

Abstract
The cluster of differentiation 36 (CD36) is implicated in the intake of long-chain fatty acids and fat storage in various cell types including the pancreatic beta cell, thus contributing to the pathogenesis of metabolic stress and diabetes. Recent evidence indicates that CD36 undergoes post-translational modifications such as acetylation-deacetylation. However, putative roles of such modifications in its functional activation and onset of beta cell dysregulation under the duress of glucolipotoxicity (GLT) remain largely unknown. Using pharmacological approaches, we validated, herein, the hypothesis that acetylation-deacetylation signaling steps are involved in CD36-mediated lipid accumulation and downstream apoptotic signaling in pancreatic beta (INS-1 832/13) cells under GLT. Exposure of these cells to GLT resulted in significant lipid accumulation without affecting the CD36 expression. Sulfo-n-succinimidyl oleate (SSO), an irreversible inhibitor of CD36, significantly attenuated lipid accumulation under GLT conditions, thus implicating CD36 in this metabolic step. Furthermore, trichostatin A (TSA) or valproic acid (VPA), known inhibitors of lysine deacetylases, markedly suppressed GLT-associated lipid accumulation with no discernable effects on CD36 expression. Lastly, SSO or TSA prevented caspase 3 activation in INS-1 832/13 cells exposed to GLT conditions. Based on these findings, we conclude that acetylation-deacetylation signaling step might regulate CD36 functional activity and subsequent lipid accumulation and caspase 3 activation in pancreatic beta cells exposed to GLT conditions. Identification of specific lysine deacetylases that control CD36 function should provide novel clues for the prevention of beta-cell dysfunction under GLT.
Keywords: CD36, Caspase 3, Glucolipotoxicity, Histone deacetylases, Pancreatic beta cells
1. Introduction
Type 2 diabetes (T2D) is a chronic metabolic disorder, which is manifested by abnormal glucose and lipid homeostasis. Insulin resistance and pancreatic beta-cell dysfunction are the two key contributors for the pathogenesis of T2D. Diabetic patients frequently exhibit hyperlipidemia in addition to hyperglycemia [1]. Chronic exposure of cells to elevated glucose and unsaturated fatty acids (often referred to as glucolipotoxicity; GLT), is a major contributor to the beta-cell dysfunction and progression of T2D [2, 3]. GLT causes excess fat accumulation and functional impairment in several metabolic pathways in adipose tissue as well as pancreas, liver, heart and muscle [2, 4]. Increased circulating lipids and the metabolic alterations in fatty acid utilization and downstream intracellular signaling, have also been implicated in beta-cell dysfunction [1, 3]. Lastly, long-term exposure of primary islets to elevated fatty acid has been shown to cause insulin secretory defects, loss of beta cell mass and apoptosis [5, 6]. Numerous mechanisms have been proposed for GLT-induced beta-cell dysregulation, and these include alterations in lipid metabolism, endoplasmic reticulum stress and mitochondrial dysfunction [7–9]. Interestingly, increased intracellular generation of reactive oxygen species and associated oxidative stress mediated by phagocyte-like NADPH oxidase (Nox2) have also been implicated in metabolic dysfunction and demise of the pancreatic beta cell [10–12].
Cluster of differentiation 36 (CD36), also known as fatty acid translocase (FAT), is a membrane glycoprotein present on many cell types including pancreatic beta-cells. CD36 is a multifunctional protein that facilitates lipid/fat intake, fat taste perception, fat absorption, immunity, and angiogenesis [13]. CD36 is also a receptor for fatty acids and is involved in lipid utilization and storage, thus contributing in the pathogenesis of metabolic disorders including obesity and diabetes [14]. It is noteworthy that CD36 undergoes a variety of post-translational modifications, such as ubiquitination, glycosylation, palmitoylation, acetylation and phosphorylation [15]. Such modifications are likely to exert regulatory effects on subcellular localization and function of CD36. However, little is known with regard to regulation of CD36 function in the pancreatic beta cell.
It has been shown recently that sulfosuccinimidyl oleate (SSO), an irreversible inhibitor of CD36, prevents CD36-mediated fatty acid intake in a variety of cell types. SSO inhibits CD36 function via formation of n-hydroxysuccinimidyl esters with the lysine (Lys) 164 of CD36 in the fatty acid binding pocket leading to a conformational change in CD36 thereby preventing fatty acid intake and/or fatty acid-induced signaling [16, 17]. It should also be noted that the binding pocket of CD36 contains another lysine (Lys-166), which does not bind SSO. Thus, the acetylation-deacetylation of Lys-166 and/or other Lys-52, -231, and -403 residues might dictate access of fatty acids to the binding pocket, thus contributing to regulation of CD36 and it downstream signaling events [14]. Along these lines published evidence also suggests that acetylation-deacetylation steps control function of a number of proteins involved in glucose and fatty acid metabolism potentially resulting in the beta cell dysfunction and the onset of diabetes [18–20]. In this context, published evidence suggests that acetylation-deacetylation of histone and non-histone proteins can directly or indirectly control signaling steps involved in glucose and lipid metabolism [21, 22]. Indeed, recent findings by Daneshpajooh et al., demonstrated that HDAC7 is overexpressed in human diabetic islets and contributes to impaired insulin secretion [23]. Based on the findings reviewed above, we undertook the current investigation to validate the hypothesis that acetylation-deacetylation signaling steps underlie functional activation of CD36 leading to lipid accumulation and caspase 3 activation in pancreatic beta cells under the duress of GLT conditions.
2. Materials and Methods
2.1. Materials
Sulfosuccinimidyl oleate (SSO; CAS No. 135661-44-8; purity ≥95%), valproic acid (VPA; CAS No. 1069-66-5; purity ≥95%) and trichostatin A (TSA; CAS No. 58880-19-6; purity ≥98%) were from Cayman Chemical (Ann Arbor, MI, USA). Anti-CD36 was from Santa Cruz Biotechnology (CA, USA). Antisera directed against cleaved (active) caspase 3 and mouse HRP-conjugated secondary antibodies were obtained from Cell Signaling (Danvers, MA, USA). Antibody for β-actin and all other reagents employed in the current studies were purchased from Sigma Aldrich (St. Louis, MO, USA).
2.2. Cell culture and experimental conditions
INS-1 832/13 cells (kindly provided by Prof. Chris Newgard, Duke University Medical Center) were cultured in RPMI-1640 medium containing 10% heat inactivated FBS supplemented with 100 IU/ml penicillin and 100 IU/ml streptomycin, 1 mM sodium pyruvate, 50 μM 2-mercaptoethanol and 10 mM HEPES (pH 7.4). The cultured cells were sub-cloned twice weekly following trypsinization and passages 55–61 were used for the experiments. To assess the roles of CD36 in lipid accumulation and caspase-3 activation, INS-1 832/13 cells were exposed to glucotoxic (20 mM), liopotoxic (0.5 mM palmitate) and GLT (20 mM glucose plus 0.5 mM palmitate) conditions in the absence or presence of SSO (200 μM), VPA (2.5–5.0 mM) or TSA (0.25–0.5 μM), as indicated.
2.3. Western blotting
Following incubations (above), the cells were harvested and lysed in RIPA buffer containing 1 mg/ml protease inhibitor cocktail, 1 mM NaF, 1 mM PMSF and 1 mM Na3VO4. Cellular lysate proteins (30–50 μg) were separated by SDS-PAGE and electro-transferred onto the nitrocellulose membrane. The membranes were then blocked with 1 % casein in 0.2× PBS for 1 hr. at room temperature. Blots were then incubated overnight at 4 °C with appropriate primary antibody in 0.2× PBS-T containing 0.1 % casein. The membranes were washed three times for 15 min each with PBS-T and probed with appropriate HRP-conjugated secondary antibody in 0.1 % casein in PBS-T at room temperature for 1 hr. After washing, the immune complexes comprised of the target proteins, were detected on X-ray film using Pierce-ECL western blotting substrate Thermo Fisher Scientific (Waltham, MA, USA). The intensity of bands was quantified using Carestream® Molecular Imaging Software.
2.4. Quantification of lipid accumulation
INS-1 832/13 cells were cultured in chamber slides and incubated in a medium containing the low glucose (Con) and GLT (20 mM glucose plus 0.5 mM palmitate) in the absence and presence of SSO (200 μM), VAP (2.5–5.0 mM) or TSA (0.25–0.5 μM) for 24 hrs. At the end of the incubation, media was discarded and cells were washed twice with ice-cold 1× PBS. The cells were then fixed with 10% buffer formalin at room temperature for 1 hrs. Then lipid (Oil Red O) staining was performed using a staining kit (MAK194-1KT, Sigma) as per the manufacturer’s instructions. Slides were mounted with aqueous media using glass cover-slip and stored at 4°C until observed under microscope (Olympus, XI71) at 40x and 100x objectives. Images were captured for each treatment condition. For quantitative analysis 12–15 images were process and % ORO stained area (lipid accumulation) was measured by Image J software (https://imagej.nih.gov/ij/).
2.5. Statistical analysis
All the data were presented as mean ± standard error of mean (SEM) of three independent experimental conditions unless otherwise mentioned. The statistical significance of the differences between the experimental conditions was determined by ANOVA. The p values <0.05 was considered as statistically significant.
3. Results
3.1. Lipotoxic or glucolipotoxic conditions promote lipid accumulation in INS-1 832/13 cells without affecting CD36 expression
At the outset we assessed the effects of gluco-, lipo-, or glucolipotoxic conditions on lipid accumulation in insulin-secreting INS-1 832/13 cells. Data depicted in Figure 1A and S1 indicate that lipotoxic, but not glucotoxic conditions significantly augmented lipid accumulation within 24 hrs. of exposure. It is noteworthy that the lipid accumulation was much more significant in the combined presence of both glucose and palmitate (glucolipotoxicity). We next assessed if increased lipid accumulation is, in part, due to increased expression of CD36, which is known mediate the uptake and storage of lipids in various cell types, including the islet β-cell. Data in Figure 1B and S2 indicate no significant effects of GLT conditions on CD36 expression, especially under conditions of increased lipid accumulation as shown in Figure 1.
Figure 1. Lipotoxic and glucolipotoxic conditions promote lipid accumulation in INS-1 832/13 cells without affecting CD36 expression.
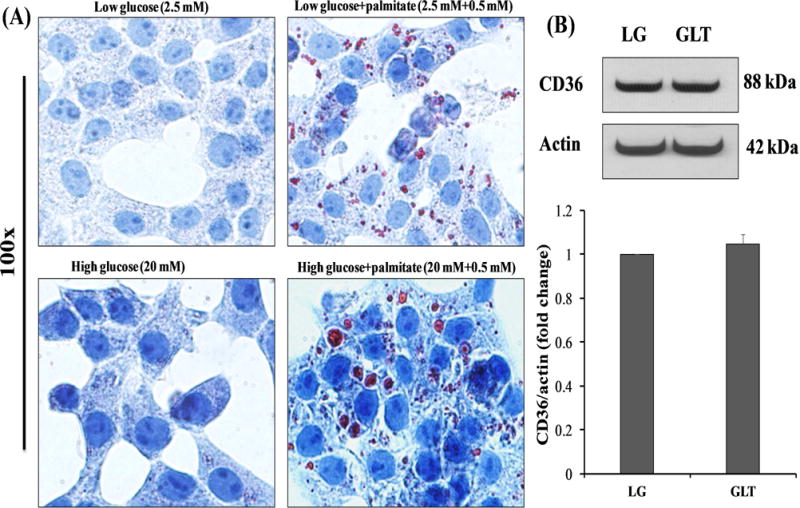
Panel A: INS-1 832/13 cells were cultured in a medium containing low glucose (2.5 mM), high glucose (20 mM) or palmitate (2.5 mM glucose plus 0.5 mM palmitate or 20 mM glucose plus 0.5 mM palmitate) for 24 hrs. Lipid accumulation was determined by Oil-Red-O (ORO) staining in fixed cells and observed under microscope.
Panel B: INS-1 832/13 cells were serum starved (12-15 hours) and then incubated in a medium containing low glucose (2.5 mM) with 3.75% BSA (Con), GLT (20 mM glucose plus 0.5 mM palmitate) for 24 hrs. CD36 expression was quantified by Western blotting and densitometry. β-actin was used as loading control. Data are expressed as mean ± SEM from four independent experiments.
3.2. Sulfo-n-succinimidyl oleate (SSO), an irreversible inhibitor of CD36, attenuates lipid accumulation in pancreatic beta-cells under GLT conditions without affecting the CD36 expression
As a logical extension to the above studies, we asked the question if functional activation (not precisely its expression) of CD36 is requisite for lipid accumulation in INS-1 832/13 cells under conditions of GLT. To address this, we quantified the effects of SSO, a known irreversible inhibitor of CD36 activity, on GLT-mediated lipid accumulation. Data shown in Figure 2 indicated that GLT-mediated lipid accumulation was significantly inhibited in INS-1 832/13 cells following exposure to SSO. Furthermore, compatible with findings in Figure 1, no significant effects of GLT singly or in combination with SSO on CD36 expression were noted in INS-1 832/13 cells (Figure 2; Panel B). Based on these findings, we conclude that functional inactivation of CD36 by SSO leads to GLT-induced lipid accumulation.
Figure 2. SSO, an irreversible inhibitor of CD36, reduces the lipid accumulation without affecting the CD36 expression.
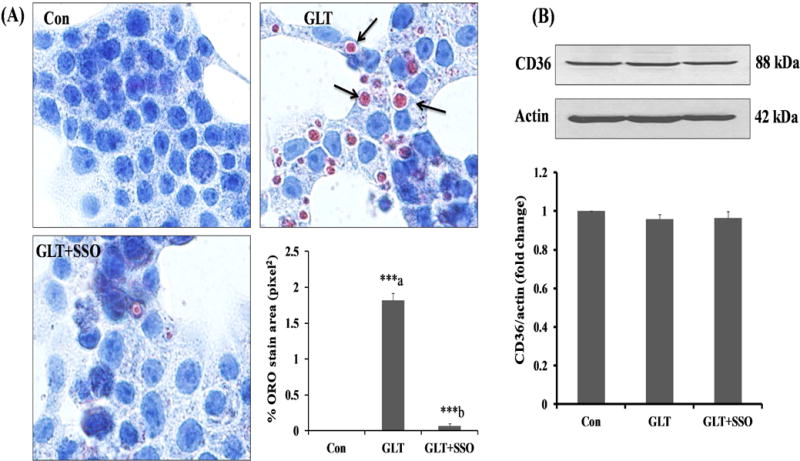
Panel A: INS-1 832/13 cells were incubated in a medium containing low glucose (2.5 mM; Con), GLT (20 mM glucose plus 0.5 mM palmitate) in the absence and presence of SSO (200 μM) for 24 hrs. The cells were treated with SSO for 1 hour before the exposure to GLT. Lipid accumulation was determined by Oil-Red-O (ORO) staining in fixed cells and quantified with Image J software. The arrows indicate the lipid droplets accumulated under GLT and stained intense red color with ORO, whereas treatment with SSO decreased the lipids droplets. Data represent mean ± SEM from 12–15 images of each condition from three independent experiments, and are expressed as % ORO stain area in pixel2. ***p < 0.001 ‘a’ vs. con and ‘b’ vs. GLT.
Panel B: INS-1 832/13 cells were incubated in a medium containing low glucose (2.5 mM) with 3.75% BSA (Con), GLT (20 mM glucose plus 0.5 mM palmitate) in the absence and presence of SSO (200 μM) for 24 hrs. CD36 expression was quantified by Western blotting and densitometry. β-actin is used as loading control. Data are expressed as mean ± SEM from three independent experiments.
3.3. Pharmacologic inhibition of deacetylases prevents GLT-induced lipid accumulation in INS-1 832/13 cells
As stated above, CD36 undergoes posttranslational acetylation-deacetylation [14, 15]. Therefore, we asked if inhibition of lysine deacetylation signaling steps by TSA or VPA prevents GLT-induced lipid accumulation in INS-1 832/13 cells. Data represented in Figure 3 demonstrate that both VPA (2.5 and 5 mM; Panels A) and TSA (0.25 and 0.5 μM; Panels C) significantly reduced the GLT-induced lipid accumulation in these cells. Further, quantitative image analysis indicated that both VPA (Panel B) and TSA (Panel D) significantly reduced the % ORO stained area as compared to GLT condition. Lastly, no significant effects of these two inhibitors on CD36 expression were noted (Panels E and F). These finding indicated that GLT conditions promote deacetylation and functional efficiency of CD36, and inhibition of deacetylation by VPA or TSA leads to inhibition of these signaling steps culminating in inhibition of lipid accumulation, and likely cell dysfunction induced by GLT conditions.
Figure 3. HDAC inhibitors markedly reduce GLT-induced lipid accumulation without affecting the CD36 expression in INS-1 832/13 cells.
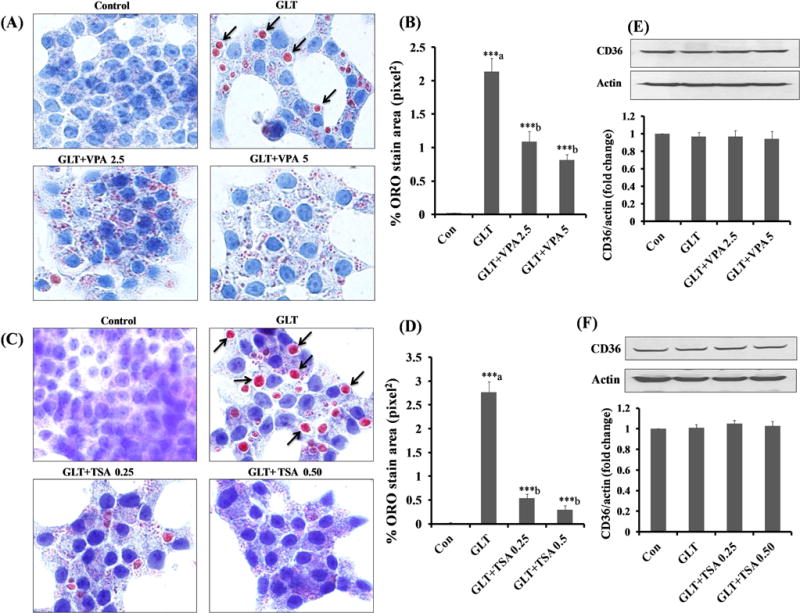
Panel A–D: INS-1 832/13 cells were incubated in a medium containing low glucose (2.5 mM) with 3.75% BSA (Con), glucolipotoxicity (20 mM glucose plus 0.5 mM palmitate) in the absence and presence of VPA (2.5 or 5 mM) and TSA (0.25 and 0.50 μM) for 24 hrs. Lipid accumulation was determined by Oil-Red-O (ORO). The arrows indicate the lipid droplets accumulated under GLT and stained intense red color with ORO, whereas treatment with VPA and TSA decreased the lipids droplets. Data represent mean ± SEM of 12–15 images of each condition from three independent experiments, and are expressed as % ORO stain area in pixel2. ***p < 0.001 ‘a’ vs. con and ‘b’ vs. GLT.
Panel E and F: INS-1 832/13 cells were incubated in a medium containing low glucose (2.5 mM) with 3.75% BSA (Con), GLT (20 mM glucose plus 0.5 mM palmitate) in the absence and presence of VPA (2.5 and 5 mM) and TSA (0.25 and 0.50 μM) for 24 hrs. CD36 expression under these conditions was quantified by densitometry. Data are expressed as mean ± SEM from three independent experiments.
3.4. Functional inactivation of CD36 prevents GLT-induced caspase 3 activation in INS-1 832/13 cells
In the last set of experiments, we asked if pharmacological inhibition of CD36 and lipid accumulation by SSO (Figure 2) or TSA (Figure 3) attenuates GLT-induced caspase 3 activation in INS-1 832/13 cells. Data shown in Figure 4 indicate significant increase in caspase 3 activation in INS-1 832/13 cells under GLT conditions, and that such an increase in caspase 3 activation is sensitive to (or inhibited by) TSA or SSO.
Figure 4. TSA or SSO attenuate GLT-induced caspase 3 activation in INS-1 832/13 cells.
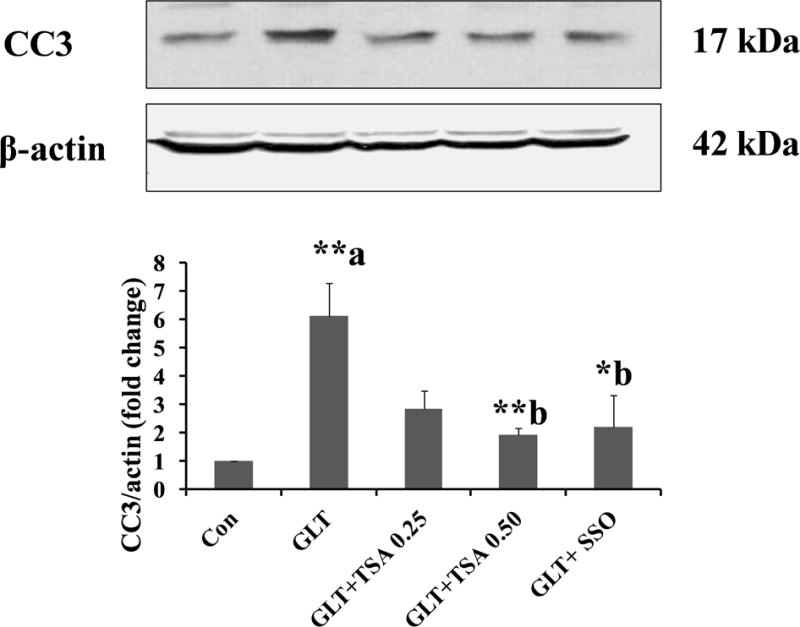
INS-1 832/13 cells were incubated in a medium containing low glucose (2.5 mM; Con), GLT (20 mM glucose plus 0.5 mM palmitate) in the absence and presence of SSO (200 μM) and TSA (0.25 or 0.50 μM) for 24 hrs. as indicated. The cells were treated with SSO for 1 hour before the exposure to GLT. Abundance of cleaved caspase 3 (active) in cell lysates was quantified by Western blotting and densitometry. Data are expressed as mean ± SEM (n=3). **p < 0. 01 and *p < 0.05 ‘a’ vs. con and ‘b’ vs. GLT.
Based on these observations, we conclude that CD36 plays regulatory roles in lipid accumulation and metabolic dysregulation (caspase 3 activation) in pancreatic beta cells under the duress of GLT. We also conclude that deacetylation of CD36 could contribute to GLT-mediated effects on lipid uptake, accumulation and cell dysregulation (see below).
4. Discussion
One of the main objectives of our current investigation was to identify putative mechanisms underlying intracellular lipid accumulation and cell dysregulation in insulin-secreting INS-1 832/13 cells under the duress of GLT conditions. Salient findings from our studies are: [i] GLT conditions promote lipid accumulation without significantly affecting CD36 expression; [ii] SSO, an irreversible inhibitor of CD36, significantly attenuated GLT-mediated lipid accumulation with no discernable effects on CD36 expression; [iii] two structurally distinct inhibitors of lysine deacetylation (TSA or VPA) markedly suppressed GLT-induced lipid accumulation without affecting CD36 expression; and (iv) TSA or SSO also suppressed GLT-induced caspase 3 activation. Based on these observations, we surmise that a signaling step that requires lysine deacetylation mediates CD36 catalytic function, which, in turn, leads to lipid uptake and accumulation and downstream caspase 3 activation in pancreatic beta-cells following exposure to GLT conditions.
CD36 is a multifunctional protein, which is involved in the lipid/fat intake, fat taste perception, immunity, inflammation, and angiogenesis [13, 16]. Emerging evidence also implicates regulatory roles for CD36 in lipid utilization and storage leading to the onset of beta-cell dysfunction, obesity and pathogenesis of diabetes [14]. Furthermore, CD36 expression is regulated by PPARα or γ in liver and adipose tissue, respectively [24]. Thus, therapeutics effects of PPAR-α and γ class of drugs may, in part, be due to their ability to regulate CD36 function leading to reduced hyperglycemia and dyslipidemia. Recently, Motojima et al. reported that fenofibrate, a PPAR-α agonist, and lipid lowering drug, attenuates fatty acid-induced islet beta-cell dysfunction and apoptosis via inhibiting the NF-kB/MIF-dependent pathway [25]. Studies by Mansor and associates suggested that pharmacological inhibition of CD36, by SSO, rapidly restores cardiac function following hypoxia/reoxygenation in the diabetic rats [26].
It has been shown that AMP-activated protein kinase (AMPK) plays an important role in the lipid and glucose homeostasis via stimulation of skeletal muscle fatty acid oxidation and glucose uptake, inhibition of cholesterol and triglyceride synthesis, adipocyte lipolysis, and modulation of insulin secretion from pancreatic beta-cells [27–29]. Recent studies by Samovski et al have provided novel CD36 mediated regulation of AMPK to promote fatty acid uptake and β-oxidation in skeletal and cardiac muscles of mice [30]. Liraglutide, a GLP-1 analogue, regulates pancreatic beta-cell proliferation and also prevents GLT-induced beta cell apoptosis by activating the AMPK/mTOR signaling pathway [31]. In addition, AMPK activation prevents palmitate-induced apoptosis and lipid accumulation in cardiomyocytes [28]. VPA has been shown to suppress hepatic fat accumulation and serum glucose in obese mice [29]. Together, above observations raise a potential possibility that inhibition of CD36 activity by VPA and TSA or SSO (current study) might promote activation of the AMPK signaling pathway in beta-cells, resulting in reduced lipid accumulation and caspase 3 activation under GLT conditions. Additional studies are needed to validate this postulation.
It is well established that acetylation-deacetylation signaling steps mediate functional regulation of several proteins involved in glucose and fatty acid metabolism, beta-cell dysfunction and obesity [18–20]. Findings from our current studies suggested that co-provision of TSA or VPA prevented GLT-mediated lipid accumulation and caspase 3 activation in pancreatic beta cells. It should be noted that similar degrees of inhibition of GLT-induced lipid accumulation were noticeable in cells treated with TSA or SSO. Since SSO inhibits the CD36 by formation of n-hydroxysuccinimidyl with the Lys-164 in its fatty acid binding pocket and inhibits CD36-mediate fatty acid intake signaling steps [16, 17], it is likely that acetylation-dacetylation of a different lysine residue(s) of CD36, distinct from the one in fatty acid binding pocket, might regulate its functional activity in beta-cells. Available evidence also suggests that HDAC expression and actives are increased in different tissues in diabetic condition including the beta-cells [23, 32–34]. Therefore, it is likely that increased HDAC activity in beta cells under GLT conditions might, in turn, deacetylate putative lysine residues of CD36 in the fatty acid binding pocket (Lys-164 and Lys-166), thereby promoting lipid intake in the beta-cells. Indeed, inhibition of deacetylases (TSA or VPA) leads to inhibition of CD36 activity and decreased lipid accumulation, and downstream caspase 3 activation. It is noteworthy to mention that HDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta-cells, whereas inhibiting HDAC7 activity with TSA or siRNA-mediated knockdown restored glucose-stimulated insulin secretion in beta-cells [23]. Further, inhibition of HDAC3 improves glycaemia and insulin secretion in obese diabetic rats [35]. Along these lines, Plaisance et al reported that the class I HDACs, particularly HDAC3 inhibitor, MS-275 also prevents palmitate-induced pancreatic beta-cell death [36]. Taken together, based on observations from extant studies in the field and our own findings from the current study, we propose that a signaling step that requires lysine deacetylation mediates CD36 catalytic function, which, in turn, leads to lipid uptake and accumulation and downstream caspase 3 activation in pancreatic beta-cells following exposure to GLT conditions. Identification of specific lysine deacetylases that control CD36 functional activity should provide novel clues for the prevention of beta-cell dysfunction under GLT.
Supplementary Material
Acknowledgments
This research is supported from the US Department of VA and the National Institutes of Health. AK received a Senior Research Career Scientist Award from the Department of VA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests
The authors declare that no conflict of interests.
References
- 1.Bardini G, Rotella CM, Giannini S. Dyslipidemia and diabetes: reciprocal impact of impaired lipid metabolism and Beta-cell dysfunction on micro- and macrovascular complications. Rev Diabet Stud. 2012;9:82–93. doi: 10.1900/RDS.2012.9.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prentki M, Madiraju SR. Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes, Mol. Cell Endocrinol. 2012;353:88–100. doi: 10.1016/j.mce.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Roomp K, Kristinsson H, Schvartz D, et al. Combined lipidomic and proteomic analysis of isolated human islets exposed to palmitate reveals time-dependent changes in insulin secretion and lipid metabolism. PLoS ONE. 2017;12:e0176391. doi: 10.1371/journal.pone.0176391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nolan CJ, Ruderman NB, Kahn SE, et al. Insulin Resistance as a Physiological Defense Against Metabolic Stress: Implications for the Management of Subsets of Type 2 Diabetes. Diabetes. 2015;64:673–686. doi: 10.2337/db14-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kristinsson H, Smith DM, Bergsten P, et al. FFAR1 is involved in both the acute and chronic effects of palmitate on insulin secretion. Endocrinology. 2013;154:4078–4088. doi: 10.1210/en.2013-1352. [DOI] [PubMed] [Google Scholar]
- 6.Staaf J, Ubhayasekera SJ, Sargsyan E, et al. Initial hyperinsulinemia and subsequent beta-cell dysfunction is associated with elevated palmitate levels. Pediatr Res. 2016;80:267–274. doi: 10.1038/pr.2016.80. [DOI] [PubMed] [Google Scholar]
- 7.Lin N, Chen H, Zhang H, et al. Mitochondrial reactive oxygen species (ROS) inhibition ameliorates palmitate-induced INS-1 beta cell death. Endocrine. 2012;42:107–117. doi: 10.1007/s12020-012-9633-z. [DOI] [PubMed] [Google Scholar]
- 8.Hong J, Jeppesen PB, Hermansen K. Effects of elevated fatty acid and glucose concentrations on pancreatic islet function in vitro. Diabetes Obes Metab. 2009;11:397–404. doi: 10.1111/j.1463-1326.2008.00971.x. [DOI] [PubMed] [Google Scholar]
- 9.Vernier S, Chiu A, Schober J, et al. β-cell metabolic alterations under chronic nutrient overload in rat and human islets. Islets. 2012;4:379–392. doi: 10.4161/isl.22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kowluru A, Kowluru RA. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem Pharmacol. 2014;88:275–283. doi: 10.1016/j.bcp.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowluru A. Role of G-proteins in islet function in health and diabetes. Diabetes Obes Metab. 2017;19(Suppl 1):63–75. doi: 10.1111/dom.13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elumalai S, Karunakaran U, Lee IK, et al. Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol. 2017;11:126–134. doi: 10.1016/j.redox.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pepino MY, Kuda O, Samovski D, et al. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr. 2014;34:281–303. doi: 10.1146/annurev-nutr-071812-161220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luiken JJ, Chanda D, Nabben M, et al. Post-translational modifications of CD36 (SR-B2): Implications for regulation of myocellular fatty acid uptake. Biochim Biophys Acta. 2016;1862:2253–2258. doi: 10.1016/j.bbadis.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Kuda O, Jenkins CM, Skinner JR, et al. CD36 protein is involved in store-operated calcium flux, phospholipase A2 activation, and production of prostaglandin E2. J Biol Chem. 2011;286:17785–17795. doi: 10.1074/jbc.M111.232975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuda O, Pietka TA, Demianova Z, et al. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J Biol Chem. 2013;288:15547–15555. doi: 10.1074/jbc.M113.473298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iyer A, Fairlie DP, Brown L. Lysine acetylation in obesity, diabetes and metabolic disease. Immunol Cell Biol. 2012;90:39–46. doi: 10.1038/icb.2011.99. [DOI] [PubMed] [Google Scholar]
- 19.Lundby A, Lage K, Weinert BT, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012;2:419–431. doi: 10.1016/j.celrep.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lundh M, Christensen DP, Rasmussen DN, et al. Lysine deacetylases are produced in pancreatic beta cells and are differentially regulated by proinflammatory cytokines. Diabetologia. 2010;53:2569–2578. doi: 10.1007/s00125-010-1892-8. [DOI] [PubMed] [Google Scholar]
- 21.Ferrari A, Fiorino E, Giudici M, et al. Linking epigenetics to lipid metabolism: focus on histone deacetylases. Mol Membr Biol. 2012;29:257–266. doi: 10.3109/09687688.2012.729094. [DOI] [PubMed] [Google Scholar]
- 22.Khan S, Kumar S, Jena G. Valproic acid reduces insulin-resistance, fat deposition and FOXO1-mediated gluconeogenesis in type-2 diabetic rat. Biochimie. 2016;125:42–52. doi: 10.1016/j.biochi.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 23.Daneshpajooh M, Bacos K, Bysani M, et al. HDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta cells. Diabetologia. 2017;60:116–125. doi: 10.1007/s00125-016-4113-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sato O, Kuriki C, Fukui Y, et al. Dual promoter structure of mouse and human fatty acid translocase/CD36 genes and unique transcriptional activation by peroxisome proliferator-activated receptor alpha and gamma ligands. J Biol Chem. 2002;277:15703–15711. doi: 10.1074/jbc.M110158200. [DOI] [PubMed] [Google Scholar]
- 25.Zheng S, Ren X, Han T, et al. Fenofibrate attenuates fatty acid-induced islet beta-cell dysfunction and apoptosis via inhibiting the NF-kappaB/MIF dependent inflammatory pathway. Metabolism. 2017 doi: 10.1016/j.metabol.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Mansor LS, Sousa Fialho MDL, Yea G, et al. Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc Res. 2017;113:737–748. doi: 10.1093/cvr/cvx045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia D, Shaw RJ. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell. 2017;66:789–800. doi: 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adrian L, Lenski M, Todter K, et al. AMPK Prevents Palmitic Acid-Induced Apoptosis and Lipid Accumulation in Cardiomyocytes. Lipids. 2017;52:737–750. doi: 10.1007/s11745-017-4285-7. [DOI] [PubMed] [Google Scholar]
- 29.Avery LB, Bumpus NN. Valproic Acid Is a Novel Activator of AMP-Activated Protein Kinase and Decreases Liver Mass, Hepatic Fat Accumulation, and Serum Glucose in Obese Mice. Mol Pharmacol. 2014;85:1–10. doi: 10.1124/mol.113.089755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samovski D, Sun J, Pietka T, et al. Regulation of AMPK Activation by CD36 Links Fatty Acid Uptake to β-Oxidation. Diabetes. 2015;64:353–359. doi: 10.2337/db14-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miao XY, Gu ZY, Liu P, et al. The human glucagon-like peptide-1 analogue liraglutide regulates pancreatic beta-cell proliferation and apoptosis via an AMPK/mTOR/P70S6K signaling pathway. Peptides. 2013;39:71–79. doi: 10.1016/j.peptides.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Khan S, Jena G. Sodium butyrate reduces insulin-resistance, fat accumulation and dyslipidemia in type-2 diabetic rat: A comparative study with metformin. Chem Biol Interact. 2016;254:124–134. doi: 10.1016/j.cbi.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Khan S, Jena G. Valproic Acid Improves Glucose Homeostasis by Increasing Beta-Cell Proliferation, Function, and Reducing its Apoptosis through HDAC Inhibition in Juvenile Diabetic Rat. J Biochem Mol Toxicol. 2016;30:438–446. doi: 10.1002/jbt.21807. [DOI] [PubMed] [Google Scholar]
- 34.Sathishkumar C, Prabu P, Balakumar M, et al. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin Epigenetics. 2016;8:125. doi: 10.1186/s13148-016-0293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lundh M, Galbo T, Poulsen SS, et al. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes Metab. 2015;17:703–707. doi: 10.1111/dom.12470. [DOI] [PubMed] [Google Scholar]
- 36.Plaisance V, Rolland L, Gmyr V, et al. The class I histone deacetylase inhibitor MS-275 prevents pancreatic beta cell death induced by palmitate. J Diabetes Res. 2014;2014:195739. doi: 10.1155/2014/195739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.