

Summary
Recent integrative epigenome analyses highlight the importance of functionally distinct chromatin states for accurate cell function. How these states are established and maintained is a matter of intense investigation. Here, we present evidence for DNA damage as an unexpected means to shape a protective chromatin environment at regions of recurrent replication stress (RS). Upon aberrant fork stalling, DNA damage signaling and concomitant H2AX phosphorylation coordinate the FACT-dependent deposition of macroH2A1.2, a histone variant that promotes DNA repair by homologous recombination (HR). MacroH2A1.2, in turn, facilitates the accumulation of the tumor suppressor and HR effector BRCA1 at replication forks to protect from RS-induced DNA damage. Consequently, replicating primary cells steadily accrue macroH2A1.2 at fragile regions, whereas macroH2A1.2 loss in these cells triggers DNA damage signaling-dependent senescence, a hallmark of RS. Altogether, our findings demonstrate that recurrent DNA damage contributes to the chromatin landscape to ensure the epigenomic integrity of dividing cells.
eTOC Blurb
Kim et al. show that replication stress associated DNA damage can help create a protective chromatin environment to ensure efficient repair of fragile genomic regions in subsequent cell divisions. This involves the histone variant macroH2A1.2 and depends on both DNA damage signaling and replication fork-associated chromatin remodeling.
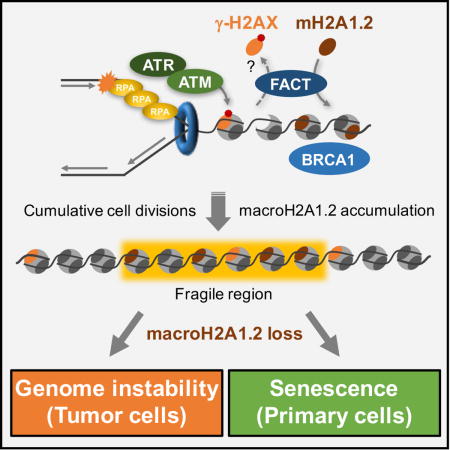
Introduction
The tightly controlled organization of genomic DNA is essential for nuclear transactions including gene expression, DNA replication and accurate genome maintenance. This process involves a diverse and in many cases evolutionarily conserved set of factors that maintain metazoan higher order chromatin structure (Ho et al., 2014). Recent integrative analyses have identified a range of distinct chromatin domains, which are centrally defined by differences in nucleosome density as well as histone variant composition or modification (Roadmap Epigenomics et al., 2015). Dynamic alterations in the resulting chromatin landscape are closely linked to cellular differentiation, aging and malignant transformation (Booth and Brunet, 2016). Dissecting the processes that help establish, shape and maintain chromatin organization is, thus, vital for our understanding of both normal and aberrant cell function.
Over the past decade, DNA damage has emerged as an important modulator of epigenetic integrity (Dabin et al., 2016; Price and D’Andrea, 2013). However, chromatin organization is generally restored upon repair (Soria et al., 2012), and the potential for DNA damage to shape chromatin may thus be most relevant in the context of chronic genotoxic stress. A major source of chronic DNA damage is replication stress (RS), which results from impaired DNA polymerase progression and occurs naturally during each cell division at genomic regions that are inherently difficult to replicate, including common and early replicating fragile sites (CFSs, ERFSs) (Barlow et al., 2013; Durkin and Glover, 2007). The recognition of RS-associated single-stranded or double-stranded DNA lesions involves activation of the DNA damage response (DDR) via ATR and ATM kinases, respectively (Zeman and Cimprich, 2014). The resolution of arrested forks, in turn, critically depends on the DDR-induced recruitment of the tumor suppressors and DNA repair effectors BRCA1 and BRCA2, among other factors (Schlacher et al., 2011; Schlacher et al., 2012; Willis et al., 2014). Moreover, both normal replication and RS are tightly linked to fork-proximal chromatin remodeling (Alabert and Groth, 2012), which is emerging as an important modulator of the repair of RS-associated DNA lesions (Pellegrino et al., 2017; Saredi et al., 2016) and has the potential to change replication fork-associated nucleosome composition (Alabert and Groth, 2012; Jasencakova et al., 2010; Khurana and Oberdoerffer, 2015). Consistent with this, RS was reported to cause local gene deregulation in model organisms by altering the abundance of repressive epigenetic marks at stalled replication forks (Dubarry et al., 2011; Sarkies et al., 2010). On the other hand, overall nucleosome turnover is suppressed at RS-prone genomic regions in yeast (Taneja et al., 2017). Moreover, post-replicative chromatin appears to be largely reset during the S/G2 phase of the cell cycle in mammalian cells (Alabert et al., 2014; Khurana and Oberdoerffer, 2015; Saredi et al., 2016), highlighting a remarkable capacity of dividing cells to maintain accurate epigenetic inheritance. The extent and persisting impact of RS on epigenome integrity, therefore, remains to be determined (Jasencakova et al., 2010; Khurana and Oberdoerffer, 2015).
Defects in the molecular pathways that control RS can cause DNA breakage, which may ultimately result in genomic aberrations and malignant transformation (Durkin and Glover, 2007; Zeman and Cimprich, 2014). Moreover, RS was found to promote aberrant DDR signaling, which can trigger irreversible cell cycle arrest in primary cells (Bartkova et al., 2006; Di Micco et al., 2006). While the resulting senescent phenotype protects from excessive cell growth upon oncogene activation, it is frequently associated with features of age-associated organ decline and can further indirectly contribute to malignancy (Campisi, 2013; He and Sharpless, 2017). Altogether, these observations point to an intricate, yet poorly understood interplay between DNA replication, the DDR and (epi)genome maintenance, which is likely to control the functional integrity of dividing cells.
Here, we uncover a protective epigenetic response to RS as an unexpected means to demarcate expansive, RS-prone regions across the genome. This process involves the targeted deposition of the macro-histone variant macroH2A1.2 at sites of RS-induced DNA damage, which requires the FACT (facilitates chromatin transcription) complex and ensures a BRCA1-permissive and hence repair-promoting chromatin environment. Three major conclusions arise from this work: (i) DNA damage plays an important role in shaping a protective chromatin landscape at fragile genomic regions; (ii) macroH2A1.2 emerges as a critical modulator of RS and cellular senescence; (iii) manipulating macroH2A1.2 abundance may provide a means to interfere with malignant transformation and tumor growth.
Results
MacroH2A1.2 accumulates at sites of replication stress
We recently identified macroH2A1.2 as a mediator of homology-directed DNA repair (HR) by promoting a chromatin environment that facilitates BRCA1 retention at DNA double-strand breaks (DSBs) (Khurana et al., 2014). Given the essential role for BRCA1 in response to RS, we hypothesized that macroH2A1.2 may be implicated in RS-associated chromatin reorganization. To test this, we performed genome-wide mapping of macroH2A1.2 occupancy using chromatin-immunoprecipitation followed by deep sequencing (ChIP-Seq) in K562 erythroleukemia cells in the presence or absence of the DNA polymerase inhibitor aphidicolin (Aph) (Fig. 1A). Sites of RS-induced DNA damage were identified based on S139 phosphorylation of histone H2AX (γ-H2AX). We observed a significant and reproducible overlap of macroH2A1.2 peaks with ~ 40% of Aph-induced γ-H2AX domains (Fig. 1A, Fig. S1A, B). Non-random colocalization of macroH2A1.2 and γ-H2AX was confirmed using the Jaccard Index, a statistic for measuring sample set similarity, comparing the overlap between observed and randomly shuffled (permuted) peaks of equal size (Fig. 1B). No correlation was observed between macroH2A1.2 and H2AZ or H3K9me3, two unrelated chromatin marks with genomic coverage comparable to Aph-induced γ-H2AX peaks, ruling out high genomic peak coverage as a major contributing factor for peak colocalization (Fig. 1C). Of note, macroH2A1.2 peak coverage increased upon RS, with newly formed macroH2A1.2 domains being significantly enriched near pre-existing macroH2A1.2 peaks (Fig. 1D, Fig. S1D, E). Moreover, compared to macroH2A1.2 regions devoid of detectable DNA damage, γ-H2AX-associated macroH2A1.2 domains were enriched for replication origins as well as repetitive DNA elements, indicative of active DNA replication and DNA fragility, respectively (Barlow et al., 2013; Fungtammasan et al., 2012; Shah and Mirkin, 2015) (Fig. 1E, Fig. S1F). Consistent with the latter, macroH2A1.2 was more abundant at CFSs than non-fragile regions of the genome. Perhaps more importantly, macroH2A1.2 peak coverage directly correlated with CFS break susceptibility, as did γ-H2AX peak coverage (Fig. 1F). Of note, macroH2A1.2 occupancy at CFSs was already apparent in the absence of damage, albeit to a lower extent (Fig. 1D, F), pointing to macroH2A1.2 as an epigenetic mark of fragile regions, the continued deposition of which may be functionally linked to RS. Unlike macroH2A1.2, H3K9me3 and H2AZ showed little to no enrichment at break-prone CFSs, demonstrating that the latter was not simply an artifact of abundant peak coverage (Fig. 1G). Confirming RS-mediated accumulation of macroH2A1.2, ChIP qPCR revealed increased macroH2A1.2 levels at select fragile sites in response to both Aph and the ribonucleotide reductase inhibitor hydroxyurea (HU), two independent sources of RS (Zeman and Cimprich, 2014). No change in macroH2A1.2 occupancy was observed at two non-fragile control loci (Fig. 1H, Fig. S2A C). Moreover, RS did not alter overall macroH2A1.2 protein levels (Fig. S3A, B) or fragile site-associated enrichment of two unrelated H2A variants, H2AX and H2AZ (Fig. S2D). RS-induced macroH2A1.2 accumulation at CFSs was an active consequence of DNA damage signaling, as it was ablated by inhibition of ATR or ATM kinases (Fig. 1H, I, Fig. S2E, F). Of note, three of four CFSs tested were exclusively responsive to ATR inhibition (Fig. 1H, Fig. S2E), whereas recruitment to the remaining CFS was predominantly dependent on ATM (Fig. 1I). These findings indicate that, in the context of RS, macroH2A1.2 accumulation can occur both in the presence and absence of DSB formation.
Figure 1. RS promotes macroH2A1.2 accumulation at fragile sites.
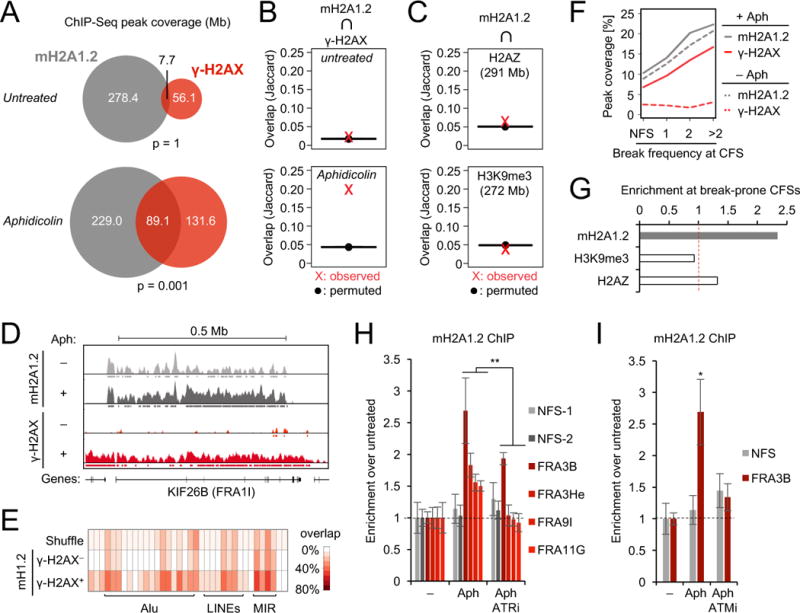
(A) Overlap in megabase pairs between macroH2A1.2 and γ-H2AX peaks, for untreated and Aph-treated cells. The indicated p-values are the result of permutation tests (n = 1,000 trials). MacroH2A1.2 antibody specificity was confirmed by macroH2A1.2 knockdown (Fig. S1C). (B) Jaccard Index for γ-H2AX coverage of observed macroH2A1.2 peaks and randomly shuffled (permuted) peaks of equal size, in the absence (top) or presence of Aph (bottom). Values on the y-axis represent the intersection divided by the union in base pairs. (C) Jaccard Index for H2AZ (top) or H3K9me3 coverage (bottom) of observed and permuted macroH2A1.2 peaks from Aph-treated samples in (A). H2AZ and H3K9me3 data were from the Roadmap Epigenomics Consortium. (D) Browser view of macroH2A1.2 and γ-H2AX ChIP-Seq data within the FRA1I CFS. Data are log2 ChIP/Input and tracks are displayed on scales from 0 to 1.8. (E) Overlap of macroH2A1.2 peaks with various repeat classes, when overlapping γ-H2AX (γ-H2AX+) or not (γ-H2AX−). A randomly shuffled set of peaks is presented as a control. (F) Peak coverage of macroH2A1.2 and γ-H2AX within CFSs partitioned by break/gap frequency per 10 metaphases in Aph-treated K562 cells (Table S1). Non-fragile sites (NFS) are given as control. (G) Fold enrichment of macroH2A1.2 and chromatin marks in (C) at CFSs with >2 breaks/10 metaphases compared to NFSs. (H) ChIP analysis for macroH2A1.2 in untreated or Aph-treated K562 cells in the presence or absence of ATR inhibitor (ATRi). Enrichment relative to untreated cells is shown at the indicated CFSs, two NFSs served as control. (I) ChIP analysis as in (H) in the presence or absence of ATM inhibitor (ATMi). Values in (H) and (I) are expressed as mean and SEM (n = 3). “* p < 0.05, ** p < 0.01 based on Student’s two-tailed t-test.
To independently validate the impact of RS on the macroH2A1.2 chromatin landscape at single cell resolution, we took advantage of a tractable, imaging-based approach to follow replication fork blockage at Lac operon arrays (LacO × 256) stably integrated into U2OS osteosarcoma cells (Fig. 2A) (Burgess et al., 2014). LacO arrays pose an obstacle to DNA polymerase progression when bound by the Lac repressor (LacR), resulting in S phase-specific, localized DNA damage accumulation (Beuzer et al., 2014). Consistent with this, expression of an mCherry-LacR fusion protein, which simultaneously allowed for fork blockage and LacO array detection, resulted in robust accumulation of BRCA1 at the arrays of actively replicating cells, which were identified based on incorporation of the thymidine analog ethyl-deoxy-uridine (EdU) (Fig. 2B). Using this approach, we observed array-specific accumulation of macroH2A1.2 predominantly in EdU+ cells (Fig. 2C). Together, we thus identify RS as a driver of macroH2A1.2 deposition at fragile genomic regions.
Figure 2. MacroH2A1.2 promotes BRCA1 accumulation at blocked replication forks.
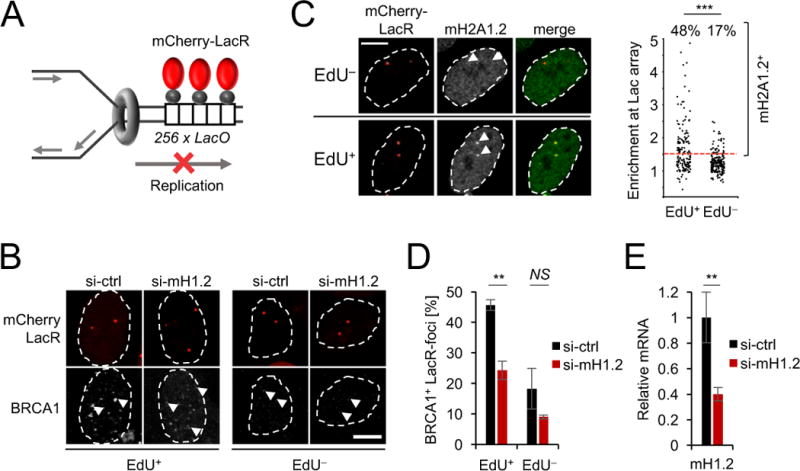
(A) Schematic for LacR-induced replication fork block at transgenic LacO array. (B) BRCA1 colocalization at LacR-occupied LacO arrays in the presence of the indicated siRNAs in EdU+ and EdU− U2OS cells. Arrow heads depict LacO arrays. Note that the EdU− subset contains both G1 and G2 phase cells. (C) MacroH2A1.2 enrichment at LacR-occupied LacO arrays determined by IF in EdU+ and EdU− cells. The percentage of macroH2A1.2+ foci was determined based on enrichment of macroH2A1.2 intensity at the array relative to total nuclear intensity. *** denotes p<0.001 by Mann-Whitney U test. (D) The percentage of BRCA1+ foci in (B), determined based on enrichment of BRCA1 intensity at the array relative to total nuclear intensity. Values represent mean and SEM (n = 3). (E) MacroH2A1.2 expression in the presence of the indicated siRNAs in cells from (B), mRNA levels were normalized to rpl13a and β-actin, samples were analyzed in triplicate, values are expressed as mean and SD. Scale bars: 10 μm. ** p < 0.01 based on Student’s two-tailed t-test.
MacroH2A1.2 promotes BRCA1 accumulation at replication forks
We next asked if RS-induced macroH2A1.2 accumulation is required to protect fragile genomic elements from RS-associated DNA damage. Consistent with its previously described role in DSB repair (Khurana et al., 2014; Leung et al., 2017), siRNA-mediated depletion of macroH2A1.2 resulted in a significant decrease of BRCA1 levels at sites of LacR-associated replication blockage, particularly in S phase cells (Fig. 2B, D, E). Extending this observation to endogenous RS, deletion of macroH2A1.2 (in this case via CRISPR/Cas9, see Fig. S3B) impaired BRCA1 accumulation on chromatin upon treatment with either HU or Aph (Fig. 3A). Of note, isolation of proteins on nascent DNA (iPOND) revealed that macroH2A1.2 ablation impairs BRCA1 accumulation directly at the replication fork, even in the absence of exogenous RS. MacroH2A1.2 loss did not decrease overall fork abundance, as measured by the DNA sliding clamp proliferating nuclear antigen A (PCNA), nor did it alter BRCA1 protein levels (Fig. 3B). Providing a molecular basis for macroH2A1.2-assisted BRCA1 loading, immunoprecipitation (IP) of BRCA1 in 293 cells revealed a robust interaction with Flag-tagged macroH2A1.2 expressed form a single, stably integrated transgene (Fig. 3C). Using GST-pulldown of BRCA1 fragments (Scully et al., 1997), we further found that macroH2A1.2 specifically associates with a poorly characterized BRCA1 N-terminal region, whereas no interaction was observed for H2A or H2AZ (Fig. 3D, E). GST-BRCA1 association with macroH2A1.2 was independent of DNA damage (Fig. 3C, E), in agreement with a model wherein, rather than enhancing protein-protein interactions, RS increases macroH2A1.2 abundance at fragile elements to promote BRCA1 localization and, consequently, fork repair. Altogether, these findings identify macroH2A1.2 as a modulator of BRCA1 recruitment in the context of RS.
Figure 3. MacroH2A1.2 interacts with and promotes BRCA1 accumulation on chromatin.
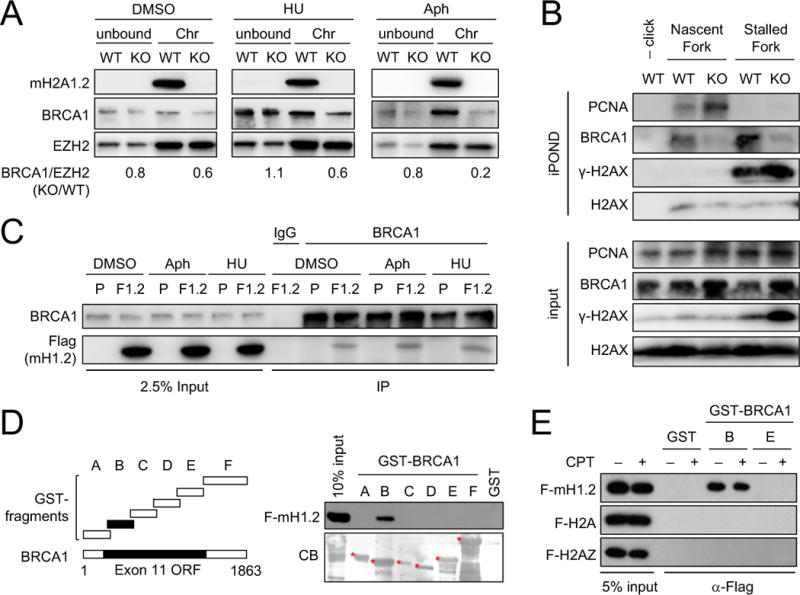
(A) Western blot analysis of chromatin-bound (Chr) and non-chromatin-bound (unbound) protein fractions from K562 WT or macroH2A1.2 knockout (KO) cells treated with HU, Aph or vehicle (DMSO). BRCA1 levels were normalized to EZH2 and are expressed relative to respective WT samples. (B) Western blot analysis of input and iPOND samples from macroH2A1.2 WT or KO K562 cells in the presence or absence of HU. (C) BRCA1 IP in 293 cells with (F1.2) or without (parental, P) stably integrated Flag-macroH2A1.2 in the presence or absence of RS. IgG served as a control IP. (D) Western blot of GST-pulldown of the indicated BRCA1-GST fusion fragments with 293T nuclear lysates overexpressing Flag-macroH2A1.2 (F-mH1.2). A Commassie Blue (CB) staining of input protein is shown. (E) Western blot of GST-pulldown using the indicated BRCA1 fragments with nuclear extracts expressing F-mH1.2, Flag-H2A (F-H2A) or Flag-H2AZ (F-H2AZ).
MacroH2A1.2 protects from RS
Based on the above observations, we sought to assess the impact of macroH2A1.2 loss on the resolution of RS-associated DNA damage. Analysis of RS-induced DDR signaling revealed a dose-dependent increase in the phosphorylation of ATM, RPA (serine 4/8) and H2AX (Fig. S3A) upon macroH2A1.2 knockdown, indicative of increased DSB formation upon macroH2A1.2 loss (Zeman and Cimprich, 2014). Similar effects were observed upon CRISPR/Cas9-mediated deletion of macroH2A1.2 (Fig. S3B). Increased DNA damage signaling was not the result of altered S phase progression, as macroH2A1.2 deletion had only a minor effect on cell cycle profiles in K562 cells (Fig. S3C-F). The moderate extent of overall DDR activation was not unexpected given the CFS-specific accumulation of macroH2A1.2 upon RS (Fig. 1F–I). To determine if macroH2A1.2 loss-associated DNA damage is similarly restricted to sites of RS, we performed ChIP-Seq to assess γ-H2AX accumulation genome-wide in control or macroH2A1.2 KO cells. Indeed, deletion of macroH2A1.2 resulted in an increase in H2AX phosphorylation that was most pronounced at break-prone CFSs and further aggravated upon RS, while little DNA damage accumulation was observed in non-fragile control regions (Fig. 4A, Fig. S4A). Moreover, the majority of RS-induced DNA damage occurred in genomic regions normally occupied by macroH2A1.2 in WT cells (Fig. S4B). The CFS-associated increase in γ-H2AX signal was not the result of altered H2AX abundance (Fig. S2D), was independent of the source of RS and was observed both in macroH2A1.2 KO and knockdown cell lines (Fig. 4A, B, Fig. S4A, C). Exacerbated, RS-dependent DNA breakage upon macroH2A1.2 depletion was further confirmed using the alkaline comet assay (Fig. 4C).
Figure 4. MacroH2A1.2 protects from RS-induced DNA damage.
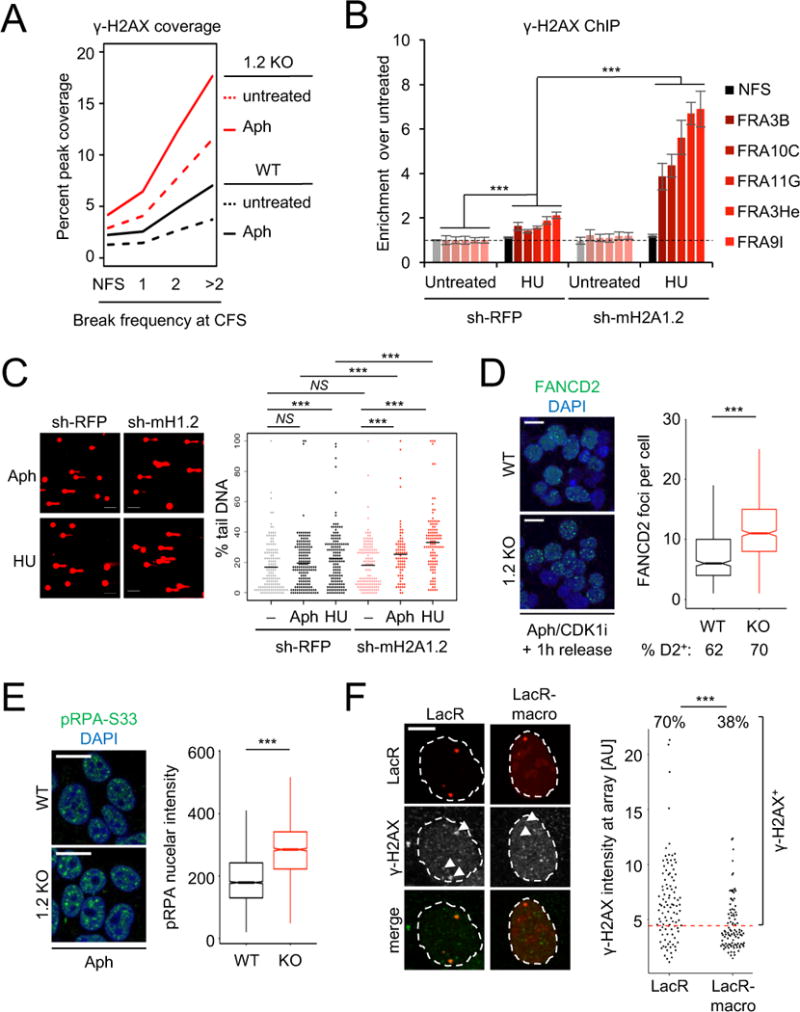
(A) Peak coverage of γ-H2AX within CFSs in macroH2A1.2 WT or KO K562 cells in the presence or absence of Aph. CFSs are partitioned by break frequency (see Fig. 1F). (B) ChIP analysis for γ-H2AX in presence or absence of HU in macroH2A1.2 knock-down and control K562 cells. Values are expressed as mean and SEM (n = 3). “*** p < 0.001 based on Student’s two-tailed t-test. (C) Comet assay assessing DNA damage following HU or Aph treatment in the presence or absence of macroH2A1.2 KD. Percent tail DNA for each cell is shown, black bars represent the mean. *** denotes p < 0.001 by Student’s two-tailed t-test. Scale bar: 100 μM. (D) IF analysis of FANCD2 foci in WT (n = 917 cells) or KO K562 cells (n = 1156 cells) treated with 0.3 μM Aph for 16 h, arrested in late G2 by CDK1 inhibition (CDK1i) and released for 1 h prior to analysis. Representative images are shown. Cells were combined from three independent experiments. The fraction of cells with 1 or more FANCD2 foci is indicated (% D2+). Box plots depict the number of foci in FANCD2+ cells, whiskers extend to 1.5 × the interquartile range (IQR), values outside of the 1.5 IQR are not shown. Scale bar: 20 μm. (E) IF analysis of pRPA-S33 in 293T cells treated with 0.3 μM Aph for 16 h. Box plots depict total pRPA-S33 nuclear intensities in WT (n = 2917 cells) or KO cells (n = 1968 cells). Representative images are shown, cells were combined from three independent experiments. Scale bar: 20 μm. (F) γ-H2AX IF at LacO arrays upon tethering of LacR-macro or LacR alone. The percentage of γ-H2AX+ foci was determined based on total γ-H2AX intensity at the array, data were combined from three independent experiments. Scale bar: 10 μm. *** in (D-F) denotes p < 0.001 by Mann-Whitney U test.
To assess the impact of macroH2A1.2 loss on CFS fragility beyond DSB-associated H2AX phosphorylation, we performed IF analyses for FANCD2, a Fanconi Anemia repair factor that specifically associates with under-replicated CFSs in late G2/M, irrespective of chromosome breakage (Chan et al., 2009; Madireddy et al., 2016; Minocherhomji et al., 2015). To enrich for FANCD2-associated CFSs, Aph-treated cells were arrested in late G2 by CDK1 inhibition followed by release into mitosis (Minocherhomji et al., 2015). Consistent with aggravated CFS-associated DNA damage, macroH2A1.2 loss resulted in an ~ 2-fold increase in FANCD2 foci (Fig. 4D, Fig. S4D). Further supporting a role for macroH2A1.2 upstream of RS-associated chromosome breakage, loss of this variant caused a pronounced increase in the Aph-induced phosphorylation of RPA on serine 33 (pRPA-S33), an ATR-dependent, early marker of RS-associated ssDNA (Fig. 4E, Fig. S4E, F). Altogether, these findings demonstrate a protective role for macroH2A1.2 at sites of RS, including but not necessarily limited to CFSs. To determine if the latter involves repair of stalled replication forks in cis, we asked if macroH2A1.2 can counteract RS-associated DDR signaling induced by LacR-induced fork blockage. To mimic macroH2A1.2 accumulation specifically at blocked forks, we fused mCherry-LacR to the macro-domain of macroH2A1.2 (LacR-macro). Tethering of LacR-macro resulted in a significant reduction in γ-H2AX signal at Lac arrays when compared to mCherry-LacR alone (Fig. 4F), supporting a site-specific role for macroH2A1.2 in counteracting RS-associated DNA damage.
FACT promotes RS-specific macroH2A1.2 deposition
We next sought to identify the molecular basis for RS-induced macroH2A1.2 deposition. Although macro-histones were discovered more than two decades ago, their incorporation into chromatin remains enigmatic (Buschbeck and Hake, 2017). Given the RS-dependent nature of CFS-associated macroH2A1.2 accumulation, we focused on the FACT histone chaperone complex, which associates with the replicative helicase MCM, promotes removal and reassembly of histone H2A/H2B dimers at replication forks, and protects from RS sensitivity in yeast and human cells (Herrera-Moyano et al., 2014; Kurat et al., 2017; Tan et al., 2006; Yang et al., 2016). To assess a potential role for FACT in macroH2A1.2 variant deposition, we analyzed macroH2A1.2 levels at LacR-associated RS foci in the presence or absence of either of the two FACT subunits, SUPT16H or SSRP1, and observed a pronounced decrease in macroH2A1.2 accumulation specifically in EdU+ cells (Fig. 5A, B, Fig. S5A). Next, we performed macroH2A1.2 ChIP to assess macroH2A1.2 accumulation at endogenous sites of RS upon depletion of SUPT16H with two independent shRNAs. Confirming our Lac array data, SUPT16H was strictly required for both Aph- and HU-induced macroH2A1.2 deposition at CFSs, whereas SUPT16H depletion had little to no effect on overall macroH2A1.2 levels in the absence of RS or at a non-fragile control region (Fig. 5C, Fig. S5B-E). Of note, FACT depletion did not alter overall nucleosome density, as H2B levels remained unchanged (Fig. S5F). Moreover, in contrast to macroH2A1.2, RS-dependent γ-H2AX abundance was moderately increased in cells depleted of SUPT16H (Fig. 5C), consistent with previous work demonstrating a role for FACT in fork protection (Herrera-Moyano et al., 2014). To determine if FACT can directly act on macroH2A1.2, we compared FACT binding to H2A/H2B and macroH2A1.2/H2B histone dimers, using a fluorescence polarization assay that permits precise quantification of dissociation constants (Kd) for FACT:histone interactions in vitro (Winkler et al., 2011). These analyses demonstrate that FACT can bind macroH2A1.2/H2B histone dimers with an affinity that is only moderately weaker than that for major-type histones (Fig. 5D, E). To determine if FACT can interact with macroH2A1.2 in vivo, we performed co-IP experiments in 293 cells with or without a stably integrated single copy of Flag-macroH2A1.2. Expression of Flag-macroH2A1.2 was comparable to that of endogenous macroH2A1.2 and Flag IP efficiently co-purified both SSRP1 and SUPT16H in Flag-macroH2A1.2 expressing cells but not parental control cells (Fig. 5F).
Figure 5. FACT and H2AX phosphorylation are required for macroH2A1.2 accumulation at sites of RS.
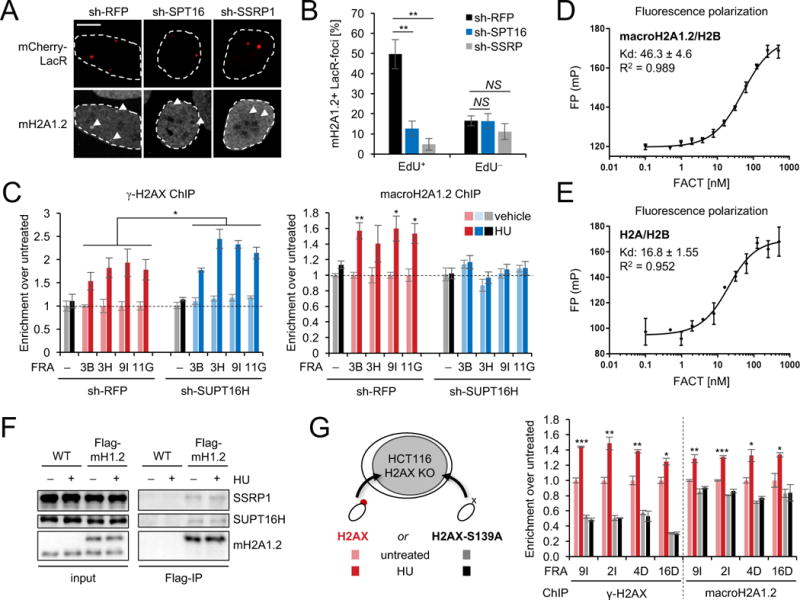
(A) MacroH2A1.2 recruitment to LacR-occupied LacO arrays in the presence or absence of shRNA-mediated depletion of SUPT16H (SPT16) or SSRP1 (SSRP). (B) The percentage of macroH2A1.2+ foci in (A), determined as described in Fig. 2C. Scale bar: 10 μM. (C) ChIP analysis for γ-H2AX (left) or macroH2A1.2 (right) in K562 cells expressing the indicated shRNAs in the presence (dark bars) or absence of HU (light bars). Enrichment at CFSs was normalized to untreated cells, a NFS served as control. (D, E) Fluorescence polarization measurements of FACT interaction with macroH2A1.2/H2B (D) or H2A/H2B dimers (E). Representative binding curves are shown. Analyses were performed in quadruplicate, Kd values represent mean and SEM (n = 3). mP: milliPolarization. (F) Flag-IP of the indicated proteins in 293 cells with (Flag-mH1.2) or without (WT) stably integrated Flag-macroH2A1.2. (G) ChIP analysis for γ-H2AX or macroH2A1.2 in H2AX WT (H2AX) or S139A mutant (H2AX-S139A) HCT116 cells at the indicated CFSs in the presence or absence of HU. Values are expressed as mean and SEM (n = 3), * p<0.05, ** p<0.01, *** p<0.001 by Student’s two-tailed t-test.
Given the co-dependence of macroH2A1.2 deposition on both FACT and ATM/ATR activity, we next asked if the two processes are functionally linked. Of note, FACT was previously found to replace nucleosomal H2AX with canonical H2A in an exchange process facilitated by H2AX phosphorylation on S139 (Heo et al., 2008). We, thus, assessed if macroH2A1.2 deposition similarly depends on the phosphorylation of H2AX, using H2AX-deficient HCT116 cells reconstituted with either WT H2AX or an S139A phospho-mutant (Weyemi et al., 2016). While RS-induced macroH2A1.2 accumulation at CFSs was readily observed in HU-treated WT H2AX expressing cells, this process was blunted in H2AX KO cells as well as two independent KO clones expressing the S139A mutant (Fig. 5G, Fig. S5G). The absence of H2AX phosphorylation upon RS in S139A mutants was confirmed by γ-H2AX ChIP (Fig. 5G). Taken together, these findings are consistent with a model wherein H2AX phosphorylation coordinates FACT-dependent macroH2A1.2 deposition at sites of RS, likely involving γ-H2AX exchange upon DDR activation. FACT is, to our knowledge, the first histone chaperone to be implicated in macroH2A1.2 incorporation on chromatin. However, as neither mechanism nor structure of the FACT complex are known, it is not possible to separate its putative assembly activity from its putative disassembly activity, both of which have been invoked to explain the role of FACT during replication fork progression (Kurat et al., 2017; Yang et al., 2016). Given that FACT generally prevents non-productive histone interactions with DNA to facilitate nucleosomal histone-DNA interactions, we conclude that FACT either directly or indirectly contributes to the assembly of macroH2A1.2-containing nucleosomes at stalled replication forks.
RS shapes macroH2A1.2 domains with replicative age
The observation that RS promotes protective macroH2A1.2 accumulation raises the intriguing possibility that the genome-wide overrepresentation of macroH2A1.2 at fragile sites (see Fig. 1F) may be a consequence of repeated genomic insults to better deal with inherent CFS susceptibility to RS. To determine if cumulative cell divisions can cause an accrual of macroH2A1.2 at CFSs, we took advantage of human primary fibroblasts, which can be cultured for a finite number of population doublings (PDs) and exhibit extensive, yet poorly understood chromatin changes in late passage cells (Cruickshanks et al., 2013; Shah et al., 2013). In agreement with our findings in tumor cells, RS caused robust, CFS-specific macroH2A1.2 accumulation in BJ fibroblasts, which correlated with γ-H2AX induction (Fig. 6A, Fig. S6A). To assess the consequences of repeated rounds of replication in the absence of exogenous RS, we next compared macroH2A1.2 accumulation at eight RS-prone CFSs and two non-fragile control loci in mid (PD 50) and late passage BJ cells (PD 70). While control loci exhibited stable macroH2A1.2 levels, we observed a significant, PD-dependent increase in macroH2A1.2 at all eight CFSs (Fig. 6B). The latter was not the result of altered nucleosome occupancy as H2B levels remained unaltered (Fig. S6B). To establish a causal relationship between cell division and macroH2A1.2 accumulation at CFSs, we exposed PD 50 BJ cells to serum starvation for the time equivalent of 20 PDs, which resulted in minimal replication (5 PDs) without altering chronological age. Demonstrating dependence on continued cycling, macroH2A1.2 accumulation was significantly reduced in starved compared to replicating cells (Fig. 6B). The latter was not due to differences in cell cycle distribution between starved and cycling cells, as short-term starvation in PD 70 BJ cells did not alter macroH2A1.2 enrichment when compared to non-starved PD 70 cells (Fig. S6C, D). The observed 40 – 60% increase in macroH2A1.2 levels in late passage cells is consistent with a cumulative, 1 – 2% increase in CFS-associated macroH2A1.2 per S phase. We thus conclude that, even though macroH2A1.2 accumulation upon RS is largely transient (Fig. S2A), consistent with a prime-access-restore model of DNA repair (Alabert and Groth, 2012; Soria et al., 2012), a small fraction of RS-induced macroH2A1.2 is retained after resolution of RS to continously shape RS-associated chromatin.
Figure 6. Continuous replication shapes macroH2A1.2 domains in primary cells to protect from DDR-induced senescence.
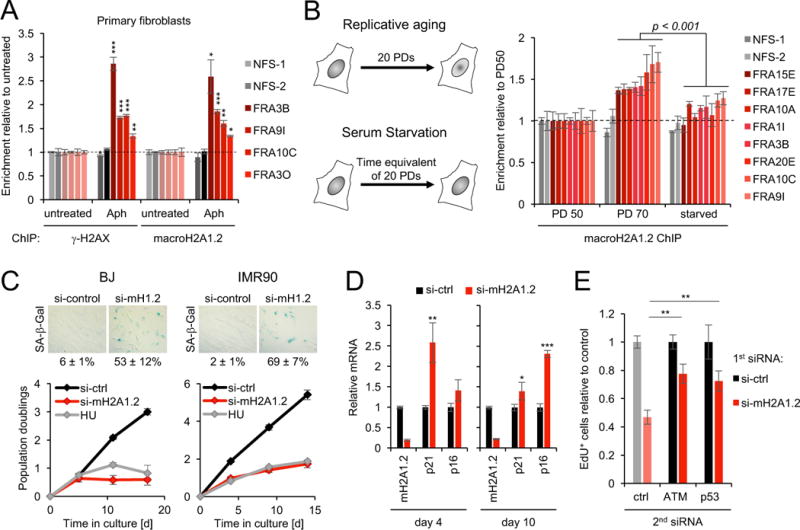
(A) ChIP analysis for γ-H2AX or macroH2A1.2 in primary BJ fibroblasts in the presence or absence of Aph. Enrichment over untreated samples is shown for the indicated CFSs or NFS controls. (B) ChIP analysis for macroH2A1.2 in mid (PD 50) or late passage fibroblasts (PD 70) or PD 50 cells that were starved for the equivalent of 20 PDs (see schematic). Enrichment relative to PD 50 is shown for the indicated CFSs and two NFS control loci. (C) PD analysis of BJ and IMR90 fibroblasts in the presence or absence of macroH2A1.2 knockdown or HU, values are expressed as mean and SEM (n ≥ 4). Representative SA-β-Gal stainings from 12 days post siRNA transfection are shown. Scale bar: 100 μm. (D) Relative expression of macroH2A1.2, p21 and p16 in IMR90 cells at the indicated days after siRNA transfection. Samples were analyzed in triplicate, values are expressed as mean and SD. (E) EdU incorporation in IMR90 cells upon macroH2A1.2 knockdown in the presence or absence of a second siRNA directed against ATM or p53. Unless noted otherwise, values are expressed as mean and SEM (n = 3). * p < 0.05, ** p<0.01, *** p<0.001 by Student’s two-tailed t-test.
MacroH2A1.2 protects from RS-induced senescence in primary cells
RS and the concomitant DDR activation were found to cause irreversible growth arrest (senescence) in primary cells, which has in turn been linked to tissue degeneration, modulation of malignant transformation and aging (Bartkova et al., 2006; Di Micco et al., 2006; He and Sharpless, 2017). We, thus, asked if the RS-protective role of macroH2A1.2 identified above has implications for cellular senescence. Consistent with this notion, siRNA-mediated macroH2A1.2 depletion resulted in a pronounced growth arrest in both BJ and IMR90 fibroblasts, which coincided with the expression of senescence-associated β-galactosidase (SA-β-Gal) (Fig. 6C). A similar growth defect was observed with a second, independent siRNA (Fig. S6E). The senescent phenotype was further corroborated by the induction of the cyclin-dependent kinase inhibitor p16 in macroH2A1.2-depleted IMR90 fibroblasts (He and Sharpless, 2017) (Fig. 6D). Of note, macroH2A1.2 inactivation mirrored the effect of the replication poison HU on cell growth and further resulted in increased expression of the p53 target p21, supporting a link to RS-associated DNA damage signaling (Figs. 6C, D). DDR signaling via ATM/ATR and the concomitant activation of p53 are central mediators of RS-induced senescence, both upon oncogene-induced hyper-replication and upon loss of RS response mediators, including BRCA1 (Bartkova et al., 2006; Cao et al., 2006; Di Micco et al., 2006). We thus asked if activation of ATM and/or p53 are similarly implicated in macroH2A1.2 loss-induced cell cycle arrest. In support of this notion, depletion of either protein was able to partially rescue the effect of macroH2A1.2 loss, as measured by an increased ability to incorporate EdU (Fig. 6E, Fig. S6F, G). Together, these findings identify a role for macroH2A1.2 in preventing irreversible cell cycle arrest, which can be at least in part attributed to protection from excessive, RS-associated DDR activation. However, more work is needed to understand the full extent of macroH2A1.2 function in the context of cellular senescence.
Discussion
Here, we identify the macroH2A1.2 histone variant as an epigenetic mediator of the RS response. MacroH2A1.2 accumulates preferentially at fragile genomic regions in a manner that depends on DNA damage-induced, FACT-dependent chromatin remodeling. Loss of macroH2A1.2 results in impaired BRCA1 accumulation at stalled forks and a concomitant increase in RS-associated damage load, which in turn causes increased genome instability in tumor cells and aberrant senescence in primary cells. Altogether, our findings are consistent with a model wherein cumulative RS is necessary to shape a chromatin environment that facilitates an improved RS response in subsequent cell divisions.
Despite much effort, the physiological relevance of macroH2A variants has remained enigmatic. Recent findings point to a role in chromatin reorganization that can affect both DNA repair outcome and the control of gene expression and cellular differentiation (Buschbeck and Hake, 2017; Gamble and Kraus, 2010). In agreement with previous work, the genome-wide mapping of macroH2A1.2 described here highlights a “superdomain” appearance, with regions of enrichment often spanning several hundred kilobases (Chen et al., 2014) (Fig. 1D). The latter is reminiscent of the spread of γ-H2AX domains upon DNA damage and fits well with the notion that macroH2A1.2 domains function beyond the modulation of gene expression (Gamble and Kraus, 2010; Iacovoni et al., 2010). We now elucidate a molecular rationale for the formation of at least a subset of these macroH2A1.2 superdomains. Together with previous work identifying FACT-mediated removal of γ-H2AX from nucleosomes (Heo et al., 2008), our findings suggest a model in which RS triggers an exchange of some of the DNA damage-induced γ-H2AX variants for macroH2A1.2, thereby bookmarking damage-prone regions within the more broadly distributed H2AX domains (Iacovoni et al., 2010).
Like H2AX, macroH2A1.2 serves as a platform for repair factor accumulation, with BRCA1 being a key effector (Khurana et al., 2014; Leung et al., 2017), thus providing a chromatin environment optimized for the resolution of RS. It will be of interest to determine if other RS effectors similarly depend on macroH2A1.2 and conversely, if macroH2A1.2-associated proteins identified in the context of DSB repair are involved in the RS response. Consistent with the latter, loss of ZMYM3, a DSB repair mediator that physically links BRCA1 to H2A variants including macroH2A1.2, was recently found to cause increased sensitivity to the replication poison CPT (Leung et al., 2017). Of note, the defect in BRCA1 accumulation upon macroH2A1.2 loss is most apparent upon RS (Fig. 3A, B), consistent with the notion that BRCA1 binding to chromatin is controlled by various additional chromatin modifications and remodeling events, which ensure efficient BRCA1 recruitment to sites of damaged DNA (Price and D’Andrea, 2013). Given that macroH2A1 was recently identified as an E3 ligase target for BRCA1, it is further tempting to speculate that BRCA1 by itself may be able to modulate its interaction with macroH2A1.2 in vivo (Kim et al., 2017).
Importantly, our findings indicate that repeated DNA replication results in a cumulative increase in macroH2A1.2 at CFSs, particularly in late passage fibroblasts. The latter is consistent with the observation that, unlike most core histones, macroH2A1.2 levels do not decline with replicative age (Feser et al., 2010; O’Sullivan et al., 2010; Sporn et al., 2009). Moreover, macroH2A1.2 has been linked to the formation of senescence-associated heterochromatin foci (SAHFs), a phenotypic hallmark of some senescent cells that involves the condensation of large chromatin domains (Zhang et al., 2005). Given that SAHF formation is frequently observed upon oncogene-induced RS (Narita et al., 2003), it is tempting to speculate that excessive, RS-associated macroH2A1.2 deposition at fragile genomic regions may be causally implicated in this process.
Beyond its potential impact on SAHFs and, hence, senescent state maintenance, we show here that macroH2A1.2 is essential for the prevention of senescence in cycling, primary cells. Senescence induction upon macroH2A1.2 loss can at least in part be attributed to aberrant DDR signaling involving p53 activation and is remarkably similar to the impact of BRCA1 depletion reported previously (Bartkova et al., 2006; Cao et al., 2006; Di Micco et al., 2006). Together, these observations further emphasize the role of macroH2A1.2 as a modulator of RS-associated BRCA1 function and underline the physiological importance of macroH2A1.2-containing chromatin domains. The p53 dependence of macroH2A1.2 loss-induced cell cycle arrest in primary cells may at least in part explain the lack of a growth defect in macroH2A1.2-depleted K562 cells, which are deficient in p53 (Law et al., 1993). However, we cannot exclude the selection of additional compensatory pathways able to counteract RS-associated cell cycle arrest in macroH2A1.2 deficient tumor cells. Of further note, an alternative macroH2A1 splice variant, macroH2A1.1, which differs in 33 aa within the macro-domain, was recently found to have the opposite effect on cell growth. Overexpression of macroH2A1.1 but not macroH2A1.2 was shown to induce senescence by controlling senescence-associated secretory phenotype (SASP) gene expression (Chen et al., 2015). While these findings point to pronounced functional differences between the two macroH2A1 splice variants, their only known structural distinction is the unique ability of macroH2A1.1 to bind (poly−)ADP-ribose (Kustatscher et al., 2005). Dissecting the molecular characteristics as well as physiological implications associated with macroH2A1 splice isoforms will, thus, be an important subject of future investigations. Of note, these differences may have a significant impact on the interpretation of analyses involving the simultaneous inactivation of both isoforms and may at least in part account for the modest phenotypes observed in mice harboring a complete inactivation of the macroH2A1 gene (Pehrson et al., 2014).
Finally, this work is likely to be of relevance for our understanding of malignant transformation, as both genome maintenance defects and cellular senescence are tightly linked to the latter (Campisi, 2013; He and Sharpless, 2017; Tubbs and Nussenzweig, 2017). Consistent with the RS-protective and, hence, growth-promoting impact of macroH2A1.2, its expression is frequently increased in tumor tissue, while macroH2A1.1 is reciprocally decreased (Novikov et al., 2011). This points to a splicing-regulated, proto-oncogenic role for the macroH2A1.2 variant and suggests manipulation of macroH2A1.2 expression as a potential therapeutic means to interfere with tumorigenesis. Altogether, our findings establish RS as a driver of a macroH2A1.2-dependent, genome-protective chromatin reorganization, which may be exploited to manipulate both cell growth and genome integrity in health and disease.
STARS Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-macroH2A1.2 | Millipore | Millipore Cat# MABE61, RRID:AB_10807977 |
| Anti-macroH2A1 | Millipore | Millipore Cat# 07-219, RRID:AB_11214187 |
| Anti-phospho-H2A.X (S139A) | Millipore | Millipore Cat# 05-636, RRID:AB_309864 |
| Anti-H2A.X | Abcam | Abcam Cat# ab20669, RRID:AB_445689 |
| Anti-H2B | Abcam | Abcam Cat# ab52484, RRID:AB_1139809 |
| Normal mouse IgG | Millipore | (Millipore Cat# 12-371, RRID:AB_145840 |
| Anti-BRCA1 | Santa Cruz | Santa Cruz Biotechnology Cat# sc-6954, RRID:AB_626761 |
| Anti-PCNA | Santa Cruz | Santa Cruz Biotechnology Cat# sc-56, RRID:AB_628110 |
| Anti-GAPDH | Santa Cruz | Santa Cruz Biotechnology Cat# sc-32233, RRID:AB_627679 |
| Anti-SPT16 | Santa Cruz | Santa Cruz Biotechnology Cat# sc-28734, RRID:AB_661341 |
| Anti-EZH2 | Cell Signaling Technologies | Cell Signaling Technology Cat# 5246, RRID:AB_10694683 |
| Anti-H2A | Cell Signaling Technologies | Cell Signaling Technology Cat# 12349, RRID:AB_2687875 |
| Anti-H2A | Abcam | Abcam Cat# ab15653, RRID:AB_732909 |
| Anti-SSRP1 | Cell Signaling Technologies | Cell Signaling Technology, Cat# 13421, RRID:AB_2714160 |
| Anti-phospho-ATM (Ser1981) | Cell Signaling Technologies | Cell Signaling Technology Cat# 4526, RRID:AB_2062663 |
| Anti-phospho-Chk2 (Thr68) | Cell Signaling Technology | Cell Signaling Technology Cat# 2661, RRID:AB_331479 |
| Anti-H2A.Z | Abcam | Abcam Cat# ab4174, RRID:AB_304345) |
| goat anti-mouse IgG-HRP | Santa Cruz | Santa Cruz Biotechnology Cat# sc-2005, RRID:AB_631736 |
| goat anti-rabbit IgG-HRP | Santa Cruz | Santa Cruz Biotechnology Cat# sc-2004, RRID:AB_631746 |
| Anti-phospho-H2A.X (S139A) | Abcam | (Abcam Cat# ab11174, RRID:AB_297813 |
| Anti-phospho-RPA32 (S33) | Bethyl | Bethyl Cat# A300-246A, RRID:AB_2180847 |
| Anti-phospho-RPA32 (S4/S8) | Bethyl | Bethyl Cat# A300-245A, RRID:AB_210547 |
| Anti-RPA2 | Millipore | Millipore Cat# NA19L, RRID:AB_565123 |
| Anti-FANCD2 | Novus | Novus Cat# NB100-182, RRID:AB_10002867 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| MNase (Micrococcal nuclease) | Life Technologies | 88216 |
| Aphidicolin | Sigma | A0781 |
| Hydroxyurea | Sigma | H8627 |
| KU-55933 | Millipore | 118500 |
| VE-821 | AdooQ Bioscience | A11605 |
| EdU (5-ethynyl-2’-deoxyuridine) | Life Technologies | E10187 |
| Biotin Azide (PEG4 carboxamide-6-Azidohexanyl Biotin) | Life Technologies | B10184 |
| Streptavidin-MyOne C1 beads | Life Technologies | 65001 |
| Alexa Fluor 488-azide | Life Technologies | A10266 |
| 7-AAD (7-Amino-Actinomycin D) | BD Pharmingen | 51-68981E |
| PI/RNase staining buffer | BD Pharmingen | 550825 |
| Hoechst 33342 | Life Technologies | H3570 |
| cOmplete Mini EDTA-free protease inhibitor | Roche | 11836170001 |
| Critical Commercial Assays | ||
| Click-iT EdU Alexa Fluor 647 Imaging Kit | Life Technologies | C10340 |
| FITC BrdU Flow Kit | BD Bioscience | 559619 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling Technology | 9860S |
| Phosphatase inhibitor cocktail set II | Millipore | 524625 |
| Lipofectamine2000 | Life Technologies | 11668019 |
| SuperscriptIII | Life Technologies | 18080044 |
| TURBO DNase | Life Technologies | AM1907 |
| Deposited Data | ||
| Raw and processed data (ChIP-Seq) | This study | GSE104800 |
| Experimental Models: Cell Lines | ||
| K-562 | ATCC | ATCC Cat# CCL-243, RRID:CVCL_0004 |
| BJ | ATCC | ATCC Cat# CRL-2522, RRID:CVCL_3653 |
| IMR-90 | Coriell | Coriell Cat# I90-15, RRID:CVCL_0347 |
| HCT116 H2A.X KO cells | W. Bonner | |
| U2OS-LacO cells | T. Misteli | |
| T-REx™-293 Cell Line | Thermo Fisher Scientific | Thermo Fisher Scientific Cat#: R78007, RRID:CVCL_U427 |
| HEK293T | The Broad Institute | |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| shRNA targeting sequences, see Table S2 | This study | N/A |
| siRNA targeting sequences, see Table S2 | This study | N/A |
| gRNA for CRISPR/Cas9 KO, see Table S2 | This study | N/A |
| Primers for ChIP-qPCR, see Table S3 | This study | N/A |
| Primers for RT-PCR, see Table S3 | This study | N/A |
| Primers for CRISPR screening, see Table S3 | This study | N/A |
| Recombinant DNA | ||
| Cherry-LacRep | Addgene | 18985 |
| Cherry-LacR-macro1.2 | This study | N/A |
| pSpCas9n(BB)-2A-Puro (PX459) V2.0 | Addgene | 62987 |
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | 48138 |
| GST-BRCA1 fragments | D. Livingston | Scully et al., 1997 |
| Flag-macroH2A1.2 | A. Ladurner | |
| Flag-H2A | T. Sixma | |
| Software and Algorithms | ||
| UCSC genome browser | Kent et al., 2002 (PMID: 12045153) | https://genome.ucsc.edu |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| CometScore | TriTek | |
| FlowJo | FlowJo | https://www.flowjo.com |
| R | R Core Team, 2016 | https://www.r-project.org/ |
| SICER | Zang et al., 2009 (PMID: 19505939) | http://home.gwu.edu/~wpeng/Software.htm |
| Bedtools | Quinlan and Hall, 2010 (PMID: 20110278) | https://github.com/arq5x/bedtools2 |
| bacl2fastq2 | Illumina, San Diego, USA | https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-19.html |
| deeptools | Ramirez et al., 2016 (PMID: 24799436) | https://github.com/fidelram/deepTools |
| Genomation | Akalin et al., 2015 (PMID: 25417204) | http://bioconductor.org/packages/genomation/ |
| Gplots | Warnes et al., 2016 (R package) | https://cran.r-project.org/web/packages/gplots/index.html |
| Bowtie2 | Langmead et al., 2009 (PMID: 19261174) | http://bowtie-bio.sourceforge.net/ |
| Samtools | Li et al., 2009 (PMID: 19505943) | http://samtools.sourceforge.net |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philipp Oberdoerffer (Philipp.Oberdoerffer@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
K562 cells (American Type Culture Collection [ATCC], female) were cultured in RPMI-1640 (Gibco) with 10% BCS (Hyclone), BJ (ATCC, male) and IMR90 fibroblasts (Coriell, female) were cultured in MEM (Gibco), with 10% FBS (Gemini), 2 mM L-Glutamine (Gibco), 1 mM sodium pyruvate (Sigma), 10 mM Non-Essential Amino Acids (NEAA, Gibco), with or without 1% penicillin-streptomycin (Gibco). HCT116 H2AX KO cell lines (male) (Weyemi et al., 2016) were cultured in DMEM (Gibco) with 10% FBS and 1% penicillin-streptomycin. U2OS cells (female) with stably integrated LacO arrays (Burgess et al., 2014) were cultured in DMEM with 10% FBS and 200 μg/ml hygromycin. 293T cells (female, gift from the Broad Institute, Cambridge, MA) were cultured in DMEM supplemented with 10% FBS. Flag-mH2A1.2 expressing T-REx™ 293 cells were generated using the Flp-In system (Thermo Fisher Scientific), macroH2A1.2 cDNA N-terminally conjugated with 3 × Flag was inserted into the pcDNA5/FRT vector and co-transfected into T-REx™ 293 cells with pOG44 plasmid encoding Flp recombinase. Single clones were selected with and 100 μg/ml hygromycin B. All cells were cultured at 37 °C in a humidified incubator containing 5% CO 2. Cell lines were negative for mycoplasma.
METHOD DETAILS
Cell culture treatments
Plasmid transfections were performed using Lipofectamine 2000 transfection reagent (Life Technologies), cells were analyzed 24 h to 48 h post transfection. Lentiviral infection was carried out by spin infection (2250 rpm, 90 min, Beckman-Coulter Allegra X-12R centrifuge) with 8 μg/ml polybrene (Sigma), cells were incubated overnight prior to virus removal and selection with puromycin (1–2 μg/ml). Individual MISSION shRNA-expressing lentiviral vectors were from Sigma (Table S2). SiRNAs (Dharmacon ON-TARGET, Table S2) were transfected using DF-1 reagent following the manufacturer’s instructions (Dharmacon) and analyzed 48 h – 96 h post transfection. For population doubling analyses, mid-passage BJ and IMR90 cells were serially transfected with 25 nM siRNA to ensure persistent knockdown over the course of the experiment. Cells were harvested and counted every four to five days post transfection and re-transfected on the following day. Where indicated, HU (300 μM) was added upon transfection with control siRNA. For macroH2A1.2 CRISPR/Cas9 knockouts, K562 or 293T cells were transfected with a combination of two guide RNAs (gRNAs) flanking the macroH2A1.2 variant-specific exon 6a to ablate macroH2A1.2 but not macroH2A1.1. 48 h post transfection, cells were treated with 1 μg/mL puromycin for 24 h to select for gRNA #1, followed by FACS sorting of GFP+ cells to select for gRNA #2. Single cell clones were obtained by limited dilution and screened by PCR for exon 6a deletion, gRNAs were designed using the CRISPR Design tool (http://crispr.mit.edu) (Table S2). Drug treatments were performed as follows: unless noted otherwise, HU (Sigma) and Aph (Sigma) were administered for 24 h at 0.3 mM and 0.5 μM, respectively. For a subset of analyses, Aph treatment was performed at 0.3 μM for 16 h, as indicated in the figure legends. For FANCD2 foci analyses in late G2/M cells, cells were treated with 9 μM of the CDK1 inhibitor RO3306 (Millipore) for 16 h in the presence of 0.3 μM Aph and released into normal medium for 1 h (Minocherhomji et al., 2015). For iPOND, cells were treated with 4 mM HU for 2 h. ATM inhibitor KU-55933 (Calbiochem) and ATR inhibitor VE-821 (AdooQ Bioscience) were added at a concentration of 10 μM 30 min prior to Aph or HU treatment.
Antibodies
The following Abs were used for ChIP: α-macroH2A1.2 (Millipore MABE61), α-macroH2A1 (Millipore 07–219), α-phospho-H2AX (Millipore 05–636), α-H2AX (Abcam ab20669), α-H2AZ (Abcam ab4174), α-H2B (Abcam ab52484), normal mouse IgG (Millipore 12–371). Antibodies for IP were α-Flag M2 (Sigma F1804) and α-BRCA1 (Santa Cruz sc-6954). Primary antibodies for western blotting were: α-macroH2A1.2 (Millipore MABE61), α-phospho-H2AX (S139) (Millipore 05–636), α-BRCA1 (Santa Cruz sc-6954), α-PCNA (Santa Cruz sc-56), α-GAPDH (Santa Cruz sc-32233), α-SPT16 (Santa Cruz sc-28734), α-EZH2 (CST 5246s), α-SSRP1 (CST 134215), α-phospho-ATM (CST 4526s), α-phospho-CHK2 (CST 2661s), α-phospho-RPA S4/8 (Bethyl A300-245A), α-RPA2 (Millipore NA19L), α-H2AX (Abcam ab20669), α-H2AZ (Abcam ab4174), α-H2A (CST 12349s, Abcam ab15653). Secondary antibodies were goat anti-mouse IgG-HRP (Santa Cruz sc-2005, Invitrogen 31430) and goat anti-rabbit IgG-HRP (Santa Cruz sc-2004, Invitrogen 31460, CST 7074). Primary antibodies for IF were: α-γ-H2AX (Abcam ab11174, Milipore 05–636), α-BRCA1 (Santa Cruz, sc-6954), α-macroH2A1.2 (Millipore, MABE61), α-pRPA S33 (Bethyl, A300-246A), α-FANCD2 (Novus NB100-182). Secondary antibodies were from Life Technologies (goat-α-mouse or goat-α-rabbit conjugated to Alexa Fluor 488, 568, or 647).
Chromatin immunoprecipitation and real time PCR
Cells were treated as indicated, crosslinked with 1% formaldehyde in PBS for 10 min, followed by quenching with 125 mM glycine. Cells were then resuspended in lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% nonidet P-40) to isolate nuclei. Nuclei were resuspended in micrococcal nuclease (MNase) digestion buffer (10 mM Tris pH 7.4, 15 mM NaCl, 60 mM KCl, 0.15 mM spermine, 0.5 mM spermidine) and 1.2 U/μl MNase was added for 30–45 min at 37 °C. The reaction was stop ped by adding 50 mM EDTA and nuclear pellets were resuspended in 10mM Tris-HCl (pH8.0), 100 mM NaCl, 1mM EDTA, 0.5mM EGTA, 0.1% Na-deoxycholate, 0.5% N-lauroylsarcosine. Lysates were sonicated briefly to disrupt nuclear membranes using an ultra sonicator water bath (Bioruptor, Diagenode). Diluted lysates were incubated o/n at 4 °C with the indicated antibodies after addition of 1% Triton X-100. IPs were performed using 30 μl Protein A/G magnetic beads (Pierce). Eluted DNA was purified using the QIAquick PCR purification kit (Qiagen) according to the manufacturer instructions. Purified ChIP DNA was analyzed by qPCR using a LightCycler 480 II (Roche), see Table S3 for primer sequences. All ChIP samples were normalized to input. In ChIP-PCR quantification, the standard comparative cycle threshold method was used to measure the amount of DNA. Where indicated, input-corrected ChIP DNA content was further normalized to a control treatment.
RNA isolation and RT-PCR
Total RNA was extracted using the RNeasy Mini Kit according to the manufacturer’s instructions (QIAGEN). cDNA was synthesized from 0.5–1 μg of total RNA using the Superscript III RT-PCR system (Invitrogen), and expression of the indicated genes was analyzed by quantitative RT-PCR using a LightCycler 480 II (Roche) (see Table S3 for primer sequences).
Immunoblotting, cellular fractionation and co-immunoprecipitation
For whole cell lysates, cells were lysed in RIPA lysis buffer (150 mM NaCl, 1% NP-40, 1% Na-deoxycholate, 0.1% SDS, 25 mM Tris-HCl pH 7.5) supplemented with protease inhibitors (Sigma) and phosphatase inhibitors (Calbiochem). Lysates were sonicated, centrifuged, and heated with reducing sample buffer (375 mM Tris-HCl (pH 6.8), 9% SDS, 50% glycerol, 9% β-mercaptoethanol, 0.03% bromophenol blue). For chromatin fractionation, cells were resuspended in 0.5 ml lysis buffer containing 10 mM HEPES (pH 7.4), 10 mM KCl, 0.5% NP-40 with phosphatase inhibitors (Calbiochem), and protease inhibitors and incubated on ice for 20 min. The cytosolic fraction was separated from the nuclei by centrifugation at 14,000 rpm at 4 °C for 10 min. The nuclear pellet was washed in lysis buffer, pelleted and resuspended in 250 μl low salt buffer, containing 10 mM Tris-HCl (pH 7.4), 0.2 mM MgCl2, 1% Triton X-100 with phosphatase inhibitors and protease inhibitors, and incubated on ice for 15 min. The nucleoplasmic fraction was separated by centrifugation at 14,000 rpm at 4 °C f or 10 min. Chromatin-bound proteins were released from the DNA by addition of 0.2 N HCl and incubation for 10 min on ice, followed by centrifugation at 14,000 rpm at 4 °C fo r 10 min and neutralization with 1M Tris-HCl. For co-IP assays, cells were trypsinized, PBS-washed and lysed in hypotonic buffer (20 mM HEPES [pH 8.0], 10 mM KCl, 2 mM MgCl2, 10% glycerol, 320 mM sucrose, 0.5% NP-40 and protease inhibitors). Nuclei were collected and resuspended in MNase digestion buffer (20 mM HEPES [pH 8.0], 150 mM, KCl, 10% glycerol, 3 mM CaCl2 and protease inhibitors), and treated with MNase (Thermo Fisher Scientific) at 4 °C for 1h. The reaction was terminated with 20 mM EGTA, samples were passed through 27 G needle and centrifuged to remove debris. Supernatant was collected and used for subsequent IP. Nuclear lysate was incubated with Anti-Flag M2 Magnetic Beads (Sigma Aldrich) at 4°C for 1h, and beads were washed with wash buffer (20 mM HEPES [pH 8.0], 150 mM KCl and 0.1% NP-40) 4 times and with 50 mM HEPES (pH 8.0) once. Proteins were eluted by addition of 2 × SDS sample buffer (0.125 M Tris-HCl [pH 6.8], 4% SDS, 20% glycerol and 0.01% bromophenol blue, 5% 2-β-mercaptoethanol, protease/phosphatase inhibitors) and incubation at 95°C for 5 min. Lysates of equal protein amount based on BCA assay were separated by SDS-PAGE and subjected to western blotting. Immunoblotting for SSRP1 and SUPT16H was performed under non-reducing conditions. HRP-conjugated secondary antibodies were used for signal detection by enhanced chemiluminescence (Amersham). Band intensities were quantified using ImageJ software.
In vitro protein-protein interaction assays
Glutathione S-transferase (GST) fusion proteins were expressed in E. coli BL21 strain, affinity-purified, and immobilized on glutathione sepharose 4B beads. Immobilized, purified GST and GST-BRCA1 fragment fusion proteins were incubated for 1 h with nuclear extracts prepared from 293T cells transiently expressing Flag-macroH2A1.2 and treated with or without 4 mM HU for 1 h. After extensive washing with NETN buffer (100 mM NaCl, 1 mM EDTA, 10 mM Tris-Cl, pH 8.0, 0.1% Nonidet P-40, 10% glycerol, and 1 mM dithiothreitol), SDS-PAGE sample buffer was added to the beads, boiled, and separated by SDS-PAGE. Western blots were probed with anti-Flag antibodies and the immobilized GST and GST fusion proteins were visualized by Coomassie Brilliant Blue staining. For nuclear extract preparation, cells were lysed in low salt buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40 with protease inhibitors) and subjected to flash freezing. The cells were thawed at room temperature and spun down at 10,000 g for 10 min. The supernatant was saved as the cytoplasmic fraction. The nuclear pellets were resuspended in low salt buffer and sonicated using an ultra sonicator water bath (Bioruptor, Diagenode). After sonication, cells were pelleted at 10,000 g for and the supernatant was used as nuclear extract for GST pull-down assays.
Fluorescence polarization
Histones were purified from E. coli cells and labeled as previously described (Dyer et al., 2004). H2B was labeled with Alexa488 on mutated residue T112C prior to refolding with the histone partner. FACT was expressed in SF21 cells and purified using a HisTrap column (GE) in buffer containing 20 mM TRIS 8.0, 600 mM NaCl, 5% glycerol, 0.01% n-octyl-β-D-glucoside, 0.01% CHAPS, and 2 mM TCEP in presence of protease inhibitors (Sigma), DNase I, 3 mM CaCl2 and 3 mM MgCl2. FACT was then eluted using the same buffer with 250 mM imidazole, loaded on a Hitrap Q column (GE) in buffer A (20 mM TRIS 8.0, 200 mM NaCl, 0.01% n-octyl-β-D-glucoside, 0.01% CHAPS, 2 mM TCEP) and eluted with buffer B containing 1 M NaCl. The protein was then injected onto a size exclusion column (Superdex 200, GE) in 20 mM TRIS pH 8.0, 600 mM NaCl, 0.01% n-octyl-β-D-glucoside, 0.01% CHAPS, 2 mM TCEP. Protein was concentrated to ~2.5 mg/ml and stored at −80 °C in 20 mM TRIS pH 8.0, 150 mM NaCl, 0.01% n-octyl-β-D-glucoside, 0.01% CHAPS 1 mM TCEP. Fluorescence polarization assays were carried out by combining 5 or 10 nM (macro)H2A/H2B dimers with increasing amounts of FACT (0.1 – 1000 nM) in 20 mM TRIS pH 8.0, 150 mM NaCl, 5% glycerol, 1 mM TCEP, 0.01% n-octyl-β-D-glucoside, 0.01% CHAPS. Fluorescence polarization was measured using a BMG Clariostar plate reader. Data was analyzed using Graphpad Prism, R2 values correspond to a quadratic single site binding model.
Isolation of Proteins on Nascent DNA (iPOND)
iPOND was essentially performed as described previously (Sirbu et al., 2011). In brief, 2 × 108 cells were labelled with 10 μM EdU (Life Technologies) for 10 min to detect nascent forks. For stalled forks, cells were subsequently treated with HU for 2 h in the continued presence of EdU. Cells were crosslinked with 1% formaldehyde for 10 min at room temperature and quenched with 0.125 M glycine. For EdU conjugation with biotin azide, cells were permeabilized with 0.25% Triton X-100 in PBS buffer for 30 min, and then subjected to click–iT reaction buffer (10 mM sodium-L-ascorbate, 20 μM biotin azide [Life Technologies] and 2 mM CuSO4) for 2 h at room temperature. Cells were resuspended in lysis buffer (50 mM Tris-HCl (pH 8.0) and 1% SDS) supplemented with protease inhibitors (Roche), followed by sonication at 4°C using a Bioruptor (Diagenode). After centrifugation, EdU-labeled DNA was immunoprecipitated from supernatants by incubation with streptavidin-MyOne C1 beads (Life Technologies). Beads were washed and captured proteins were eluted by boiling beads in 2 × NuPAGE LDS (Lithium dodecyl sulfate) sample buffer (Invitrogen) containing 200 mM dithiothreitol for 35 min at 95 °C. Proteins were resolved by electrophoresis and d etected by western blot with the indicated antibodies.
Immunostaining and imaging
U2OS cells were fixed in 2% PFA/PBS, followed by permeabilization in 0.1% TritonX-100/PBS. Cells were subsequently blocked in 5% BSA/PBS + 0.1% Tween-20 (PBS-T). 293T cells were plated on glass coverslips coated with poly-L-lysine (Sigma) prior to treatment and staining. K562 cells were harvested, washed, fixed and permeabilized in PTEMF buffer (20 mM PIPES (pH 6.8), 10 mM EGTA, 0.2% Triton X-100, 1 mM MgCl2 and 4% paraformaldehyde) at room temperature on a shaker. Fixed cells were washed twice in PBS and blocked for 1 h at room temperature (3% BSA, 0.5% Triton X-100 in PBS). Following blocking, cells were washed twice and resuspended with water. Cells were layered onto glass coverslips coated with 0.1% gelatin at 37 °C until dry prior to antibody incubation. Primary and secondary antibody stainings were carried out in PBS-T. Confocal z-stacks were acquired using a Zeiss LSM 780 microscope and a 40X oil objective (NA=1.4). Images were displayed and analyzed as maximum intensity projections. Cells were analyzed 24 h (Fig. 2B) or 48 h post mCherry-LacR transfection. For S-phase discrimination, cells were pulsed with 10 μM EdU (Life Technologies) at 37°C, 30 mins prior to fixation. Incorporated EdU was Click-labeled using azide-linked Alexa Fluor-647 (Life Technologies).
SA-β-Galactosidase staining
SA-β-Gal staining was performed on day 12 of population doubling analyses in siRNA transfected BJ or IMR90 fibroblasts using the Beta-galactosidase staining kit (Cell signalling technology) according to manufacturer’s instructions. After color development, cells were layered using 70% glycerol, mounted using cover slips and imaged using a Zeiss AxioObserver Z1 wide-field microscope. The percentage of SA-β-Gal+ cells was calculated as the fraction of blue cells, a minimum of 300 cells were analyzed per sample.
Comet assay
Alkaline comet assay was performed according to the manufacturer’s protocol (Trevigen). Briefly, cells were resuspended in molten agarose and distributed onto CometSlides (Trevigen). Slides were immersed in cold lysis solution (Trevigen) for 60 min followed by immersion in alkaline unwinding solution (0.2 M NaOH, 1 mM EDTA) for 20 min. The processed slides were subjected to gel electrophoresis in alkaline electrophoresis buffer (0.2 M NaOH, 500 mM EDTA pH 8.2). Slides were dried overnight after washing with water and 70% ethanol. Cells were stained with 100 μl of 1X SYBR gold (Trevigen) for 30 minutes. Slides were rinsed and dried, images were acquired on the Delta Vision Core system (GE Healthcare) consisting of a Olympus IX70 inverted microscope (Olympus America) with 100X NA 1.4 oil immersion objective and a CoolSnap HQ 12-bit camera (Photometrics). Comet tail DNA was quantified using CometScore IV (Perceptive Instruments). To visualize low frequency breaks, images were subjected to auto-threshold using CometScore IV.
Cell cycle analyses
For propidium-iodide (PI) based cell cycle analyses, cells were treated as indicated and fixed in 70% ethanol at −20 °C overnight. Fixed cells were resuspended in PI/RN ase staining buffer (BD Pharmingen) at RT for 20 min. DNA content was analyzed by flow cytometry (FACS Calibur, BD). For flow cytometry-based analyses of DNA synthesis, cells were treated with EdU (10 μM) or BrdU (10 μM, BD Pharmingen) for 4 h, fixed with Cytofix/Cytoperm (BD) and permeabilized with Perm/Wash buffer (BD). EdU was detected following incubation with EdU Click-iT reaction buffer (10 mM sodium-L-ascorbate, 5 μM Alexa Fluor 488-azide [Invitrogen] and 2 mM CuSO4) at RT for 30 min. BrdU was detected using a FITC-conjugated antibody (BD Pharmingen #51-2354AK) and DNA content was determined by 7-AAD. For IF-based analyses, IMR90 fibroblasts were treated 10 μM EdU for 16 h, fixed in 4% PFA/PBS, followed by permeabilization in 0.1% TritonX-100/PBS. Incorporated EdU was Click-labeled using azide-linked Alexa-Fluor-488 and total DNA was visualized using Hoechst 33342 (Life Technologies). EdU mean fluorescence intensity was quantified following Hoechst-labeled nuclear segmentation. EdU+ cells were determined based on the distribution of EdU fluorescence intensity.
CFS mapping
A list of common fragile sites (CFSs) and their cytobands was obtained from (Mrasek et al., 2010). The cytoband coordinate file from the UCSC genome browser was used to translate cytobands to genomic coordinates: (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/cytoBand.txt.gz). CFSs where cytoband nomenclature was not consistent between data sources were manually curated. Details of coordinate conversion are provided in Table S1. CFSs were further screened for potential alignment artifacts, by representation in the ENCODE “Blacklist regions”: http://www.broadinstitute.org/~anshul/projects/encode/rawdata/blacklists/hg19-blacklist-README.pdf. To create a set of non-fragile sites (NFSs) for comparison, we took genomic regions that were not identified as fragile in (Mrasek et al., 2010), or overlapping ENCODE blacklist regions. The percent of the respective region covered by ChIP-Seq peaks was calculated using the genomeCoverage method in bedtools (Quinlan and Hall, 2010). Regions within the CFS for which sequencing data are not available were removed by intersecting with hg19 sequencing gaps. CFS-associated break and gap frequencies were determined based on 10 metaphase spreads from K562 cells treated with 0.5 μM Aph for 24 h (Table S1).
ChIP-Seq analysis
Library construction and sequencing
Sequencing libraries were constructed from DNA samples (ChIP and control samples) with the Illumina TruSeq V3 library construction protocol (Illumina, San Diego, USA). Sequencing runs were performed on an Illumina HiSeq 2000 for 51 cycles in single-end mode using Illumina TruSeq V3 chemistry (flowcells C56J3ACXX and C56KBACXX), or on an Illumina NextSeq 500 for 76 cycles in single-end mode using NextSeq V4 chemistry (flowcells H0Y1PBGXX, H11P6BGXX, and H13V2BGXX) (Table S4).
Read processing and alignment
Reads were adapter trimmed during the demultiplexing stage by the bacl2fastq2 software (v2.15, Illumina). Processed reads were aligned to the hg19 assembly with Bowtie2 v2.1.0 (Flowcell H0Y1PBGXX) or v2.2.3 (all others). Default alignment parameters were used, reporting a single best alignment where there are multiple alignments possible.
Peak calling and analysis
Peak calling was performed with SICER (v1.1) (Zang et al., 2009), with a window size of 200 bp and a gap size of 600 bp. For each ChIP sample, peak calling was performed relative to the respective input sample. We generated sequencing depth and input normalized genome coverage tracks for each experiment in bigWig format with deepTools. Proximity and overlap analysis between peaks and other features was performed using the Bedtools intersectBed and coverageBed functions. Venn diagrams were generated from intersection results with the VennDiagram Bioconductor package. Peak overlap between two ChIP-Seq data is presented as the number of bases common between the peaks from each experiment. We used the Jaccard Index as a measure of similarity between peak sets, as it provides a metric that is comparable across sets of varying size, Jaccard Indices were calculated using bedtools (Quinlan et al., 2010), and are defined as the bases in the intersection divided by the bases in the union minus the intersection, producing values bounded by zero and 1 (no overlap and complete overlap, respectively). To determine whether this metric was different than expected by chance, we performed random shuffles of both peak sets (N=1,000 shuffles) and collected the permuted Jaccard values. When performing random shuffles, centromeres and assembly gaps were excluded from placement of random peaks. Summaries of signal within coordinate ranges were extracted from bigWig files using custom wrappers with UCSC executables. Summaries of ChIP enrichment over genes (“metagenes”), and over peak subsets, were computed with deepTools. Correlations of peak subsets with repeat elements or replication origins were performed with Genomation (Akalin et al., 2015). Repeat locations were retrieved from the UCSC genome browser, selecting repeat classes with at least 10% overlap with any peak subset to display. Replication origins were identified based on (Smith et al., 2016). Heatmaps were generated using heatmaps.2 function within the R package gplots.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details, including definition of significance, statistical methods used, exact n values of sample size and definition of center, and dispersion and precision measures are indicated in the figure legends.
DATA AND SOFTWARE AVAILABILITY
ChIP-Seq data have been deposited on Gene Expression Omnibus (GEO), accession number GSE104800.
Supplementary Material
Table S1, related to Figures 1, 4: CFS break-frequency in Aph-treated K562 cells (see attached Excel file)
Highlights.
DNA damage shapes the epigenome at sites of recurrent replication stress (RS)
The macroH2A1.2 histone forms a BRCA1-permissive, protective chromatin environment
The DDR and FACT coordinate RS-associated macroH2A1.2 accumulation at fragile sites
MacroH2A1.2 loss triggers excessive RS, DNA damage and cellular senescence
Acknowledgments
We thank M. Aladjem, T. Misteli and M. Seidman for critical reading of the manuscript, T. Karpova for imaging support, A. Ladurner, D. Livingston, T. Misteli and T. Sixma for reagents. G. E. acknowledges support from the NIH/CU Molecular Biophysics Training Program. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov) and was supported by the Intramural Research Program of the National Institutes of Health (NIH), NCI, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
J. K., S. K. and P. O. conceived the project. J. K. performed ChIP, western blot, cell cycle analyses, flow cytometry, RT-PCR and iPOND. D. S. performed bioinformatics analyses. S. K. and R. S. performed PD and SA-β-Gal analyses. R. S. performed EdU incorporation and RT-PCR analyses in primary fibroblasts. A. T. and A. K. generated CRISPR KO cell lines, A.T. performed IF analyses with help from W. H. E. H. generated Flag-macroH2A1.2 293T-REx™ cells and performed Co-IP experiments. G. E. and K. L. performed fluorescence polarization assays. S. B. performed metaphase analyses. U. W. and W. B. generated HCT116 H2AX KO cells. P. O. wrote the manuscript with help from J. K., R. S and S.K.
References
- Akalin A, Franke V, Vlahovicek K, Mason CE, Schubeler D. Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics. 2015;31:1127–1129. doi: 10.1093/bioinformatics/btu775. [DOI] [PubMed] [Google Scholar]
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, Groth A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol. 2014;16:281–293. doi: 10.1038/ncb2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, Groth A. Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol. 2012;13:153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, et al. Identification of early replicating fragile sites that contribute to genome instability. Cell. 2013;152:620–632. doi: 10.1016/j.cell.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Beuzer P, Quivy JP, Almouzni G. Establishment of a replication fork barrier following induction of DNA binding in mammalian cells. Cell Cycle. 2014;13:1607–1616. doi: 10.4161/cc.28627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth LN, Brunet A. The Aging Epigenome. Mol Cell. 2016;62:728–744. doi: 10.1016/j.molcel.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RC, Burman B, Kruhlak MJ, Misteli T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014;9:1703–1717. doi: 10.1016/j.celrep.2014.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschbeck M, Hake SB. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol. 2017;18:299–314. doi: 10.1038/nrm.2016.166. [DOI] [PubMed] [Google Scholar]
- Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Kim S, Xiao C, Wang RH, Coumoul X, Wang X, Li WM, Xu XL, De Soto JA, Takai H, et al. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. EMBO J. 2006;25:2167–2177. doi: 10.1038/sj.emboj.7601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol Cell. 2015;59:719–731. doi: 10.1016/j.molcel.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ruiz PD, Novikov L, Casill AD, Park JW, Gamble MJ. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat Struct Mol Biol. 2014;21:981–989. doi: 10.1038/nsmb.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, et al. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013;15:1495–1506. doi: 10.1038/ncb2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabin J, Fortuny A, Polo SE. Epigenome Maintenance in Response to DNA Damage. Mol Cell. 2016;62:712–727. doi: 10.1016/j.molcel.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Dubarry M, Loiodice I, Chen CL, Thermes C, Taddei A. Tight protein-DNA interactions favor gene silencing. Genes Dev. 2011;25:1365–1370. doi: 10.1101/gad.611011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK. Elevated histone expression promotes life span extension. Mol Cell. 2010;39:724–735. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD. A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res. 2012;22:993–1005. doi: 10.1101/gr.134395.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble MJ, Kraus WL. Multiple facets of the unique histone variant macroH2A: from genomics to cell biology. Cell Cycle. 2010;9:2568–2574. doi: 10.4161/cc.9.13.12144. [DOI] [PubMed] [Google Scholar]
- He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017;169:1000–1011. doi: 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo K, Kim H, Choi SH, Choi J, Kim K, Gu J, Lieber MR, Yang AS, An W. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell. 2008;30:86–97. doi: 10.1016/j.molcel.2008.02.029. [DOI] [PubMed] [Google Scholar]
- Herrera-Moyano E, Mergui X, Garcia-Rubio ML, Barroso S, Aguilera A. The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014;28:735–748. doi: 10.1101/gad.234070.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn KA, Minoda A, Tolstorukov MY, Appert A, et al. Comparative analysis of metazoan chromatin organization. Nature. 2014;512:449–452. doi: 10.1038/nature13415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, Legube G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29:1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasencakova Z, Scharf AN, Ask K, Corpet A, Imhof A, Almouzni G, Groth A. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol Cell. 2010;37:736–743. doi: 10.1016/j.molcel.2010.01.033. [DOI] [PubMed] [Google Scholar]
- Khurana S, Kruhlak MJ, Kim J, Tran AD, Liu J, Nyswaner K, Shi L, Jailwala P, Sung MH, Hakim O, et al. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014;8:1049–1062. doi: 10.1016/j.celrep.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Oberdoerffer P. Replication Stress: A Lifetime of Epigenetic Change. Genes (Basel) 2015;6:858–877. doi: 10.3390/genes6030858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Chan DW, Jung SY, Chen Y, Qin J, Wang Y. The Histone Variant MacroH2A1 Is a BRCA1 Ubiquitin Ligase Substrate. Cell Rep. 2017;19:1758–1766. doi: 10.1016/j.celrep.2017.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurat CF, Yeeles JT, Patel H, Early A, Diffley JF. Chromatin Controls DNA Replication Origin Selection, Lagging-Strand Synthesis, and Replication Fork Rates. Mol Cell. 2017;65:117–130. doi: 10.1016/j.molcel.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG. Splicing regulates NAD metabolite binding to histone macroH2A. Nat Struct Mol Biol. 2005;12:624–625. doi: 10.1038/nsmb956. [DOI] [PubMed] [Google Scholar]
- Law JC, Ritke MK, Yalowich JC, Leder GH, Ferrell RE. Mutational inactivation of the p53 gene in the human erythroid leukemic K562 cell line. Leuk Res. 1993;17:1045–1050. doi: 10.1016/0145-2126(93)90161-d. [DOI] [PubMed] [Google Scholar]
- Leung JW, Makharashvili N, Agarwal P, Chiu LY, Pourpre R, Cammarata MB, Cannon JR, Sherker A, Durocher D, Brodbelt JS, et al. ZMYM3 regulates BRCA1 localization at damaged chromatin to promote DNA repair. Genes Dev. 2017;31:260–274. doi: 10.1101/gad.292516.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madireddy A, Kosiyatrakul ST, Boisvert RA, Herrera-Moyano E, Garcia-Rubio ML, Gerhardt J, Vuono EA, Owen N, Yan Z, Olson S, et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol Cell. 2016;64:388–404. doi: 10.1016/j.molcel.2016.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015;528:286–290. doi: 10.1038/nature16139. [DOI] [PubMed] [Google Scholar]
- Mrasek K, Schoder C, Teichmann AC, Behr K, Franze B, Wilhelm K, Blaurock N, Claussen U, Liehr T, Weise A. Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int J Oncol. 2010;36:929–940. doi: 10.3892/ijo_00000572. [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Novikov L, Park JW, Chen H, Klerman H, Jalloh AS, Gamble MJ. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol Cell Biol. 2011;31:4244–4255. doi: 10.1128/MCB.05244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol. 2010;17:1218–1225. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehrson JR, Changolkar LN, Costanzi C, Leu NA. Mice without macroH2A histone variants. Mol Cell Biol. 2014;34:4523–4533. doi: 10.1128/MCB.00794-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino S, Michelena J, Teloni F, Imhof R, Altmeyer M. Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative Chromatin. Cell Rep. 2017;19:1819–1831. doi: 10.1016/j.celrep.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152:1344–1354. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saredi G, Huang H, Hammond CM, Alabert C, Bekker-Jensen S, Forne I, Reveron-Gomez N, Foster BM, Mlejnkova L, Bartke T, et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature. 2016;534:714–718. doi: 10.1038/nature18312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P, Reams C, Simpson LJ, Sale JE. Epigenetic instability due to defective replication of structured DNA. Mol Cell. 2010;40:703–713. doi: 10.1016/j.molcel.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- Shah KA, Mirkin SM. The hidden side of unstable DNA repeats: Mutagenesis at a distance. DNA Repair (Amst) 2015;32:106–112. doi: 10.1016/j.dnarep.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–1799. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith OK, Kim R, Fu H, Martin MM, Lin CM, Utani K, Zhang Y, Marks AB, Lalande M, Chamberlain S, et al. Distinct epigenetic features of differentiation-regulated replication origins. Epigenetics Chromatin. 2016;9:18. doi: 10.1186/s13072-016-0067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria G, Polo SE, Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell. 2012;46:722–734. doi: 10.1016/j.molcel.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Sporn JC, Kustatscher G, Hothorn T, Collado M, Serrano M, Muley T, Schnabel P, Ladurner AG. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene. 2009;28:3423–3428. doi: 10.1038/onc.2009.26. [DOI] [PubMed] [Google Scholar]
- Tan BC, Chien CT, Hirose S, Lee SC. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 2006;25:3975–3985. doi: 10.1038/sj.emboj.7601271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja N, Zofall M, Balachandran V, Thillainadesan G, Sugiyama T, Wheeler D, Zhou M, Grewal SI. SNF2 Family Protein Fft3 Suppresses Nucleosome Turnover to Promote Epigenetic Inheritance and Proper Replication. Mol Cell. 2017;66:50–62 e56. doi: 10.1016/j.molcel.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell. 2017;168:644–656. doi: 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyemi U, Redon CE, Choudhuri R, Aziz T, Maeda D, Boufraqech M, Parekh PR, Sethi TK, Kasoji M, Abrams N, et al. The histone variant H2A.X is a regulator of the epithelial-mesenchymal transition. Nat Commun. 2016;7:10711. doi: 10.1038/ncomms10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, Scully R. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature. 2014;510:556–559. doi: 10.1038/nature13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DD, Muthurajan UM, Hieb AR, Luger K. Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J Biol Chem. 2011;286:41883–41892. doi: 10.1074/jbc.M111.301465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhang X, Feng J, Leng H, Li S, Xiao J, Liu S, Xu Z, Xu J, Li D, et al. The Histone Chaperone FACT Contributes to DNA Replication-Coupled Nucleosome Assembly. Cell Rep. 2016;14:1128–1141. doi: 10.1016/j.celrep.2015.12.096. [DOI] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–1958. doi: 10.1093/bioinformatics/btp340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figures 1, 4: CFS break-frequency in Aph-treated K562 cells (see attached Excel file)