

Abstract
Ischemic stroke is a complex brain injury caused by a thrombus or embolus obstructing blood flow to parts of the brain. This leads to deprivation of oxygen and glucose, which causes energy failure and neuronal death. After an ischemic stroke insult, astrocytes become reactive and proliferate around the injury site as it develops. Under this scenario, it is difficult to study the specific contribution of astrocytes to the brain region exposed to ischemia. Therefore, this article introduces a methodology to study primary astrocyte reactivity and proliferation under an in vitro model of an ischemia-like environment, called oxygen glucose deprivation (OGD). Astrocytes were isolated from 1-4 day-old neonatal rats and the number of non-specific astrocytic cells was assessed using astrocyte selective marker Glial Fibrillary Acidic Protein (GFAP) and nuclear staining. The period in which astrocytes are subjected to the OGD condition can be customized, as well as the percentage of oxygen they are exposed to. This flexibility allows scientists to characterize the duration of the ischemic-like condition in different groups of cells in vitro. This article discusses the timeframes of OGD that induce astrocyte reactivity, hypertrophic morphology, and proliferation as measured by immunofluorescence using Proliferating Cell Nuclear Antigen (PCNA). Besides proliferation, astrocytes undergo energy and oxidative stress, and respond to OGD by releasing soluble factors into the cell medium. This medium can be collected and used to analyze the effects of molecules released by astrocytes in primary neuronal cultures without cell-to-cell interaction. In summary, this primary cell culture model can be efficiently used to understand the role of isolated astrocytes upon injury.
Keywords: Neuroscience, Issue 128, Oxygen Glucose Deprivation (OGD), astrocytes, GFAP, Ischemic Stroke, in vitro model, rat, PCNA
Introduction
Stroke is defined as "an acute neurological dysfunction of vascular origin with either sudden or rapid development of symptoms and signs, corresponding to the involvement of focal areas in the brain"1,2. There are two types of stroke: hemorrhagic and ischemic. When vascular dysfunction is caused either by an aneurysm or an arteriovenous malfunction, accompanied by weakening with posterior rupture of an artery, this is termed hemorrhagic stroke3 which, in most cases, leads to death. When a thrombus or an embolus obstructs blood flow, causing a temporary deprivation of oxygen and glucose to a brain region, it is called ischemic stroke4. Failure to nourish cells around the affected area or ischemic core leads to a homeostatic and metabolic imbalance, energetic dysfunction, neuronal death, and inflammation5, which can induce a life-long disability for patients6.
Ischemic stroke is a multifactorial injury involving several types of cells that react and exert their effects at different time points. Many interactions create a difficult environment to study the behavior of individual cells. So, how do we study the contribution of a specific cell type under such a complex environment? An accepted in vitro model of ischemia consists of exposing cells to oxygen and glucose deprivation (OGD), for a certain period, followed by the restoration of cells to a normoxic environment. This system simulates an ischemic stroke followed by blood reperfusion. In this method, cells or tissues are exposed to a glucose-free media in an environment purged of oxygen, using a specialized hypoxic chamber. The OGD incubation time can vary from a few minutes up to 24 h, depending on the hypothesis that wants to be tested. Studies have shown that depending on the times of OGD and normoxic environment, specific phenotypes of stroke (i.e., acute or subchronic) can be achieved. Primary isolated astrocytes, exposed to OGD with posterior restoration to normoxic conditions, is a well-studied cellular model to mimic stroke in vitro7. Using OGD is possible to reveal the independent molecular mechanisms of isolated cells under a stroke-like environment.
As our knowledge of astrocyte biology increases, it has become evident that they are crucial for maintaining synapses and sustaining neural repair, development, and plasticity8. Under normal conditions, astrocytes release and respond to cytokines, chemokines, growth factors and gliotransmitters, keeping metabolic balance and homeostasis within synapses5,9. In acute neuroinflammation, such as ischemic stroke, these cells can become reactive, show a long-term overexpression of Glial Fibrillary Acidic Protein (GFAP), and show hypertrophy in their morphology5,10,11,12. As the ischemic infarct develops, the homeostasis provided by astrocytes becomes affected, regarding normal glutamate uptake, energy metabolism, exchange of active molecules, and antioxidant activity13.
Reactivated astrocytes proliferate around the infarct tissue while leukocytes migrate towards the lesioned area14. Astrocytic proliferation can be measured using markers such as proliferating cell nuclear antigen (PCNA), Ki67, and bromodeoxyuridine (BrdU)15. This proliferative response is generated in a time-dependent manner and it helps forming the glial scar, an array of irreversibly reactive astrocytes along the parenchyma of the damaged site after an injury9. One of the initial functions of this scar is to limit the immune cell extravasation from this area. However, studies have shown that the scar becomes a physical impediment for axons to extend, as they release molecules inhibiting axonal growth, and create a physical barrier preventing axons from extending around the injured area16. Nevertheless, there is scientific evidence showing that after a spinal cord injury, completely preventing glial scar formation can impair the regeneration of axons17. Thus, the context in which the specific astrocytic response is measured, must be considered upon the framework of the injury studied.
The presented methodology can be applied to study the individualized function of astrocytes after oxygen glucose deprivation and it can be modified depending on the questions that the investigator wants to answer. For example, besides the morphological change and the markers expressed at different OGD times, the supernatants from astrocytes exposed to OGD can be further analyzed to identify soluble factors released by these cells, or used as a conditioned media to assess its effect in other brain cells. This approach enables studies on astrocyte reactivity that could lead to the elucidation of the factors that govern and modulate their response in an ischemic-stroke scenario.
Protocol
Postnatal rats (Sprague Dawley) 1-4 days old are used to isolate cortices. The method of euthanasia is decapitation, as approved by NIH guidelines.
1. Preparation of Instruments and Materials for Surgery
Sterilize instruments in an autoclave (temperature: 121 ºC, pressure: 15 psi, time: 30 mins) using a steel box or an instant sealing sterilization pouch. See materials in Table of Materials.
2. Complete DMEM Preparation
In a one liter beaker containing 700 mL autoclaved water, add Dulbecco's Modified Eagle's Medium powder at room temperature.
Add 3.7 g/L sodium bicarbonate, while the solution is stirred with a magnetic stirrer.
Adjust the pH to 7.4, with autoclaved water bring volume up to 1 L, then filter. For culture purposes, supplement the desired volume of media with 10% FBS and 1% Penicillin/Streptomycin, and warm at 37 ˚C. See materials in Table of Materials. Note: The pH was adjusted to 7.4 via inserting the pH meter electrode into the media. The media must be stirring, then slowly add 1M NaOH using a dropper.
3. Primary Astrocyte Culture
Note: After seeding, cell media must be changed every three days and cells can be grown up to confluency (11-13 days). On the third day, tap the flask several times to lift microglial cells and oligodendrocyte progenitor cells off the culture, remove all the 'old' media, wash twice using 10 mL of PBS, aspirate the PBS, then add fresh new media. See materials in Table of Materials.
- Dissection of newborn rat brain
- Perform the dissection in a tissue culture hood. Obtain 1-4 days old newborn rat pups (2 rat brains are used per 75 cm2 flask).
- Prepare three 60 mm petri dishes and pipette 5 mL of complete DMEM on each. Note: They can be divided as follows: #1 for each rat brain, #2 for all the cortices of the brains, and #3 for all the peeled cortices (without meninges).
- Grasp the pup and spray with 70% ethanol. Decapitate it and discard the body in a small biohazard waste container.
- Using the micro-dissecting tweezers, hold the head and cut the skin on top of the head to expose the cranium.
- With another pair of scissors, make a "T" incision starting from the back of the skull towards the nose. Use a pair of forceps to gently remove the skull.
- Remove the exposed brain with a pair of tweezers and place the brain in one of the petri dishes previously filled with medium.
- Use another set of sterile instruments for the subsequent steps.
- Use micro-dissecting forceps to gently place one brain on the inverted lid of a 60 mm petri dish. When the petri dish is placed on sterile gauze, it is possible to view the brain clearly and it is easier to dissect.
- Steady the brain with micro-dissecting forceps and separate the cerebral hemisphere by gently teasing along the midline fissure with the sharp end of the micro-dissecting forceps.
- Deflect and peel the cortices, leaving behind the white matter, and transfer them to a petri dish, previously labeled #2. Repeat the procedure for all the brains, combining all the dissected cortices in the petri dish labeled #2.
- Take out the hippocampus from underneath each cortex and discard it.
- Use the micro-dissecting tweezers and a 60 mm petri to gently peel the meninges from the individual cortical lobes, and place them into the petri dish labeled #3.
- Tissue dissociation: homogenizer blender method
- Pour tissue/medium suspension (peeled cortices of the brain) into the sterile blender bag and add enough medium to bring the total volume in the bag to 5 mL.
- Place the bag into the homogenizer blender, leaving approximately 2 cm of the bag visible above the closed door. The cell dissociation is done during 2.5 min at high speed. Note: Alternatively, this can be done by closing the bag and gently pounding it with a beaker in the hood. Make sure to use a cloth in between the bag and beaker to avoid friction.
- Pour the cell suspension into a number 60 mesh sieve and then pour the filtered flow through onto the number 100 sieve, allowing it to filter by gravity.
- Using 15 mL tubes, centrifuge the cell suspension at 200 x g in a clinical centrifuge (preferably swing bucket rotor) for 5 min.
- Pour off the supernatant in a beaker, then using a serological pipette, re-suspend and dissociate the pellet of cells in a maximum of 5 mL of media.
- Cell counting
- With a micropipette, collect 100 µL of cell suspension in a 1.5 mL microcentrifuge tube and add 100 µL of trypan blue.
- Insert 10 µL of the collected suspension in the hemocytometer and count all the cells that are viable in the four corners of the square.
- The formulae for calculating the density of the cells before preparing the flasks are:
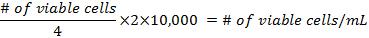
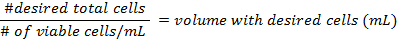
- Add at least 500,000 cells per 25 cm2 flask. Pipette up to 5 mL of DMEM into each flask. The mixture should not reach the neck of the flask. Place them in a 37 °C incubator at 5% CO2, for 3 days, then change the media.
- Astrocyte purification techniques: media change, mechanical, and chemical strategiesNote: The metabolic rate of astrocytes is high compared to other cell types, therefore, in nutritionally deprived conditions, these cells show low survival rates after a few days. Unlike astrocytes, microglia are not affected by nutritional deprivation and proliferate in such environments18. To maintain a reduced number of microglial cells, the authors change the astroglial cell culture media every 3 days. This constant change in cell media will also decrease the microglial population that can grow on top of the astrocytic cell monolayer in the flask19.
- Use a Pasteur pipette to aspirate the media with non-adherent microglial cells from each of the flasks. Wash any debris left behind using sterile-filtered PBS to each of the flasks in a dropwise manner (5 mL/flask).
- Add the sterile-filtered DMEM (with antibiotic and fetal bovine serum) dropwise to each flask (5 mL/flask). Gently agitate in a circular motion to cover the entire surface.
- Place cells in a 37 °C incubator at 5% CO2 for 8-10 days until the flasks reach confluency (>95% plate coverage). Note: Several protocols have shown that when astrocytes reach confluency , mechanical methods, such as shaking the culture from 2 to 24 h, leads to the detachment of cells that lie on top of these astrocytes, mainly microglia and precursor cells19,20.
- Non-astrocytic cells are reduced in culture by performing orbital shaking at 180 rpm for 30 min. After removing cells in supernatant, fresh media is pipetted into the flask (~15 mL).
- To eliminate the oligodendrocyte progenitor cells, increase shaking speed to 200 rpm for 6 h21,22,23 , aspirate supernatant, and pipet fresh media. Note: To decrease the presence of microglia in the cultures, two chemical methods can be used sequentially (not simultaneously) in the primary cell culture methodology: Arabinose cytosine (Ara-C) and L-leucine methyl ester (LME)24.
- Immediately when cells reach confluency (100%), 3-4 days after the mechanical removal of microglia, these cells can be treated with 8 µM of the antimitotic compound Arabinose cytosine (Ara-C) in DMEM for 5 days (change to fresh Ara-C in DMEM daily). Note: The time point to add Ara-C is critical since this compound is an antimitotic drug, that reduces the number of rapidly-dividing cells such as microglia19,25 and fibroblasts26. In contrast to microglia, when confluent, astrocytes stop proliferating via contact inhibition27.
- Allow 2 days for cells to recover from Ara-C treatment.
- To further reduce the microglial contamination, after cells reach confluency, use 50 mM of L-leucine methyl ester (LME) in DMEM fixed at pH 7.4, for 1 h at 37 ˚C. Note: LME crosses the membrane of cells and organelles, and after being hydrolyzed, the L-leucine produced accumulates in the lysosomes of these cells. The accumulation of this amino acid leads to an osmotic swelling and rupture of lysosomes28,29. LME is highly effective at eliminating microglia, compared to other cells20,30.
4. Cultivating Astrocytes in 6-Well Plates
- Coating coverslips with poly-D-lysine
- Put one coverslip in each 35 mm petri dish, then add 100 µL of the diluted Poly-D-Lysine solution to each coverslip and wait 1 h.
- Remove the Poly-D-Lysine and wash twice with 2 mL sterile water.
- Remove water and wait for coverslips to dry inside the hood. Store the coverslips at -20 °C for up to two weeks. Note: Freeze the diluted Poly-D-Lysine solution; it can be stored for up to two weeks.
- Trypsinization
- Using a Pasteur pipette, aspirate the medium from the flasks. Add 5 mL/flask of sterile-filtered PBS to gently rinse the cells.
- Aspirate off the PBS, and then add 5 mL/flask of sterile-filtered 0.25% Trypsin and 0.5 mM EDTA solution. Place the flask in the 37 ˚C, 5% CO2 incubator for 10 min. Note: While the cells are incubating, warm 5 mL/flask of fresh complete DMEM at 37 °C.
- After 10 min of incubation, agitate to make sure all the cells are lifted off the flask. Quickly add the 5 mL of warm complete DMEM to each flask, to neutralize the trypsin.
- Gently pipette the cells up and down several times to break up any "clumps". Transfer suspended cells from two flasks to a 15 mL conical centrifuge tube.
- Centrifuge for 5 min at 200 x g to collect the cell pellet. Re-suspend the pellet in 500 µL of medium and proceed to place cells in the flasks. Note: Examples of analyses to be made with cells: 1) Reverse transcription polymerase chain reaction (RT-PCR) to analyze gene expression; 2) Western blot to evaluate protein expression; 3) Flow cytometry analysis to quantify the number of neural progenitor cells and proteins of interest; 4) Immunofluorescence to analyze protein expression and localization.
- Seeding astrocytes
- After treating the primary astrocytes with trypsin (refer to 4.2), collect 100 μL of cell suspension in a 1.5 mL microcentrifuge tube and add 100 μL of 0.5% trypan blue.
- Add 10 μL of the collected suspension in trypan blue to the hemocytometer and count all the cells that are viable.
- Use the formulae in step 3.3 to calculate the density of the cells before preparing the flasks.
- Prepare the multi-well dishes by adding one sterile, poly-D-Lysine coated coverslip to each well. Pipette the 50,000 cells and fill the rest of the volume in each multi-well dish with fresh medium (approximately 2 mL).
- Place the multi-well dishes into the 37 ˚C incubator at 5% CO2 and, after 5-7 days, astrocytes may grow and cover the entire surface of the coverslips.
5. OGD Protocol for Primary Astrocytes
- OGD medium preparation
- Supplement Dulbecco's modified Eagle's medium-free glucose with 3.7 g/L sodium bicarbonate, and 1% Penicillin/Streptomycin. See materials in Table 4.
- Purge 20 mL of the glucose free medium by bubbling with 95% N2/5% CO2 for 10 min at a flow of 15 L/min (indicated with the flow meter) by inserting a serological pipette to the gas system in the medium. Note: After purging the medium of oxygen, adjust the pH at 7.4 (refer to note on step 2.1).
- Filter the purged glucose-free medium using a 50 mL tube filter system. Note: Make fresh OGD medium for every experiment, bubbling with 95% N2/5% CO2 before using, to ensure that cells are exposed to an environment purged of oxygen.
- Experimental procedure
- Inside the tissue culture hood, remove astrocyte media from previously prepared coverslips (refer to step 4.3.5) and wash with cellular PBS twice to remove any glucose on the cells.
- Add 2 mL of the OGD media to each well and place the multi-well dishes inside the hypoxia chamber with their lids off.
- Connect the gas mixture to the entrance valve and leave the exit valve open. Flush the chamber with the gas mixture of 95% N2/5% CO2, at 15 L/min for 5 min.
- Stop the gas flow, and first close the clamp on the exit valve, then close the clamp on the gas entrance valve tubing. Note: Ensure there is no gas leakage by covering the seal of the chamber with soapy water. If the seal is secure, no bubbles should form.
- Place the entire chamber into the cell culture incubator at 37 °C for 1 h or 6 h in OGD.
- After incubation time is over, aspirate the OGD media and carefully pipet 2 mL of complete DMEM to each 4.67 cm2 well for the normoxic process 24 h.
6. Protocol for Primary Astrocyte Immunofluorescence Preparation
Note: To assess astrocyte purity, different brain cell types such as neurons, microglia, and oligodendrocytes can be detected by immunofluorescence using different cell markers. Proliferation markers, PCNA, and propidium iodide (PI) can be used on primary astrocytes exposed to OGD followed by normoxic conditions. See materials in Table of Materials.
- Immunofluorescence preparationNote: This is a general immunofluorescence protocol, minor modifications can be performed to achieve an optimal signal (e.g., longer incubation periods, incubation temperature). Each marker from Table 1 can be used according to the manufacturer's protocol.
- To evaluate cell viability, cells can be incubated for 5 min at 37 °C with PI (5 µM in complete DMEM) and wash twice with 2 mL of PBS for 5 min. Cells that show PI staining are less viable. Note: Use the cells treated in step 5.2.6.
- To fix cells after PI staining, add 2 mL of 4% formalin per 4.67 cm2 well, for 20 min at 4 ˚C. For samples labeled with PCNA, the preferred fixing solution is methanol for 10 min at 4 ˚C. Caution: Formalin and methanol are toxic. Note: Always check specific antibody protocols from each company.
- Wash cells with 2 mL of PBS for 5 min and repeat this step 3 times.
- Incubate cells in blocking solution (0.5% non-ionic surfactant, 10% FBS in PBS) for 30 min at room temperature with gentle shaking.
- Wash the cells with 2 mL of PBS for 5 min, repeat wash 3 times.
- Incubate at 4 °C, overnight (16 h), with primary antibody (PCNA, GFAP) in a solution of 1% FBS in PBS.
- Remove the primary antibody solution, wash the cells with 2 mL of PBS for 5 min, and repeat this step 3 times.
- Incubate with secondary antibody conjugated with fluorophore in a solution of 1% FBS in PBS, for 1 h, at room temperature.
- Wash the cells with 2 mL of PBS for 5 min, repeat this step 3 times. Add 1 µg/mL of DAPI (4',6'-diamidino-2-phenylindole) for 5 min to stain cell nuclei.
- Wash cells with 2 mL of PBS 5 min, repeat this step twice and keep in PBS.
- Prepare the coverslips on a microscope slide using mounting medium.
- Confocal microscope
- After setting up the microscope, place the coverslip side down on the microscope stage.
- Using the software, select a laser beam at 543nm for GFAP-Cy3 conjugated antibody.
7. Cell Viability Assay
Trypsinize and quantify astrocytes by following steps 4.3 and 4.4.
Add 1x104 cells/well to 96-well plates and grow them to confluency.
Using the methodology from step 5, expose cultured 96-well plates to either normoxic 1 h OGD or 6 h OGD.
After OGD treatment, normoxic media is added to all cells for 24 h and viability is measured using MTT reagent assay (use as manufacturer's instructions indicate).
From a 5 mg/mL MTT solution, add 10% of the total volume to each well and incubate for 3 h at 37 ˚C with the MTT reagent.
Remove media and resuspend crystals using 200 µL dimethyl sulfoxide. Read the plate in a spectrophotometer at 570 nm. Note: A decreased absorbance relative to control cells will correlate with less viability.
Representative Results
One of the main concerns of primary astrocytic culture is the presence of other cells such as neurons, oligodendrocytes, fibroblasts, and microglia. In Figure 1, isolated cells from rat cortices had media changes every 3 days and were either untreated or treated with added LME for 1 h. 24 h later, cells were immunostained for GFAP and counterstained with DAPI. Untreated cells showed an average of 39% non-GFAP positive cells, while LME-treated cells showed 8%. These results show how LME treatment and media change effectively decreased non-GFAP positive cells by 4.75 fold, producing an enriched-astrocyte culture. To determine if 1 h or 6 h OGD affects astrocyte viability after this methodology, Figure 2 shows an MTT assay of astrocyte cultures, 24 h after the insult. No significant difference in viability was found in any condition, showing that both times can be used to study astrocyte reactivity. Cell proliferation is one of the effects triggered by OGD in astrocytes. Proliferating cell nuclear antigen (PCNA) in Figure 3 increased from 10% in normoxic cells, to 30% after 1 h OGD, showing the expected in vivo astrocytic response15. Finally, astrocytes were exposed to 6 h of OGD followed by 24 h normoxic conditions. After this period of time, immunofluorescence against GFAP was performed. OGD-exposed cells were compared to astrocytes under normoxic conditions. Astrocytes without OGD show the traditional stellate morphology (Figure 4A) while cells that underwent OGD clearly present the characteristic hypertrophy of reactive astrocytes (Figure 4B). This hypertrophy can be visualized in the corresponding differential interference contrast (DIC) image of cortical rat astrocytes under normoxic and OGD conditions (Figure 5).
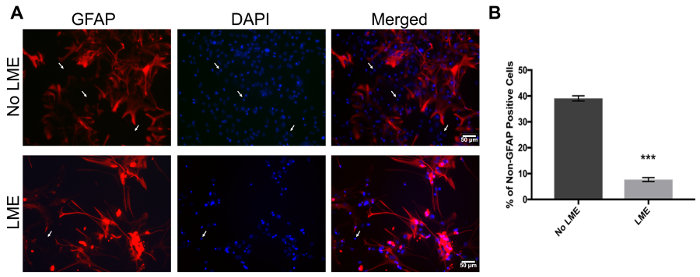
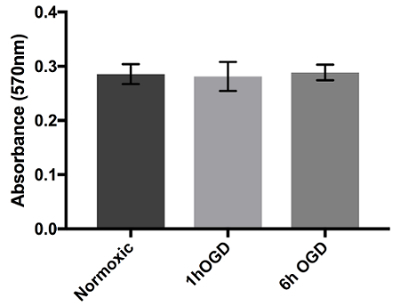
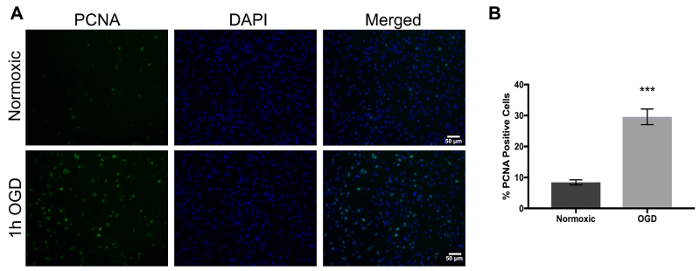
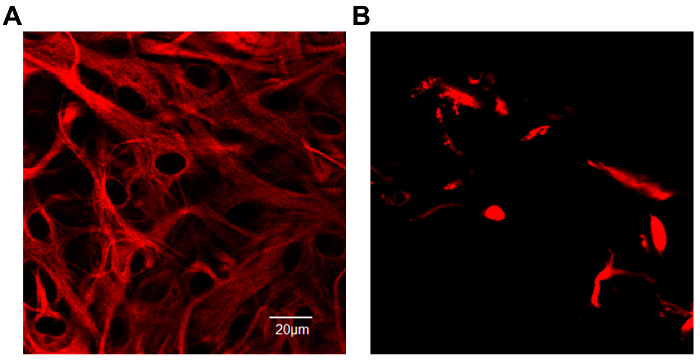
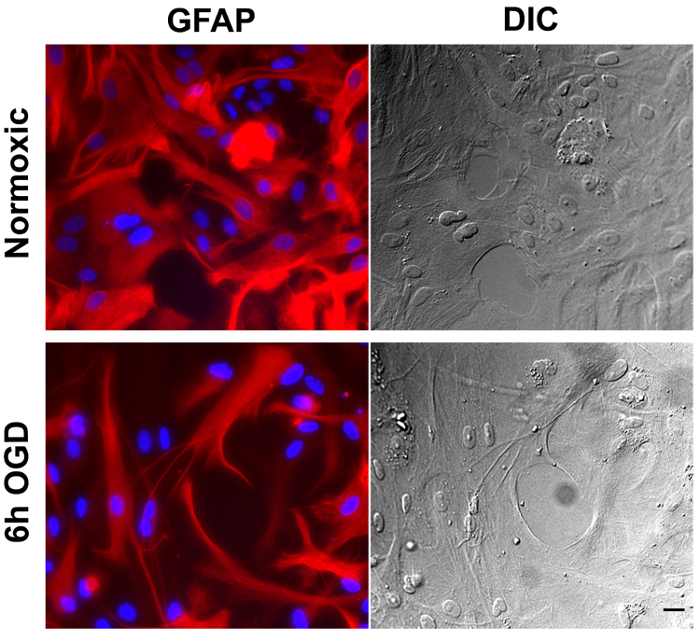
Figure 1. Immunofluorescence to validate primary cortical rat astrocytes culture purity with GFAP and DAPI. (A) The left panel represents GFAP positive cells (red), the middle panel shows nuclei (DAPI, blue), and the right panel shows merged images of non-LME-treated (upper panel) and LME-treated (lower panel) cells. White arrows show non-GFAP-positive stained cells. (B) Quantification of non-GFAP positive nuclei in LME and non-LME treated cells. An unpaired t-test was performed (p<0.0001) and the error bars represent the mean with SEM. Scale bars represent 50 microns and pictures were taken at 20x magnification. Please click here to view a larger version of this figure.
Figure 2. Viability assay of astrocytes with 24 h exposure to OGD. Astrocytes were cultured in 96-well plates and exposed to either normoxic, 1 h OGD, or 6 h OGD conditions. After treatment, cells were added to normoxic media for 24 h and viability was measured using MTT assay. Absorbance at 570 nm shows the cell viability of OGD-exposed cells in culture when compared to cells in normoxic conditions. Data was analyzed with one-way ANOVA (p=0.9669) and presented with error bars that represent the mean with SEM. Please click here to view a larger version of this figure.
Figure 3. Proliferation in rat astrocytes increases upon exposure to oxygen and glucose deprivation and can be detected using PCNA as a marker. (A) The left panel represents PCNA positive cells (green), the middle panel shows nuclei (DAPI, blue), and the right panel shows merged images of normoxic cells (upper panel) and 1 h OGD treated cells (lower panel). (B) Percent of PCNA positive cells in normoxic versus OGD treated cells. An unpaired-test was performed (p<0.0001) and the error bars represent the mean with SEM. Scale bars represent 50 microns and pictures were taken at 20x magnification. Please click here to view a larger version of this figure.
Figure 4. Expression of Glial Fibrillary Acidic Protein (GFAP) conjugated with Cy3 in primary rat cortical astrocyte culture. (A) Astrocytes in a normoxic environment show the characteristic stellate morphology. The intermediate filament protein, GFAP, fills the cell bodies and extends into the thin cytoplasmic processes. (B) After 6 h of OGD exposure astrocytes adopted a hypertrophic morphology. Scales represent 20 microns and pictures were taken at 40x magnification in a confocal microscope. Please click here to view a larger version of this figure.
Figure 5. Cortical rat astrocytes under normoxic and oxygen-glucose deprivation. Immunofluorescence of astrocytes under normoxic conditions (upper left panel) or 6 h OGD (lower left panel), stained using a Cy3-conjugated antibody against GFAP. The corresponding differential interference contrast (DIC) image is shown on the right panel. Scale bar represents 20 microns and pictures were taken at 20x magnification. Please click here to view a larger version of this figure.
| Purpose | Antibody/Marker | Description |
| Astrocyte Culture Validation | Iba-1 | Detects microglial cells |
| NeuN | Identifies mature neurons | |
| Olig1 | Detects mature oligodendrocytes | |
| Olig2, NG2 | Detects oligodendrocyte precursor cells | |
| GalC | Identifies immature or mature oligodendrocytes | |
| Proliferation | PCNA | Binds p36 protein, expressed at high levels in proliferating cells |
| Cell viability | Propidium iodide (PI) | Binds to the DNA, this marker indicates lack of cell viability |
| MTT | Measures mitochondrial functionality | |
| Reactivity | GFAP | Indicator of astrocyte reactivity |
Table 1. List of reagents used to stain various types of cells and cellular processes
Discussion
This protocol describes the isolation of astrocytes from rat cortices. In this method, it is critical to decrease contamination with other cellular types such as microglia, oligodendrocytes, and fibroblasts. To reduce the number of microglia, several steps can be taken: changing the media, orbital shaking, and chemical treatments. Once culture purity is confirmed by immunofluorescence using selective cellular markers or for the most prominent cell contaminants, experiments can be performed. For instance, antibody against ionized calcium binding adaptor molecule 1 (Iba-1) can be used to detect microglia. Neuronal death starts early in the culture and these cells are eliminated together with oligodendrocyte precursor cells (OPCs) after flask tapping and media change at day 331. Oligodendrocytic precursor cells can be identified using antibodies such as olig2 or NG2. GalC can be used to identify immature or mature oligodendrocytes, but at this culture stage these are unlikely to be found. Alternatively, non-GFAP positive cells can be quantified and contaminants can be estimated. This last methodology does not discern between the specific cells, but will be a faster and more economic methodology to determine the percentage of non-astrocytic cells in the culture.
Once an astrocyte enriched culture is produced, OGD experiments can be conducted to study astrocyte reactivity. Purging a solution with nitrogen by bubbling to substitute oxygen is a commonly used protocol32. In our methodology, we use the bubbling method to purge oxygen with nitrogen gas (95% N2, 5% CO2) into the glucose-free media at a flow rate of 15 L/min, for 10 min, for a total volume of 0.025 L. To confirm that the oxygen is purged, we measured the oxygen concentration remaining in the media by using an oxygen meter for liquids and air. The oxygen concentration remaining in the media after purging for 10 min decreased from 7.9 mg/L to 0.3 mg/L. To determine how long the chamber must be purged to replace oxygen, purging with N2 was performed using no less than the chamber manufacturer's suggestion (20 L/min, 2-5 min). After 5 min of purging at 15 L/min, the oxygen concentration remaining in the chamber was 0%. Other methodologies using similar chambers have purged at an 8-20 L/min flow rate, and times of 5-10 min to substitute air oxygen with nitrogen33,34.
Like other cellular models, the OGD has limitations and results should be interpreted carefully. The OGD can be harmful to neurons, however, astrocytes can withstand these conditions for up to 24 h35. Another limitation is that astrocytes are isolated in this system, lacking signaling from other cells. One way to overcome this limitation is to use conditioned media from other cells under the same ischemic-like conditions. Alternatively, cytokines or other factors can be added to the media after OGD36.
An alternative method to study the effects of energy deficiency in cells, is to add ouabain to the media. This compound mainly inhibits the sodium/potassium pump, which depletes the intracellular production of ATP, similar to the effects of OGD37,38. However, this method has the disadvantage that all its mechanisms are not fully understood and it induces off-target effects, which introduces variables which constitute an unrealistic ischemic stroke scenario.
In summary, the OGD method is an isolated cell system which allows us to directly test the effects of different astrocytic drug targets in vitro that could further contribute to neuroprotection. It also provides a tool to study basic astrocyte biology. This model can create a strong basis of evidence to justify experimental conditions for the development of drugs using an in vivo model of stroke.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors want to thank Paola López Pieraldi for the technical assistance. A.H.M. is grateful for the grants 8G12MD007600 and U54-NS083924 that supported this publication. We thank NIH-NIMHD-G12-MD007583 grant for the facility support. D.E.R.A. is grateful for the fellowship provided by NIHNIGMS-R25GM110513.We are grateful for the use of the Common Instrumentation Area and the aid of Dr. Priscila Sanabria for the use of the Optical Imaging Facility of the RCMI program by grant G12MD007583. In addition, we want to thank Jose Padilla for his outstanding role in filming and editing the visual protocol.
References
- Goldstein LB, Bertels C, Davis JN. Interrater reliability of the NIH stroke scale. Arch Neurol. 1989;46(6):660–662. doi: 10.1001/archneur.1989.00520420080026. [DOI] [PubMed] [Google Scholar]
- Hinkle JL, Guanci MM. Acute ischemic stroke review. J Neurosci Nurs. 2007;39(5):285–293. doi: 10.1097/01376517-200710000-00005. [DOI] [PubMed] [Google Scholar]
- Kassner A, Merali Z. Assessment of Blood-Brain Barrier Disruption in Stroke. Stroke. 2015;46(11):3310–3315. doi: 10.1161/STROKEAHA.115.008861. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Haim L, Carrillo-de Sauvage MA, Ceyzeriat K, Escartin C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci. 2015;9:278. doi: 10.3389/fncel.2015.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick J, et al. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage in Adults 2007 Update: A Guideline From the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38(6):2001–2023. doi: 10.1161/STROKEAHA.107.183689. [DOI] [PubMed] [Google Scholar]
- Wang R, et al. Oxygen-glucose deprivation induced glial scar-like change in astrocytes. PLoS One. 2012;7(5):e37574. doi: 10.1371/journal.pone.0037574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. The Neuroscientist. 2005;11(5):400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta neuropathologica. 2010;119(1):7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza DG, Bellaver B, Souza DO, Quincozes-Santos A. Characterization of adult rat astrocyte cultures. PLoS One. 2013;8(3):e60282. doi: 10.1371/journal.pone.0060282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puschmann TB, et al. HB-EGF affects astrocyte morphology, proliferation, differentiation, and the expression of intermediate filament proteins. J Neurochem. 2014;128(6):878–889. doi: 10.1111/jnc.12519. [DOI] [PubMed] [Google Scholar]
- Robinson C, Apgar C, Shapiro LA. Astrocyte Hypertrophy Contributes to Aberrant Neurogenesis after Traumatic Brain Injury. Neural Plast. 2016. p. 1347987. [DOI] [PMC free article] [PubMed]
- Brekke E, Berger HR, Wideroe M, Sonnewald U, Morken TS. Glucose and Intermediary Metabolism and Astrocyte-Neuron Interactions Following Neonatal Hypoxia-Ischemia in Rat. Neurochem Res. 2016. [DOI] [PubMed]
- Cekanaviciute E, et al. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 2014;62(8):1227–1240. doi: 10.1002/glia.22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, et al. Inhibiting cell cycle progression reduces reactive astrogliosis initiated by scratch injury in vitro and by cerebral ischemia in vivo. Glia. 2007;55(5):546–558. doi: 10.1002/glia.20476. [DOI] [PubMed] [Google Scholar]
- Bovolenta P, Wandosell F, Nieto-Sampedro M. Neurite outgrowth over resting and reactive astrocytes. Restor Neurol Neurosci. 1991;2(4):221–228. doi: 10.3233/RNN-1991-245609. [DOI] [PubMed] [Google Scholar]
- Anderson MA, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532(7598):195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao C, Richardson A, Fedoroff S. Macrophage-like cells originate from neuroepithelium in culture: characterization and properties of the macrophage-like cells. Int J Dev Neurosci. 1991;9(1):1–14. doi: 10.1016/0736-5748(91)90067-v. [DOI] [PubMed] [Google Scholar]
- Saura J. Microglial cells in astroglial cultures: a cautionary note. J Neuroinflammation. 2007;4:26. doi: 10.1186/1742-2094-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6(8):2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. J Vis Exp. 2013. [DOI] [PMC free article] [PubMed]
- Armstrong RC. Isolation and characterization of immature oligodendrocyte lineage cells. Methods. 1998;16(3):282–292. doi: 10.1006/meth.1998.0685. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85(3):890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont-Lezica L, Colasse S, Bessis A. Depletion of microglia from primary cellular cultures. Methods Mol Biol. 2013;1041:55–61. doi: 10.1007/978-1-62703-520-0_7. [DOI] [PubMed] [Google Scholar]
- Svensson M, Aldskogius H. Synaptic density of axotomized hypoglossal motorneurons following pharmacological blockade of the microglial cell proliferation. Exp Neurol. 1993;120(1):123–131. doi: 10.1006/exnr.1993.1046. [DOI] [PubMed] [Google Scholar]
- Wong VK, Shapourifar-Tehrani S, Kitada S, Choo PH, Lee DA. Inhibition of rabbit ocular fibroblast proliferation by 5-fluorouracil and cytosine arabinoside. J Ocul Pharmacol. 1991;7(1):27–39. doi: 10.1089/jop.1991.7.27. [DOI] [PubMed] [Google Scholar]
- Nakatsuji Y, Miller RH. Density dependent modulation of cell cycle protein expression in astrocytes. J Neurosci Res. 2001;66(3):487–496. doi: 10.1002/jnr.1240. [DOI] [PubMed] [Google Scholar]
- Reeves JP. Accumulation of amino acids by lysosomes incubated with amino acid methyl esters. J Biol Chem. 1979;254(18):8914–8921. [PubMed] [Google Scholar]
- Thiele DL, Kurosaka M, Lipsky PE. Phenotype of the accessory cell necessary for mitogen-stimulated T and B cell responses in human peripheral blood: delineation by its sensitivity to the lysosomotropic agent, L-leucine methyl ester. J Immunol. 1983;131(5):2282–2290. [PubMed] [Google Scholar]
- Hamby ME, Uliasz TF, Hewett SJ, Hewett JA. Characterization of an improved procedure for the removal of microglia from confluent monolayers of primary astrocytes. J Neurosci Methods. 2006;150(1):128–137. doi: 10.1016/j.jneumeth.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Corvalan V, Cole R, de Vellis J, Hagiwara S. Neuronal modulation of calcium channel activity in cultured rat astrocytes. Proc Natl Acad Sci U S A. 1990;87(11):4345–4348. doi: 10.1073/pnas.87.11.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler IB, Schoonen MA, Rickard DT. Removal of dissolved oxygen from water: A comparison of four common techniques. Talanta. 1994;41(2):211–215. doi: 10.1016/0039-9140(94)80110-x. [DOI] [PubMed] [Google Scholar]
- Tasca CI, Dal-Cim T, Cimarosti H. In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol Biol. 2015;1254:197–210. doi: 10.1007/978-1-4939-2152-2_15. [DOI] [PubMed] [Google Scholar]
- Wu D, Yotnda P. Induction and testing of hypoxia in cell culture. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Rivera-Aponte D, et al. Hyperglycemia reduces functional expression of astrocytic Kir4. 1 channels and glial glutamate uptake. Neuroscience. 2015;310:216–223. doi: 10.1016/j.neuroscience.2015.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger R, Garnier Y, Pfeiffer D, Jensen A. Lipopolysaccharides do not alter metabolic disturbances in hippocampal slices of fetal guinea pigs after oxygen-glucose deprivation. Pediatric research. 2000;48(4):531–535. doi: 10.1203/00006450-200010000-00018. [DOI] [PubMed] [Google Scholar]
- Anderson TR, Jarvis CR, Biedermann AJ, Molnar C, Andrew RD. Blocking the anoxic depolarization protects without functional compromise following simulated stroke in cortical brain slices. Journal of neurophysiology. 2005;93(2):963–979. doi: 10.1152/jn.00654.2004. [DOI] [PubMed] [Google Scholar]
- Jarvis CR, Anderson TR, Andrew RD. Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cerebral Cortex. 2001;11(3):249–259. doi: 10.1093/cercor/11.3.249. [DOI] [PubMed] [Google Scholar]