

Publisher's Note: There is an Inside Blood Commentary on this article in this issue.
Key Points
FKBP12 suppresses hepcidin by interaction with the BMP receptor ALK2.
Disruption of FKBP12–ALK2 interaction increases hepcidin and renders the receptor responsive to the inflammatory ligand Activin A.
Abstract
The expression of the key regulator of iron homeostasis hepcidin is activated by the BMP-SMAD pathway in response to iron and inflammation and among drugs, by rapamycin, which inhibits mTOR in complex with the immunophilin FKBP12. FKBP12 interacts with BMP type I receptors to avoid uncontrolled signaling. By pharmacologic and genetic studies, we identify FKBP12 as a novel hepcidin regulator. Sequestration of FKBP12 by rapamycin or tacrolimus activates hepcidin both in vitro and in murine hepatocytes. Acute tacrolimus treatment transiently increases hepcidin in wild-type mice. FKBP12 preferentially targets the BMP receptor ALK2. ALK2 mutants defective in binding FKBP12 increase hepcidin expression in a ligand-independent manner, through BMP-SMAD signaling. ALK2 free of FKBP12 becomes responsive to the noncanonical inflammatory ligand Activin A. Our results identify a novel hepcidin regulator and a potential therapeutic target to increase defective BMP signaling in disorders of low hepcidin.
Visual Abstract
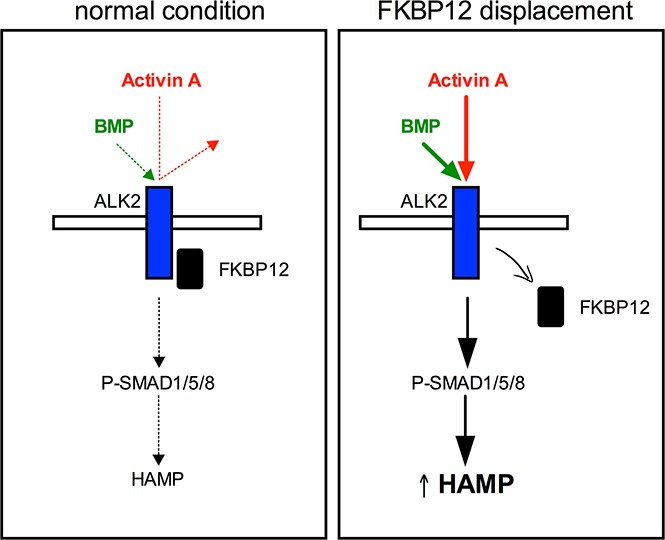
Introduction
Hepcidin, the key regulator of iron homeostasis, is a liver-secreted peptide that binds the sole cellular iron exporter ferroportin, triggering its internalization and degradation.1 Through this mechanism, hepcidin reduces circulating iron by blocking iron absorption by duodenal enterocytes and macrophage iron release. Because iron is essential for multiple cell functions such as energy production, DNA synthesis, metabolic pathways, and oxygen transport, hepcidin expression is tightly regulated in response to multiple stimuli such as body iron concentration, erythropoiesis, inflammation, gluconeogenesis, hormones, and drugs, including the mTOR inhibitor rapamycin.2
Hepcidin synthesis is mainly regulated by the BMP-SMAD pathway. In the presence of appropriate ligands, such as BMP2 for basal activation3 and BMP6 for the iron-dependent response,4-7 BMP type II receptors (BMPR-IIs), which are constitutively active, phosphorylate type I receptors (BMPR-Is). Type I receptors ALK2 and ALK3 have a crucial role8 in hepatocytes, whereas type II receptors (BMPR2 and ACVR2A) have a redundant function in hepcidin regulation.9 Activated type I receptors phosphorylate SMAD1/5/8 that, after SMAD4 binding, translocate to the nucleus as a multiprotein complex that interacts with the hepcidin promoter, inducing its expression.
Decreased production of hepcidin leads to iron overload. In hereditary hemochromatosis, defective hepcidin synthesis is caused by mutations in genes that regulate the liver BMP-SMAD pathway, as the BMP coreceptor hemojuvelin (HJV), hepcidin (HAMP) itself, or its activators TFR2 and HFE.10 In β-thalassemia, the expanded ineffective erythropoiesis, resulting from defective β-globin chain synthesis, downregulates hepcidin11 through the erythroid regulator erythroferrone.12
Rapamycin, an immunosuppressive drug that inhibits mTOR in complex with the immunophilin FK506-binding protein 1A (FKBP12), activates hepcidin in vitro2 through an unknown mechanism. Interestingly, patients treated with rapamycin may develop mild anemia and microcytosis,13 conditions compatible with high hepcidin, suggesting that mTOR might modulate hepcidin in vivo.
Here we investigate the molecular mechanism of hepcidin activation by rapamycin, showing that it is mediated by FKBP12, which interacts with BMPR-I14 to avoid uncontrolled activation of the pathway.15 In hepatocytes, FKBP12 preferentially binds ALK2. ALK2 mutants with impaired binding to FKBP12 constitutively activate the BMP-SMAD signaling and increase hepcidin in vitro in a ligand-independent manner. Tacrolimus (FK506), a drug that inhibits calcineurin in complex with FKBP12, activates hepcidin in vitro, ex vivo in primary hepatocytes, and in vivo in mice. Interestingly, genetic or pharmacologic FKBP12 displacement renders ALK2 responsive to Activin A, a transforming growth factor-β (TGF-β) ligand released in inflammation.
Our results clarify hematologic effects of rapamycin, identify a novel mechanism of hepcidin regulation, indicate FKBP12 as a potential target for disorders with insufficient hepcidin production, and suggest a possible contributory role for Activin A in hepcidin control in inflammation.
Methods
Detailed materials and methods are described in the supplemental Data on the Blood Web site.
Cell culture treatment
Rapamycin, Torin1, and cyclosporin A were from Tocris (Tocris Bioscience, Bristol, UK). Tacrolimus/FK506, GPI-1046 and LDN212854 were from Cayman Chemicals (Ann Arbor, MI). DMH1, β-cyclodextrin, and dorsomorphin were from Sigma-Aldrich (Milan, Italy). BMP2, BMP6, and Activin A were from R&D Systems (Minneapolis, MN). When indicated, cells were serum starved (2% or 0% fetal bovine serum) for 3 to 8 hours and treated with rapamycin (100 nM), Torin1 (100 nM), tacrolimus/FK506 (1 μg/mL), cyclosporin A (1 μg/mL), GPI-1046 (100 μg/mL), DMH1 (0.5 μg/mL), LDN212854 (150 nM) or dorsomorphin (10 μM). BMP2, BMP6, and Activin A were used at increasing concentrations: 0.1 to 10 ng/mL for BMP2 and Activin A and 0.1 to 100 ng/mL for BMP6.
Luciferase assay
Hepcidin promoter activation was studied by using pGL2-HAMP-Luc. SMAD1/5/8 and SMAD2/3 activation was measured by using pGL3-BMP-responsive element (BRE)-Luc and the pGL3-CAGA-Luc plasmids, respectively.16
qRT-PCR
Total RNA was extracted from cells and murine tissues with the UPzol reagent (Biotechrabbit, Düsseldorf, Germany) and complementary DNA (cDNA) was synthetized with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Framingham, MA). Gene expression levels were measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) using the SybrGreen or the TaqMan Gene Expression Master Mix (Applied Biosystems). Hprt1 or GAPDH were used as housekeeping genes. Primers for qRT-PCR are in supplemental Table 1. RNA expression data were plotted as mean (ΔCt values) ± standard deviation (SD) obtained after a change of origin, ie, after subtracting the mean ΔCt in untreated samples. Gene expression fold change was calculated as 2–ΔΔCt, where –ΔΔCt = mean (ΔCt)ut − mean(ΔCt)t (ut, untreated; t, treated).
Mice treatment
Wild-type (WT) C57BL/6N male mice (7 weeks old) were from Charles River. Mice were housed under a standard 12-hour light/dark cycle with water and chow ad libitum in a pathogen-free animal facility at San Raffaele Scientific Institute, in accordance with the European Union guidelines. The study was approved by the Institutional Animal Care and Use. A single dose of tacrolimus (10 mg/kg in dimethyl sulfoxide [DMSO]) was administered by subcutaneous injections. Mice were euthanized at 3, 6, 9, and 18 hours postinjection. Control mice were injected with DMSO and euthanized at 3 and 18 hours postinjection. Mice were anesthetized and then euthanized by cervical dislocation. Liver was dissected and immediately snap-frozen for RNA analysis. Liver and spleen samples were dried for iron quantification (supplemental Data).
Serum hepcidin quantification
Serum hepcidin was measured using the Hepcidin-Murine Compete enzyme-linked immunosorbent assay kit (Intrinsic Lifescience, La Jolla, CA), according to the manufacturer’s protocol and calculated against a standard curve using the 4-parameter logistic model (GraphPad Prism v7).
Statistical analysis
Data are shown as mean ± SD and compared with 2-tailed Student t test or 2-way analysis of variance (ANOVA). Statistical analyses were performed using Prism v5.
Results
The mTOR inhibitor rapamycin increases hepcidin expression activating the BMP-SMAD pathway
To investigate whether hepcidin activation by rapamycin was mTOR dependent, human hepatoma-derived cells were incubated with either mTOR complex 1 (mTORC1) inhibitor rapamycin, or Torin1, an adenosine triphosphate–competitive inhibitor of mTORC1 and mTORC2. Both drugs efficiently inhibit mTOR signaling, as demonstrated by decreased phosphorylation of the mTOR target S6 ribosomal protein (S6RP) (Figure 1A). Rapamycin upregulates endogenous HAMP expression in Hep3B cells (Figure 1B) and, as reported previously,2 in murine primary hepatocytes (Figure 1C), On the contrary, Torin1 is ineffective, suggesting that hepcidin modulation is not dependent on mTOR inhibition but, rather, a rapamycin-specific effect.
Figure 1.

Rapamycin upregulates hepcidin through activation of the BMP-SMAD pathway. (A) Hep3B cells, treated with rapamycin (RAPA, 100 nM) or Torin1 (T1, 100 nM) for 15 hours, were lysed and whole-cell extract loaded onto a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) for western blot analysis. mTOR activation was detected by following Ser240/244 and Ser235/236 phosphorylation of the mTOR target protein S6RP. Total S6RP and actin were analyzed for normalization of gel loading. Molecular weight markers are indicated on the left. A representative western blot, made in triplicate, is shown. (B,E) Hep3B cells were treated with RAPA or T1 as described in panel A. Total RNA was isolated and analyzed by qRT-PCR for hepcidin (HAMP) (B) and ID1 (E) expression. GAPDH was used as a housekeeping gene. A representative experiment, made in triplicate, is shown. (C) Primary murine hepatocytes were treated with RAPA (100 nM) or T1 (100 nM) for 5 hours. RNA was isolated and qRT-PCR was performed to analyze hepcidin (Hamp) expression. Hprt1 was used as housekeeping gene. A representative experiment, made in triplicate, is shown. Mean ΔCt values in each group were subjected to a change of origin by subtracting the mean ΔCt of vehicle-treated cells (B: 7.1; C: −0.83; E: 0.34). (D) Hep3B cells were transfected with the BRE-Luc reporter vector and treated with RAPA (100 nM) as described in panel A. Cells were lysed and analyzed for the luciferase activity that was normalized to an untreated mean value of 1. A representative experiment, made in triplicate, is shown. Error bars indicate SD. *P < .05; **P < .01; ***P < .001; ****P < .0001. Estimates of the fold changes in gene expression (2−ΔΔCt) are shown in the graphs. RQ, relative quantification; ns, not significant.
Next, we asked whether rapamycin modulates the BMP-SMAD pathway. Rapamycin treatment upregulates the luciferase activity in hepatoma cells transfected with the BRE-Luc vector (Figure 1D) that expresses the luciferase under the control of an element activated by the SMAD1/5/8-SMAD4 complex. In addition, rapamycin, but not Torin1, increases the endogenous expression of the BMP-SMAD target gene inhibitor of DNA binding 1 (ID1), both in Hep3B cells (Figure 1E) and in murine primary hepatocytes (data not shown). Overall, these data demonstrate that rapamycin increases hepcidin through the activation of the BMP-SMAD pathway.
Drugs targeting FKBP12 activate hepcidin through the BMP-SMAD pathway
To inhibit mTOR, rapamycin complexes with FKBP12, an immunophilin reported to interact with BMPR-I to avoid ligand-independent activation of the pathway.14,17,18 To explore the potential role of FKBP12 in hepcidin activation, we modulated FKBP12 binding by using tacrolimus (FK506), which interacts with the same FKBP12-binding pocket that binds rapamycin.19 Because tacrolimus exerts its immunosuppressive effect by inhibiting calcineurin, cells were also treated with cyclosporine A, a calcineurin inhibitor that acts through a different mechanism. Tacrolimus upregulates the luciferase activity of Hep3B cells transfected with the BRE-Luc vector, indicating a SMAD1/5/8-SMAD4–dependent signaling (Figure 2A). In agreement, both endogenous hepcidin (Figure 2B) and ID1 (Figure 2C) are upregulated in Hep3B cells treated with tacrolimus, but not in cells treated with cyclosporine A. A similar effect on hepcidin (Figure 2D) and Id1 (Figure 2E) is observed in murine primary hepatocytes, where the effect of cyclosporine A on hepcidin is even suppressive.
Figure 2.

Tacrolimus upregulates hepcidin through BMP-SMAD pathway activation. (A) Luciferase activity was analyzed in Hep3B cells transfected with the BRE-Luc reporter vector and treated with tacrolimus (TAC, 1 μg/mL) or vehicle for 15 hours. The luciferase activity of treated cells was normalized to an untreated mean value of 1. A representative experiment, made in triplicate, is shown. (B,C) Hep3B cells, treated with TAC (1 μg/mL), the calcineurin inhibitor cyclosporine A (CA, 1 μg/mL), or vehicle for 15 hours, were processed for RNA purification. Hepcidin (HAMP) and ID1 expression were quantified by qRT-PCR and normalized to the housekeeping gene GAPDH. A representative experiment, made in triplicate, is shown. (D,E) Murine primary hepatocytes were isolated and treated with TAC (1 μg/mL), CA (1 μg/mL), or vehicle for 18 hours. Hepcidin (Hamp) and Id1 expression were evaluated by qRT-PCR and normalized to the housekeeping gene Hprt1. Mean ΔCt values in each group were subjected to a change of origin by subtracting the mean ΔCt of vehicle-treated cells (B: 7.1; C: −3.0; D: 3.0; E: 4.2). A representative experiment, made in triplicate, is shown. Error bars indicate SD. *P < .05; **P < .01; ***P < .001; ****P < .0001. Estimates of the fold changes in gene expression (2−ΔΔCt) are shown in the graphs.
To further exclude any contribution of the immunosuppression on hepcidin regulation, we used GPI-1046. This synthetic compound interacts with FKBP12 but lacks immunosuppressive activity.20,21 GPI-1046 increases the luciferase activity in Hep3B cells transfected with both BRE-Luc (supplemental Figure 1A) and HAMP-Luc (supplemental Figure 1B) vectors, with the latter expressing the luciferase under the control of the hepcidin promoter.22 In addition, GPI-1046 upregulates endogenous hepcidin (supplemental Figure 1C) and Id1 (supplemental Figure 1D) expression in murine primary hepatocytes. Overall, these data indicate that FKBP12 sequestration is sufficient to activate the BMP-SMAD pathway and to increase hepcidin expression.
FKBP12 functionally interacts with the BMPR-I ALK2
Hepcidin activation is dependent on BMPR-I ALK2 and ALK3. ALK3 is crucial for basal activation of the pathway, whereas both ALK2 and ALK3 regulate hepcidin in response to iron and BMP ligands.8 To discriminate which of the 2 receptors binds FKBP12 to suppress hepcidin expression, we took advantage of the inhibitor DMH1 (4-(6-(4-isopropoxyphenyl)pyrazolo[1,5-a]pyrimidin-3-yl) quinolone), which blocks BMP signaling by mainly targeting the intracellular kinase domain of ALK2.23 If FKBP12 preferentially binds ALK2, its pharmacologic displacement in the presence of DMH1 will not activate hepcidin. On the contrary, if FKBP12 binds ALK3, DMH1 treatment will not interfere with hepcidin activation by rapamycin/tacrolimus. As shown in supplemental Figure 2, DMH1 strongly inhibits hepcidin upregulation by rapamycin (supplemental Figure 2A) and tacrolimus (supplemental Figure 2B), indicating that the effect is mediated by FKBP12 displacement from ALK2. These data were further confirmed by using the selective ALK2 inhibitor LDN212854.23 HuH7 cells were transfected with the HAMP-Luc reporter vector in the presence or absence of ALK2wt, the constitutively active form of ALK2, ALK2R206H, or ALK3, and luciferase activity was evaluated. Although LDN212854 is effective in inhibiting luciferase activation in cells overexpressing ALK2R206H, it does not interfere with the ALK3 activity, confirming its selectivity for ALK2 (supplemental Figure 2C). In agreement, coimmunoprecipitation experiments excluded a physical interaction between ALK3 and FKBP12 (supplemental Figure 2D). Moreover, LDN212854 selectively reduced the HAMP-Luc promoter activation in tacrolimus-treated HuH7, confirming that FKBP12 binds preferentially ALK2 in hepatoma-derived cells (supplemental Figure 2E).
To further demonstrate that FKBP12 binds ALK2, the human hepatoma cell line HuH7, transfected with ALK2wt and FKBP12, was treated with rapamycin, tacrolimus, or GPI-1046; the interaction between ALK2 and FKBP12 was assessed by immunoprecipitation. We observed a strong interaction between FKBP12 and ALK2 that is completely abrogated by tacrolimus and significantly reduced by rapamycin and GPI-1046 (Figure 3A). The different efficiency shown by the 3 drugs on FKBP12 displacement is probably related to their different half-life or FKBP12 affinity.
Figure 3.

Displacement of FKBP12 from ALK2 increases hepcidin through BMP-SMAD pathway activation. (A) HuH7 cells were transiently transfected with FKBP12MYC-FLAG and ALK2wt-MYC and treated with TAC (1 μg/mL), rapamycin (RAPA, 100 nM), GPI-1046 (100 μg/mL), or vehicle for 15 hours. Protein extracts were immunoprecipitated with an anti-FLAG M2 affinity gel (Sigma-Aldrich). Total extract and immunoprecipitated proteins were loaded onto a 12% SDS-PAGE and analyzed by western blot. ALK2 and FKBP12 were detected by using anti-MYC and anti-FKBP12 antibodies, respectively. Molecular weight markers are indicated on the right. (B) HuH7 cells were transfected with FKBP12MYC-FLAG in the presence of ALK2wt-MYC, ALK2R206H-MYC or ALK2Q207E-MYC, ALK2R258S-MYC, or empty vector. When indicated, cells were treated for 15 hours with BMP6 (100 ng/mL). Whole-cell extract was immunoprecipitated and analyzed as described in panel A. Molecular weight markers are indicated on the right. (C) HuH7 cells were transfected with the Smad1FLAG expressing vector in the presence of ALK2wt-MYC, ALK2R206H-MYC, ALK2Q207E-MYC, ALK2R258S-MYC, or empty vector (mock). When indicated, transfected cells were treated with BMP6 (50 ng/mL) for 30 minutes or 1.5 hours. Cells were lysed, loaded onto 10% SDS-PAGE, and analyzed by western blot. Activation of the BMP-SMAD pathway was detected by using an antibody recognizing phospho-SMAD1/5/8 and total SMAD1. ALK2 was detected by using anti-MYC antibody. Molecular weight markers are indicated on the right. (D,E) RNA was isolated from HuH7 cells transfected with ALK2wt-MYC-, ALK2R206H-MYC-, ALK2Q207E-MYC-, or ALK2R258S-MYC-expressing vector. Hepcidin (HAMP) (D) and ID1 (E) expression levels were quantified by qRT-PCR and normalized to the housekeeping gene GAPDH. Mean ΔCt values in each group were subjected to a change of origin by subtracting the mean ΔCt of ALK2wt-transfected cells (D: −1.2; E: −3.9). Estimates of the fold changes in gene expression (2−ΔΔCt) are shown on the graphs. (F) Hep3B cells were transfected with hepcidin promoter firefly luciferase reporter (HAMP-Luc) and increasing concentration of ALK2wt-MYC-FLAG-, ALK2R206H-MYC-FLAG-, ALK2Q207E-MYC-FLAG-, or ALK2R258S-MYC-FLAG-expressing vector. Cells transfected with the highest concentration of ALK2 cDNA were treated with dorsomorphin (DM, 10 μM). Cells were lysed and analyzed for the luciferase activity that was normalized to an untreated mean value of 1. (G) Hep3B cells were transfected with hepcidin promoter firefly luciferase reporter (HAMP-Luc), ALK2wt-MYC, ALK2R206H-MYC, ALK2R258S-MYC, or empty vector (mock) and increasing concentration of FKBP12. Luciferase activity was normalized to an untreated mean value of 1. (A-F) Representative results of experiments performed in triplicate. Error bars indicate SD. The 2-way ANOVA was used in panel F (ALK2 wt vs ALK2 mutants). *P < .05; **P < .01; ***P < .001; ****P < .0001. Western blot results are representative of 3 independent experiments.
We then replaced amino acid residues within (R206, Q207) or close to (R258) the glycine-serine-rich domain of ALK2, essential for FKBP12 binding.24,25 We introduced R206H, Q207E, and R258S ALK2 substitutions reported in patients affected by fibrodysplasia ossificans progressiva (OMIM #135100), a dominant disorder characterized by heterotopic ossification of soft tissues secondary to high and uncontrolled activity of the BMP-SMAD pathway.17 These mutations are partially resistant to the suppressive effect of FKBP12.26 Both R206H and Q207E have defective binding to FKBP12, and R258S fails to interact with the immunophilin (Figure 3B). To further investigate the FKBP12 binding ability of ALK2, Hep3B cells, transfected with the HAMP-Luc vector and WT or mutants ALK2, were treated with increasing concentration of tacrolimus to displace FKBP12. We expected a reduced effect of tacrolimus in mutants ALK2 because of the low amount of associated FKBP12. The fold change of hepcidin activation by tacrolimus is lower in cells transfected with mutants than with WT ALK2, suggesting that the former have a reduced FKBP12 binding capacity (supplemental Figure 3A). Consistent with this finding, SMAD1/5/8 phosphorylation is higher in cells transfected with mutants than with WT ALK2 (Figure 3C). This experiment confirms that defective FKBP12-ALK2 interaction increases BMP-SMAD signaling, causing hepcidin (Figure 3D) and ID1 (Figure 3E) upregulation. Activation of hepcidin by ALK2 mutants is dose-dependent and is blunted by the BMP-SMAD pathway inhibitor dorsomorphin (Figure 3F). Interestingly, WT ALK2 overexpression in hepatoma-derived cells does not activate hepcidin, even at high concentrations, suggesting that endogenous FKBP12 blocks ALK2 activity in the absence of ligands (data not shown).
To further confirm the role of FKBP12 as a modulator of ALK2 and hepcidin expression, Hep3B cells were transfected with the HAMP-Luc vector, WT or mutant ALK2, and increasing concentrations of FKBP12. As shown in Figure 3G, overexpression of FKBP12 partially inhibits the activity of ALK2 mutants on hepcidin promoter, whereas it has no effect on WT ALK2, suggesting that in the absence of the ligand ALK2 is likely blocked by endogenous FKBP12. Because Hep3B cells express endogenous ALK2 and ALK3, these results indirectly confirm that FKBP12 acts preferentially through ALK2 binding.
Hemojuvelin is dispensable for FKBP12-dependent hepcidin regulation
The main activator of hepcidin, with an in vivo role, is the BMP coreceptor HJV. Inactivation of HJV causes juvenile hemochromatosis in humans and severe iron overload in mice resulting from low hepcidin levels. To investigate whether HJV is essential for FKBP12-dependent hepcidin regulation, murine hepatocytes isolated from Hjv knockout (HjvKO) mice were treated with rapamycin or tacrolimus or GPI-1046. All drugs upregulate hepcidin, demonstrating that the BMP-coreceptor Hjv is dispensable for hepcidin activation mediated by loss of FKBP12-ALK2 interaction (Figure 4A). Irrespective of the different basal hepcidin levels the fold change of hepcidin activation by tacrolimus is the same in WT and HjvKO hepatocytes (Figure 4B). Activation of hepcidin by tacrolimus decreases at the highest concentrations (Figure 4C), likely because calcineurin inhibits hepcidin in both WT (Figure 2D) and HjvKO hepatocytes (Figure 4A).
Figure 4.

The BMP coreceptor HJV is dispensable for FKBP12-dependent hepcidin activation. (A) Primary hepatocytes from Hjv KO mice were isolated and treated for 18 hours with rapamycin and Torin1 (RAPA 100 nM, red bar, and T1 100 nM, light red bar respectively), TAC (1 μg/mL, blue bar) and CA (1 μg/mL, light blue bar) and GPI-1046 (100 μg/mL, green bar). RNA was isolated and hepcidin (Hamp) levels measured by qRT-PCR. Hprt1 was used as housekeeping gene. Mean ΔCt values in each group have been subjected to a change of origin by subtracting the mean ΔCt of vehicle-treated hepatocytes [vehicle(1): 5.1; vehicle(2): 5.6; vehicle(3): 3.7]. (B) TAC-dependent Hamp increase was evaluated in WT and Hjv KO hepatocytes. Mean ΔCt values in each group were subjected to a change of origin by subtracting the mean ΔCt of vehicle-treated hepatocytes (WT, 1.7; Hjv KO, 4.1). A representative experiment, made in triplicate, is shown. (C) Primary hepatocytes from WT and Hjv KO mice were isolated and treated with increasing concentrations of TAC. Hamp ΔCt values in each group were subjected to a change of origin by subtracting the mean ΔCt of vehicle-treated WT hepatocytes, as in panel B. A representative experiment, made in triplicate, is shown. Error bars indicate SD. *P < .05; **P < .01; ***P < .001; ****P < .0001. Estimates of the fold changes in gene expression (2−ΔΔCt) are shown in the graphs.
Acute tacrolimus treatment upregulates hepcidin in vivo
To explore whether FKBP12 is functional in vivo, adult C57BL/6N male WT mice were treated with a single subcutaneous injection of tacrolimus (10 mg/kg) or vehicle (DMSO) for different time points (from 3 to 18 hours) (Figure 5A). To normalize for variation of liver iron, we expressed hepcidin as the hepcidin/liver iron concentration ratio (hepcidin messenger RNA [mRNA] levels and liver iron concentration are shown in supplemental Figure 4A-B). Both hepcidin mRNA (Figure 5B) and serum hepcidin (Figure 5C) were significantly increased by tacrolimus at 6 hours postinjection. This effect was accompanied by an increased spleen iron content (Figure 5D) and a trend toward serum iron reduction (supplemental Figure 4C), pointing out that the drug modulates hepcidin and iron homeostasis in vivo, at least in an acute setting.
Figure 5.

Tacrolimus increases hepcidin expression in vivo. (A) Schematic representation of the experimental design: C57BL/6 WT male mice (n = 3-6 mice/group) were treated with vehicle or 10 mg/kg TAC via subcutaneous injection and sacrificed at different time points. (B) Liver hepcidin (Hamp) expression and (C) serum hepcidin were quantified by qRT-PCR, as described in supplemental Figure 4A, and competitive enzyme-linked immunosorbent assay, respectively. Hepcidin mRNA was normalized to the housekeeping gene Hprt1 and expressed as a ratio compared with total liver iron content (LIC). (D) Spleen iron content (SIC). Error bars indicate SD. *P < .05; **P < .01. Estimates of the fold changes in gene expression (2−ΔΔCt) are shown in the graphs.
FKBP12 regulates the ALK2 ligand responsiveness to Activin A
Because ALK2 is responsible for the ligand-dependent hepcidin activation,8 we investigated the response of WT and mutant ALK2 to different ligands in hepatoma cells. We tested BMP2, which is highly expressed in mouse liver endothelial cells and involved in hepcidin regulation,3 and BMP6, which is increased by liver iron content.
Mutant ALK2 activates hepcidin in a dose-dependent manner and with the same efficiency of WT ALK2 when stimulated with both BMP2 (Figure 6A) and BMP6 (Figure 6B), suggesting that mutations do not affect the hepcidin response to the iron-related ligands.
Figure 6.

ALK2-FKBP12–resistant mutants activate hepcidin through Activin A. Hep3B cells were transfected with the hepcidin promoter luciferase reporter vector (HAMP-Luc) and ALK2wt-MYC (purple line), ALK2R206H-MYC (red line), or ALK2R258S-MYC (green line) and treated for 15 hours with increasing concentrations of BMP2 (A), BMP6 (B), and Activin A (C). Cells were lysed and analyzed for the luciferase activity that was normalized to an untreated ALK2wt-MYC mean value of 1. Hep3B cells were transfected with the SMAD2/3 reporter vector (CAGA-Luc) (D) or the SMAD1/5/8 reporter vector (BRE-Luc) (E) in the presence of ALK2wt-MYC, ALK2R206H-MYC, or ALK2R258S-MYC. When indicated, cells were incubated for 15 hours with BMP6 (1 ng/mL) or Activin A (10 ng/mL). Luciferase activity was normalized to an untreated-ALK2wt-MYC mean value of 1. (F) SMAD1/5/8 phosphorylation was analyzed in HuH7 transfected with the Smad1FLAG-expressing vector and 5 or 10 μg of ALK2wt-MYC, ALK2R206H-MYC, or ALK2R258-MYC. When indicated, cells were treated for 15 hours with 10 ng/mL Activin A. Whole cell extract, loaded onto a 10% SDS-PAGE, was analyzed by western blot. SMAD1 and phospho-SMAD1/5/8 were detected using anti-SMAD1 and anti-phosphoSMAD1/5/8 antibodies. A representative western blot, made in triplicate, is shown. Molecular weight markers are indicated on the right. Error bars indicate SD. Two-way ANOVA was used in panels A-C (ALK2 wt vs ALK2 mutants). *P < .05; ***P < .001; ****P < .0001.
We also tested Activin A, a proposed ligand of ALK2 mutants in fibrodysplasia ossificans progressiva.24,25 As shown in Figure 6C, only mutant ALK2 upregulates hepcidin in response to Activin A, whereas WT ALK2 is unresponsive.
To investigate the signaling pathway of hepcidin regulation induced by Activin A, we analyzed both the TGF-β and the BMP-SMAD pathways by measuring the luciferase activity under the control of CAGA and BRE responsive elements, respectively. In basal conditions, the TGF-β pathway activation is comparable among ALK2wt-, ALK2R206H-, and ALK2R258S-transfected cells and is increased by Activin A with the same efficiency in both WT and mutant ALK2 (Figure 6D). The BMP-SMAD signaling increased in cells transfected with mutants ALK2 and, following BMP6 treatment, remains unchanged in WT ALK2 transfected cells treated with Activin A (Figure 6E). On the contrary, the BRE-Luc activity increases in cells transfected with mutants ALK2 in the presence of Activin A (Figure 6E). As further confirmation of this finding, SMAD1/5/8 phosphorylation level, high in cells overexpressing mutants ALK2, is further increased by Activin A (Figure 6F), and hepcidin activation is strongly decreased by DMH1 (supplemental Figure 3B). Thus Activin A upregulates the BMP-SMAD pathway only in the presence of mutants ALK2 that show impaired binding and are resistant to the effect of FKBP12. To investigate whether modulation of the FKBP12-ALK2 interaction may affect the receptor responsiveness, Hep3B cells transfected with ALK2wt were treated with tacrolimus to displace FKBP12 and with increasing concentrations of Activin A. In this case, ALK2wt becomes responsive to Activin A, increasing the HAMP-Luc activity (Figure 7A). The effect of tacrolimus on Activin A response is maintained in primary murine hepatocytes (Figure 7B) and is mediated by the BMP-SMAD pathway, as shown by increased Id1 expression in primary hepatocytes (supplemental Figure 5A) and by upregulation of the BRE-Luc activity in Hep3B cells (Figure 7C). As expected, the TGF-β signaling, measured by CAGA-Luc activity, is increased by Activin A and unaffected by tacrolimus (Figure 7D).
Figure 7.

Pharmacologic displacement of FKBP12 from ALK2 leads to Activin A–dependent SMAD1/5/8 activation and hepcidin upregulation. (A) Hep3B cells, transfected with the hepcidin promoter luciferase reporter vector (HAMP-Luc) and ALK2wt-MYC-expressing vector, were pretreated with 1 μg/mL TAC or vehicle for 3 hours and then treated with increasing concentrations of Activin A, in the presence or absence of TAC, for 15 hours. Cells were lysed and analyzed for the luciferase activity that was normalized to an untreated mean value of 1. (B) Primary murine hepatocytes from WT mice were pretreated for 3 hours with 1 μg/mL TAC and incubated for 5 hours with Activin A (10 ng/mL) in the presence or absence of TAC. Hepcidin (Hamp) mRNA expression was quantified by qRT-PCR and normalized to the housekeeping gene Hprt1. Mean ΔCt values in each group were subjected to a change of origin by subtracting the mean ΔCt (−1.2) of untreated cells. A representative experiment, made in triplicate, is shown. SMAD1/5/8 and SMAD2/3 signaling pathways were analyses in Hep3B cells transfected with the BRE-Luc (C) and the CAGA-Luc (D) reporter vectors. Transfected cells were incubated with TAC (1 μg/mL) ± Activin A (10 ng/mL) and then lysed for analysis of the luciferase activity that was normalized to an untreated mean value of 1. A representative experiment, made in triplicate, is shown. (E) Murine primary hepatocytes were pretreated with 100 nM rapamycin (RAPA) for 3 hours and treated with Activin A (10 ng/mL) for 5 hours, in the presence or absence of RAPA. As control, cells were treated with Torin1 (T1, 100 nM). Cells were processed and analyzed as described in panel B. Mean ΔCt values in each group were subjected to a change of origin by subtracting the mean ΔCt (7.1) of untreated cells. (F) Hep3B cells, transfected with the hepcidin promoter luciferase reporter vector (HAMP-Luc), were treated for 15 hours with increasing concentration of BMP6 in presence or absence of 10 ng/mL of Activin A. Luciferase activity was analyzed and normalized to an untreated mean value of 1. A representative experiment, made in triplicate, is shown. Two-way ANOVA was used in panels A,B,F (ALK2 wt vs ALK2 mutants). **P < .01; ***P < .001; ****P < .0001. Estimates of the fold changes in gene expression (2−ΔΔCt) are shown in the graphs.
The change of the ligand responsiveness is due to FKBP12 sequestration, because endogenous hepcidin (Figure 7E) and Id1 (supplemental Figure 5B) expression is further enhanced in murine primary hepatocytes treated with rapamycin and Activin A. In addition, the acquisition of Activin A responsiveness is independent from mTOR and calcineurin because it also occurs in cells treated with GPI-1046 (supplemental Figure 5C) through the BMP-SMAD pathway activation (supplemental Figure 5D). Overall, these results indicate that FKBP12 binding to ALK2 modulates the Activin A responsiveness of the receptor.
BMP6 synergizes with Activin A to activate hepcidin
Because BMP6 is upregulated by iron, we asked how the ALK2-FKBP12 interaction is regulated in this context. HuH7 cells, transfected with ALK2 and FKBP12, were treated with BMP6-increasing concentration and their interaction studied by immunoprecipitation. FKBP12 binding to ALK2 is reduced in the presence of BMP6 (Figure 3B), suggesting that phosphorylation of ALK2 induced by the ligand negatively interferes with FKBP12 binding. In agreement, Activin A synergizes with high BMP6 and further increases hepcidin activation (Figure 7F). Overall, these results suggest that high levels of BMP6, as in iron overload, favor Activin A responsiveness of ALK2.
Discussion
Here we characterize a new level of hepcidin regulation contributed by FKBP12 binding to the BMPR-I ALK2 in hepatocytes, a finding that provides novel insights into the control of systemic iron homeostasis and a potential pharmacologic target for treatment of iron overload/low hepcidin disorders.
Our study started by analyzing the effect of rapamycin on hepcidin expression. To be functional, rapamycin interacts with FKBP12, a peptidyl-prolyl-cis-trans cytosolic isomerase that belongs to the immunophilin superfamily. FKBP12 targets mTOR and calcineurin in complex with rapamycin and tacrolimus, respectively, with an immunosuppressive effect in both cases. It also modulates other signaling pathways,14,27-31 including BMPRs-I.17,18 FKBP12 binds to their glycine-serine–rich domain to avoid uncontrolled activation of the pathway.
Here we demonstrate that hepcidin is activated by rapamycin and by other compounds that bind and sequester the immunophilin independently from mTOR or calcineurin inhibition. We show that efficient upregulation of hepcidin expression by BMPR ligands requires the disruption of FKBP12-ALK2 interaction. In agreement, overexpression of ALK2 mutants with defective binding to FKBP12, responsible of fibrodysplasia ossificans progressiva, mirrors rapamycin effect on hepcidin upregulation. The mechanism, characterized in vitro in hepatoma cells, is conserved in vivo, as shown by the transient hepcidin increase and spleen iron retention observed in WT mice treated with a single dose of tacrolimus. The mechanism is also active in humans, as shown by an informative patient affected by iron refractory iron deficiency anemia (IRIDA), a condition characterized by high hepcidin levels, who was compound heterozygous for TMPRSS6 (the IRIDA causative gene) and ALK2 mutations.32 Overall, our results indicate that ALK2 is the BMPR-I targeted by FKBP12 in hepatocytes that activates hepcidin only when not bound to the immunophilin.
Then we asked what is the physiologic significance of ALK2-FKBP12 interaction. Both BMP2 and BMP6 are expressed in liver endothelial cells and regulate hepcidin in hepatocytes in a paracrine manner. Only BMP6 is upregulated in response to iron increase,4 whereas BMP2 maintains the hepcidin basal activation.3 In several cell types, as mesenchymal stem cells and endothelial cells, BMP6 binds both ALK2 and ALK3, whereas BMP2 preferentially interacts with ALK3.33-35 In agreement, silencing ALK3 in hepatoma cells impairs both BMP2- and BMP6-dependent hepcidin activation, whereas ALK2 downregulation affects only the BMP6 response.36 In accordance with the in vitro data, Alk3 liver-conditional inactivation in mice causes a stronger repression of hepcidin and a more severe iron overload than conditional inactivation of Alk2 8. ALK2 seems to have a marginal role in basal hepcidin activation likely because it interacts with FKBP12.
We showed that, as reported for other tissues,24,25 ALK2 mutants may signal through the noncanonical ligand Activin A in hepatocytes. The mechanism is activated by loss of FKBP12 binding because it also occurs in WT ALK2 after treatment with FKBP12-sequestering drugs. We speculate that this result has implications on hepcidin regulation in inflammation. It is well known that inflammatory cytokines, as interleukin 6, increase hepcidin expression through the JAK2-STAT3 signaling and that, for a full hepcidin expression, a concomitant activation of the BMP-SMAD1/5/8 pathway is required.37,38 In accordance, suppression of the BMP pathway by LDN 193189 decreases hepcidin levels and has been proposed as a potential treatment of anemia of chronic disease.38 However, the ligand that activates the BMP pathway in chronic inflammation remained unknown. Interestingly, in mouse models of acute (lipopolysaccharide [LPS]) and chronic (Brucella abortus) inflammation, treatment with follistatin, an activin inhibitor that binds at high efficiency both Activin A and Activin B, reverts hepcidin activation.39 Activin B, a member of the TGF-β superfamily released in inflammation, initially proposed as a potential ligand,40 was recently dismissed as hepcidin regulator.41 Activin A, a critical component of the inflammatory response, secreted in the circulation, was not previously reported to take part in hepcidin activation.39,42 Activin A binds BMPR-II, ACVR2A and ACVR2B, and the BMPR-I ALK4.25,43 The complex signals through phosphorylation and nuclear translocation of the transcription factors SMAD2 and SMAD3 (supplemental Figure 6A).
We show that when ALK2-FKBP12 interaction is impaired, as in the case of ALK2 mutants or in the presence of FKBP12-binding drugs, the receptor becomes responsive to Activin A and triggers hepcidin activation through SMAD1/5/8 (supplemental Figure 6B).
Although these data have been obtained in vitro, we speculate that a mechanism that reduces FKBP12 or interferes with its ALK2 binding may facilitate SMAD1/5/8 activation. In vivo, the administration of the ALK2 inhibitor momelotinib strongly suppresses hepcidin activation in rats with anemia of inflammation,44 with documented increased hepatic SMAD1/5/8 phosphorylation,38 whereas it is ineffective in basal condition.44 These results suggest that in inflammation ALK2 is functional and not bound to FKBP12. In this condition, hepcidin activation might occur either by preformed BMPRs that signal independently from the ligand concentration45 or through BMPRs response to noncanonical ligands.25
Activin A, in contrast to BMP6, probably has very weak affinity for ALK2, insufficient to reduce the FKBP12-ALK2 interaction enough to activate the pathway. High concentrations of BMP6 reduce the FKBP12-ALK2 interaction (supplemental Figure 6B), as reported for other TGF-β ligands on the corresponding receptors.46 Thus disruption of the FKBP12-ALK2 interaction might be relevant in conditions of iron overload characterized by increased BMP6. When functional, ALK2 becomes sensitive to noncanonical ligands such as Activin A that contributes to activate hepcidin. We speculate that the synergism we observed between Activin A and high concentration of BMP6 might be relevant when inflammation/infections occur in severe iron loading by further enhancing hepcidin expression in the attempts of achieving the protective condition of hypoferremia.47
Our results suggest that hepcidin production undergoes multiple control levels to coordinate systemic iron homeostasis and are in agreement with the recently proposed model of 2 BMP-regulated pathways that additively contribute to hepcidin activation.48 In the first (that we speculate involves ALK3), BMP2 and BMP6 interact with the coreceptor HJV and the other hemochromatosis proteins HFE and TFR2 to allow endocytosis and signaling, as proposed.49 The second (ALK2-dependent) would signal after BMP6 binding to type 1/type 2 preformed receptor complexes without requiring the HJV coreceptor (supplemental Figure 6A). In agreement with the latter model, we showed that the BMP-coreceptor HJV is dispensable for the ALK2 effect in HjvKO primary hepatocytes treated with tacrolimus, although, because of the extremely low basal levels, the maximal hepcidin expression achieved is about half that of WT hepatocytes.
As expected, HJV further potentiates the hepcidin activation induced by mutant ALK2 in vitro (data not shown). HJV undergoes a posttranslational control by serine-protease matriptase-2, encoded by TMPRSS6, which cleaves the coreceptor from the cell membrane50 (supplemental Figure 6). High hepcidin levels resulting from TMPRSS6 recessive mutations cause IRIDA.51 The rare patient we reported, a carrier of both ALK2R258S and TMPRSS6I212T heterozygous mutations, was affected by both IRIDA and fibrodysplasia ossificans progressiva.32 In patients with the latter disease who have constitutively active ALK2 mutants, hepcidin is not upregulated at a level able to induce IRIDA, unless the activity of the inhibitor TMPRSS6 is impaired and some BMP coreceptor (HJV) function is preserved. Digenic inheritance in this patient is also compatible with the 2 BMP pathways model (supplemental Figure 6B).
Finally, our results indicate that FKBP12 may become a novel target to treat iron overload conditions resulting from low hepcidin. Rescue of the BMP-SMAD activity by tacrolimus has been achieved in pulmonary artery hypertension, a disease caused by reduction of BMPR2.52 Chronic treatment with low-dose tacrolimus that does not trigger immunosuppression rescues endothelial dysfunction of pulmonary hypertension in mice with conditional deletion of Bmpr2 in endothelial cells.18 More recently, a pilot study with the same drug ameliorated the clinical condition of 3 patients with end-stage pulmonary hypertension.53 Considering that, in our hands, tacrolimus upregulates hepcidin in HjvKO hepatocytes and that in vivo Hjv is dispensable for hepcidin upregulation by iron,54 tacrolimus-like drugs might be proposed for disorders of impaired hepcidin production, such as hemochromatosis and β-thalassemia. It remains to be defined whether the upregulation of hepcidin obtained by tacrolimus treatment may counteract severe iron overload.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Martina Muckenthaler for discussion of the results, criticism, and suggestions; Sandro Altamura for serum hepcidin measurement; Stefano Piccolo, University of Padua, Italy, for providing the pGL3-BRE and pGL3-CAGA vectors; and Takenobu Katagiri, Division of Pathophysiology, Research Center for Genomic Medicine, Saitama Medical University, Japan, for the gift of the pcDEF/FLAG-mSmad1 vector.
This work was supported in part by Telethon Fondazione Onlus grant GGP15064 (L.S.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.C. conceived and performed the experiments and drafted the manuscript; A.P., M.P., I.A., and A.N. performed experiments; L.S. and C.C. conceived the experiments and wrote the manuscript. L.S. secured funding.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for S.C. is Department of Pediatric Hematology, Oncology and Immunology, University of Heidelberg, INF 350, 69120 Heidelberg, Germany.
Correspondence: Laura Silvestri, Division of Genetics and Cell Biology, San Raffaele Scientific Institute, Via Olgettina, 58 20132, Milan, Italy; e-mail: silvestri.laura@hsr.it.
References
- 1.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090-2093. [DOI] [PubMed] [Google Scholar]
- 2.Mleczko-Sanecka K, Roche F, da Silva AR, et al. Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood. 2014;123(10):1574-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koch PS, Olsavszky V, Ulbrich F, et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood. 2017;129(4):415-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kautz L, Meynard D, Monnier A, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112(4):1503-1509. [DOI] [PubMed] [Google Scholar]
- 5.Andriopoulos B Jr, Corradini E, Xia Y, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41(4):482-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canali S, Zumbrennen-Bullough KB, Core AB, et al. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood. 2017;129(4):405-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41(4):478-481. [DOI] [PubMed] [Google Scholar]
- 8.Steinbicker AU, Bartnikas TB, Lohmeyer LK, et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood. 2011;118(15):4224-4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayeur C, Leyton PA, Kolodziej SA, Yu B, Bloch KD. BMP type II receptors have redundant roles in the regulation of hepatic hepcidin gene expression and iron metabolism. Blood. 2014;124(13):2116-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717. [DOI] [PubMed] [Google Scholar]
- 11.Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92(5):583-588. [DOI] [PubMed] [Google Scholar]
- 12.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sofroniadou S, Goldsmith D. Mammalian target of rapamycin (mTOR) inhibitors: potential uses and a review of haematological adverse effects. Drug Saf. 2011;34(2):97-115. [DOI] [PubMed] [Google Scholar]
- 14.Wang T, Li BY, Danielson PD, et al. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell. 1996;86(3):435-444. [DOI] [PubMed] [Google Scholar]
- 15.Song GA, Kim HJ, Woo KM, et al. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem. 2010;285(29):22542-22553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inui M, Manfrin A, Mamidi A, et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell Biol. 2011;13(11):1368-1375. [DOI] [PubMed] [Google Scholar]
- 17.Chaikuad A, Alfano I, Kerr G, et al. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem. 2012;287(44):36990-36998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spiekerkoetter E, Tian X, Cai J, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123(8):3600-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson KP, Yamashita MM, Sintchak MD, et al. Comparative x-ray structures of the major binding protein for the immunosuppressant FK506 (tacrolimus) in unliganded form and in complex with FK506 and rapamycin. Acta Crystallogr D Biol Crystallogr. 1995;51(Pt 4):511-521. [DOI] [PubMed] [Google Scholar]
- 20.Sich C, Improta S, Cowley DJ, et al. Solution structure of a neurotrophic ligand bound to FKBP12 and its effects on protein dynamics. Eur J Biochem. 2000;267(17):5342-5355. [DOI] [PubMed] [Google Scholar]
- 21.Sun F, Li P, Ding Y, et al. Design and structure-based study of new potential FKBP12 inhibitors. Biophys J. 2003;85(5):3194-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pagani A, Silvestri L, Nai A, Camaschella C. Hemojuvelin N-terminal mutants reach the plasma membrane but do not activate the hepcidin response. Haematologica. 2008;93(10):1466-1472. [DOI] [PubMed] [Google Scholar]
- 23.Mohedas AH, Xing X, Armstrong KA, Bullock AN, Cuny GD, Yu PB. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem Biol. 2013;8(6):1291-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatsell SJ, Idone V, Wolken DM, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med. 2015;7(303):303ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hino K, Ikeya M, Horigome K, et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci USA. 2015;112(50):15438-15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haupt J, Deichsel A, Stange K, et al. ACVR1 p.Q207E causes classic fibrodysplasia ossificans progressiva and is functionally distinct from the engineered constitutively active ACVR1 p.Q207D variant. Hum Mol Genet. 2014;23(20):5364-5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell. 1995;83(3):463-472. [DOI] [PubMed] [Google Scholar]
- 28.Dargan SL, Lea EJ, Dawson AP. Modulation of type-1 Ins(1,4,5)P3 receptor channels by the FK506-binding protein, FKBP12. Biochem J. 2002;361(Pt 2):401-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals. 2008;16(4):318-325. [DOI] [PubMed] [Google Scholar]
- 30.Ahearn IM, Tsai FD, Court H, et al. FKBP12 binds to acylated H-ras and promotes depalmitoylation. Mol Cell. 2011;41(2):173-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathea S, Li S, Schierhorn A, Jahreis G, Schiene-Fischer C. Suppression of EGFR autophosphorylation by FKBP12. Biochemistry. 2011;50(50):10844-10850. [DOI] [PubMed] [Google Scholar]
- 32.Pagani A, Colucci S, Bocciardi R, et al. A new form of IRIDA due to combined heterozygous mutations of TMPRSS6 and ACVR1A encoding the BMP receptor ALK2. Blood. 2017;129(25):3392-3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Upton PD, Long L, Trembath RC, Morrell NW. Functional characterization of bone morphogenetic protein binding sites and Smad1/5 activation in human vascular cells. Mol Pharmacol. 2008;73(2):539-552. [DOI] [PubMed] [Google Scholar]
- 34.Lavery K, Swain P, Falb D, Alaoui-Ismaili MH. BMP-2/4 and BMP-6/7 differentially utilize cell surface receptors to induce osteoblastic differentiation of human bone marrow-derived mesenchymal stem cells. J Biol Chem. 2008;283(30):20948-20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hurst LA, Dunmore BJ, Long L, et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat Commun. 2017;8:14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia Y, Babitt JL, Sidis Y, Chung RT, Lin HY. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood. 2008;111(10):5195-5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pagani A, Nai A, Corna G, et al. Low hepcidin accounts for the proinflammatory status associated with iron deficiency. Blood. 2011;118(3):736-746. [DOI] [PubMed] [Google Scholar]
- 38.Theurl I, Schroll A, Sonnweber T, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood. 2011;118(18):4977-4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canali S, Core AB, Zumbrennen-Bullough KB, et al. Activin B induces noncanonical SMAD1/5/8 signaling via BMP type I receptors in hepatocytes: evidence for a role in hepcidin induction by inflammation in male mice. Endocrinology. 2016;157(3):1146-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Besson-Fournier C, Latour C, Kautz L, et al. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood. 2012;120(2):431-439. [DOI] [PubMed] [Google Scholar]
- 41.Besson-Fournier C, Gineste A, Latour C, et al. Hepcidin upregulation by inflammation is independent of Smad1/5/8 signaling by activin B. Blood. 2017;129(4):533-536. [DOI] [PubMed] [Google Scholar]
- 42.Kanamori Y, Sugiyama M, Hashimoto O, Murakami M, Matsui T, Funaba M. Regulation of hepcidin expression by inflammation-induced activin B. Sci Rep. 2016;6:38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrison CA, Gray PC, Fischer WH, Donaldson C, Choe S, Vale W. An activin mutant with disrupted ALK4 binding blocks signaling via type II receptors. J Biol Chem. 2004;279(27):28036-28044. [DOI] [PubMed] [Google Scholar]
- 44.Asshoff M, Petzer V, Warr MR, et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood. 2017;129(13):1823-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ehrlich M, Horbelt D, Marom B, Knaus P, Henis YI. Homomeric and heteromeric complexes among TGF-β and BMP receptors and their roles in signaling. Cell Signal. 2011;23(9):1424-1432. [DOI] [PubMed] [Google Scholar]
- 46.Huse M, Muir TW, Xu L, Chen YG, Kuriyan J, Massagué J. The TGF beta receptor activation process: an inhibitor- to substrate-binding switch. Mol Cell. 2001;8(3):671-682. [DOI] [PubMed] [Google Scholar]
- 47.Arezes J, Jung G, Gabayan V, et al. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe. 2015;17(1):47-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Latour C, Besson-Fournier C, Meynard D, et al. Differing impact of the deletion of hemochromatosis-associated molecules HFE and transferrin receptor-2 on the iron phenotype of mice lacking bone morphogenetic protein 6 or hemojuvelin. Hepatology. 2016;63(1):126-137. [DOI] [PubMed] [Google Scholar]
- 49.Healey EG, Bishop B, Elegheert J, Bell CH, Padilla-Parra S, Siebold C. Repulsive guidance molecule is a structural bridge between neogenin and bone morphogenetic protein. Nat Struct Mol Biol. 2015;22(6):458-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8(6):502-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finberg KE, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet. 2008;40(5):569-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deng Z, Morse JH, Slager SL, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spiekerkoetter E, Sung YK, Sudheendra D, et al. Low-dose FK506 (tacrolimus) in end-stage pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(2):254-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gkouvatsos K, Fillebeen C, Daba A, Wagner J, Sebastiani G, Pantopoulos K. Iron-dependent regulation of hepcidin in Hjv-/- mice: evidence that hemojuvelin is dispensable for sensing body iron levels. PLoS One. 2014;9(1):e85530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.