibit the growth and development of cells and that show greater biocompatibility.
5.2. Fluorescent Metallic Nanoclusters
Among the different fluorescent metallic nanoclusters (FNCs), fluorescent gold clusters (FGCs) are gradually emerging as promising imaging probes, because of their tunable emission in the visible‐to‐NIR range.260, 261 The FGC probes currently available have several limitations, including low fluorescence quantum yields, unstable fluorescence, low synthesis yields, and poor functionalization options. Research on improving FGC probes continues, and these probes could potentially be used for sensing enzyme‐related reactions because of their size‐dependent emission wavelength, satisfactory water solubility, and satisfactory photostability.262 One of these examples has recently been reported by Ke and co‐workers.263 The researchers describe the synthesis and characterization of dual‐emission probe 188 for the fluorescent ratiometric sensing of H2O2 concentration and pH change (Figure 34).
Figure 34.
a) Step‐by‐step illustration of the procedure used to prepare FITC/BSA gold nanoclusters (188). b) MALDI‐TOF mass spectra of a) FITC‐BSA and b) BSA. c) Native‐PAGE image of BSA (lane 1), FITC‐BSA (lane 2), BSA gold nanoclusters (lane 3), and FITC/BSA gold nanoclusters (lane 4) after staining with Coomassie Brilliant Blue. Native‐PAGE image of BSA gold nanoclusters (lane 5) and FITC/BSA gold nanoclusters (lane 6) under λ=488 nm excitation (laser‐based gel scanners) without Coomassie brilliant blue. Reproduced with permission from Ref. 263. Copyright 2015 Elsevier B. V.
Probe 188 contains a pH‐sensitive dye, fluorescein‐5‐isothiocyanate (FITC), the emission intensity of which diminishes with increasing concentration. FITC is conjugated to the amino groups of BSA protein. This FITC/BSA conjugate is used as a template to synthesize red‐emitting gold nanoclusters under alkaline conditions, and thus, probe 188 (FITC/BSA‐stabilized gold nanoclusters) is formed. Using λ=488 nm as the excitation wavelength, the fluorescence spectrum of 188 shows two bands at λ=525 and 670 nm. The band at λ=525 nm is sensitive to pH changes (0.1 pH‐unit change, pH 5.0–8.5), and the band at λ=670 nm is sensitive to changes in H2O2 concentration. Thus, this dual‐emission probe is able to detect changes in pH and H2O2 concentration separately.
5.3. Semiconductor Nanocrystals
Semiconductor nanocrystals (NCs) possess unique photoluminescent properties that make them excellent candidates for the design of fluorescence probes for chemo/biosensing applications. NCs offer distinct advantages over organic dyes such as high photoluminescence efficiency, broad absorption, narrow and symmetric emission, and good photostability.264 Zhang et al.265 have developed probe 189 for determination of the organophosphate compound diethylphosphorothioate (DEP). Probe 189 is an intrinsic dual‐emitting Mn‐doped ZnS nanocrystal‐based probe. In the presence of DEP, the electron‐transfer pathway is switched off and red emission of the probe is enhanced, whereas the blue emission is almost unchanged. By varying the concentration of DEP, the intensity ratio of the two emissions gradually varies and displays color changes from dark‐blue to purple to red (Figure 35). Thus, this probe can be used for the quantitative and visual detection of DEP with a limit of detection (LOD) of 1.8 μm. Significantly, the researchers have also successfully prepared paper‐based test strips with 189 that allow the rapid and visual detection of DEP residues. Probes based on ratiometric measurements present more advantages than NC‐based sensors, which use either turn‐off or turn‐on fluorescence intensity as the sole responsive signal. Ratiometric measurements can eliminate perturbations by experimental factors, such as fluctuation in probe concentration and instrumental efficiency, and can provide more precise measurements because of their self‐referencing capability, which derives from the use of the intensity ratio of the two emissions as the measurement tool.
Figure 35.
Illustration for the synthesis of the dual‐emitting probe 189 and the mechanism for fluorescence turn‐on and ratiometric detection of diethylphosphorothioate (DEP). (The bottom panel shows the fluorescent spectral changes of the dual‐emitting probe upon exposure to DEP and the corresponding fluorescence photographs of the probe solution taken under UV illumination). Reprinted with permission from Ref. 265. Copyright (2014) American Chemical Society.
6. Final Remarks and Perspectives
The aim of this review was to provide an in‐depth overview of key developments in the synthesis of green and red fluorescent probes over the last few years. The importance of taking a rational approach to the design of probes that can selectively detect and/or visualize a range of analytes has been emphasized.
As a matter of fact, green and red fluorescent dyes are the most‐common fluorophores to sense several analytes in biological media, because of their excellent optical properties, such as long excitation wavelengths, high excitation coefficients, and high fluorescent quantum yields. Such properties make these probes very appealing for use in cells and tissues being irradiated with light without cell damage. However, the use of these chromophores, besides their low cost in some cases and low toxicity, has some limitations, such as high rate of photobleaching, pH‐sensitive fluorescence, tendency to self‐quench, and broad fluorescence emission spectra, all of which limit the efficiency in multicolor applications. On the other hand, the probability of self‐quenching increases in red‐fluorescent probes due to the low solubility and high degree of substitution of these compounds, and this leads to a decrease in the fluorescence quantum yield. To overcome such issues, several authors have developed and designed fluorophores containing several water‐soluble groups, such as amino acids, phosphorus‐containing groups, sulfur‐containing groups, and vitamin units.
The aim is to design new selective, sensitive, and biocompatible fluorescent probes with increasingly more complex and sophisticated structures that would allow additional fine‐tuning of their properties and expand the range of translational applications. Indeed, the excellent optical properties of green and red chromophores make them very valuable for working with biological tissues and cells. The amazing work conducted by Lippard et al. shows how it is possible to design biocompatible fluorescent “turn‐on” selective probes for the detection of metal ions and anions for cellular applications, and much work has been done in this direction. Modifications to the chromophore skeleton by the introduction of hetero groups such as carboxylic acids, sulfonic acids, and carbonyl, amino, nitro, and hydroxy groups can strongly contribute to enhancing the intensity of color and to improving solubility. Modification of the receptor unit, by changing the nature and position of the donor atoms, plays a crucial role in improving selectivity and sensitivity towards the analyte. Some of the probes mentioned are only able to detect analytes in organic solvents; however, the tendency is to design new sophisticated materials that are soluble in aqueous solutions, such as nanomaterials and modified proteins, to push the detection limits further and to improve the brightness and efficacy of these probes to detect particular targets. Further innovations in probe design, the use of multichannel techniques, and streamlined toxicity screening will allow the development of safer and more versatile probes. With further systematic experimental and theoretical advances, we believe fascinating new probes based on green and red dyes will be used to address important issues related to a wide range of fields, including translational applications in environment monitoring, medicine, ad medical diagnosis.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Elisabete Oliveira graduated in 2006 in Applied Chemistry from FCT‐University NOVA of Lisbon (Portugal), obtained her Master's degree in Biotechnology in 2007, and completed a Ph.D. degree in Biotechnology in 2010 at the same university. In 2013, she obtained a second Ph.D. degree in Food Science and Technology from the Science Faculty of Ourense Campus in the University of Vigo (Spain). Her scientific interests are focused on the synthesis of new bioinspired emissive peptide as fluorescence chemosensors; supramolecular chemistry (photophysics and photochemistry); multifunctional application of chemosensors in vitro (solution and solid studies) and in vivo (cell‐imaging studies); the synthesis of new emissive nanomaterials, such as quantum dots and silica for drug delivery; and biomarker discovery in biological samples.

Biographical Information
Emilia Bértolo is a Principal Lecturer (Chemistry) at Canterbury Christ Church University (CCCU, UK) and previously worked in the Department of Materials, Imperial College London (UK). She obtained her Ph.D. and B.Sc. degrees in Chemistry (Inorganic) from the University of Santiago de Compostela (Spain). Additionally, she obtained her M.Sc. degree in Environmental Technology from Imperial College London (UK). She also has a Postgraduate Certificate in Learning and Teaching in Higher Education (CCCU, UK) and a Postgraduate Certificate in Online and Distance Education (Open University). Her main focus is on the synthesis and applications of fluorescence chemosensors. Her other research interests are in the field of pedagogical research, with special focus on enhancing the teaching‐research nexus in the undergraduate curriculum (undergraduate students as producers of research and scholarship).

Biographical Information
Cristina Núñez obtained her Ph.D. degree in Chemistry in 2009 from the University of Santiago de Compostela (Spain). She spent short periods of time performing research at the Universidade NOVA de Lisboa, REQUIMTE (Portugal) during 2007 and 2008. In 2010, she started her postdoctoral research period between the University NOVA of Lisbon (UNL, Portugal) and the Canterbury Christ Church University (UK), finishing in 2015. Dr. Núñez is a researcher at the Research Unit of the Hospital Universitario Lucus Augusti (HULA, Spain).

Biographical Information
Viviane Pilla received her Ph.D. degree in Applied Physics Sciences in 2001 from the University of São Paulo (Brazil). She performed postdoctoral research in Applied Physics in Nonlinear Optics at the Federal University of Pernambuco (Brazil) and State University of Campinas (Brazil). She later completed a postdoctoral period in Physical Chemistry Applications in the BIOSCOPE research group at the University NOVA of Lisbon (Portugal). Currently, she is Adjunct Physics Professor IV at the Physics Institute in the Federal University of Uberlândia UFU (Brazil) and researcher of the group of Optical and Thermal Properties of Materials of the Physics Institute UFU (Brazil). Dr. Pilla has experience in the optical and spectroscopic properties of materials, acting mainly on the following subjects: photothermal effects and thermo‐optical characterization of different materials as quantum dots for biological applications; crystal and glasses; and biomaterials (biofluids, natural dyes, and dental resin composites).

Biographical Information
Hugo M. Santos graduated in Applied Chemistry from University NOVA of Lisbon (Portugal) and completed a Ph.D. degree in Biochemistry from the same university in 2010. During his time as a Ph.D. candidate, he spent six months at the Turku Centre for Biotechnology (Finland) working with state‐of‐the‐art mass spectrometry (MS) instrumentation for biomedical research. He worked as a postdoctoral researcher at the University of Vigo (Spain) followed by a move to the Institute of Biomedicine and Biotechnology (Barcelona, Spain) to advance biomedical applications of mass spectrometry and translational research. In 2011, Dr. Santos joined FCT NOVA (Portugal) to continue his research in biological MS. Currently, he is Assistant Researcher—FCT Investigator Programme at UCIBIO‐REQUIMTE FCT NOVA (Portugal). His scientific interests focus on the identification of molecules involved in complex biological processes, characterizing their structure and monitoring how their abundance may change during these processes to gain insight into the underlying molecular mechanisms; nanoproteomics and nanomedicine; application of chemosensors to the detection/quantification of metals; and MS analysis of organic molecules, metal complexes, and supramolecular systems.

Biographical Information
Javier Fernández‐Lodeiro received his Ph.D. degree in 2012 from the University of Vigo (Spain). In 2013, he was a postdoctoral researcher at the Faculty of Science and Technology at the University NOVA of Lisbon (Portugal) in the REQUIMTE‐UCIBIO, working in the BIOSCOPE research group. He then moved to the Institute of Chemistry at the University of Sao Paulo (Brazil), working in the LOCSIN research group and focusing on the synthesis and application of chalcogen molecules for construction of fluorescence nanoprobes. Since September 2014, he has been working in the Faculty of Science and Technology University NOVA of Lisbon (Portugal) at the REQUIMTE‐UCIBIO in the BIOSCOPE research group as a postdoctoral researcher. His research interests focus on the synthesis of new nanoparticles of Au, Ag, Pt, Fe, and quantum dots; application of new synthetic methodologies in nanomaterials using chalcogen atoms (Se and Te); and new molecular probes for biochemical and proteomics applications.

Biographical Information
Adrian Fernández‐Lodeiro graduated in Chemistry in 2013 from the University of Santiago de Compostela (Spain) and is now a Ph.D. candidate in the Green Chemistry Ph.D. Program at the REQUIMTE‐UCIBIO, BIOSCOPE Group, Faculty of Science and Technology of the University NOVA of Lisbon (Portugal). His scientific interests are focused on nanosynthesis; metallic and polymeric nanoparticles, including rods and quantum dots, nano‐biomedicine; and fluorescent and colorimetric dyes for sensing.

Biographical Information
Jamila Djafari graduated in 2013 in Chemical Biology from the University Paris‐Saclay (France). In 2015, she obtained a double Master's degree in Chemical Science and Engineering at the Ecole Nationale Supérieure de Chimie de Paris (ENSCP, France) and in Molecular Chemistry from the University Pierre et Marie‐Curie, Paris (France). She is now working towards a Ph.D. degree in Chemistry focusing on antibiotic‐functionalized nanomaterials at the REQUIMTE‐UCIBIO, BIOSCOPE Group, Faculty of Science and Technology of the University NOVA of Lisbon (Portugal). Her scientific interests are centered around the synthesis of nanomaterials, organic chemistry, and biochemistry.

Biographical Information
José L. Capelo received his Ph.D. degree from the University of Vigo (2002), completed postdoctoral research at the IST in Lisbon (Portugal), and was later appointed as Researcher at REQUIMTE (FCT‐UNL, Portugal). He then moved to the University of Vigo (Spain) as IPP (Isidro Parga Pondal) Research Lecturer. He was appointed Assistant Professor at FCT‐UNL (Portugal) in 2012, where he is currently based. Prof. Capelo has developed research on the following topics: quantification of metal and metals species in environmental and food samples, new methods to speed protein identification by using mass spectrometry based workflows, accurate bottom‐up protein quantification, bacterial identification through mass spectrometry, fast determination of steroids in human samples, biomarker discovery, application of sensors and chemosensors to the detection/quantification of metals, and nanoproteomics and nanomedicine.

Biographical Information
Carlos Lodeiro graduated in Chemistry in 1995 and received his Ph.D. degree in Chemistry in 1999 from the University of Santiago de Compostela (Spain). In 1999, he moved to the University NOVA of Lisbon (UNL, Portugal) as European Marie Curie Postdoctoral Researcher in a project concerning molecular devices and machines; in 2004, he became a Fellow Researcher and Invited Assistant Lecturer at the REQUIMTE‐CQFB, Chemistry Department (UNL, Portugal). In 2008, he received habilitation in Chemistry in Spain, and a year later he moved to the University of Vigo, Faculty of Sciences of Ourense (FCOU, Spain) as IPP (Isidro Parga Pondal) Researcher–Lecturer. He is currently an Assistant Professor in the Chemistry Department UCIBIO‐REQUIMTE Laboratory in the Faculty of Science and Technology, University NOVA of Lisbon (Portugal). In 2017, he received habilitation in Inorganic Analytical Chemistry in Portugal at the FCT‐UNL. His research interests include physical organic and physical inorganic chemistry of fluorescence chemosensors; the synthesis of functionalized nanoparticles, nanocomposites, and nanomaterials; applications of nanomaterials in environmental research; application of nanomaterials in biomedical research; supramolecular analytical proteomics; and onco‐ and nanoproteomics.

Acknowledgements
The authors thank PROTEOMASS Scientific Society (Portugal) for funding. All authors also thank LAQV/REQUIMTE (UID/QUI/50006/2013) and UCIBIO/REQUIMTE (UID/Multi/04378/2013) for funding. H.M.S. is funded by the FCT 2015 Investigator Program (IF/00007/2015). E.O. and J.F.L. are grateful for the postdoctoral grants SFRH/BPD/108660/2015 and SFRH/BPD/93982/2013, respectively. A.F.L. is thankful for the doctoral grant SFRH/BD/52528/2014 from Fundação para a Ciência e a Tecnologia (FCT‐MEC) (Portugal). J.D thanks the Proteomass Scientific Society and a BI grant from FCT Project PTDC/QEQMED/2118/2014. C.N. was funded by the Miguel Servet Program (CP16/00139), Instituto de Salud Carlos III, Ministerio de Economía, Industria y Competitividad (Spain). E.O. thanks the Foundation L'Oreal (UNESCO and FCT) for the Prize For Women in Science 2015, “Medalhas de Honra L'Oréal Portugal para as Mulheres na Ciência”, healthy Sciences area. All authors thank FCT‐MECT for grant PTDC/QEQ‐MED/2118/2014. E.O. (green dyes) and C.N. (red dyes) have been involved equally in the redaction of the paper.
E. Oliveira, E. Bértolo, C. Núñez, V. Pilla, H. M. Santos, J. Fernández-Lodeiro, A. Fernández-Lodeiro, J. Djafari, J. L. Capelo, C. Lodeiro, ChemistryOpen 2018, 7, 9.
References
- 1. Vendrell M., Zhai D., Er J. C., Chang Y. T., Chem. Rev. 2012, 112, 4391–4420. [DOI] [PubMed] [Google Scholar]
- 2. Nidheesh P. V., Gandhimathi R., Ramesh S. T., Environ. Sci. Pollut. Res. 2013, 20, 2099–2132. [DOI] [PubMed] [Google Scholar]
- 3. Moussavi G., Mahmoudi M., J. Hazard. Mater. 2009, 168, 806–812. [DOI] [PubMed] [Google Scholar]
- 4. dos Santos A. B., Cervantes F. J., van Lier J. B., Bioresour. Technol. 2007, 98, 2369–2385. [DOI] [PubMed] [Google Scholar]
- 5. Leung B. O., Chou K. C., Appl. Spectrosc. 2011, 65, 967–980. [DOI] [PubMed] [Google Scholar]
- 6. Santi P. A., J. Histochem. Cytochem. 2011, 59, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang J., Campbell R. E., Ting A. Y., Tsien R. Y., Nat. Rev. Mol. Cell Biol. 2002, 3, 906–918. [DOI] [PubMed] [Google Scholar]
- 8. Zheng Q., Juette M. F., Jockusch S., Wasserman M. R., Zhou Z., Altman R. B., Blanchard S. C., Chem. Soc. Rev. 2014, 43, 1044–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alamudi S. H., Satapathy R., Kim J., Su D., Ren H., Das R., Hu L., Alvarado-Martínez E., Lee J. Y., Hoppmann C., Peña-Cabrera E., Ha H.-H., Park H.-S., Wang L., Chang Y.-T., Nat. Commun. 2016, 7, 11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neto B. A. D., Carvalho P. H. P. R., Correa J. R., Acc. Chem. Res. 2015, 48, 1560–1569. [DOI] [PubMed] [Google Scholar]
- 11. Beija M., Afonso C. A. M., Martinho J. M. G., Chem. Soc. Rev. 2009, 38, 2410–2433. [DOI] [PubMed] [Google Scholar]
- 12. Lavis L. D., Raines R. T., ACS Chem. Biol. 2014, 9, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verma A. K., Dash R. R., Bhunia P., J. Environ. Manage. 2012, 93, 154–168. [DOI] [PubMed] [Google Scholar]
- 14. Chen X., Pradhan T., Wang F., Kim J. S., Yoon J., Chem. Rev. 2012, 112, 1910–1956. [DOI] [PubMed] [Google Scholar]
- 15. Heek T., Nikolaus J., Schwarzer R., Fasting C., Welker P., Licha K., Herrmann A., Haag R., Bioconjugate Chem. 2013, 24, 153–158. [DOI] [PubMed] [Google Scholar]
- 16. Montalti M., Battistelli G., Cantelli A., Genovese D., Chem. Commun. 2014, 50, 5326–5329. [DOI] [PubMed] [Google Scholar]
- 17. Weißenstein A., Würthner F., Chem. Commun. 2015, 51, 3415–3418. [DOI] [PubMed] [Google Scholar]
- 18. Sun M., Müllen K., Yin M., Chem. Soc. Rev. 2016, 45, 1513–1528. [DOI] [PubMed] [Google Scholar]
- 19. Würthner F., Saha-Möller C. R., Fimmel B., Ogi S., Leowanawat P., Schmidt D., Chem. Rev. 2016, 116, 962–1052. [DOI] [PubMed] [Google Scholar]
- 20. Kim H. N., Lee M. H., Kim H. J., Kim J. S., Yoon J., Chem. Soc. Rev. 2008, 37, 1465–1472. [DOI] [PubMed] [Google Scholar]
- 21. Sun Y. Q., Liu J., Lv X., Liu Y., Zhao Y., Guo W., Angew. Chem. Int. Ed. 2012, 51, 7634–7636; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7752–7754. [Google Scholar]
- 22. Campbell W. M., Burrell A. K., Officer D. L., Jolley K. W., Coord. Chem. Rev. 2004, 248, 1363–1379. [Google Scholar]
- 23. Hagfeldt A., Boschloo G., Sun L., Kloo L., Pettersson H., Chem. Rev. 2010, 110, 6595–6663. [DOI] [PubMed] [Google Scholar]
- 24. Flamigni L., Gryko D. T., Chem. Soc. Rev. 2009, 38, 1635–1646. [DOI] [PubMed] [Google Scholar]
- 25. Voloshchuk R., Gryko D. T., Chotkowski M., Ciuciu A. I., Flamigni L., Chem. Eur. J. 2012, 18, 14845–14859. [DOI] [PubMed] [Google Scholar]
- 26. Fornalé F., Vaglio S., Spiezio C., Previde E. P., Commun. Integr. Biol. 2012, 5, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schliep M., Cavigliasso G., Quinnell R. G., Stranger R., Larkum A. W. D., Plant Cell Environ. 2013, 36, 521–527. [DOI] [PubMed] [Google Scholar]
- 28.H. Scheer, J. J. Katz, Nuclear Magnetic Resonance Spectroscopy of Porphyrins and Metalloporphyrins, 1975 in J. E. Falk, K. M. Smith (Eds.): Porphyrins and Metalloporphyrins Amsterdam, Elsevier pp. 399-524 .
- 29. Woodward H. V. R. B., Ayer W. A., Beaton J. M., Bickelhaupt F., Bonnett R., Buchschacher P., Closs G. L., Dutler H., Hannah J., Hauck F. P., Itô S., Langemann A., Le Goff E., Leimgruber W., Lowwski W., Sauer J., Valenta Z., Tetrahedron 1990, 46, 7599–7659. [Google Scholar]
- 30. Gust D., Moore T. A., Moore A. L., Acc. Chem. Res. 2001, 34, 40–48. [DOI] [PubMed] [Google Scholar]
- 31. Parkinson T. M., Brown J. P., Ann. Rev. Nutr. 1981, 1, 175–205. [DOI] [PubMed] [Google Scholar]
- 32. Wrolstad R. E., Culver C. A., Annu. Rev. Food Sci. Technol. 2012, 3, 59–77. [DOI] [PubMed] [Google Scholar]
- 33. Song Y., Wei W., Qu X., Adv. Mater. 2011, 23, 4215–4236. [DOI] [PubMed] [Google Scholar]
- 34. Carvalho E., Fraser P. D., Martens S., Food Chem. 2013, 139, 744–752. [DOI] [PubMed] [Google Scholar]
- 35. Luterotti S., Marković K., Franko M., Bicanic D., Madžgalj A., Kljak K., Food Chem. 2013, 140, 390–397. [DOI] [PubMed] [Google Scholar]
- 36. Grotewold E., Annu. Rev. Plant Biol. 2006, 57, 761–780. [DOI] [PubMed] [Google Scholar]
- 37. He J., Giusti M. M., Annu. Rev. Food Sci. Technol. 2010, 1, 163–187. [DOI] [PubMed] [Google Scholar]
- 38. Yoshida K., Mori M., Kondo T., Nat. Prod. Rep. 2009, 26, 884–915. [DOI] [PubMed] [Google Scholar]
- 39. Tanaka Y., Sasaki N., Ohmiya A., Plant J. 2008, 54, 733–749. [DOI] [PubMed] [Google Scholar]
- 40. Han J., Burgess K., Chem. Rev. 2010, 110, 2709–2728. [DOI] [PubMed] [Google Scholar]
- 41. Formica M., Fusi V., Giorgi L., Micheloni M., Coord. Chem. Rev. 2012, 256, 170–192. [Google Scholar]
- 42. Bianchi A., Delgado-Pinar E., García-España E., Giorgi C., Pina F., Coord. Chem. Rev. 2014, 260, 156–215. [Google Scholar]
- 43. Kaur M., Choi D. H., Chem. Soc. Rev. 2015, 44, 58–77. [DOI] [PubMed] [Google Scholar]
- 44. Ding Y. B., Zhu W. H., Xie Y. S., Chem. Rev. 2017, 117, 2203–2256. [DOI] [PubMed] [Google Scholar]
- 45. Carter K. P., Young A. M., Palmer A. E., Chem. Rev. 2014, 114, 4564–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu H., Mack J., Yang Y., Shen Z., Chem. Soc. Rev. 2014, 43, 4778–4823. [DOI] [PubMed] [Google Scholar]
- 47. Prodi L., New J. Chem. 2005, 29, 20–31. [Google Scholar]
- 48. Mameli M., Aragoni M. C., Arca M., Caltagirone C., Demartin F., Farruggia G., De Filippo G., Devillanova F. A., Garau A., Isaia F., Lippolis V., Murgia S., Prodi L., Pintus A., Zaccheroni N., Chem. Eur. J. 2010, 16, 919–30. [DOI] [PubMed] [Google Scholar]
- 49. Clark M. A., Duffy K., Tibrewala J., Lippard S. J., Org. Lett. 2003, 5, 2051–2054. [DOI] [PubMed] [Google Scholar]
- 50. Retting W., Strehmel B., Schrader S., Seifert H. Applied Fluorescence in Chemistry, Biology and Medicine, Springer, Heidelberg, 1999. [Google Scholar]
- 51. Rao J., Dragulescu-Andrasi A., Yao H., Curr. Opin. Biotechnol. 2007, 18, 17–25. [DOI] [PubMed] [Google Scholar]
- 52. Jiao Y., Zhu B. C., Chen J. H., Duan X. H., Theranostics 2015, 5, 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rossi F. M., Kao J. P. Y., Bioconjugate Chem. 1997, 8, 495–497. [DOI] [PubMed] [Google Scholar]
- 54. Sun W. C., Gee K. R., Klaubert D. H., Haugland R. P., J. Org. Chem. 1997, 62, 6469–6475. [Google Scholar]
- 55. Jiang P., Guo Z., Coord. Chem. Rev. 2004, 248, 205–229. [Google Scholar]
- 56. Nolan E. M., Lippard S. J., Acc. Chem. Res. 2009, 42, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chang P. V., Bertozzi C. R., Chem. Commun. 2012, 48, 8864–8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oliveira E., Lorenzo J., Cid A., Capelo J. L., Lodeiro C., J. Photochem. Photobiol. A 2013, 269, 17–26. [Google Scholar]
- 59. Ueno T., Urano Y., Setsukinai K. I., Takakusa H., Kojima H., Kikuchi K., Ohkubo K., Fukuzumi S., Nagano T., J. Am. Chem. Soc. 2004, 126, 14079–14085. [DOI] [PubMed] [Google Scholar]
- 60. Sameiro M., Gonçalves T., Chem. Rev. 2009, 109, 190–212. [DOI] [PubMed] [Google Scholar]
- 61. Choi M. G., Cha S., Park J. E., Lee H., Jeon H. L., Chang S.-K., Org. Lett. 2010, 12, 1468–1471. [DOI] [PubMed] [Google Scholar]
- 62. Margulies D., Melman G., Shanzer A., J. Am. Chem. Soc. 2006, 128, 4865–4871. [DOI] [PubMed] [Google Scholar]
- 63. Rurack K., Spectrochim. Acta Part A 2001, 57, 2161–2195. [DOI] [PubMed] [Google Scholar]
- 64. Frederickson C. J., Int. Rev. Neurobiol. 1989, 31, 145–238. [DOI] [PubMed] [Google Scholar]
- 65. Burdette S. C., Walkup G. K., Spingler B., Tsien R. Y., Lippard S. J., J. Am. Chem. Soc. 2001, 123, 7831–7841. [DOI] [PubMed] [Google Scholar]
- 66. Burdette S. C., Frederickson C. J., Bu W., Lippard S. J., J. Am. Chem. Soc. 2003, 125, 1778–1787. [DOI] [PubMed] [Google Scholar]
- 67. Goldsmith C. R., Lippard S. J., Inorg. Chem. 2006, 45, 6474–6478. [DOI] [PubMed] [Google Scholar]
- 68. Nolan E. M., Ryu J. W., Jaworski J., Feazell R. P., Sheng M., Lippard S. J., J. Am. Chem. Soc. 2006, 128, 15517–15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nolan E. M., Jaworski J., Okamoto K.-I., Hayashi Y., Sheng M., Lippard S. J., J. Am. Chem. Soc. 2005, 127, 16812–16823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McQuade L. E., Lippard S. J., Inorg. Chem. 2010, 49, 7464–7471. [DOI] [PubMed] [Google Scholar]
- 71. Buccella D., Horowitz J. A., Lippard S. J., J. Am. Chem. Soc. 2011, 133, 4101–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Radford R. J., Chyan W., Lippard S. J., Chem. Sci. 2014, 5, 4512–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Abebe F. A., Eribal C. S., Ramakrishna G., Sinn E., Tetrahedron Lett. 2011, 52, 5554–5558. [Google Scholar]
- 74. Muthuraj B., Deshmukh R., Trivedi V., Iyer P. K., ACS Appl. Mater. Interfaces 2014, 6, 6562–6569. [DOI] [PubMed] [Google Scholar]
- 75. Abebe F. A., Sinn E., Tetrahedron Lett. 2011, 52, 5234–5237. [Google Scholar]
- 76. Chawla H. M., Shukla R., Pandey S., Tetrahedron Lett. 2013, 54, 2063–2066. [Google Scholar]
- 77. Wei G., Wang L., Jiao J., Hou J., Cheng Y., Zhu C., Tetrahedron Lett. 2012, 53, 3459–3462. [Google Scholar]
- 78. Swamy K. M. K., Kim H. N., Soh J. H., Kim Y., Kim S.-J., Yoon J., Chem. Commun. 2009, 1234–1236. [DOI] [PubMed] [Google Scholar]
- 79. Lin D.-S., Lai J.-P., Sun H., Yang Z., Zuo Y., Anal. Methods 2014, 6, 1517–1522. [Google Scholar]
- 80. Nolan E. M., Lippard S. J., J. Am. Chem. Soc. 2003, 125, 14270–14271. [DOI] [PubMed] [Google Scholar]
- 81. Nolan E. M., Racine M. E., Maryann, Lippard S. J., Inorg. Chem. 2006, 45, 2742–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fernández-Lodeiro J., Nuñez C., Lodeiro A. F., Oliveira E., Rodríguez-González B., Dos Santos A. A., Capelo J. L., Lodeiro C., J. Nanopart. Res. 2014, 16, 2315. [Google Scholar]
- 83. Choi M. G., Ryu D. H., Jeon H. L., Cha S., Cho J., Joo H. H., Hong K. S., Lee C., Ahn S., Chang S.-K., Org. Lett. 2008, 10, 3717–3720. [DOI] [PubMed] [Google Scholar]
- 84. Wang H., Zhou G., Mao C., Chen X., Dye. Pigm. 2013, 96, 232–236. [Google Scholar]
- 85. Wang X., Miao Q., Song T., Yuan Q., Gao J., Liang G., Analyst 2014, 139, 3360–3364. [DOI] [PubMed] [Google Scholar]
- 86. Lim M. H., Wong B. A., W. H. Pitcock, Jr. , Mokshagundam D., Baik M. H., Lippard S. J., J. Am. Chem. Soc. 2006, 128, 14364–14373. [DOI] [PubMed] [Google Scholar]
- 87. Tonzetich Z. J., McQuade L. E., Lippard S. J., Inorg. Chem. 2010, 49, 6338–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zheng F., Zeng F., Yu C., Hou X., Wu S., Chem. Eur. J. 2013, 19, 936–942. [DOI] [PubMed] [Google Scholar]
- 89. Zhang X., Shiraishi Y., Hirai T., Tetrahedron Lett. 2007, 48, 8803–8806. [Google Scholar]
- 90. Yang X.-F., Ye S.-J., Bai Q., Wang X.-Q., J. Fluoresc. 2007, 17, 81–87. [DOI] [PubMed] [Google Scholar]
- 91. Swamy K. M. K., Lee Y. J., Lee H. N., Chun J., Kim Y., Kim S. J., Yoon J., J. Org. Chem. 2006, 71, 8626–8628. [DOI] [PubMed] [Google Scholar]
- 92. Yang X.-F., Wang L., Xu H., Zhao M., Anal. Chim. Acta 2009, 631, 91–95. [DOI] [PubMed] [Google Scholar]
- 93. Zhang P., Li C., Zhang H., Li Y., Yu X., Geng L., Wang Y., Zhen X., Ma Z., J. Incl. Phenom. Macrocycl. Chem. 2015, 81, 295–300. [Google Scholar]
- 94. Asthana S. K., Kumar A., Neeraj, Upadhyay K. K., Tetrahedron Lett. 2014, 55, 5988–5992. [Google Scholar]
- 95. Kwon S. K., Kou S., Kim H. N., Chen X., Hwang H., Nam S. W., Kim S. H., Swamy K. M. K., Park S., Yoon J., Tetrahedron Lett. 2008, 49, 4102–4105. [Google Scholar]
- 96. Lv X., Liu J., Liu Y., Zhao Y., Chen M., Wang P., Guo W., Org. Biomol. Chem. 2011, 9, 4954–4958. [DOI] [PubMed] [Google Scholar]
- 97. Chung S.-Y., Nam S.-W., Lim J., Park S., Yoon J., Chem. Commun. 2009, 4, 2866. [DOI] [PubMed] [Google Scholar]
- 98. Yin W., Zhu H., Wang R., Dye. Pigment. 2014, 107, 127–132. [Google Scholar]
- 99. Ma X., Liu C., Shan Q., Wei G., Wei D., Du Y., Sens. Actuators B 2013, 188, 1196–1200. [Google Scholar]
- 100. Song J. H., Zhang D., Liu Y. Q., Zhao Y., Ye Y., New J. Chem. 2015, 39, 6284–6288. [Google Scholar]
- 101. Hou F., Huang L., Xi P., Cheng J., Zhao X., Xie G., Shi Y., Cheng F., Yao X., Bai D., Zeng Z., Inorg. Chem. 2012, 51, 2454–2460. [DOI] [PubMed] [Google Scholar]
- 102. Zhang X., Shiraishi Y., Hirai T., Org. Lett. 2007, 9, 5039–5042. [DOI] [PubMed] [Google Scholar]
- 103. Park J., Rao B. A., Son Y.-A., Fibers Polym. 2015, 16, 953–960. [Google Scholar]
- 104. Lee J.-Y., Rao B. A., Hwang J.-Y., Son Y.-A., Sens. Actuators B 2015, 220, 1070–1085. [Google Scholar]
- 105. Lozano V. A., Escandar G. M., Mahedero M. C., de la Peña A. M., Anal. Methods 2012, 4, 2002–2008. [Google Scholar]
- 106. Sakabe M., Asanuma D., Kamiya M., Iwatate R. J., Hanaoka K., Terai T., Nagano T., Urano Y., J. Am. Chem. Soc. 2013, 135, 409–414. [DOI] [PubMed] [Google Scholar]
- 107. Oliveira E., Santos S. M., Núñez C., Capelo J. L., Lodeiro C., Dalton Trans. 2016, 45, 1254–1258. [DOI] [PubMed] [Google Scholar]
- 108. Karp N. A., Lilley K. S., Proteomics 2005, 5, 3105–3115. [DOI] [PubMed] [Google Scholar]
- 109. Fegan A., Shirude P. S., Balasubramanian S., Chem. Commun. 2008, 2, 2004–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Peng X., Yang Z., Wang J., Fan J., He Y., Song F., Wang B., Sun S., Qu J., Qi J., Yan M., J. Am. Chem. Soc. 2011, 133, 6626–6635. [DOI] [PubMed] [Google Scholar]
- 111. Zanotti K. J., Silva G. L., Creeger Y., Robertson K. L., Waggoner A. S., Berget P. B., Armitage B. A., Org. Biomol. Chem. 2011, 9, 1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hamer F. M. (Ed.), The Chemistry of Heterocyclic Compounds: The Cyanine Dyes and Related Compounds, Vol. 18, John Wiley & Sons, Hoboken, NJ, 1964. [Google Scholar]
- 113. Ferguson L. R., Denny W. A., Mutat. Res. Mol. Mech. Mutagen. 2007, 623, 14–23. [DOI] [PubMed] [Google Scholar]
- 114. Mahmood T., Wu Y., Loriot D., Kuimova M., Ladame S., Chemistry 2012, 18, 12349–12356. [DOI] [PubMed] [Google Scholar]
- 115. Bohländer P. R., Wagenknecht H.-A., Eur. J. Org. Chem. 2014, 7547–7551. [Google Scholar]
- 116. Xiong K., Huo F., Yin C., Yang Y., Chao J., Zhang Y., Xu M., Sens. Actuators B 2015, 220, 822–828. [Google Scholar]
- 117. Li Q., Guo Y., Shao S., Sens. Actuators B 2012, 171–172, 872–877. [Google Scholar]
- 118. Guo H., Jing Y., Yuan X., Ji S., Zhao J., Li X., Kan Y., Org. Biomol. Chem. 2011, 9, 3844–3853. [DOI] [PubMed] [Google Scholar]
- 119. Singh R. S., Gupta R. K., Paitandi R. P., Misra A., Pandey D. S., New J. Chem. 2015, 39, 2233–2239. [Google Scholar]
- 120. Wang L., Fang G., Cao D., Sens. Actuators B 2015, 207, 849–857. [Google Scholar]
- 121. Barba-Bon A., Calabuig L., Costero A. M., Gil S., Martínez-Máñez R., Sancenón F., RSC Adv. 2014, 4, 8962–8965. [Google Scholar]
- 122. Li Q., Guo Y., Shao S., Analyst 2012, 137, 4497–4501. [DOI] [PubMed] [Google Scholar]
- 123. Xu J., Li Q., Yue Y., Guo Y., Shao S., Biosens. Bioelectron. 2014, 56, 58–63. [DOI] [PubMed] [Google Scholar]
- 124. Gotor R., Gaviña P., Ochando L. E., Chulvi K., Lorente A., Martínez-Máñez R., Costero A. M., RSC Adv. 2014, 4, 15975–15982. [Google Scholar]
- 125. Pan Z.-H., Luo G.-G., Zhou J.-W., Xia J.-X., Fang K., Wu R.-B., Dalton Trans. 2014, 43, 8499–8507. [DOI] [PubMed] [Google Scholar]
- 126. Xu Z., Kim G.-H., Han S. J., Jou M. J., Lee C., Shin I., Yoon J., Tetrahedron 2009, 65, 2307–2312. [Google Scholar]
- 127. Fu Y., Zeng X., Mu L., Jiang X.-K., Deng M., Zhang J.-X., Yamato T., Sens. Actuators B 2012, 164, 69–75. [Google Scholar]
- 128. Liu S.-R., Wu S.-P., J. Fluoresc. 2011, 21, 1599–1605. [DOI] [PubMed] [Google Scholar]
- 129. Kim J. H., Noh J. Y., Hwang I. H., Lee J. J., Kim C., Tetrahedron Lett. 2013, 54, 4001–4005. [Google Scholar]
- 130. Bin Ruan Y., Maisonneuve S., Xie J., Dye. Pigm. 2011, 90, 239–244. [Google Scholar]
- 131. Tsui Y.-K., Devaraj S., Yen Y.-P., Sens. Actuators B 2012, 161, 510–519. [Google Scholar]
- 132. Niu L.-Y., Zheng H.-R., Chen Y.-Z., Wu L.-Z., Tung C.-H., Yang Q.-Z., Analyst 2014, 139, 1389–1395. [DOI] [PubMed] [Google Scholar]
- 133. Kand D., Saha T., Talukdar P., Sens. Actuators B 2014, 196, 440–449. [Google Scholar]
- 134. Chen Y.-H., Tsai J.-C., Cheng T.-H., Yuan S.-S., Wang Y.-M., Biosens. Bioelectron. 2014, 56, 117–123. [DOI] [PubMed] [Google Scholar]
- 135. Hanani M., J. Cell. Mol. Med. 2012, 16, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Xu L., Xu Y., Zhu W., Yang C., Han L., Qian X., Dalton Trans. 2012, 41, 7212–7217. [DOI] [PubMed] [Google Scholar]
- 137. Huang C.-B., Li H.-R., Luo Y., Xu L., Dalton Trans. 2014, 43, 8102–8108. [DOI] [PubMed] [Google Scholar]
- 138. Wentland E. J., Stewart P. S., Huang C. T., McFeters G. A., Biotechnol. Prog. 1996, 12, 316–321. [DOI] [PubMed] [Google Scholar]
- 139. Millard P. J., Roth B. L., Thi H. P. T., Yue S. T., Haugland R. P., Appl. Environ. Microbiol. 1997, 63, 2897–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. The Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies (Eds.: I. Johnson, M. T. Z. Spence), Life Technologies, New York, 2010. [Google Scholar]
- 141. Hilderbrand S. A., Weissleder R., Curr. Opin. Chem. Biol. 2010, 14, 71–79. [DOI] [PubMed] [Google Scholar]
- 142. Binda F. S. M., Agostinelli T., Caironi M., Natali D., Sampietro M., Beverina L., Ruffo R., Org. Electron. 2009, 10, 1314–1319. [Google Scholar]
- 143. Bura T., Leclerc N., Fall S., Lévêque P., Heiser T., Retailleau P., Rihn S., Mirloup A., Ziessel R., J. Am. Chem. Soc. 2012, 134, 17404–17407. [DOI] [PubMed] [Google Scholar]
- 144. Buschmann V., Weston K. D., Sauer M., Bioconjugate Chem. 2003, 14, 195–204. [DOI] [PubMed] [Google Scholar]
- 145. Gómez-Hens A., Aguilar-Caballos M.., TrAC Trends Anal. Chem. 2004, 23, 127–136. [Google Scholar]
- 146. Yao Y.-S., Zhou Q.-X., Wang X.-S., Wang Y., Zhang B.-W., Adv. Funct. Mater. 2007, 17, 93–100. [Google Scholar]
- 147. Kotani M., Kikuta J., Klauschen F., Chino T., Kobayashi Y., Yasuda H., Tamai K., Miyawaki A., Kanagawa O., Tomura M., Ishii M., J. Immunol. 2013, 190, 605–612. [DOI] [PubMed] [Google Scholar]
- 148. Tynan C. J., Clarke D. T., Coles B. C., Rolfe D. J., Martín-Fernández M. L., Webb S. E. D., PLoS One 2012, 7, 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Mitronova G. Y., Belov V. N., Bossi M. L., Wurm C. A., Meyer L., Medda R., Moneron G., Bretschneider S., Eggeling C., Jakobs S., Hell S. W., Chem. Eur. J. 2010, 16, 4477–4488. [DOI] [PubMed] [Google Scholar]
- 150. Kessler F., Watanabe Y., Sasabe H., Katagiri H., Nazeeruddin M. K., Grätzel M., Kido J., J. Mater. Chem. C 2013, 1, 1070–1075. [Google Scholar]
- 151. Ni Y. Z. Y. R., Su H. Q., Huang W., Zhao J. F., Qian Y., Xie L. H., Xie G. H., Appl. Mech. Mater. 2013, 331, 503–507. [Google Scholar]
- 152. Rich R. M., Stankowska D. L., Maliwal B. P., Sørensen T. J., Laursen B. W., Krishnamoorthy R. R., Gryczynski Z., Borejdo J., Gryczynski I., Fudala R., Anal. Bioanal. Chem. 2013, 405, 2065–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Hong G., Lee J. C., Robinson J. T., Raaz U., Xie L., Huang N. F., Cooke J. P., Dai H., Nat. Med. 2012, 18, 1841–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Krasovitskii B. M., Bolotin B. M., Organic Luminescent Materials, VCH, Weinheim, 1988. [Google Scholar]
- 155. Patra A., Chandaluri C. G., Radhakrishnan T. P., Nanoscale 2012, 4, 343–359. [DOI] [PubMed] [Google Scholar]
- 156. Fery-Forgues S., Nanoscale 2013, 5, 8428–8442. [DOI] [PubMed] [Google Scholar]
- 157. Patonay G., Salon J., Sowell J., Strekowski L., Molecules 2004, 9, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Guo Z., Kim G.-H., Yoon J., Shin I., Nat. Protoc. 2014, 9, 1245–1254. [DOI] [PubMed] [Google Scholar]
- 159. Yang L., Li X., Qu Y., Qu W., Zhang X., Hang Y., Ågren H., Hua J., Sens. Actuators B 2014, 203, 833–847. [Google Scholar]
- 160. Berridge M. J., Lipp P., Bootman M. D., Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [DOI] [PubMed] [Google Scholar]
- 161. Zhu B., Jia H., Zhang X., Chen Y., Liu H., Tan W., Anal. Bioanal. Chem. 2010, 397, 1245–1250. [DOI] [PubMed] [Google Scholar]
- 162. Boens N., Leen V., Dehaen W., Chem. Soc. Rev. 2012, 41, 1130–1172. [DOI] [PubMed] [Google Scholar]
- 163. Ozlem S., Akkaya E. U., J. Am. Chem. Soc. 2009, 131, 48–49. [DOI] [PubMed] [Google Scholar]
- 164. Ziessel R., Ulrich G., Harriman A., New J. Chem. 2007, 31, 496–501. [Google Scholar]
- 165. Bouit P. A., Kamada K., Feneyrou P., Berginc G., Toupet L., Maury O., Andraud C., Adv. Mater. 2009, 21, 1151–1154. [Google Scholar]
- 166. Lee C. Y., Hupp J. T., Langmuir 2010, 26, 3760–3765. [DOI] [PubMed] [Google Scholar]
- 167. Hesari M., Lu J.-S., Wang S., Ding Z., Chem. Commun. 2015, 51, 1081–1084. [DOI] [PubMed] [Google Scholar]
- 168. Ulrich G., Ziessel R., Harriman A., Angew. Chem. Int. Ed. 2008, 47, 1184–1201; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1202–1219. [Google Scholar]
- 169. Thivierge C., Bandichhor R., Burgess K., Org. Lett. 2007, 9, 2135–2138. [DOI] [PubMed] [Google Scholar]
- 170. Umezawa K., Nakamura Y., Makino H., Citterio D., Suzuki K., J. Am. Chem. Soc. 2008, 130, 1550–1551. [DOI] [PubMed] [Google Scholar]
- 171. Wu D., O'Shea D. F., Org. Lett. 2013, 15, 3392–3395. [DOI] [PubMed] [Google Scholar]
- 172. Palao E., Agarrabeitia A. R., Bañuelos-Prieto J., Lopez T. A., Lopez-Arbeloa I., Armesto D., Ortiz M. J., Org. Lett. 2013, 15, 4454–4457. [DOI] [PubMed] [Google Scholar]
- 173. Shao J., Sun H., Guo H., Ji S., Zhao J., Wu W., Yuan X., Zhang C., James T. D., Chem. Sci. 2012, 3, 1049–1061. [Google Scholar]
- 174. Hiruta Y., Koiso H., Ozawa H., Sato H., Hamada K., Yabushita S., Citterio D., Suzuki K., Org. Lett. 2015, 17, 3022–3025. [DOI] [PubMed] [Google Scholar]
- 175. Hirata T., Terai T., Komatsu T., Hanaoka K., Nagano T., Bioorganic Med. Chem. Lett. 2011, 21, 6090–6093. [DOI] [PubMed] [Google Scholar]
- 176. Fu L., Wang F.-F., Gao T., Huang R., He H., Jiang F.-L., Liu Y., Sens. Actuators B 2015, 216, 558–562. [Google Scholar]
- 177. Poirel A., Retailleau P., De Nicola A., Ziessel R., Chem. Eur. J. 2014, 20, 1252–1257. [DOI] [PubMed] [Google Scholar]
- 178. Zhu X., Huang H., Liu R., Jin X., Li Y., Wang D., Wang Q., Zhu H., J. Mater. Chem. C 2015, 3, 3774–3782. [Google Scholar]
- 179. Chen J.-B., Zhang H.-X., Guo X.-F., Wang H., Zhang H.-S., Anal. Bioanal. Chem. 2013, 405, 7447–7456. [DOI] [PubMed] [Google Scholar]
- 180. Krzeszewski M., Vakuliuk O., Gryko D. T., Eur. J. Org. Chem. 2013, 5631–5644. [Google Scholar]
- 181. Pashkova A., Chen H. S., Rejtar T., Zang X., Giese R., Andreev V., Moskovets E., Karger B. L., Anal. Chem. 2005, 77, 2085–2096. [DOI] [PubMed] [Google Scholar]
- 182. Signore G., Nifosì R., Albertazzi L., Storti B., Bizzarri R., J. Am. Chem. Soc. 2010, 132, 1276–1288. [DOI] [PubMed] [Google Scholar]
- 183. Ciga M., Donovalova J., Szo V., Gas J., Jakusova K., Ga A., J. Phys. Chem. A 2013, 117, 4870–4883. [DOI] [PubMed] [Google Scholar]
- 184. Khemakhem K., Soulié M., Brousses R., Ammar H., Abid S., Fery-Forgues S., Chem. Eur. J. 2015, 21, 7927–7937. [DOI] [PubMed] [Google Scholar]
- 185. Hou P., Chen S., Wang H., Wang J., Voitchovsky K., Song X., Chem. Commun. 2014, 50, 320–322. [DOI] [PubMed] [Google Scholar]
- 186. Ma D.-L., He H.-Z., Leung K.-H., Zhong H.-J., Chan D. S.-H., Leung C.-H., Chem. Soc. Rev. 2013, 42, 3427–3440. [DOI] [PubMed] [Google Scholar]
- 187. Biffi G., Tannahill D., McCafferty J., Balasubramanian S., Nat. Chem. 2013, 5, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Yan J.-W., Ye W.-J., Chen S.-B., Wu W.-B., Hou J.-Q., Ou T.-M., Tan J.-H., Li D., Gu L.-Q., Huang Z.-S., Anal. Chem. 2012, 84, 6288–6292. [DOI] [PubMed] [Google Scholar]
- 189. Yan J.-W., Chen S.-B., Liu H.-Y., Ye W.-J., Ou T.-M., Tan J.-H., Li D., Gu L.-Q., Huang Z.-S., Chem. Commun. 2014, 50, 6927–6930. [DOI] [PubMed] [Google Scholar]
- 190. Schäfer F. P., Ed., Dye Lasers, Springer, Berlin, 1973. [Google Scholar]
- 191. Kolmakov K., Belov V. N., Bierwagen J., Ringemann C., Müller V., Eggeling C., Hell S. W., Chem. Eur. J. 2010, 16, 158–166. [DOI] [PubMed] [Google Scholar]
- 192. Cardone R. A., Casavola V., Reshkin S. J., Nat. Rev. Cancer 2005, 5, 786–795. [DOI] [PubMed] [Google Scholar]
- 193. Sun R., Liu X.-D., Xun Z., Lu J.-M., Xu Y.-J., Ge J.-F., Sens. Actuators B 2014, 201, 426–432. [Google Scholar]
- 194. Nakanishi J., Nakajima T., Sato M., Ozawa T., Tohda K., Umezawa Y., Anal. Chem. 2001, 73, 2920–2928. [DOI] [PubMed] [Google Scholar]
- 195. Tang C., Zheng Q., Zong S., Wang Z., Cui Y., Sens. Actuators B 2014, 202, 99–104. [Google Scholar]
- 196. Bhosale S. V., Jani C. H., Langford S. J., Chem. Soc. Rev. 2008, 37, 331–342. [DOI] [PubMed] [Google Scholar]
- 197. Doria F., Oppi A., Manoli F., Botti S., Kandoth N., Grande V., Manet I., Freccero M., Chem. Commun. 2015, 51, 9105–9108. [DOI] [PubMed] [Google Scholar]
- 198. Bordeau G., Lartia R., Metge G., Fiorini-Debuisschert C., Charra F., Teulade-Fichou M.-P., J. Am. Chem. Soc. 2008, 130, 16836–16837. [DOI] [PubMed] [Google Scholar]
- 199. Xie X., Choi B., Largy E., Guillot R., Granzhan A., Teulade-Fichou M. P., Chem. Eur. 2013, 19, 1214–1226. [DOI] [PubMed] [Google Scholar]
- 200. Taki M., Ogasawara H., Osaki H., Fukazawa A., Sato Y., Ogasawara K., Higashiyama T., Yamaguchi S., Chem. Commun. 2015, 51, 11880–11883. [DOI] [PubMed] [Google Scholar]
- 201. Lin W., Mohandas B., Fontaine C. P., Colvin R. A., Biometals 2007, 20, 891–901. [DOI] [PubMed] [Google Scholar]
- 202. Lin W., Buccella D., Lippard S. J., J. Am. Chem. Soc. 2013, 135, 13512–13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Kim H. N., Ren W. X., Kim J. S., Yoon J., Chem. Soc. Rev. 2012, 41, 3210–3244. [DOI] [PubMed] [Google Scholar]
- 204. Lodeiro C., Capelo J. L., Mejuto J. C., Oliveira E., Santos H. M., Pedras B., Nuñez C., Chem. Soc. Rev. 2010, 39, 2948–2976. [DOI] [PubMed] [Google Scholar]
- 205. Moura N. M. M., Nuñez C., Santos S. M., Faustino M. A. F., Cavaleiro J. a. S., Neves M. G. P. M. S., Capelo J. L., Lodeiro C., ChemPlusChem 2013, 78, 1230–1243. [DOI] [PubMed] [Google Scholar]
- 206. Moura N. M. M., Núñez C., Santos S. M., Faustino M. A. F., Cavaleiro J. A. S., Almeida Paz F. A., Neves M. G. P. M. S., Capelo J. L., Lodeiro C., Chem. Eur. J. 2014, 20, 6684–6692. [DOI] [PubMed] [Google Scholar]
- 207. Moura N. M. M., Nunez C., Faustino M. A. F., Cavaleiro J. A. S., Neves M. G. P. M. S., Capelo J. L., Lodeiro C., J. Mater. Chem. C 2014, 2, 4772–4783. [Google Scholar]
- 208. Moura N. M. M., Núñez C., Santos S. M., Faustino M. A. F., Cavaleiro J. a. S., Neves M. G. P. M. S., Capelo J. L., Lodeiro C., Inorg. Chem. 2014, 53, 6149–6158. [DOI] [PubMed] [Google Scholar]
- 209. Han Z. X., Luo H. Y., Zhang X. B., Kong R. M., Shen G. L., Yu R. Q., Spectrochim. Acta Part A 2009, 72, 1084–1088. [DOI] [PubMed] [Google Scholar]
- 210. Santos C. I. M., Oliveira E., Barata J. F. B., Faustino M. A. F., Cavaleiro J. A. S., Neves M. G. P. M. S., Lodeiro C., J. Mater. Chem. 2012, 22, 13811–13819. [Google Scholar]
- 211. Santos C. I. M., Oliveira E., Barata J. F. B., Faustino M. A. F., Cavaleiro J. A. S., Neves M. G. P. M. S., Lodeiro C., Inorg. Chim. Acta 2014, 417, 148–154. [Google Scholar]
- 212. Santos C. I. M., Oliveira E., Menezes J. C. J. M. D. S., Barata J. F. B., Faustino M. A. F., Ferreira V. F., Cavaleiro J. A S., Neves M. G. P. M. S., Lodeiro C., Tetrahedron 2014, 70, 3361–3370. [Google Scholar]
- 213. Santos C. I. M., Oliveira E., Santos H. M., Menezes J. C. J. M. D. S., Faustino M. A. F., Cavaleiro J. A. S., Capelo J. L., Neves M. D. G. P. M. S., Lodeiro C., Photochem. Photobiol. Sci. 2015, 14, 757–764. [DOI] [PubMed] [Google Scholar]
- 214. Josefsen L. B., Boyle R. W., Theranostics 2012, 2, 916–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Wójcik J., Peszke J., Ratuszna A., Kuś P., Wrzalik R., Phys. Chem. Chem. Phys. 2013, 15, 19651–19658. [DOI] [PubMed] [Google Scholar]
- 216. MacDonald I. J., Dougherty T. J., J. Porphyrins Phthalocyanines 2001, 5, 105–129. [Google Scholar]
- 217. Dougherty T. J., Gomer C. J., Henderson B. W., Jori G., Kessel D., Korbelik M., Moan J., Peng Q., J. Natl. Cancer Inst. 1998, 90, 889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Benson D. M., Bryan J., Plant A. L., Gotto A. M., Smith L. C., J. Cell Biol. 1985, 100, 1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.“Fluorophores for Confocal Microscopy: Photophysics and Photochemistry”: Tsien A. S., Waggoner R. Y. in Handbook of Biological Confocal Microscopy, Plenum Press, New York, 1995. [Google Scholar]
- 220. Xia H., Li J., Zou G., Zhang Q., Jia C., J. Mater. Chem. A 2013, 1, 10713. [Google Scholar]
- 221. Han Y.-H., Tian C.-B., Li Q.-H., Du S.-W., J. Mater. Chem. C 2014, 2, 8065–8070. [Google Scholar]
- 222. He C., Lu K., Lin W., J. Am. Chem. Soc. 2014, 136, 5181–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Xu W., Zhou Y., Huang D., Su M., Wang K., Xiang M., Hong M., J. Mater. Chem. C 2015, 3, 2003–2015. [Google Scholar]
- 224. Parker D., Dickins R. S., Puschmann H., Crossland C., Howard J. A. K., Chem. Rev. 2002, 102, 1977–2010. [DOI] [PubMed] [Google Scholar]
- 225. Zhang X., Jiao Y., Jing X., Wu H., He G., Duan C., Dalton Trans. 2011, 40, 2522–2527. [DOI] [PubMed] [Google Scholar]
- 226. Chi Y., Chou P.-T., Chem. Soc. Rev. 2010, 39, 638–655. [DOI] [PubMed] [Google Scholar]
- 227. You Y., Nam W., Chem. Soc. Rev. 2012, 41, 7061–7084. [DOI] [PubMed] [Google Scholar]
- 228. Farinola G. M., Ragni R., Chem. Soc. Rev. 2011, 40, 3467–3482. [DOI] [PubMed] [Google Scholar]
- 229. Ma D.-L., Chan D. S.-H., Leung C., Acc. Chem. Res. 2014, 47, 3614–3631. [DOI] [PubMed] [Google Scholar]
- 230. Ma X., Jia J., Cao R., Wang X., Fei H., J. Am. Chem. Soc. 2014, 136, 17734–17737. [DOI] [PubMed] [Google Scholar]
- 231. Zhang S., Hosaka M., Yoshihara T., Negishi K., Iida Y., Tobita S., Takeuchi T., Cancer Res. 2010, 70, 4490–4498. [DOI] [PubMed] [Google Scholar]
- 232. Maggioni D., Galli M., D'Alfonso L., Inverso D., Dozzi M. V., Sironi L., Iannacone M., Collini M., Ferruti P., Ranucci E., D'Alfonso G., Inorg. Chem. 2015, 54, 544–553. [DOI] [PubMed] [Google Scholar]
- 233. Kando A., Hisamatsu Y., Ohwada H., Itoh T., Moromizato S., Kohno M., Aoki S., Inorg. Chem. 2015, 54, 5342–5357. [DOI] [PubMed] [Google Scholar]
- 234. Fischer L. H., Stich M. I. J., Wolfbeis O. S., Tian N., Holder E., Schäferling M., Chem. Eur. J. 2009, 15, 10857–10863. [DOI] [PubMed] [Google Scholar]
- 235. Balzani V., Juris A., Coord. Chem. Rev. 2001, 211, 97–115. [Google Scholar]
- 236. Schmittel M., Lin H.-W., Angew. Chem. Int. Ed. 2007, 46, 893–896; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 911–914. [Google Scholar]
- 237. Nazeeruddin M. K., Di Censo D., Humphry-Baker R., Grätzel M., Adv. Funct. Mater. 2006, 16, 189–194. [Google Scholar]
- 238. Boricha V. P., Patra S., Chouhan Y. S., Sanavada P., Suresh E., Paul P., Eur. J. Inorg. Chem. 2009, 1256–1267. [Google Scholar]
- 239. Berni E., Gosse I., Badocco D., Pastore P., Sojic N., Pinet S., Chem. Eur. J. 2009, 15, 5145–5152. [DOI] [PubMed] [Google Scholar]
- 240. Schmittel M., Lin H., J. Mater. Chem. 2008, 18, 333–343. [Google Scholar]
- 241. Ji S., Guo H., Yuan X., Li X., Ding H., Gao P., Zhao C., Wu W., Wu W., Zhao J., Org. Lett. 2010, 12, 2876–2879. [DOI] [PubMed] [Google Scholar]
- 242. McClenaghan N. D., Leydet Y., Maubert B., Indelli M. T., Campagna S., Coord. Chem. Rev. 2005, 249, 1336–1350. [Google Scholar]
- 243. Williams J. A. G., Wilkinson A. J., Whittle V. L., Dalton Trans. 2008, 2081. [DOI] [PubMed] [Google Scholar]
- 244. Schmittel M., Lin H., Inorg. Chem. 2007, 46, 9139–9145. [DOI] [PubMed] [Google Scholar]
- 245. Bard A. J., Electrogenerated Chemiluminescence, Marcel Dekker, New York, 2004. [Google Scholar]
- 246. Miao W., Chem. Rev. 2008, 108, 2506–2553. [DOI] [PubMed] [Google Scholar]
- 247. High B., Bruce D., Richter M. M., Anal. Chim. Acta 2001, 449, 17–22. [Google Scholar]
- 248. Il Kim J., Shin I.-S., Kim H., Lee J.-K., J. Am. Chem. Soc. 2005, 127, 1614–1615. [DOI] [PubMed] [Google Scholar]
- 249. Lin H., Cinar M. E., Schmittel M., Dalton Trans. 2010, 39, 5130–5138. [DOI] [PubMed] [Google Scholar]
- 250. Xie H.-Y., Liang J.-G., Liu Y., Zhang Z.-L., Pang D.-W., He Z.-K., Lu Z.-X., Huang W.-H., J. Nanosci. Nanotechnol. 2005, 5, 880–886. [DOI] [PubMed] [Google Scholar]
- 251. Zheng F.-F., Wu J.-F., Zhao G.-C., Anal. Methods 2012, 4, 3932–3936. [Google Scholar]
- 252. Kim G., Park S. B., Moon J.-H., Lee S., Anal. Methods 2013, 5, 5717–5723. [Google Scholar]
- 253. Joshi K. V., Joshi B. K., Harikrishnan U., Patel M. B., Menon S. K., Anal. Methods 2013, 5, 4973–4977. [Google Scholar]
- 254. Lidke D. S., Nagy P., Heintzmann R., Arndt-Jovin D. J., Post J. N., Grecco H. E., Jares-Erijman E. A., Jovin T. M., Nat. Biotechnol. 2004, 22, 198–203. [DOI] [PubMed] [Google Scholar]
- 255. Zhang Q., Li Y., Tsien R. W., Science 2009, 323, 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Dahan M., Lévi S., Luccardini C., Rostaing P., Riveau B., Triller A., Science 2003, 302, 442–445. [DOI] [PubMed] [Google Scholar]
- 257. Wang Y., Chen L., Nanomedicine: NBM 2011, 7, 385–402. [DOI] [PubMed] [Google Scholar]
- 258. Zamaleeva A. I., Collot M., Bahembera E., Tisseyre C., Rostaing P., Yakovlev A. V., Oheim M., de Waard M., Mallet J.-M., Feltz A., Nano Lett. 2014, 14, 2994–3001. [DOI] [PubMed] [Google Scholar]
- 259. Yu Y., Li R.-Y., Wu S.-S., Lin B.-X., Cao Y.-J., Hu X.-G., Wu J.-Z., Anal. Methods 2014, 6, 2331–2337. [Google Scholar]
- 260. Wang Y., Chen J., Irudayaraj J., ACS Nano 2011, 5, 9718–9725. [DOI] [PubMed] [Google Scholar]
- 261. Yu M., Zhou C., Liu J., Hankins J. D., Zheng J., J. Am. Chem. Soc. 2011, 133, 11014–11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Lu Y., Chen W., Chem. Soc. Rev. 2012, 41, 3594–3623. [DOI] [PubMed] [Google Scholar]
- 263. Ke C.-Y., Wu Y.-T., Tseng W.-L., Biosens. Bioelectron. 2015, 69, 46–53. [DOI] [PubMed] [Google Scholar]
- 264. Wu P., Yan X.-P., Chem. Soc. Rev. 2013, 42, 5489–5521. [DOI] [PubMed] [Google Scholar]
- 265. Zhang K., Yu T., Liu F., Sun M., Yu H., Liu B., Zhang Z., Jiang H., Wang S., Anal. Chem. 2014, 86, 11727–11733. [DOI] [PubMed] [Google Scholar]