

Abstract
Understanding the mechanisms underlying autism spectrum disorders (ASD) is a challenging goal. Here we review recent progress on several fronts, including genetics, proteomics, biochemistry and electrophysiology, that raise motivation for forming a viable pathophysiological hypothesis. In place of a traditionally unidirectional progression, we put forward a framework that extends homeostatic hypotheses by explicitly emphasizing autoregulatory feedback loops and known synaptic biology. The regulated biological feature can be neuronal electrical activity, the collective strength of synapses onto a dendritic branch, the local concentration of a signaling molecule, or the relative strengths of synaptic excitation and inhibition. The sensor of the biological variable (which we have termed the homeostat) engages mechanisms that operate as negative feedback elements to keep the biological variable tightly confined. We categorize known ASD-associated gene products according to their roles in such feedback loops, and provide detailed commentary for exemplar genes within each module.
Introduction and plan of presentation
Understanding the combined dysfunction of social communication and repetitive behavior of individuals with autism spectrum disorders (ASD) presents a formidable challenge. Despite the uncertainties of success for discovering therapeutic treatments for ASD (Mullard, 2015), even limited progress could be leveraged into the broader realm of other neuropsychiatric diseases. Further, we can gain great insights into the normal function of the brain by studying it in cases of dysfunction. Pursuing hopes of ameliorating brain disorders, investigators are presented with an unprecedented chance to “get under the hood” of cognition in a mechanistic way.
Current work on ASD is extremely multidisciplinary. The field offers an abundance of data that is intriguing but often difficult to synthesize. Relating genetic underpinnings to normal or dysfunctional behavior requires connecting synapses, cells, circuits and networks. To support this effort, we aim this review at a broad audience that includes readers interested in genetics, neurophysiology, and behavior. As one of us has worked with individuals with ASD, another studies neuronal development, and a third explores synaptic signaling, the diversity of our experience reflects the breadth of areas involved in this disorder.
Current high-water marks of thinking in the field are represented by the following categories of papers, each with both value and limitation:
Research publications that focus on a mouse model of monogenic ASD, often followed up by attempted amelioration of ASD-like behavior with pharmacological intervention (Aguilar-Valles et al., 2015; Bear et al., 2004; Braat et al., 2015; Chevere-Torres et al., 2012; Choi et al., 2011; Gkogkas et al., 2014; Nageshappa et al., 2015). This approach admirably takes direct aim at issues of necessity and sufficiency, but by definition is only credible for a subset of ASD, even when the scope is broadened by considering the pathways that may be affected (Mullard, 2015).
Reviews of ASD that categorize genes according to properties of the proteins they encode, such as their anatomical location (Ebert and Greenberg, 2013; Toro et al., 2010; Uzunova et al., 2014), their protein-protein interactions with other gene products (Sakai et al., 2011), or their transcript levels in correlation with other mRNAs (Voineagu et al., 2011). Each provides valuable groupings of proteins, but not an explicit hypothesis about their mechanistic relationship to the disorder as a whole.
Perhaps most influential are reviews that emphasize a relatively novel concept or particularly vulnerable process: (1) ASD arises from too high a ratio of excitation/inhibition (E:I imbalance) (Rubenstein and Merzenich, 2003), as recently reviewed (Nelson and Valakh, 2015; Rubenstein, 2010); (2) ASD originates from an excess of local protein translation (Darnell and Klann, 2013; Kelleher and Bear, 2008); (3) ASD is caused by a defect in neuronal homeostasis (Bourgeron, 2015; Ramocki and Zoghbi, 2008; Toro et al., 2010; Wondolowski and Dickman, 2013); (4) ASD stems from a dysfunction of activity-dependent gene expression, particularly regulation of nuclear transcription (Ebert and Greenberg, 2013). These ideas have energized research efforts, but it is presently unclear whether they fully differentiate between cause and effect, or how one possible mechanism relates to another.
Researchers can try to make sense of the diversity of hypotheses by subdividing ASD into distinct disorders, or by trying to unify the disparate elements, as attempted here. In line with the advice of George Box, “all models are wrong but some are useful,” attempts to organize genes and signaling pathways, aided by concepts from neurophysiology, neural plasticity, autoregulatory mechanisms, Ca2+ signaling, and neurotransmission could help provoke argument and encourage fresh experimentation.
In this effort, we will advance a framework for uniting current themes about ASD under an overriding pathophysiological tent that explicitly emphasizes feedback regulation, building upon an oft-mentioned unidirectional chain of events:
Genetics → altered proteins → pathophysiology → circuit dysfunction → ASD behavior
This one-way street of causality is likely true in part, but does not fully capture the theme of neural adaptability, which must involve feedback in some way. To lay the foundation for our scheme we briefly review genetic findings and set out our reasons for putting genes and gene products into functional rather than anatomical categories. In turn, this organization spurs us to offer some fresh perspective to multiple themes in ASD research. With respect to signaling from synapse to nucleus (Ebert and Greenberg, 2013), we reopen the question of how cascades of ASD-related proteins (Ma et al., 2014) may control the expression of other ASD-related genes (Tian et al., 2014). Regarding synaptic plasticity by local regulation of protein synthesis (Aguilar-Valles et al., 2015; Fernandez et al., 2013; Kelleher and Bear, 2008; Kelleher et al., 2004; Niere et al., 2012), we meld the concept of coordination amongst neighboring dendritic synapses (Bourne and Harris, 2012; Rabinowitch and Segev, 2008) with ideas about ASD as dysfunctional homeostasis (Huguet et al., 2013; Ramocki and Zoghbi, 2008; Toro et al., 2010). Along the way, we combine morphological (Gilman et al., 2011; Jiang et al., 2013; Penzes et al., 2011; Varea et al., 2015; Zoghbi, 2003) and functional (Bateup et al., 2011) representations of synaptic plasticity, and attempt to broaden thinking about “E:I balance” to a more flexible view of “E:I coordination.” We then place each of these themes into a framework of feedback loops for autoregulation of neurons and circuits. Defects in any component of these control systems can lead to related patterns of dysfunction and ASD, which nicely meshes with the disparate mechanisms suggested by others.
Rapid progress in ASD genetics
The challenge and promise of ASD genetics can be telegraphed by a series of numbers. The reported prevalence of ASD is ∼1% of the total U.S. population, with estimates abruptly jumping from 1/88 to 1/68 based on surveillance studies conducted in 2008 and 2010. ASD is highly heritable, as judged by heightened risk for individuals with an affected twin or sibling (Bailey et al., 1995; Folstein and Rutter, 1977a, b). Quantitative measures of heritability are derived from rates of concordance for monozygotic twins, dizygotic twins, and siblings, with some evidence coming from a 2014 survey of >2 million Swedish families, the largest population-based study of ASD so far (Sandin et al., 2014). With one notable exception (Hallmayer et al., 2011), heritability estimates range from 52-90% (Chen et al., 2015; De Rubeis and Buxbaum, 2015a, b; Gaugler et al., 2014; Rosenberg et al., 2009; Sandin et al., 2014; Toro et al., 2010). Thus, genetic factors contribute substantially, and likely combine with environmental effects to guide clinical outcome (Bourgeron, 2015).
The genetic landscape of ASD is extremely complex, involving some 400-1200 genes according to current best estimates (De Rubeis and Buxbaum, 2015a; Geschwind and State, 2015; Ronemus et al., 2014; Willsey and State, 2015). In a handful of rare syndromes, autistic features arise from dysfunction of only one gene, as exemplified by Rett (MECP2), Angelman (UBE3A), Timothy (CACNA1C), Fragile X (FMR1) and Tuberous sclerosis (TSC1,2) syndromes. While highly penetrant and instructive, these syndromic forms are rare; pooling them together accounts for less than 5% of individuals on the ASD spectrum. This leaves geneticists with the daunting challenge of sifting through the 3 billion nucleotides of the human genome, looking amongst the 1% that differ between two randomly selected people (Geschwind and State, 2015) and determining which differences, or combinations thereof, are causal to ASD amidst the vast sea of those that are not (McCarroll and Hyman, 2013). Thus, it seems remarkable that within just the last few years, genetic studies have identified a sizeable fraction of the sought-after ASD genes, roughly 20-30% according to most estimates (Bourgeron, 2015; Krumm et al., 2014; Schaaf and Zoghbi, 2011).
A wide array of approaches
Multiple genetic efforts have been mounted and have contributed to varying extents. Genome Wide Association Studies (GWAS) have not yet reached desired reproducibility, most likely because they have been underpowered (typically 2,000-5,000 patients, far fewer than the ∼50,000 that drove a highly successful GWAS in schizophrenia (Cross-Disorder Group of the Psychiatric Genomics et al., 2013)). Much recent progress has come with the intensive genetic analysis of ASD families, including the finding of recessive genes with powerful effects (Jamain et al., 2003; Morrow et al., 2008). Exome sequencing has uncovered de novo single nucleotide variations (SNVs) and insertion/deletion (indel) mutations that segregate with ASD. Four independent studies, all published in the same watershed year (Iossifov et al., 2012; Neale et al., 2012; O'Roak et al., 2012b; Sanders et al., 2012), advanced our knowledge of the genetic complexity of ASD and greatly improved the reliability of gene discovery. Identification of additional genes has accelerated with recent sequencing papers (De Rubeis et al., 2014; Iossifov et al., 2014; O'Roak et al., 2012a). Together, these studies have yielded gene lists with stringent false-discovery rates and suggested possible interrelationships between genes. Further progress in genetics will no doubt continue as the cost of sequencing decreases and the size of cohorts increases.
Genetic Variants and Networks
How newly discovered genetic variations affect gene expression remains a pressing issue. Mouse models are readily available for highly penetrant syndromic forms of ASD (Bader et al., 2011; Crawley, 2012; Guy et al., 2001; Horev et al., 2011; 1994; Yu et al., 2006), but many more studies will be needed to explore how different mutations affect function at the level of protein, neuron, circuit and whole animal. We can expect important advances in the use of model organisms (mouse, zebrafish, fruitfly and possibly even yeast)(Kozol et al., 2015), and in new molecular shortcuts to explore the full range of genetic variants through emerging genome editing methods (Zhang, 2015). Given the effort involved in exploring even one biological hypothesis, distinguishing between high and low confidence hits is essential.
Various approaches have recently been developed to query networks of functionally related genes. For example, systematic study has uncovered hundreds of direct protein-protein interactions amongst products of genes previously implicated in ASD, including a network involving SHANK and TSC1 (Sakai et al., 2011). Direct protein-protein interactions are informative but challenging to interpret because of the diversity, and in some cases, promiscuity of such interactions.
In a complementary approach, transcriptomic studies in postmortem tissue have uncovered clusters of genes (so-called modules) whose transcripts co-vary. The co-variation is revealed agnostically by a clustering procedure known as weighted gene co-expression network analysis (WGCNA) (Zhang and Horvath, 2005), and is used to infer a functional relationship. The co-regulation may occur among individual ASD probands and controls (Voineagu et al., 2011), among multiple brain regions (Ben-David and Shifman, 2012; Voineagu et al., 2011), in various cortical layers (Parikshak et al., 2013; Willsey et al., 2013), and over the course of development (Parikshak et al., 2013; Willsey et al., 2013). Once the transcripts are grouped into modules, their behavior is compactly summarized by the module eigengenes (first principal component). Voineagu et al. performed comparative transcriptomic analysis of post-mortem ASD brain samples and found two modules highly correlated with ASD status (Voineagu et al., 2011). One of these modules (M12) was downregulated in ASD cases, contained genes known for involvement in synaptic function, vesicular transport, and neuronal projection, and was inferred to be causally involved in ASD pathophysiology.
Progressing from gene discovery to functional gene groupings to pathophysiology
With the identification of a substantial fraction (20-30%) of the full panoply of ASD-related genes, researchers are now striving to use putative gene groups to understand the pathophysiology of ASD. Traditionally, genes are grouped according to Gene Ontology (GO) terms (e.g., ion channels), by cellular location (e.g., in the postsynaptic spine), by developmental expression (e.g. during early circuit formation), or by compatibility with a prevalent hypothesis (e.g. E:I balance). Strategies based on grouping genes and gene products offer promise for inferring mechanism and would be further boosted by the addition of a more physiological view of neuronal autoregulation, as described below.
Dysfunctional feedback regulation as a unifying theme in ASD
Here we expand upon previous work by providing a general framework for ASD that combines genetic information with ideas about the pathophysiology of homeostatic feedback loops. The starting point is a hypothetical scheme for the autoregulation of neuronal firing, proposed by Abbott, Golowasch, Marder, and Turrigiano at Brandeis in a prescient series of papers two decades ago (LeMasson et al., 1993; Marder et al., 1996; Siegel et al., 1994) (Fig. 1). We have adapted this computational model to diagram neuronal firing as the controlled variable whose variations are delimited by a feedback system. Changes in a neuron's firing rate result in alterations in Ca2+ channel activity, which serves as a sensor that drives an effector mechanism. The effector mechanism works through a feedback connection to counteract variation of the sensed variable from its designated set point. The feedback loop can be likened to a temperature-control system in which the controlled variable is the temperature in a room, the sensor a thermostat, the effector mechanism a remote heating/cooling unit, and the feedback connection the ductwork between the heating/cooling unit and the room itself. In neuronal homeostasis, as in HVAC systems, the feedback connection operates to oppose change, hence its designation as negative feedback. Proper control depends upon successful operation of each of the components; failure of a component disables the entire loop, while a weakening of function can be compensated for by other elements in the loop, but only within certain limits.
Fig 1. Ca2+ regulation of gene expression in neuronal development & homeostasis.
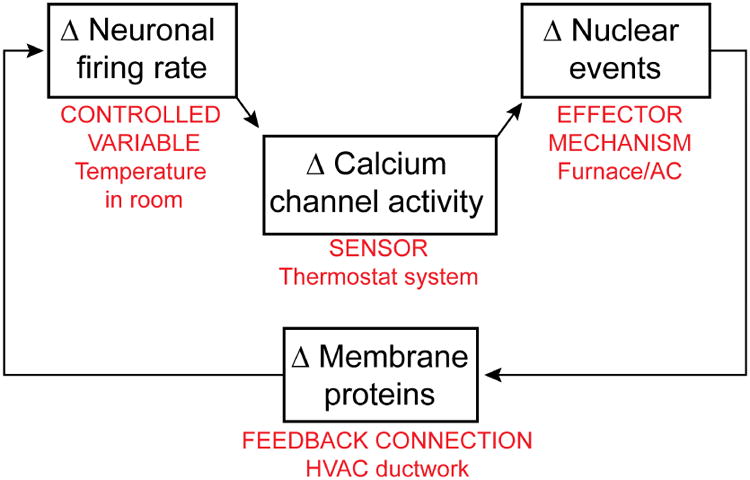
A canonical homeostatic feedback loop, proposed by Abbott, Marder et al. (LeMasson et al., 1993; Marder et al., 1996; Siegel et al., 1994), hypothesizes that Δ firing → Δ Ca2+ → Δ gene expression → Δ membrane channel proteins regulating neuronal excitability. This feedback loop is taken as key to how neuronal firing is kept away from extremes. The components of the feedback loop can be likened to elements of a temperature control system (labeled in red).
This is but one example of a servo loop mediating autoregulation in the healthy brain, whose operation can go awry in response to genetic lesions or environmental stressors. It exemplifies a variety of feedback mechanisms — not just those involved in regulating electrical activity as illustrated here, but also extending to autoregulation of neuronal biochemistry, cell biology, and microcircuit performance.
The components of this autoregulatory system provide a logical framework for grouping ASD-implicated genes (Fig. 2). Here we form a separate category in addition to those found in previous papers: a homeostatic sensor (sometimes referred to as a “homeostat”). Like the metaphorical room thermostat, it detects perturbations or dysfunction in other parts of the system, and can itself be prone to failure. In turn, the sensor triggers compensatory changes in feedback elements such as nuclear gene expression, local protein translation, and changes in synaptic function and structure. A prototypical homeostat protein would be a Ca2+ channel that couples membrane potential changes to downstream biochemical events (see Diverse systems for sensor-mediated control of gene expression). Indeed, pore-forming and auxiliary subunits of several Ca2+ channels are encoded by known autism-associated genes. Although genetic studies also implicate sodium and potassium channels, their roles in ASD are activity-related in a somewhat different way. While Ca2+ channels can send an intracellular signal, Na+ channels merely drive electrical firing, and neither Na+ nor K+ ions are established biochemical messengers; their ultimate impact is likely mediated by Ca2+ channels.
Fig 2. Homeostatic feedback through a multi-pathway loop.
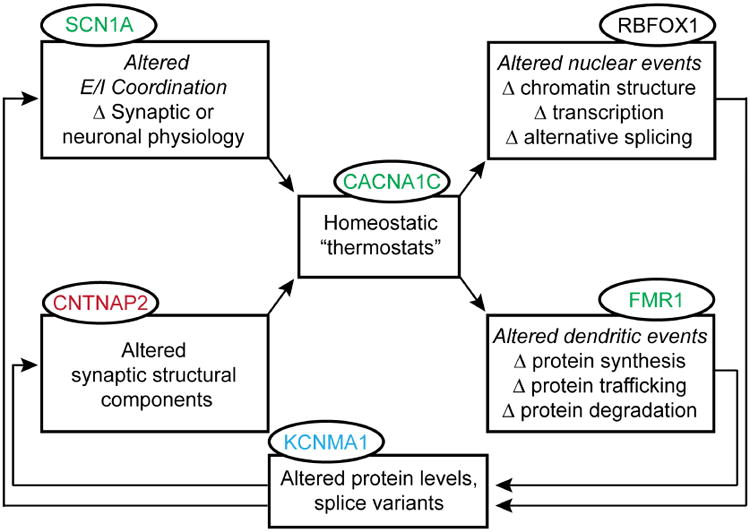
Expanding upon Figure 1 to incorporate alterations in both nuclear and dendritic events that arise during a homeostatic response. These effector mechanisms operate via feedback to alter synaptic or neuronal physiology and synaptic structure. ASD-associated mutations have been identified in genes associated with each component of this feedback loop (ovals); dysfunction in any one section can affect the operation of the entire loop to impact function at a variety of levels. This schematic representation of autoregulation appears in simplified versions in later figures (3, 4, and 6), highlighting the specific modules being discussed.
With this distinction in mind, we generalize the concept of homeostat proteins to sensors that respond to chemical signals and support downstream biochemical signaling. The chemical ligand can be a neurotransmitter like glutamate, a neurotrophin like BDNF, or a glycoprotein such as Wnt. Once again, the homeostatic sensor can detect variations in the controlled variable and trigger one or more effector mechanisms, including altered nuclear or dendritic events. The feedback can result in altered levels or properties of signaling proteins, modifying excitability or synaptic properties as part of the servo loop to oppose perturbations of the controlled variable (Fig. 2).
This scheme has several advantages for thinking about homeostatic responses in the context of both typical and atypical neuronal activity. It allows the putative assignment of genes into functional rather than anatomical groupings, as we have attempted in Table 1. Mindful of the importance of work identifying genes with high reliability, we have used a color-code to indicate the degree of confidence currently placed on individual genes (see Table 1 legend). Many of the entries do not reach genome-wide significance based on current data but are included because of their fit with a pathophysiological scenario, and are annotated with a footnote. The inclusion and classification of individual genes will likely need revision, but the overall organization conveyed by Figure 2 and Table 1 may help hypothesis-building about possible effects of mutations on downstream events. Preliminary distinctions between sensors, effectors, and feedback elements provide a bridge between gene discovery and thinking about biochemical signaling systems. In the homeostatic framework in Figure 2, a mutation in any individual element can have effects throughout the loop, and can also impact different routes through the overall loop. The scheme permits a transition between overtly successful compensation to perturbations, to outright failure when the system is challenged beyond its limits.
Table 1. Autoregulatory framework of ASD genes.
We group ASD-gene candidates into functional modules presented in Figure 2, utilizing a graded scale of confidence based on the Simons Foundation Gene Scoring Module (from red to burgundy, black, and green)(Basu et al., 2009). Gene names listed in blue are included, despite weaker or mixed evidence, based on findings of de novo mutations (see footnotes) and what we feel are interesting areas for future research. [Associated Disorder abbreviations: ADHD- Attention Deficit/Hyperactivity Disorder; BPD- Bipolar Disorder; DD- Developmental Disorder; Ep- Epilepsy; ID- Intellectual Disability; OCD- Obsessive Compulsive Disorder; MDD- Major Depressive Disorder; Scz- Schizophrenia; ALS- Amyotrophic Lateral Sclerosis; TSC- Tuberous Sclerosis].
|
| |||||
|---|---|---|---|---|---|
| Gene | Full Gene Name | Function | Chromosome Band | Associated Disorders | Primary Citation |
| Signaling to the Nucleus | |||||
| ADNP | Activity-dependent neuroprotector homeobox | Transcription factor (TF) | 20q13.13 | DD, Ep, ID | (O'Roak et al.,2012) |
| ANKRD11 | Ankyrin repeat domain 11 | Inhibits ligand dependent transcription | 16q24.3 | ADHD, DD, Ep, ID | (Marshall et al., 2008) |
| APCa | Adenomatosis polyposis coli | Tumor suppressor | 5q21-q22 | ID | (Barber et al., 1994) |
| ASH1L | Ash1 (absent, small, or homeotic)-like (Drosophila) | Histone methyltransferase | 1q22 | (Willsey et al., 2013) | |
| ASXL3 | Additional sex combs like 3 (Drosophila) | Putative polycomb group (PcG) protein | 18q11 | DD, ID | (Dinwiddie et al., 2013) |
| BCL11A | B-cell CLL/lymphoma 11A | Zinc finger protein | 2p16.1 | DD, ID, Scz | (lossifov et al., 2012) |
| CAMK2Ab | Calcium/calmodulin-dependent protein kinase alpha | Protein kinase | 5q32 I | (lossifov et al., 2014); (Stephenson et al., 2015 unpublished data) | |
| CAMK2Bc | Calcium/calmodulin-dependent protein kinase beta | Protein kinase | 7p13 | (lossifov et al., 2014) | |
| CAMK4d | Calcium/calmodulin-dependent protein kinase 4 | Protein kinase | 5q21.3 | (lossifov et al., 2014; Waltes et al., 2014) | |
| CAMKK1e | Calcium/calmodulin-dependent protein kinase kinase 1, alpha | Protein kinase | 17p13.2 | (lossifov et al., 2014) | |
| CDH8f | Cadherin 8, type 2 | Membrane protein | 16q22.1 | (Pagnamenta et al., 2011) | |
| CDKL5 | Cyclin-dependent kinase-like 5 | Protein kinase | Xp22 | ADHD, DD, Ep, ID, OCD, | (Weaving et al., 2004) |
Barber et al., 1994 reported on a patient with ASD with an interstitial deletion of chromosome 5, q15q22.3, with confirmed deletion of the APC gene.
Iossifov et al., 2014 identified a de novo variant of this gene in an intron in a male proband, and a missense mutation in a male proband and his unaffected sister (Supplementary Table 2); Stephenson et al., found changes in αCaMKII activity as a result of the Iossifov 2014 missense mutation (see- Stephenson J.R., Wang, X., Shonesy, B.C., Sutcliffe, J.S., and Colbran, R.J. (2015). Physiological effects of an ASD-associated mutation in CaMKIIalpha. Program No 58414/E17 2015 Neuroscience Meeting Planner, Chicago, IL: Society for Neuroscience, Online.)
Iossifov et al., 2014 identified a de novo missense mutation in this gene in a male proband and his unaffected sister (Supplementary Table 2).
Iossifov et al., 2014 identified a de novo missense mutation in this gene in a male proband and his unaffected brother (Supplementary Table 2); Waltes et al., 2014 reported an association between rs25925 of CAMK4 and ASD after genotyping ten SNPs in two sets of German populations.
Iossifov et al., 2014 identified a de novo mutation in an intron of this gene in a male proband and his unaffected sister (Supplementary Table 2).
Pagnamenta et al., 2011 reported rare familial 16q21 microdeletions and expression analyses implicating CDH8 in ASD and ID.
Martin et al., 2013 found a missense variant in WNT1 (S88R) that was overrepresented in ASD patients relative to controls.
Marui et al., 2010 identified three SNPs within WNT2 that showed significant associations with autism.
Neves-Pereira et al., 2009 identified and mapped a de novo chromosome breakpoint between 4q and 5q to an alternative transcript of the gene, as well as two ASD patients with single nucleotide insertions in the EIF4E promoter shown to alter activity.
Barnby et al., 2005 reported a significant association with ASD for SNPs within GRIN2A for rs1014531 and for intron 10.
Iossifov et al., 2014 identified a de novo mutation in an intron of this gene in a male proband and his unaffected sister, and a missense mutation in a female proband and her unaffected sister (Supplementary Table 2).
Laumonnier et al., 2006 identified a breakpoint in this gene in a patient with a de novo translocation, and a second patient with a sequence variation in a highly conserved region of the ion channel encoded by the gene; Neale et al., 2012 identified mutations in this gene in two individual probands, one missense and one silent.
Iossifov et al., 2014 identified a de novo missense mutation of this gene in two male probands and their unaffected sisters, and a synonymous mutation in a third male proband and his unaffected sister (Supplementary Table 2).
Feyder et al., 2010 reported a significant association between variations in two human DLG4 SNPs with neural endophenotypes related to ASD.
Iossifov et al., 2014 identified a de novo mutation in an intron of this gene in a male proband and his unaffected sister, a missense mutation in a male proband and his unaffected sister, and a frame-shift mutation in a male proband and his unaffected brother (Supplementary Table 2).
Benayed et al., 2005 replicated an association between two intronic SNPs in this gene with ASD.
In the next two sections, we take a tour of the homeostatic loop, beginning with genetic modifications in homeostat proteins and their consequences for nuclear production of mRNA, and then progressing to local protein translation of messages into synaptic proteins.
Diverse systems for sensor-mediated control of gene expression
We illustrate the importance of sensor regulation of transcription by reviewing two ASD-related signaling pathways, one responding to chemical signals, the other to electrical activity. Although rarely summarized side-by-side, these systems address similar biological challenges and are replete with components and biomarkers associated with neuropsychiatric disorders, including autism.
Wnt signaling and ASD
The Wnt signaling system has long been implicated in neuronal overgrowth, as is often observed in the autistic brain (Chenn and Walsh, 2002; Courchesne et al., 2001). Signaling downstream of the proteolipid ligands known as Wnts can proceed via the canonical Wnt/β-catenin pathway (Nelson and Nusse, 2004), or through multiple non-canonical pathways, not mediated by β-catenin (Gordon and Nusse, 2006; Rosso et al., 2005; Salinas and Zou, 2008), with some involving Ca2+ signaling (Kohn and Moon, 2005). Canonical Wnt signaling acts, indirectly, to enhance the stability of β-catenin, allowing its translocation from the cell surface to the nucleus, thereby connecting extracellular signals to the regulation of nuclear gene expression by downstream transcriptional machinery (Fig. 3). Mutation of the gene encoding β-catenin, CTNNB1, has been found among suggestive de novo ASD risk contributing mutations (de Ligt et al., 2012; O'Roak et al., 2012b); furthermore, ∼40% of those 126 mutations map to a highly interconnected β-catenin/chromatin remodeling network (O'Roak et al., 2012b). However, it is worth noting that β-catenin also participates in cell-cell interactions via cadherin (CDH8 being a candidate ASD gene, see Table 1), independent of Wnt signaling. Otherwise, the evidence favors the involvement of genes co-regulated with the canonical Wnt/β-catenin pathway. One supporting argument derives from the clear role of CHD8 (not to be confused with CDH8), an ASD gene highlighted by multiple genetic studies (Krumm et al., 2014). There is mounting evidence from work in both fly and humans that CHD8 negatively regulates β-catenin (CTNNB1) function, either through direct binding to β-catenin, or by recruitment to promoter regions of β-catenin-responsive genes (Thompson et al., 2008). This may explain why putative loss of function mutations of CHD8 and CTNNB1 exert opposite effects on brain growth in ASD probands (Chenn and Walsh, 2002; Krumm et al., 2014). CHD8 provides a leading exemplar of an autism-associated gene product that regulates other autism risk genes (Cotney et al., 2015).
Fig. 3. Canonical Wnt/β-catenin signaling pathway and ASD-related protein players.
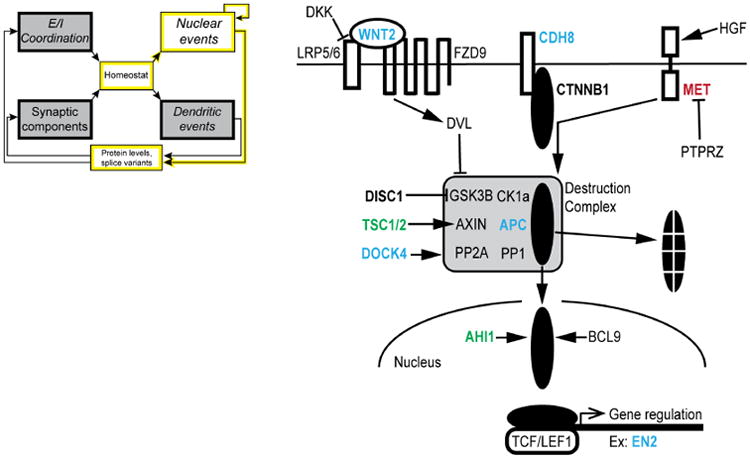
Hepatocyte growth factor (HGF) activation of the MET receptor tyrosine kinase releases β-catenin (CTNNB1) from surface cadherin molecules. β-catenin in the cytoplasm is bound by proteins that make up a destruction complex (DC). The DC marks β-catenin for proteasomal degradation. Wnt2 signaling via binding with a Frizzled receptor (FZD9) and its co-receptor LRP5/6 activates dishevelled (DVL), which saves β-catenin from degradation by inhibiting the DC. When DVL is active, β-catenin enters the nucleus, where it activates gene transcription by binding with the TCF/LEF1 family of transcription factors. Gene names are shown in bold if associated with ASD and present in Table 1 (utilizing the same color-coding scheme). The icon, simplified from Fig. 2, highlights the specific modules directly involved in the pathway illustrated by this figure (and utilized in the same manner for Figs. 4 and 6).
Surface-to-nucleus signaling by β-catenin is subject to positive and double negative (disinhibitory) regulation, both dependent on elements linked to ASD (Fig. 3, ASD-related genes shown in bold colors, matching the scale in Table 1). On one hand, MET tyrosine kinase, which has been implicated in ASD (Judson et al., 2011), responds to hepatic growth factor by liberating β-catenin from binding to surface cadherins (e.g., CDH8). On the other hand, free cytosolic β-catenin is phosphorylated by GSK3β (part of a destruction complex), and thus marked for proteosomal degradation. Finally, various species of Wnt molecules, including Wnt2, signal across the surface membrane through interaction with a frizzled receptor (mostly subtype FZD9) and an auxiliary receptor LRP5/6, which have tantalizing but at present unsubstantiated linkages to ASD. Dishevelled (DVL), which inhibits the destruction complex, was knocked out in the first mouse model of social dysfunction (Lijam et al., 1997), an early predecessor to more recent findings connecting Wnt related signaling molecules to ASD (De Ferrari and Moon, 2006; O'Roak et al., 2012a; Parikshak et al., 2013). The canonical-Wnt signaling system overlaps with multiple branches of biochemical signaling, each amply dotted with ASD-implicated proteins (Fig. 3). The availability of druggable-components in these signaling events presents intriguing opportunities for pharmacological manipulation, building upon experience with Wnt signaling and cancer therapeutics (Moon et al., 2004). However, as clearly summarized by Kalkman (Kalkman, 2012), one must recognize the multiple roles of Wnt signaling, not only in cell adhesion and dendritic remodeling but also in early brain development (Rosso et al., 2005; Salinas and Zou, 2008; Ueda et al., 2008; Wayman et al., 2006; Yu and Malenka, 2003). This is but one example of signaling proteins that participate in both neuronal development and ongoing function (Box 1; next section).
Activity-dependent gene expression in ASD involves calcium channels operating as homeostats
The idea that defects in activity-induced gene expression contribute to ASD was initially based on a provocative realization: while only 3% of the transcriptome is expressed in an activity-dependent manner, a far greater percentage of ASD implicated genes display activity-dependent expression (Kelleher and Bear, 2008; Morrow et al., 2008). Likewise, ASD-associated mutations that alter neurotransmission ultimately disrupt activity-dependent signaling to the nucleus (Ebert and Greenberg, 2013). Thus, it is widely believed that defects in activity-dependent gene expression may be a common feature of diverse forms of ASD. Just as Wnt signaling via frizzled receptors provides a mechanism for neurons to monitor their local extracellular environment, activity-dependent gene expression provides an ongoing readout of neuronal firing. Communication of neuronal depolarization proceeds by a variety of signaling cascades and regulates intranuclear events such as enhancer recruitment (Kim et al., 2010), transcription factor activation (Ebert and Greenberg, 2013) and alternative pre-mRNA splicing (Li et al., 2007). Further, multiple forms of activity-dependent regulation can operate in coordination, for example, Ca2+ channel control of nuclear gene expression by excitation-transcription (E-T) coupling or excitation-alternative splice (E-AS) coupling could act in concert with transmitter-driven alterations of local translation in dendrites (see next section).
Ca2+ channels are perfectly suited to act as sensors of electrical activity. By transducing membrane potential changes into protein conformational changes and Ca2+ influx these channels serve as homeostats, transmitting information about neuronal activity to downstream effector systems. The pore-forming α1 subunit (e.g. CACNA1C) and the auxiliary subunits β (e.g. CACNB2) and α2δ (CACNA2D3) were all prominent hits in a large-scale genetic screen across multiple neuropsychiatric diseases (Cross-Disorder Group of the Psychiatric Genomics et al., 2013). Further, there is clear evidence from Timothy syndrome (TS), a rare genetic disorder arising from mutation of either CACNA1C or CACNB2, that improper function of a Ca2+ channel can engender ASD with a penetrance as high as 60-80% (Breitenkamp et al., 2014; Splawski et al., 2005; Splawski et al., 2004). The pathogenic effects of genetically altered Ca2+ channel subunits likely revolve around their ability to generate a conformational signal in response to membrane depolarization (Li, Tadross & Tsien, accepted for publication in Science). The point mutations that give rise to ASD generally sensitize voltage-dependent gating of Ca2+ channels, shifting its activation to more negative potentials by ∼10 mV (e.g. G406R in CACNA1C, but not G402S). Thus, the neurologically penetrant mutation gives an abnormal voltage setpoint, in keeping with the premise of a disturbed regulatory feedback loop. Finally, exome sequencing of simplex libraries has repeatedly pointed to CACNA1D, the pore-forming subunit of another L-type channel, as another ASD-associated gene (De Rubeis et al., 2014). In this case, neurologically active mutations also cause a negative shift of voltage-activation (Pinggera et al., 2015), as expected from studies of CACNA1C.
What signaling pathways lie downstream of Ca2+ channel opening? The CaV1.2 and CaV1.3 proteins encoded by CACNA1C and CACNA1D are localized in postsynaptic compartments (spines and somatodendritic areas, respectively). The Ca2+ rise that matters for signaling to the nucleus must occur within 1-2 μm of the channel mouth (Deisseroth et al., 1996), but is linked to the nucleus by a signaling pathway (Ma et al., 2014), depicted in Fig. 4. Communication is initiated by a signaling complex near the dendritic CaV1 channel, which sends a shuttle protein to the nucleus upon activation. In excitatory neurons γCaMKII operates as the shuttle, gathering Ca2+/calmodulin (Ca2+/CaM) from the cytoplasm and transporting it into the nucleus. There, Ca2+/CaM is released and activates CaMKK and its substrate CaMKIV, the protein kinase responsible for CREB phosphorylation and other nuclear events. Once activated by the CaMK cascade, CaMKIV is able to phosphorylate another important nuclear factor, CREB binding protein/p300 (CBP, gene name CREBBP) (Impey et al., 2002; Kwok et al., 1994). Mutations in CREBBP give rise to Rubenstein-Taybi syndrome, a disorder with prominent autistic features (Petrij et al., 1995). Phospho-CBP acts as a requisite co-factor of multiple transcription factors, including CREB and the Wnt-dependent TCF-LEF1 complex.
Fig. 4. Linking L-type channels to nuclear events.
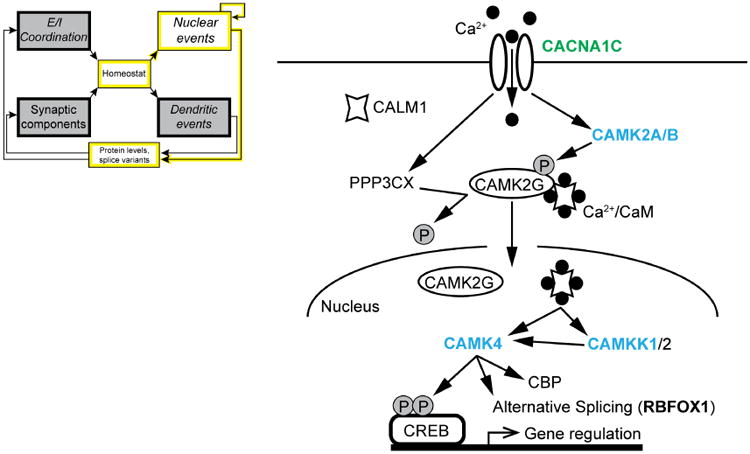
Calcium entering through CaV1.2 channels (CACNA1C) binds to calmodulin (CALM1, CaM), and triggers activation of α2/β-CaMKII (CAMK2A/B), and calcineurin (PPP3CC, CaN). The aggregation of α2/β-CaMKII at postsynaptic sites may contribute to synaptic tagging, a key step in long-lasting LTP (Frey and Morris, 1997; Hudmon et al., 2005; Redondo et al., 2010). γCaMKII (CAMK2G) is phosphorylated by α2/β-CaMKII to trap the Ca2+/CaM signal in place, and dephosphorylated at a different residue by CaN. This dephosphorylation event exposes a nuclear localization sequence on γCaMKII and triggers translocation, shuttling Ca2+/CaM into the nucleus. Nuclear Ca2+/CaM activates CaMKK (CAMKK1/2) and CaMKIV (CAMK4), which phosphorylates CREB, activating CRE-mediated gene transcription. CaMKIV also phosphorylates CREB binding protein (CBP, CREBBP), further favoring transcription, and activates alternative splicing, via splicing factors such as Rbfox1 (RBFOX1). In this scheme, both βCaMKII and CaN act in close proximity to CaV1 channels, while γCaMKII appears to operate as a carrier, not a kinase. Gene names are shown in bold if associated with ASD and present in Table 1 (utilizing the same color-coding scheme).
Strikingly, genes encoding the key molecular players have been repeatedly implicated in neuropsychiatric disorders including autism, depression, and schizophrenia (Fig. 4, ASD-associated genes in bold, colors according to Table 1). How can alterations in individual signaling or structural proteins be involved in such a diverse array of neuropsychiatric diseases? Genetic studies have revealed many disease-related alleles, but much work remains to evaluate their effects on the magnitude and even the sign of the functional differences. This effort has begun for an intronic SNP of CACNA1C (rs1006737) that crops up in multiple neuropsychiatric disorders (Ferreira et al., 2008; Green et al., 2010; Moskvina et al., 2009; Nyegaard et al., 2010), including ASD (Li et al., 2015). CaV1.2 mRNA levels and current densities were both consistently increased in induced human neurons homozygous for the risk genotype compared with non-risk genotypes (Yoshimizu et al., 2015). The consistency bodes well for therapeutic approaches, but offers no specific clues as to the differences between ASD and these other disorders. It remains possible that other alleles may cause disorder-specific effects on protein abundance or function; alternatively, those changes could be similar (Yoshimizu et al., 2015), but the final functional outcome in the brain might vary according to the genetic or environmental context. Knowledge of environmental factors is growing, as exemplified by converging evidence that fetal testosterone may contribute to the preponderance of ASD in males vs. females (∼4:1) (Baron-Cohen et al., 2011).
With overall outcome in mind, we now turn from the dysfunctional calcium channel homeostat to the altered programs of gene expression that it triggers. Abnormalities were observed in neuronal precursors derived from individuals with Timothy syndrome; the expression of >200 genes were altered, many of them CREB-dependent (Pasca et al., 2011). When subjected to an unbiased systems biology analysis (Tian et al., 2014), the pattern of differences in gene expression was compatible with involvement of known Ca2+-dependent transcriptional regulators, including CREB, NFAT, MEF2, and FOXO. The analysis uncovered Ca2+-dependent co-expression modules that reflect distinct aspects of TS, including intellectual disability and ASD-related phenotypes. The approach also uncovered several players that provide unexpected connections to other signaling modules. Thus, multiple feedback loops may interconnect (see Summary and Future Outlook). Looking at gene expression beyond transcription, activation of the nuclear CaMKIV also controls alternative splicing of transcriptionally derived pre-mRNA (E-AS coupling). For example, CaMKIV phosphorylation regulates the activity of RBFOX1, a factor that controls the alternative-splicing and stability of many mRNAs involved in synaptic and neuronal signaling, including CAMK2G, GRIN1, CNTNAP2, NRCAM and RBFOX1 itself (Gehman et al., 2011). Voineagu, Geschwind and colleagues compared normal and RBFOX1-deficient samples (therein referred to as A2BP1) and found hundreds of variations in splicing events, many of them in transcripts of ASD-linked genes (Voineagu et al., 2011). Overall, RBFOX1 target genes were enriched for synaptic proteins, actin-binding proteins and other molecules involved in cytoskeleton reorganization, processes crucial to proper neuronal structure and connectivity. Taken together, studies of E-AS coupling and E-T coupling underscore the power of activity-dependent, CaV1-regulated signaling and its impact on expression of ASD-linked genes and neuronal pathobiology.
Dysfunction of Local mRNA Translation and Synaptic Plasticity
Decades after local dendritic protein translation was first proposed (Steward and Levy, 1982), there is now wide agreement that it plays an integral part in synaptic plasticity (Schuman et al., 2006). Furthermore, consensus exists that disturbance of dendritic mRNA translation contributes to ASD (Darnell and Klann, 2013; Kelleher and Bear, 2008). Here we compare two modes of local translational regulation, each involving well-known ASD-related signaling proteins, FMRP and TSC1/2. We point out that these regulatory modes have sharply different effects on synaptic plasticity, and put forward a pathophysiological hypothesis that yokes together their dichotomous effects on both synaptic structure and activity.
Genetic alterations in the Fragile X mental retardation 1 gene (FMR1) give rise to Fragile X Syndrome (FXS) (Verkerk et al., 1991). This relatively common form of inherited intellectual disability is often associated with symptoms of ASD, including perseverative behaviors, speech and sensory difficulties, and disrupted sleep (Kidd et al., 2014). Encoded by the FMR1 gene, Fragile X mental retardation protein (FMRP) mostly operates as an mRNA-binding protein (Fig. 5B2) that represses translation of an emerging subset of mRNAs (Brown et al., 2001; Darnell et al., 2001; Darnell et al., 2011; Santoro et al., 2012), among other functions (Contractor et al., 2015). FXS is often compared to another single-gene syndromic disorder, Tuberous sclerosis (TSC). Frequently associated with deficits in long-term and working memory, along with intellectual disability, ASD, and epilepsy, TSC arises from heterozygous mutations in the TSC1 or TSC2 genes (Ehninger et al., 2009; Ehninger et al., 2008; Kong et al., 2014; Tsai et al., 2012). The TSC1/2 protein works through negative regulation of the mTOR cascade involving Rheb, TORC1, and the EIF4E/EIF4G complex to dampen translation. The net result is that TSC1/2, like FMRP, operates as a negative regulator of local translation. Strikingly, genetic manipulations that relieve inhibition by either FMRP or TSC1/2 exert sharply different effects on classic forms of synaptic plasticity such as long-term potentiation (LTP) and long-term depression (LTD) (Fig. 5). Closer examination of these disparities is illuminating.
Fig. 5. Contrasting roles of TSC1/2 and FMRP.
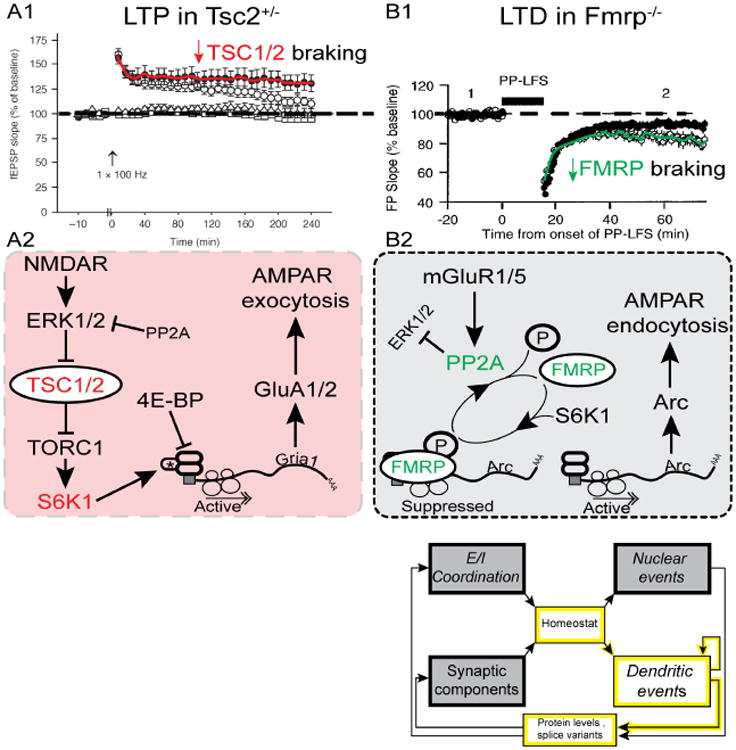
A1, LTP is exaggerated in Tsc2+/- mice. From (Ehninger et al., 2008). A2, Activation of NMDARs leads to the inhibition of TSC1/2, which disinhibits TORC1. Activation of TORC1 leads to the translation of proteins important for LTP, including GluA1/2 subunits. AMPAR exocytosis supports LTP. Loss of TSC1/2 (as seen in tuberous sclerosis) would lead to excessive TORC1 activation, and exaggerated LTP (experimentally supported by A1). B1, LTD is exaggerated in FMRP KO mice. From (Huber et al., 2002)(Copyright 2002 National Academy of Sciences, USA). B2, Putative signaling pathway linking metabotropic GluR action to local translation of Arc and LTD. Activation of mGluR1/5 activates PP2A, which dephosphorylates FMRP. This releases mRNA transcripts from FMRP repression, and allows the local translation of LTD-relevant proteins. Translation of Arc supports readout of low spine activity by β-CaMKII, Arc-dependent AMPAR endocytosis, and LTD (Okuno et al., 2012). Loss of FMRP (as seen in FXS), leads to decreased inhibition of translation, and increased LTD (see panel B1).
In hippocampal slices from Tsc2+/- mice, LTP is enhanced (Fig. 5A1); as a result, protocols that normally induce a decaying form of potentiation (E-LTP) result in the generation of long-lasting LTP (L-LTP) (Ehninger et al., 2008). Furthermore, LTD is attenuated, both in Tsc2+/- (Auerbach et al., 2011; Bateup et al., 2011), and in a loss of function mutant, Tsc2ΔRG, that weakens translational braking (Chevere-Torres et al., 2012). In accord with the consensus-signaling pathway (Fig. 5A1,A2), Ehninger et al. further demonstrated that abnormalities of LTP and deficiencies in hippocampal-dependent learning tasks in Tsc2+/- animals, in which mTOR-signaling is abnormally heightened, can be rescued by the mTOR inhibitor rapamycin.
In contrast to the loss of TSC function, deletion of FMR1 gives rise to dramatically different alterations of both potentiation and depression. The fmr1 KO spares L-LTP (Zhang et al., 2009), while LTD is famously enhanced (Huber et al., 2002) (Fig. 5B1). With either low frequency stimulation (LFS, Fig. 5B1) or application of a metabotropic glutamate receptor agonist (Huber et al., 2002), removal of FMRP braking causes an increase in the amplitude and persistence of LTD.
Taken together, these findings suggest that the physiological outcome of increased translation differs, depending on the mode of regulation. The distinction between FMRP- and TSC-mediated control and their physiological outcomes has not been emphasized in the literature. Focusing on these differences begs the question: how might various modes of elevated protein synthesis be linked to synaptic plasticity and behavior?
Relating local translation to synaptic plasticity and disease
How might malfunctioning protein synthesis impair regulation of synaptic plasticity? As a starting point, we focus on the duality between synaptic strengthening and weakening observed in long-term potentiation (LTP) and long-term depression (LTD). Textbooks show LTP and LTD as functionally opposite outcomes, either of which can occur at the level of individual synapses (all true). Sometimes LTP is presented as the inscription of a tiny piece of memory, and LTD as its erasure. This implicitly portrays memory storage as rooted in the strengths of single synapses, with each individual spine operating as a discrete unit independent of its neighbors. It is unlikely that changes in the nervous system during such events are simply a net increase or decrease in total synaptic activity as this simplified model would indicate. A more integrated view is a partnership in which LTP and LTD work in parallel, rather than in opposition, to redistribute synaptic strength in support of behavioral plasticity.
Bidirectional changes at nearby spines
In work not previously highlighted in the context of local translation, synaptic biologists postulated and observed local coordination between strengthening and weakening of synapses, advantageous for both network stability and for optimizing information storage. For example, Rabinowitch & Segev theorized about dendritic mechanisms of local regulation, where the strengthening of single spines by Hebbian plasticity is chained to compensatory mechanisms for weakening other spines nearby (Fig. 6A) (Rabinowitch and Segev, 2008). These ideas are supported by the anatomical studies of Bourne & Harris, who compared the morphology of serially reconstructed dendrites from hippocampal tissue, which had or had not undergone saturating LTP (Bourne and Harris, 2012). After LTP, single spines enlarged and the postsynaptic area increased, as expected. But remarkably, the density of spines along the dendritic length fell by almost half; as a result, the aggregate postsynaptic area over the examined stretch of dendrite was no different in LTP compared to control conditions (Fig. 6B). The implication is that spines in the neighborhood of the strengthened synapses were subject to weakening or disappearance, very much as Rabinowitch & Segev suggested. The kind of coordination Bourne & Harris observed would help keep the overall synaptic input of a dendritic segment within reasonable bounds, while allowing selective inputs to strengthen. Both theory and experiment point out the usefulness of considering spines populating a section of dendrite collectively, and reckoning with the possibility of disparate changes in synaptic strength (Fig. 6C1,C2).
Fig. 6. Competing LTD and LTP processes.
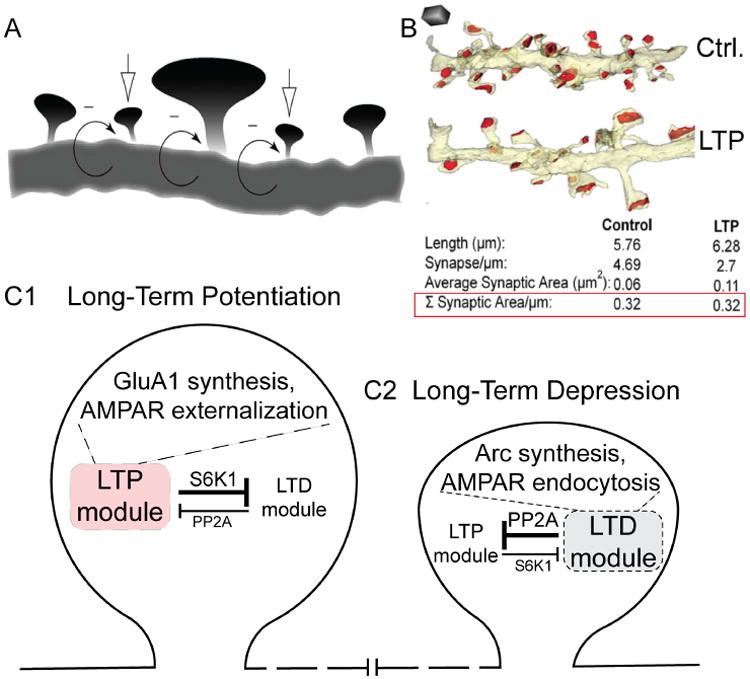
A, LTP and LTD are illustrated here as occurring within the same dendritic compartment, at neighboring spines. From (Rabinowitch and Segev, 2008). B, Following LTP-induction protocol, some spines increase in size, without the overall synaptic area changing. From (Bourne and Harris, 2012). C1,C2, LTP and LTD are seen as competing processes within single spines. PP2A activation is critical for the LTD module but inhibits the LTP module, whereas S6K1 is critical for the LTP module but inhibits the LTD module. Mutual inhibition supports a winner-take-all scenario. When the action of S6K1 predominates, the LTP module overrides LTD, and GluA1 synthesis and AMPAR externalization takes place (C1; Fig. 5A2). When the action of PP2A predominates, the LTD module prevails over LTP, and Arc synthesis and AMPAR endocytosis takes place (C2; Fig. 5B2). For clarity, this model focuses on simple aspects of multiple mutually inhibitory interactions between the FMRP and TSC1/2 pathways (Ashley and Warren, 1995; Bian et al., 2015; Darnell and Klann, 2013).
Neither Rabinowitch & Segev nor Bourne & Harris explicitly addressed how coordination between strengthening and weakening of neighboring spines might come about. Knowing that LTP is induced by a strong input and a strong release of glutamate onto a soon-to-be-strengthened spine, it becomes clear that this must be accompanied by glutamate spillover to nearby spines. By laws of diffusion, the local concentration of glutamate ([Glu]) must fall as a function of distance from the source. Thus, nearby spines must experience a decline in local [Glu] such that the more sensitive mGluRs, but not NMDARs, are strongly activated, leaving mGluR signaling to predominate in a penumbra of spines near the one destined to be strengthened via LTP. According to the mGluR scenario (Bear et al., 2004), the neighboring spines would undergo rapid PP2A-, FMRP-mediated de-repression of local translation. This in turn will increase the abundance of regulatory proteins such as Arc, CaMKIIα, PSD95, and CaMKIIβ (Darnell et al., 2011; Muddashetty et al., 2011; Niere et al., 2012; Park et al., 2008; Stefanovic et al., 2015), setting up the spine for a binary decision, to undergo LTD or LTP.
Sharp dichotomy in regulating synaptic strength
The morphological picture from Bourne and Harris suggests that individual spines experience a threshold phenomenon that determines whether they are to be strengthened or weakened, presumably guided by the inputs they receive during LTP induction. How is the decision point between one or the other determined? FMRP-regulated gene products such as Arc, βCaMKII and SAPAP (Brown et al., 2001; Darnell et al., 2011; Niere et al., 2012; Park et al., 2008) could contribute to a biochemical mode of discrimination. Okuno et al. have shown that βCaMKII serves as an activity sensor; when Ca2+ is low, CaM-free, kinase-deactivated βCaMKII interacts directly with Arc and helps facilitate Arc-mediated endocytosis of AMPARs (Okuno et al., 2012). In contrast, when Ca2+ is high, activated βCaMKII can participate in SAPAP trafficking and increase incorporation of AMPARs, enabling spine remodeling and synaptic strengthening (Shin et al., 2012). Furthermore, NMDAR-dependent mechanisms could work through αCaMKII accumulation to drive AMPAR incorporation and LTP (Huganir and Nicoll, 2013; Lisman et al., 1997). Thus, the decision between strengthening and weakening likely employs local protein synthesis as a platform, but is ultimately governed by βCaMKII as a local sensor of activity-dependent rises in Ca2+.
Sharp decision between LTP- and LTD in single spines, but coexistence along a dendrite
Published data supports that negative feedback connections between translational modes are regulated by FMRP and TSC1/2. Two possible players are a protein kinase (S6K1) and a protein phosphatase (PP2A), each known to play multiple roles (Fig. 5A2, B2). S6K1 has been shown to act downstream of TORC1 to drive EIF4E-regulated initiation of translation (Richter et al., 2015), and dampens translation of FMRP-regulated mRNAs by phosphorylating FMRP (Santoro et al., 2012). Furthermore, PP2A not only removes the FMRP brake (Niere et al., 2012) but also interferes with NMDA signaling by dephosphorylating and deactivating ERK1/2 (Alessi et al., 1995). With this negative cross-talk, individual spines are expected to select sharply between one translational program or the other (depicted schematically as mutual inhibition between LTP and LTD modules, Fig. 6C1, C2). In synapses with exaggerated LTD, as in FXS, LTD is further linked to synapse elimination via the involvement of the nuclear transcription factor MEF2 (Pfeiffer et al., 2010). Huber and colleagues showed that MEF2-mediated transcription and local FMRP-regulated translation work together to eliminate excitatory synapses. This provides a satisfying explanation for the thinning out of non-potentiated postsynaptic spines in Fig. 6B (Bourne and Harris, 2012).
To recapitulate this composite scenario for synapse weakening (Darnell et al., 2011; Gross et al., 2015; Huber et al., 2002; Niere et al., 2012):
Low, steady ↑[Glu] (or mGluR type I agonist) → mGluR activation → PP2A activation → FMRP derepression → ↑Arc;
↑Arc, working with non-active CaMKIIβ → GluA1/2 removal → synapse weakening → LTD, and possibly synapse elimination
This stands in contrast with, and in opposition to another hypothetical scenario, a composite view of synapse strengthening (Banko et al., 2005; Bateup et al., 2011; Kelleher et al., 2004):
High, brief ↑[Glu] → NMDAR act → ↑MAPK signaling → inhibition of TSC complex function → inhibition of Rheb inhibition → mTOR activation → EIF4E activation → GluA1,2 synthesis → GluA1 incorporation upon ↑ CaMKIIα,β activation → LTP → secondary morphological changes
Brief recap
Our working hypothesis depicts individual spines as veering between two kinds of activity-dependent regulation of mRNA translation. Taking advantage of the greater glutamate sensitivity of mGluR versus NMDAR, they support dichotomous changes in postsynaptic properties that favor LTD in one case and LTP in the other. We hypothesize that these processes are mutually antagonistic within single spines, but can work side by side at nearby spines. This model provides a means for putting Hebbian plasticity at one spine in close proximity to LTD at a near neighbor (Rabinowitch and Segev, 2008) and thus redistributing synaptic weights (Bourne and Harris, 2012). Proteins locally synthesized under control of FMRP or TSC1/2 are critical for LTD and LTP. Accordingly, dysfunction of either mode of regulation would be disruptive for overall LTP:LTD coordination (and potentially circuit-wide E:I coordination), and thus deleterious for behavioral plasticity.
E:I coordination and altered synapse structure
A dozen years ago, Rubenstein & Merzenich (R&M) put forth a provocative hypothesis: that an excess of excitation relative to inhibition was a major cause of autism (Rubenstein and Merzenich, 2003). As they argued, and as recent studies have reminded us, 30% of patients with an ASD also have epilepsy, and 30% of patients with epilepsy have been diagnosed with an ASD (Bolton et al., 2011; Brooks-Kayal, 2010; Tuchman et al., 2009). R&M went on to note that 50-70% of EEG or MEG sleep recordings from children with autism show evidence of sharp spike activity, indicative of unstable cortical networks. The concept of “E:I balance” has greatly stimulated studies of brain physiology and ASD pathogenesis, even though on closer examination the “balance” is neither complete nor desirable. During normal synaptic communication, the inhibitory current is typically several-fold larger than the excitatory current at the neuron's resting potential. As a result, the E:I ratio cannot be 1.0 as the word “balance” implies. In feedforward networks excitation typically occurs out-of-sync with inhibition, preceding it by milliseconds. This pattern of activity creates a compound synaptic event that drives spiking with much greater temporal precision than could be achieved by excitation alone (Pouille et al., 2009; Pouille and Scanziani, 2001). Indeed, a fixed moment-to-moment relationship between E and I would not be effective, either for throughput of single spikes, or for more complicated and powerful forms of network activity such as sharp wave ripples, which require E and I to be out of step with each other (Stark et al., 2014). In several ways then, “E:I balance” does not accurately depict the temporally asynchronous, proportional-but-not-equal, temporarily adjustable relationship between the magnitudes of E and I required for proper network activity. We utilize “E:I coordination” as a more faithful characterization of the neuronal state, amply supported by experimental evidence that neuronal circuits can enforce coordination of E and I strength (Xue et al., 2014)(Fig. 7B), even down at the level of single dendritic branches (Liu, 2004) (Fig. 7A). Terminology aside, understanding E:I coordination is a pressing issue for neuroscience in general, and for the pathogenesis of ASD in particular.
Fig. 7. E:I coordination in neuronal, circuit, and pathophysiological contexts.
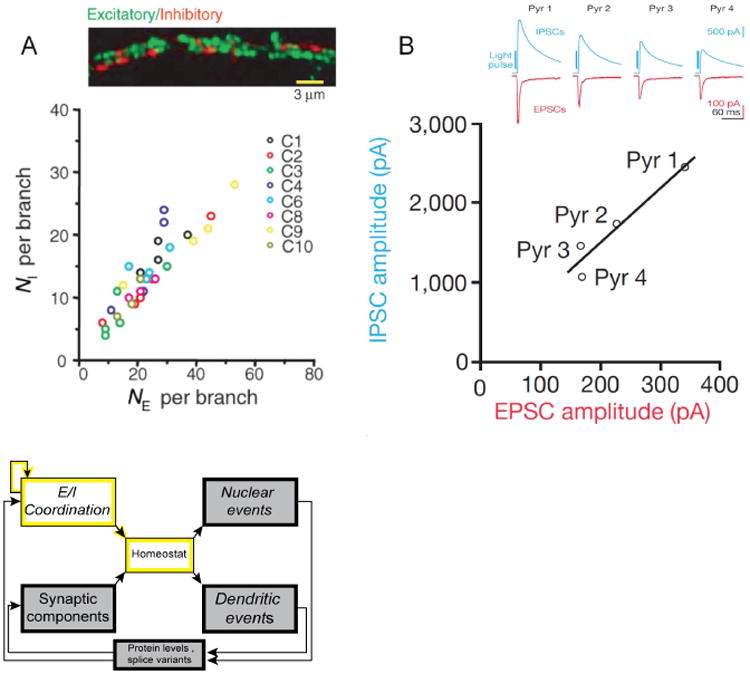
A, Coordination (but not equality) of the numbers of excitatory and inhibitory synapses per dendritic branch (NE, NI respectively). NI is ∼0.5× NE. From (Liu, 2004). B, Coordination (but not equality) of the collective strengths of E and I transmission, illustrated for 4 pyramidal neurons driven by the same optogenetic stimulation of layer IV feedforward inputs. Inset shows recordings of EPSCs (red) and IPSCs (blue) with |driving force| of ∼70 mV. |IPSC| is ∼7×|EPSC|, reflecting a ∼7-fold ratio of synaptic conductances. From (Xue et al., 2014).
So how good is the evidence that ASD results from dysfunctional E:I coordination? Does ASD arise from too much E relative to I, as R&M proposed, or is the opposite just as dangerous? These issues are addressed in a thoughtful review by Nelson and Valakh (2015). Two of their important messages are that: 1) perturbations of excitation or inhibition must be considered within the broader context of complex neuronal circuits, and 2) it is hard to disentangle the impact of a primary insult from the compensation that ensues. The first point is vividly illustrated by Rothwell, Malenka, Sudhof et al., who have uncovered a circuit basis for repetitive motor behaviors arising from ASD-based manipulations of NLGN3: altered synaptic inhibition of medium spiny inhibitory neurons in ventral striatum (Rothwell et al., 2014). The second point is extended here by putting proteins encoded by ASD genes into explicit feedback loops for neuronal autoregulation.
Regardless of the specific circuit, adjustments of inhibitory neuron firing or inhibitory transmission can operate much more rapidly than feedback by transcriptional or translational loops. As a consequence, E:I coordination can be viewed as made of the same cloth as transcription and translation autoregulation — with the control of inhibition filling in a critical kinetic gap. Thus, adjustment of inhibition provides a particularly powerful and temporally flexible form of autoregulation. Conversely, it stands to reason that dysfunction of inhibition would be a major component of ASD pathogenesis.
There is little doubt that E:I coordination would be affected by genetic perturbations of a variety of synaptic components, including postsynaptic components (PSD95, SHANK1, -2, -3; SYNGAP1), and proteins involved in trans-synaptic interaction (NRXN2, NGLN1, -3, -4). For example, the ASD-associated SHANK family are scaffolding proteins present in the postsynaptic density that serve a crucial role in synapse formation and maintenance by connecting proteins at the membrane with the actin cytoskeleton of the dendritic spine (Guilmatre et al., 2014). SHANK3 is one of the genes with the strongest statistical links to ASD, but as the protein functions postsynaptically in both excitatory and inhibitory populations, its effects on E:I coordination, while very important, are not simple to pin down. In the previous section, “Dysfunction of Local mRNA Translation and Synaptic Plasticity,” we discussed aspects of excitatory synaptic strength that can go awry in ASD. Here we briefly focus on a narrower set of examples where ASD may arises specifically from a defect in inhibition, to highlight the idea that dysfunction of E:I coordination can play a causal role in ASD.
ARX
(Aristaless related homeobox gene), an X-linked gene (Xp22.1), encodes the first transcription factor (TF) implicated in causing autism (Rubenstein and Merzenich, 2003; Turner et al., 2002). Boys that inherit mutations in ARX primarily manifest epilepsy, mental retardation or movement disorders, and some also have ASD (Turner et al., 2002). ARX mutations have pleiotropic effects (Rubenstein and Merzenich, 2003; Sherr, 2003; Turner et al., 2002), not atypical of ASD-causing genes. In the mouse, Arx is expressed in several regions and cell types within the forebrain. For ASD, its most pertinent role is in a hierarchy of TFs critical for GABAergic interneuron development. Loss of ARX severely impairs interneuron development (Vogt et al., 2014), as Arx is a repressor that functions to orchestrate interneuron differentiation. Of the 84 genes found to be abnormally regulated in Arx-/- mice (Fulp et al., 2008), many are involved in neuronal development (cell migration, axonal guidance, neurogenesis) and some have been in implicated in ASD, epilepsy, and mental retardation, befitting clinical features seen in patients with ARX mutations. While much work is needed to clarify the molecular and behavioral effects of Arx regulation (Marsh and Golden, 2012), Arx already provides a clear example of the severe impact of interneuron loss on ASD as well as epilepsy (Rubenstein and Merzenich, 2003).
SCN1A
Mutations of this sodium channel gene are most prominent in Dravet syndrome (DS), also known as severe myoclonic epilepsy in infancy (SMEI). This disorder is characterized by infantile-onset epilepsy, both febrile and afebrile seizures resistant to medication, and cognitive dysfunction. DS occurs from loss of function of SCN1A, which encodes the α-subunit of the voltage-gated sodium channel NaV1.1, prominently expressed in the soma and axon initial segment of neurons (Cheah et al., 2012). Identification of >300 mutations in SCN1A exons accounts for ∼70% of DS cases; and most mutations are truncations or deletions that disrupt voltage sensing or ion conduction, with the remaining 30% likely acting through their impact on SCN1A expression (Oakley et al., 2011). SCN1A loss-of-function impairs sodium currents and spiking in hippocampal GABAergic interneurons, without a detectable effect on excitatory pyramidal neurons. In turn, decreased inhibitory firing leads to dysregulation of inhibition, the likely cause of seizures in DS patients (Oakley et al., 2011). Closer examination of autistic features in 37 DS patients showed that ∼25% met the criteria for autism, and ∼95% presented with intellectual disability, with more severe deficits in those with autism. Interestingly, there was no apparent difference in the epileptic features of DS patients with or without autism (Li et al., 2011). Do autistic-like behaviors arise as sequelae of frequent seizures, or directly from reduced GABA transmission? To address this, Catterall's group treated Scn1a+/- mice with the benzodiazepine clonazepam, a positive allosteric modulator of GABAA receptors that enhances opening of GABA receptor chloride channels in response to presynaptically released GABA. Low doses of clonazepam completely rescued the abnormal social behaviors and deficits in fear memory typically presented by Scn1a+/- mice (Han et al., 2012), demonstrating the impact of restoring the proper coordination of inhibitory transmissions.
Overview
By reducing the number or excitability of inhibitory neurons, ARX and SCN1A weaken GABAergic transmission in distinct ways. Taken together with other genes (e.g., GABRB3, CNTNAP2 (Sanders et al., 2015), and possibly CNTNAP4 (Karayannis et al. 2014)), these ASD-associated genes illustrate the importance of E:I coordination. We note that many of the genetic actions do not encourage sharp distinctions between inhibitory neuron function or structure, or even between effects on excitatory or inhibitory neurons. Further, E:I dyscoordination is capable of influencing a variety of other neuronal functions, including plasticity and activity-dependent gene expression. How these functions may connect and interact is challenging to predict, particularly as the downstream effects will vary greatly. Nevertheless, the concept of E:I coordination will remain a pillar of ASD pathogenesis.
Summary and Future Outlook
This review extends traditional schemes that link genes → behavior in terms of a chain or tree of causality. We have emphasized a rethinking of those logical relationships: gene candidates, signaling proteins, and physiological variables, organized in a set of dynamic feedback loops (exemplified by the scheme in Fig. 2). Our emphasis on feedback was motivated in part by prevailing groupings of ASD-related genes. As categorized by putative function (activity-dependent transcription, glutamate regulation of local protein synthesis and synaptic plasticity, and alterations of E:I coordination and synaptic structure), all of these groups display a prominent autoregulatory aspect. We build upon excellent reviews on ASD that highlight the role of homeostasis (Ramocki and Zoghbi, 2008; Toro et al., 2010; Wondolowski and Dickman, 2013), the bidirectional communication between postsynaptic receptors and dendritic protein synthesis (Santini and Klann, 2014), and the influence of homeostasis on aberrant E:I coordination (Nelson and Valakh, 2015). We also take note of valuable categorizations of ASD-associated genes based on anatomical location or protein-protein interactions. The physiological framework reviewed here builds upon the recent identification of interconnected signaling hubs (Krumm et al., 2014; Parikshak et al., 2013; Voineagu et al., 2011; Willsey et al., 2013), and a venerable literature on feedback in neurons, ranging from action potential generation (Hodgkin and Huxley, 1952) to transcription-factor based neuronal differentiation (Hobert, 2011; Weinberg et al., 2013). If constellations of ASD-associated genes support the servo control of neuronal excitability, gene expression, and local synaptic strength in the healthy brain, it is reasonable to think that faulty dynamic feedback regulation will also be important for dysfunction in the autistic brain.
To introduce the power of feedback, we began with a unitary track and a specific mechanism for autoregulation of neuronal excitability (Fig. 1). However, the expanded feedback arrangement in Fig. 2 offers multiple choices for sensor, effector mechanisms, feedback connections and controlled variables, among further possibilities not enumerated. In combination, such options greatly expand the possible ways that a neuron or circuit could use feedback to keep physiological variables close to their setpoints. Servo loops for different variables may also interact (e.g., dysfunction of Wnt signaling and dendritic morphology would alter E:I coordination). Thus, the scheme provides ample opportunity for rich regulatory interactions, but adds complexity to the task of deciphering ASD.
Implications of dysfunctional autoregulation
If malfunctioning servo loops help manifest ASD, researchers will encounter a mix of good and bad news. On the positive side, stressed-out feedback systems will likely alter levels of proteins, protein phosphorylation, and signaling molecules, thus providing useful biomarkers as to the identity of the dysfunctional pathway. Furthermore, a therapeutic strategy could aim to repair the loop as a whole, rather than the specific molecular lesion itself. Confusingly, however, a specific change in cellular biochemistry could be part of the pathogenic mechanism or a beneficial homeostatic response to it. Disambiguating these possibilities would be needed to understand cause and effect, but will present the challenge of devising better experimental methods for directly and specifically varying the affected players. Analysis of genetic modifications will remain the gold standard for causative inference. Nonetheless, transcriptomic, proteomic or metabolic changes will also be useful for the identification of dysfunctional pathways well before their pathogenic or homeostatic impact is readily determined.
The hypothetical framework is put forward at the level of neurons and not any particular brain circuit. However, the expression of perturbations and autoregulatory sequelae will depend on the particular circumstances of individual circuits and their cellular components. Thus, abnormal physiological feedback would affect different circuits in varying ways, depending on their cellular and synaptic makeup (Rothwell et al., 2014). Pathology at the cellular level may even reverberate around multiple interacting circuits. The diverse impact in individual circuits likely contributes to clinical complexity, but may eventually help diagnostic efforts once underlying principles can be deciphered.
Predictions for the future
We close this review with largely optimistic predictions.
Relating the pathophysiology of ASD to the physiology of the normal brain is underexplored, but will be of mutual benefit.
Despite its current bewildering complexity, the genetics of ASD (∼500 genes) will prove useful in seeking a unifying pathophysiological scheme. Here we side with those who prefer to keep focused on the entire spectrum (like Hans Asperger and the committee that made DSM-V) rather than split it into sub-disorders (like Leo Kanner and the creators of DSM-IV). Consideration of autoregulatory loops suggests a wide diversity of specific pathophysiological sub-scenarios, possibly easing the ongoing tension between lumping and splitting (Silberman, 2015). Finding yet more causative genetic modifications might prove helpful in delineating those pathophysiological sub-scenarios.
Extrapolating from excitation-transcription coupling (Fig. 4) and local glutamate-translation coupling (Fig. 5), we expect that coupling between excitation and chromatin remodeling will be a compelling process for further investigation (Kim et al., 2010), with likely relevance to ASD. Furthermore, we anticipate further pursuit of functional synergies between the effects of LTP and LTD, aided by local translation of mRNA in dendrites.
Medium- to high-throughput methods will be developed to determine the physiological impact of genetic perturbations. This will be needed to sort the individual mutations according to their impact on signaling, which may not always be equivalent to their gene ontology class. A rare but exemplary study examined a non-coding variant in CACNA1C of wide distribution across neuropsychiatric diseases and found gain of function in transcript abundance and L-type current density in a great majority of instances (Vogt et al., 2015; Yoshimizu et al., 2015). In general, if altered function of an ASD-related protein can be traced further to changes in transcription, splicing, translation or trafficking, varied polygenic interactions are to be expected with gene products controlling those processes.
ASD-related signaling pathways that appear in genetic screens could also be targets of environmental agents. Surface signaling proteins such as calcium channels and NMDA receptors are very susceptible to toxins, drugs and antibodies (Cline et al., 1996). Research on environmental influences such as maternal inflammation (Hsiao et al., 2012; Patterson, 2011), the gut microbiome (Hsiao et al., 2013), and ASD-provoking drugs such as valproic acid may offer new understanding. Not all environmental factors will prove to be deleterious. Sleep, fever, and behavior modification each have beneficial influences that are not well understood but might influence compensatory feedback signaling.
Complementary experimental and computational approaches will be productively harnessed in studying feedback systems that interlock at cellular, circuit, and network levels. Homeostatic compensation for dysfunctional E:I coordination (Nelson and Valakh, 2015) will be an interesting proving ground for computational analysis (Rosenberg et al., 2015). The agent bumetanide, now in clinical trials for treatment of ASD (Lemonnier and Ben-Ari, 2010), enhances GABAergic inhibition and may thereby beneficially readjust activity-dependent feedback.
Network analysis will aid the development of biomarkers for dysfunctional signaling, which to be most useful, cannot be viewed statically or in isolation. As a diagnostic strategy, the time-synchronized read-out of multiple biomarkers may be a first step in designing and tuning therapeutic interventions.
Development Box
Autism spectrum disorders are often framed as the outcome of aberrant development and interconnectivity of brain structures (Berg and Geschwind, 2012; Courchesne et al., 2001; Geschwind and Levitt, 2007). This prompts the question of whether ideas about dysfunctional homeostasis in ASD might be applicable to neurodevelopment. If homeostasis provides negative feedback to minimize deviations from a fixed set point, it might appear at odds with neurodevelopment, a fundamentally transitional process. In fact, negative feedback mechanisms could also support developmental trajectories if driven by a gradually varying set point, like a graded setting on a thermostat or a changing command potential in a voltage clamp system. Alternatively, certain stages of neurodevelopment may not be driven by feedback regulation, but may harness a unidirectional chain of events like a transcription factor cascade, tuned by feedback on an evolutionary scale but not by immediate servo control.
Early neurodevelopment relies on cell-to-cell chemical signaling rather cell-autonomous, activity-dependent homeostasis like that in Fig. 1. Nonetheless, autoregulation, involving positive as well as negative feedback, is critical. Positive feedback loops generate a threshold between two discrete developmental states, enforcing all-or-none distinctions between neuronal precursors and their resultant progeny. Classic experiments have defined lateral inhibition between a neuroblast and its neuronal precursor neighbors (Doe and Goodman, 1985), the molecular players in intercellular communication (Louvi and Artavanis-Tsakonas, 2012), and downstream nuclear signaling mechanisms (Imayoshi and Kageyama, 2014). The positive feedback within a neuroblast and negative feedback to adjacent precursor cells safeguards the reservoir of precursors even as neuroblasts emerge from its midst. Analogous principles apply to the asymmetric cell division between neuroblasts and ganglion mother cells that give rise to glia and neurons (Anderson and Jan, 1997; Knoblich, 1997). In the mature state, similar positive feedback loops can operate cell autonomously to enforce sharp separation between diverse neuronal phenotypes (Hobert et al., 2010).
In later stages of circuit development, when neurons have gained excitability and synapse formation has begun, autoregulation by negative feedback is prominent (Ben-Ari, 2015; Feller, 2004; Hensch, 2005; Rosenberg and Spitzer, 2011; Turrigiano and Nelson, 2004). In some cases, the autoregulation is cell-autonomous, in others it is a distributed property of an entire circuit and its state of chemical signaling. Possibly pertinent to autism, electrical events can arise from ion fluxes through chloride, sodium, calcium, or NMDA receptor channels, and the intracellular signaling can involve Ca2+ spikes, oscillations or waves, often with the participation of internal Ca2+ stores. The various Ca2+ signals often exert effects through regulation of gene expression via E-T coupling, or regulation of local translation. The details vary among diverse brain circuits but the overall organization seems generally compatible with the schema in Fig. 2.
Transcriptomics can help define transitions between developmental stages and mechanisms
The transitions between developmental phases have been charted by tracking expression of ASD- and ID-related genes through human cortical development (Parikshak et al., 2013). An early transition (∼11 weeks post-conception) is indicated by attenuation of a gene module that opposes neuronal differentiation, giving way to one that favors it. A later transition (∼20 weeks post-conception in humans, equivalent to perinatal mice) is marked by enhancement of a gene module that supports activity-dependent synaptic development. Bioinformatics analyses suggest that these processes are connected via translational regulation by FMRP and transcriptional co-regulation by common transcription factors, mechanisms touched upon elsewhere in this review. Developmental transcriptomic data seems compatible with the hypothesis that that cascades of transcription factors and positive feedback autoregulatory loops predominate early in development, but pass the baton to activity-dependent feedback mechanisms that fine-tune neuronal morphology and synaptic connectivity (see (Fishell and Heintz, 2013; Kepecs and Fishell, 2014) for review). Genetic insults that disrupt early developmental transcriptional cascades likely result in lethality and are therefore underrepresented as proximal causes of neuropsychiatric diseases.
Involvement of ASD-associated proteins in development and mature states
There are no sharp boundaries between development and autoregulation of mature function, so proteins encoded by the same ASD-associated genes likely participate in both development and ongoing homeostasis. For example, point mutations in CACNA1C that underlie Timothy Syndrome perturb the development of neuronal dendrites (Krey and Dolmetsch, 2007), bones (Ramachandran et al., 2013), and skin (Yucel et al., 2013), as well as ongoing electrophysiological function of heart and brain (Krey and Dolmetsch, 2007; Splawski et al., 2005; Splawski et al., 2004). It remains unclear whether the same pathophysiological mechanism is responsible at all stages. Likewise, Wnt signaling is known to shape early cell-cell interactions (Nelson and Nusse, 2004), and to guide cell migration and differentiation of axons and dendrites (Salinas and Zou, 2008; Yu and Malenka, 2003); nonetheless, Wnt2 and other proteins traditionally associated with neural development may have important roles in post-critical period regulation of neuronal function (Farias et al., 2007; Moon et al., 2004; Wayman et al., 2006). The lack of temporally restricted involvement has interesting implications for discovery of ASD therapies. While some aspects of autoregulatory signaling may be complete when neural development is over, other (dys)function may carry on right through to mature brain and, with suitable caution (Kalkman, 2012; Mullard, 2015), could offer therapeutic opportunities.
Acknowledgments
We thank Ilya Bezprozvanny and Simon Sun for thoughtful comments on the manuscript, and all Tsien lab members for useful feedback and advice. R.W.T. thanks E. Einaudi, C. Feinstein, J. Hallmayer, R. Dolmetsch, L. Luo and other colleagues in the Stanford Autism Working Group. This work was supported by research grants to R.W.T. from the Institute of General Medical Sciences (GM058234), the National Institute of Neurological Disorders and Stroke (NS24067), the National Institute of Mental Health (MH071739), National Institute on Drug Abuse (DA040484), and Simons, Mathers, Druckenmiller and Burnett Family Foundations. Further support provided by research grants awarded to G.F. by the National Institute of Health, National Institute of Mental Health, National Institute of Neurological Disorders and Stroke, NYU Abu Dhabi, and the Simons Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilar-Valles A, Matta-Camacho E, Khoutorsky A, Gkogkas C, Nader K, Lacaille JC, Sonenberg N. Inhibition of Group I Metabotropic Glutamate Receptors Reverses Autistic-Like Phenotypes Caused by Deficiency of the Translation Repressor eIF4E Binding Protein 2. J Neurosci. 2015;35:11125–11132. doi: 10.1523/JNEUROSCI.4615-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P. Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr Biol. 1995;5:283–295. doi: 10.1016/s0960-9822(95)00059-5. [DOI] [PubMed] [Google Scholar]
- Alvarez Retuerto AI, Cantor RM, Gleeson JG, Ustaszewska A, Schackwitz WS, Pennacchio LA, Geschwind DH. Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum Mol Genet. 2008;17:3887–3896. doi: 10.1093/hmg/ddn291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature genetics. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, Jan YN. The determination of the neuronal phenotype. In: Cowan WM, Jessell TM, Zipursky SL, editors. Molecular and Cellular Approaches to Neural Development. New York: Oxford University Press; 1997. pp. 26–63. [Google Scholar]
- Ashley CT, Jr, Warren ST. Trinucleotide repeat expansion and human disease. Annu Rev Genet. 1995;29:703–728. doi: 10.1146/annurev.ge.29.120195.003415. [DOI] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader PL, Faizi M, Kim LH, Owen SF, Tadross MR, Alfa RW, Bett GC, Tsien RW, Rasmusson RL, Shamloo M. Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15432–15437. doi: 10.1073/pnas.1112667108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber JC, Ellis KH, Bowles LV, Delhanty JD, Ede RF, Male BM, Eccles DM. Adenomatous polyposis coli and a cytogenetic deletion of chromosome 5 resulting from a maternal intrachromosomal insertion. J Med Genet. 1994;31:312–316. doi: 10.1136/jmg.31.4.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnby G, Abbott A, Sykes N, Morris A, Weeks DE, Mott R, Lamb J, Bailey AJ, Monaco AP International Molecular Genetics Study of Autism, C. Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. American journal of human genetics. 2005;76:950–966. doi: 10.1086/430454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Cohen S, Lombardo MV, Auyeung B, Ashwin E, Chakrabarti B, Knickmeyer R. Why are autism spectrum conditions more prevalent in males? PLoS Biol. 2011;9:e1001081. doi: 10.1371/journal.pbio.1001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu SN, Kollu R, Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic Acids Res. 2009;37:D832–836. doi: 10.1093/nar/gkn835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL. Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J Neurosci. 2011;31:8862–8869. doi: 10.1523/JNEUROSCI.1617-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Is birth a critical period in the pathogenesis of autism spectrum disorders? Nat Rev Neurosci. 2015;16:498–505. doi: 10.1038/nrn3956. [DOI] [PubMed] [Google Scholar]
- Ben-David E, Shifman S. Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PLoS Genet. 2012;8:e1002556. doi: 10.1371/journal.pgen.1002556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayed R, Gharani N, Rossman I, Mancuso V, Lazar G, Kamdar S, Bruse SE, Tischfield S, Smith BJ, Zimmerman RA, et al. Support for the homeobox transcription factor gene ENGRAILED 2 as an autism spectrum disorder susceptibility locus. American journal of human genetics. 2005;77:851–868. doi: 10.1086/497705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome Biol. 2012;13:247. doi: 10.1186/gb-2012-13-7-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nature genetics. 2010;42:489–491. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- Bi C, Wu J, Jiang T, Liu Q, Cai W, Yu P, Cai T, Zhao M, Jiang YH, Sun ZS. Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum Mutat. 2012;33:1635–1638. doi: 10.1002/humu.22174. [DOI] [PubMed] [Google Scholar]
- Bian WJ, Miao WY, He SJ, Qiu Z, Yu X. Coordinated Spine Pruning and Maturation Mediated by Inter-Spine Competition for Cadherin/Catenin Complexes. Cell. 2015;162:808–822. doi: 10.1016/j.cell.2015.07.018. [DOI] [PubMed] [Google Scholar]
- Bolton PF, Carcani-Rathwell I, Hutton J, Goode S, Howlin P, Rutter M. Epilepsy in autism: features and correlates. Br J Psychiatry. 2011;198:289–294. doi: 10.1192/bjp.bp.109.076877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16:551–563. doi: 10.1038/nrn3992. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Nanoscale analysis of structural synaptic plasticity. Curr Opin Neurobiol. 2012;22:372–382. doi: 10.1016/j.conb.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braat S, D'Hulst C, Heulens I, De Rubeis S, Mientjes E, Nelson DL, Willemsen R, Bagni C, Van Dam D, De Deyn PP, et al. The GABAA receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle. 2015;14:2985–2995. doi: 10.4161/15384101.2014.989114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenkamp AF, Matthes J, Nass RD, Sinzig J, Lehmkuhl G, Nurnberg P, Herzig S. Rare mutations of CACNB2 found in autism spectrum disease-affected families alter calcium channel function. PloS one. 2014;9:e95579. doi: 10.1371/journal.pone.0095579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Kayal A. Epilepsy and autism spectrum disorders: are there common developmental mechanisms? Brain Dev. 2010;32:731–738. doi: 10.1016/j.braindev.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R, et al. A genetic variant that disrupts MET transcription is associated with autism. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16834–16839. doi: 10.1073/pnas.0605296103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nature genetics. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS, Stevens CR, Schubert CR, Collaboration AAS, Greenberg ME, et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, Rubenstein JL, Catterall WA. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14646–14651. doi: 10.1073/pnas.1211591109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JA, Penagarikano O, Belgard TG, Swarup V, Geschwind DH. The emerging picture of autism spectrum disorder: genetics and pathology. Annu Rev Pathol. 2015;10:111–144. doi: 10.1146/annurev-pathol-012414-040405. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Chevere-Torres I, Kaphzan H, Bhattacharya A, Kang A, Maki JM, Gambello MJ, Arbiser JL, Santini E, Klann E. Metabotropic glutamate receptor-dependent long-term depression is impaired due to elevated ERK signaling in the Delta RG mouse model of tuberous sclerosis complex. Neurobiol Dis. 2012;45:1101–1110. doi: 10.1016/j.nbd.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CH, Schoenfeld BP, Bell AJ, Hinchey P, Kollaros M, Gertner MJ, Woo NH, Tranfaglia MR, Bear MF, Zukin RS, et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380:106–119. doi: 10.1016/j.brainres.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline HT, Witte S, Jones KW. Low lead levels stunt neuronal growth in a reversible manner. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9915–9920. doi: 10.1073/pnas.93.18.9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, Portera-Cailliau C. Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron. 2015;87:699–715. doi: 10.1016/j.neuron.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook EH, Jr, Courchesne RY, Cox NJ, Lord C, Gonen D, Guter SJ, Lincoln A, Nix K, Haas R, Leventhal BL, et al. Linkage-disequilibrium mapping of autistic disorder, with 15q11-13 markers. American journal of human genetics. 1998;62:1077–1083. doi: 10.1086/301832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J, Yin J, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Karns CM, Davis HR, Ziccardi R, Carper RA, Tigue ZD, Chisum HJ, Moses P, Pierce K, Lord C, et al. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57:245–254. doi: 10.1212/wnl.57.2.245. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Translational animal models of autism and neurodevelopmental disorders. Dialogues Clin Neurosci. 2012;14:293–305. doi: 10.31887/DCNS.2012.14.3/jcrawley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics, C. Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, Mowry BJ, Thapar A, Goddard ME, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nature genetics. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nature neuroscience. 2013;16:1530–1536. doi: 10.1038/nn.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ferrari GV, Moon RT. The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene. 2006;25:7545–7553. doi: 10.1038/sj.onc.1210064. [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, Buxbaum JD. Genetics and genomics of autism spectrum disorder: embracing complexity. Hum Mol Genet. 2015a;24:R24–31. doi: 10.1093/hmg/ddv273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, Buxbaum JD. Recent advances in the genetics of autism spectrum disorder. Curr Neurol Neurosci Rep. 2015b;15:36. doi: 10.1007/s11910-015-0553-1. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- Dinwiddie DL, Soden SE, Saunders CJ, Miller NA, Farrow EG, Smith LD, Kingsmore SF. De novo frameshift mutation in ASXL3 in a patient with global developmental delay, microcephaly, and craniofacial anomalies. BMC Med Genomics. 2013;6:32. doi: 10.1186/1755-8794-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe CQ, Goodman CS. Early events in insect neurogenesis. II. The role of cell interactions and cell lineage in the determination of neuronal precursor cells. Dev Biol. 1985;111:206–219. doi: 10.1016/0012-1606(85)90446-4. [DOI] [PubMed] [Google Scholar]
- Dong S, Walker MF, Carriero NJ, DiCola M, Willsey AJ, Ye AY, Waqar Z, Gonzalez LE, Overton JD, Frahm S, et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 2014;9:16–23. doi: 10.1016/j.celrep.2014.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nature genetics. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493:327–337. doi: 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, de Vries PJ, Silva AJ. From mTOR to cognition: molecular and cellular mechanisms of cognitive impairments in tuberous sclerosis. J Intellect Disabil Res. 2009;53:838–851. doi: 10.1111/j.1365-2788.2009.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias GG, Valles AS, Colombres M, Godoy JA, Toledo EM, Lukas RJ, Barrantes FJ, Inestrosa NC. Wnt-7a induces presynaptic colocalization of alpha 7-nicotinic acetylcholine receptors and adenomatous polyposis coli in hippocampal neurons. J Neurosci. 2007;27:5313–5325. doi: 10.1523/JNEUROSCI.3934-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller MB. Retinal waves drive calcium transients in undifferentiated retinal cells. Focus on “spontaneous waves in the ventricular zone of developing mammalian retina”. J Neurophysiol. 2004;91:1940. doi: 10.1152/jn.01226.2003. [DOI] [PubMed] [Google Scholar]
- Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, Cook EH, Jr, Skinner C, Schwartz CE, Sommer SS. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci Lett. 2006;409:10–13. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Fernandez E, Rajan N, Bagni C. The FMRP regulon: from targets to disease convergence. Front Neurosci. 2013;7:191. doi: 10.3389/fnins.2013.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MA, O'Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nature genetics. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyder M, Karlsson RM, Mathur P, Lyman M, Bock R, Momenan R, Munasinghe J, Scattoni ML, Ihne J, Camp M, et al. Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams' syndrome. The American journal of psychiatry. 2010;167:1508–1517. doi: 10.1176/appi.ajp.2010.10040484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishell G, Heintz N. The neuron identity problem: form meets function. Neuron. 2013;80:602–612. doi: 10.1016/j.neuron.2013.10.035. [DOI] [PubMed] [Google Scholar]
- Folstein S, Rutter M. Genetic influences and infantile autism. Nature. 1977a;265:726–728. doi: 10.1038/265726a0. [DOI] [PubMed] [Google Scholar]
- Folstein S, Rutter M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977b;18:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Fulp CT, Cho G, Marsh ED, Nasrallah IM, Labosky PA, Golden JA. Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet. 2008;17:3740–3760. doi: 10.1093/hmg/ddn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J, et al. Most genetic risk for autism resides with common variation. Nature genetics. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Ares M, Jr, Mody I, Black DL. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nature genetics. 2011;43:706–711. doi: 10.1038/ng.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, State MW. Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 2015 doi: 10.1016/S1474-4422(15)00044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharani N, Benayed R, Mancuso V, Brzustowicz LM, Millonig JH. Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Molecular psychiatry. 2004;9:474–484. doi: 10.1038/sj.mp.4001498. [DOI] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Cao R, Jafarnejad SM, Prager-Khoutorsky M, Giannakas N, Kaminari A, Fragkouli A, Nader K, Price TJ, et al. Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 2014;9:1742–1755. doi: 10.1016/j.celrep.2014.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105:521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. The Journal of biological chemistry. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- Green EK, Grozeva D, Jones I, Jones L, Kirov G, Caesar S, Gordon-Smith K, Fraser C, Forty L, Russell E, et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Molecular psychiatry. 2010;15:1016–1022. doi: 10.1038/mp.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Chang CW, Kelly SM, Bhattacharya A, McBride SM, Danielson SW, Jiang MQ, Chan CB, Ye K, Gibson JR, et al. Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 2015;11:727–736. doi: 10.1016/j.celrep.2015.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozeva D, Carss K, Spasic-Boskovic O, Parker MJ, Archer H, Firth HV, Park SM, Canham N, Holder SE, Wilson M, et al. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. American journal of human genetics. 2014;94:618–624. doi: 10.1016/j.ajhg.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilmatre A, Huguet G, Delorme R, Bourgeron T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev Neurobiol. 2014;74:113–122. doi: 10.1002/dneu.22128. [DOI] [PubMed] [Google Scholar]
- Gutierrez GC, Smalley SL, Tanguay PE. Autism in tuberous sclerosis complex. J Autism Dev Disord. 1998;28:97–103. doi: 10.1023/a:1026032413811. [DOI] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature genetics. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, Joober R, Lacaille JC, Nadeau A, Milunsky JM, et al. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry. 2011;69:898–901. doi: 10.1016/j.biopsych.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA. Autistic-like behaviour in Scn1a+/-mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489:385–390. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Hobert O. Regulation of terminal differentiation programs in the nervous system. Annu Rev Cell Dev Biol. 2011;27:681–696. doi: 10.1146/annurev-cellbio-092910-154226. [DOI] [PubMed] [Google Scholar]
- Hobert O, Carrera I, Stefanakis N. The molecular and gene regulatory signature of a neuron. Trends Neurosci. 2010;33:435–445. doi: 10.1016/j.tins.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. The Journal of physiology. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M, et al. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17076–17081. doi: 10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Chow J, Mazmanian SK, Patterson PH. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:12776–12781. doi: 10.1073/pnas.1202556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci. 2005;25:6971–6983. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Kageyama R. bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron. 2014;82:9–23. doi: 10.1016/j.neuron.2014.03.018. [DOI] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Ingram JL, Stodgell CJ, Hyman SL, Figlewicz DA, Weitkamp LR, Rodier PM. Discovery of allelic variants of HOXA1 and HOXB1: genetic susceptibility to autism spectrum disorders. Teratology. 2000;62:393–405. doi: 10.1002/1096-9926(200012)62:6<393::AID-TERA6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nature genetics. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Ash RT, Baker SA, Suter B, Ferguson A, Park J, Rudy J, Torsky SP, Chao HT, Zoghbi HY, et al. Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J Neurosci. 2013;33:19518–19533. doi: 10.1523/JNEUROSCI.1745-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Eagleson KL, Levitt P. A new synaptic player leading to autism risk: Met receptor tyrosine kinase. J Neurodev Disord. 2011;3:282–292. doi: 10.1007/s11689-011-9081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkman HO. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol Autism. 2012;3:10. doi: 10.1186/2040-2392-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayannis T, Au E, Patel JC, Kruglikov I, Markx S, Delorme R, Heron D, Salomon D, Glessner J, Restituito S, et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature. 2014;511:236–240. doi: 10.1038/nature13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318–326. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerin T, Ramanathan A, Rivas K, Grepo N, Coetzee GA, Campbell DB. A noncoding RNA antisense to moesin at 5p14.1 in autism. Sci Transl Med. 2012;4:128ra140. doi: 10.1126/scitranslmed.3003479. [DOI] [PubMed] [Google Scholar]
- Kidd SA, Lachiewicz A, Barbouth D, Blitz RK, Delahunty C, McBrien D, Visootsak J, Berry-Kravis E. Fragile X syndrome: a review of associated medical problems. Pediatrics. 2014;134:995–1005. doi: 10.1542/peds.2013-4301. [DOI] [PubMed] [Google Scholar]
- Kilpinen H, Ylisaukko-Oja T, Hennah W, Palo OM, Varilo T, Vanhala R, Nieminen-von Wendt T, von Wendt L, Paunio T, Peltonen L. Association of DISC1 with autism and Asperger syndrome. Molecular psychiatry. 2008;13:187–196. doi: 10.1038/sj.mp.4002031. [DOI] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblich JA. Mechanisms of asymmetric cell division during animal development. Curr Opin Cell Biol. 1997;9:833–841. doi: 10.1016/s0955-0674(97)80085-3. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Moon RT. Wnt and calcium signaling: beta-catenin-independent pathways. Cell calcium. 2005;38:439–446. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Kong SW, Sahin M, Collins CD, Wertz MH, Campbell MG, Leech JD, Krueger D, Bear MF, Kunkel LM, Kohane IS. Divergent dysregulation of gene expression in murine models of fragile X syndrome and tuberous sclerosis. Mol Autism. 2014;5:16. doi: 10.1186/2040-2392-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozol RA, Cukier HN, Zou B, Mayo V, De Rubeis S, Cai G, Griswold AJ, Whitehead PL, Haines JL, Gilbert JR, et al. Two knockdown models of the autism genes SYNGAP1 and SHANK3 in zebrafish produce similar behavioral phenotypes associated with embryonic disruptions of brain morphogenesis. Hum Mol Genet. 2015;24:4006–4023. doi: 10.1093/hmg/ddv138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey JF, Dolmetsch RE. Molecular mechanisms of autism: a possible role for Ca2+ signaling. Curr Opin Neurobiol. 2007;17:112–119. doi: 10.1016/j.conb.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Krumm N, O'Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37:95–105. doi: 10.1016/j.tins.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SG, Green MR, Goodman RH. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- Laumonnier F, Roger S, Guerin P, Molinari F, M'Rad R, Cahard D, Belhadj A, Halayem M, Persico AM, Elia M, et al. Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. The American journal of psychiatry. 2006;163:1622–1629. doi: 10.1176/ajp.2006.163.9.1622. [DOI] [PubMed] [Google Scholar]
- LeMasson G, Marder E, Abbott LF. Activity-dependent regulation of conductances in model neurons. Science. 1993;259:1915–1917. doi: 10.1126/science.8456317. [DOI] [PubMed] [Google Scholar]
- Lemonnier E, Ben-Ari Y. The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr. 2010;99:1885–1888. doi: 10.1111/j.1651-2227.2010.01933.x. [DOI] [PubMed] [Google Scholar]
- Li BM, Liu XR, Yi YH, Deng YH, Su T, Zou X, Liao WP. Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav. 2011;21:291–295. doi: 10.1016/j.yebeh.2011.04.060. [DOI] [PubMed] [Google Scholar]
- Li J, Zhao L, You Y, Lu T, Jia M, Yu H, Ruan Y, Yue W, Liu J, Lu L, et al. Schizophrenia Related Variants in CACNA1C also Confer Risk of Autism. PloS one. 2015;10:e0133247. doi: 10.1371/journal.pone.0133247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- Lijam N, Paylor R, McDonald MP, Crawley JN, Deng CX, Herrup K, Stevens KE, Maccaferri G, McBain CJ, Sussman DJ, et al. Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell. 1997;90:895–905. doi: 10.1016/s0092-8674(00)80354-2. [DOI] [PubMed] [Google Scholar]
- Lisman J, Malenka RC, Nicoll RA, Malinow R. Learning mechanisms: the case for CaM-KII. Science. 1997;276:2001–2002. doi: 10.1126/science.276.5321.2001. [DOI] [PubMed] [Google Scholar]
- Liu G. Local structural balance and functional interaction of excitatory and inhibitory synapses in hippocampal dendrites. Nature neuroscience. 2004;7:373–379. doi: 10.1038/nn1206. [DOI] [PubMed] [Google Scholar]
- Louvi A, Artavanis-Tsakonas S. Notch and disease: a growing field. Semin Cell Dev Biol. 2012;23:473–480. doi: 10.1016/j.semcdb.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, Tsien RW. gammaCaMKII shuttles Ca(2)(+)/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell. 2014;159:281–294. doi: 10.1016/j.cell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestrini E, Pagnamenta AT, Lamb JA, Bacchelli E, Sykes NH, Sousa I, Toma C, Barnby G, Butler H, Winchester L, et al. High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L-DOCK4 gene region in autism susceptibility. Molecular psychiatry. 2010;15:954–968. doi: 10.1038/mp.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Abbott LF, Turrigiano GG, Liu Z, Golowasch J. Memory from the dynamics of intrinsic membrane currents. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13481–13486. doi: 10.1073/pnas.93.24.13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh ED, Golden JA. Aristaless-related homeobox mutations. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. Oxford University Press; 2012. [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. Structural variation of chromosomes in autism spectrum disorder. American journal of human genetics. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PM, Yang X, Robin N, Lam E, Rabinowitz JS, Erdman CA, Quinn J, Weiss LA, Hamilton SP, Kwok PY, et al. A rare WNT1 missense variant overrepresented in ASD leads to increased Wnt signal pathway activation. Transl Psychiatry. 2013;3:e301. doi: 10.1038/tp.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marui T, Funatogawa I, Koishi S, Yamamoto K, Matsumoto H, Hashimoto O, Jinde S, Nishida H, Sugiyama T, Kasai K, et al. Association between autism and variants in the wingless-type MMTV integration site family member 2 (WNT2) gene. Int J Neuropsychopharmacol. 2010;13:443–449. doi: 10.1017/S1461145709990903. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Hyman SE. Progress in the genetics of polygenic brain disorders: significant new challenges for neurobiology. Neuron. 2013;80:578–587. doi: 10.1016/j.neuron.2013.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejias R, Adamczyk A, Anggono V, Niranjan T, Thomas GM, Sharma K, Skinner C, Schwartz CE, Stevenson RE, Fallin MD, et al. Gain-of-function glutamate receptor interacting protein 1 variants alter GluA2 recycling and surface distribution in patients with autism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4920–4925. doi: 10.1073/pnas.1102233108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KJ, Axelsen MS, Sheffield VC, Patil SR, Wassink TH. Germline mosaic transmission of a novel duplication of PXDN and MYT1L to two male half-siblings with autism. Psychiatr Genet. 2012;22:137–140. doi: 10.1097/YPG.0b013e32834dc3f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskvina V, Craddock N, Holmans P, Nikolov I, Pahwa JS, Green E, Wellcome Trust Case Control, C. Owen MJ, O'Donovan MC. Gene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Molecular psychiatry. 2009;14:252–260. doi: 10.1038/mp.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O, Warren ST, Bassell GJ. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol Cell. 2011;42:673–688. doi: 10.1016/j.molcel.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. Fragile X disappointments upset autism ambitions. Nat Rev Drug Discov. 2015;14:151–153. doi: 10.1038/nrd4555. [DOI] [PubMed] [Google Scholar]
- Nageshappa S, Carromeu C, Trujillo CA, Mesci P, Espuny-Camacho I, Pasciuto E, Vanderhaeghen P, Verfaillie CM, Raitano S, Kumar A, et al. Altered neuronal network and rescue in a human MECP2 duplication model. Molecular psychiatry. 2015 doi: 10.1038/mp.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SB, Valakh V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron. 2015;87:684–698. doi: 10.1016/j.neuron.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves-Pereira M, Muller B, Massie D, Williams JH, O'Brien PC, Hughes A, Shen SB, Clair DS, Miedzybrodzka Z. Deregulation of EIF4E: a novel mechanism for autism. J Med Genet. 2009;46:759–765. doi: 10.1136/jmg.2009.066852. [DOI] [PubMed] [Google Scholar]
- Niere F, Wilkerson JR, Huber KM. Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J Neurosci. 2012;32:5924–5936. doi: 10.1523/JNEUROSCI.4650-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Roeb W, Dickel DE, Walsh T, Kusenda M, O'Connor KL, Malhotra D, McCarthy SE, Stray SM, Taylor SM, et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur J Hum Genet. 2011;19:727–731. doi: 10.1038/ejhg.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmi EL, Bradford Y, Chen Y, Hall J, Arnone B, Gardiner MB, Hutcheson HB, Gilbert JR, Pericak-Vance MA, Copeland-Yates SA, et al. Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics. 2001;77:105–113. doi: 10.1006/geno.2001.6617. [DOI] [PubMed] [Google Scholar]
- Nyegaard M, Demontis D, Foldager L, Hedemand A, Flint TJ, Sorensen KM, Andersen PS, Nordentoft M, Werge T, Pedersen CB, et al. CACNA1C (rs1006737) is associated with schizophrenia. Molecular psychiatry. 2010;15:119–121. doi: 10.1038/mp.2009.69. [DOI] [PubMed] [Google Scholar]
- O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature genetics. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012a;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012b;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley JC, Kalume F, Catterall WA. Insights into pathophysiology and therapy from a mouse model of Dravet syndrome. Epilepsia. 2011;52(2):59–61. doi: 10.1111/j.1528-1167.2011.03004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H, Akashi K, Ishii Y, Yagishita-Kyo N, Suzuki K, Nonaka M, Kawashima T, Fujii H, Takemoto-Kimura S, Abe M, et al. Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3.1 with CaMKIIbeta. Cell. 2012;149:886–898. doi: 10.1016/j.cell.2012.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnamenta AT, Khan H, Walker S, Gerrelli D, Wing K, Bonaglia MC, Giorda R, Berney T, Mani E, Molteni M, et al. Rare familial 16q21 microdeletions under a linkage peak implicate cadherin 8 (CDH8) in susceptibility to autism and learning disability. J Med Genet. 2011;48:48–54. doi: 10.1136/jmg.2010.079426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17:389–394. doi: 10.1016/j.molmed.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nature neuroscience. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persico AM, D'Agruma L, Maiorano N, Totaro A, Militerni R, Bravaccio C, Wassink TH, Schneider C, Melmed R, Trillo S, et al. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Molecular psychiatry. 2001;6:150–159. doi: 10.1038/sj.mp.4000850. [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, Cowan CW, Huber KM. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron. 2010;66:191–197. doi: 10.1016/j.neuron.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, Tuluc P, Striessnig J. CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol Psychiatry. 2015;77:816–822. doi: 10.1016/j.biopsych.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Marin-Burgin A, Adesnik H, Atallah BV, Scanziani M. Input normalization by global feedforward inhibition expands cortical dynamic range. Nature neuroscience. 2009;12:1577–1585. doi: 10.1038/nn.2441. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Rabinowitch I, Segev I. Two opposing plasticity mechanisms pulling a single synapse. Trends Neurosci. 2008;31:377–383. doi: 10.1016/j.tins.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Ramachandran KV, Hennessey JA, Barnett AS, Yin X, Stadt HA, Foster E, Shah RA, Yazawa M, Dolmetsch RE, Kirby ML, et al. Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development. J Clin Invest. 2013;123:1638–1646. doi: 10.1172/JCI66903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo RL, Okuno H, Spooner PA, Frenguelli BG, Bito H, Morris RG. Synaptic tagging and capture: differential role of distinct calcium/calmodulin kinases in protein synthesis-dependent long-term potentiation. J Neurosci. 2010;30:4981–4989. doi: 10.1523/JNEUROSCI.3140-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 2015;16:595–605. doi: 10.1038/nrn4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SJ, Wehner DE, Hagerman R. The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001;22:409–417. doi: 10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- Ronemus M, Iossifov I, Levy D, Wigler M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet. 2014;15:133–141. doi: 10.1038/nrg3585. [DOI] [PubMed] [Google Scholar]
- Roohi J, Montagna C, Tegay DH, Palmer LE, DeVincent C, Pomeroy JC, Christian SL, Nowak N, Hatchwell E. Disruption of contactin 4 in three subjects with autism spectrum disorder. J Med Genet. 2009;46:176–182. doi: 10.1136/jmg.2008.057505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg A, Patterson JS, Angelaki DE. A computational perspective on autism. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:9158–9165. doi: 10.1073/pnas.1510583112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg RE, Law JK, Yenokyan G, McGready J, Kaufmann WE, Law PA. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med. 2009;163:907–914. doi: 10.1001/archpediatrics.2009.98. [DOI] [PubMed] [Google Scholar]
- Rosenberg SS, Spitzer NC. Calcium signaling in neuronal development. Cold Spring Harb Perspect Biol. 2011;3:a004259. doi: 10.1101/cshperspect.a004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso SB, Sussman D, Wynshaw-Boris A, Salinas PC. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nature neuroscience. 2005;8:34–42. doi: 10.1038/nn1374. [DOI] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Sudhof TC. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158:198–212. doi: 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL. Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr Opin Neurol. 2010;23:118–123. doi: 10.1097/WCO.0b013e328336eb13. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE, Zoghbi HY. Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med. 2011;3:86ra49. doi: 10.1126/scitranslmed.3002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas PC, Zou Y. Wnt signaling in neural circuit assembly. Annu Rev Neurosci. 2008;31:339–358. doi: 10.1146/annurev.neuro.31.060407.125649. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA. 2014;311:1770–1777. doi: 10.1001/jama.2014.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Klann E. Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci Signal. 2014;7:re10. doi: 10.1126/scisignal.2005832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- Schaaf CP, Gonzalez-Garay ML, Xia F, Potocki L, Gripp KW, Zhang B, Peters BA, McElwain MA, Drmanac R, Beaudet AL, et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nature genetics. 2013;45:1405–1408. doi: 10.1038/ng.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Zoghbi HY. Solving the autism puzzle a few pieces at a time. Neuron. 2011;70:806–808. doi: 10.1016/j.neuron.2011.05.025. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Dynes JL, Steward O. Synaptic regulation of translation of dendritic mRNAs. J Neurosci. 2006;26:7143–7146. doi: 10.1523/JNEUROSCI.1796-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr EH. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): one gene leads to many phenotypes. Curr Opin Pediatr. 2003;15:567–571. doi: 10.1097/00008480-200312000-00004. [DOI] [PubMed] [Google Scholar]
- Shin SM, Zhang N, Hansen J, Gerges NZ, Pak DT, Sheng M, Lee SH. GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nature neuroscience. 2012;15:1655–1666. doi: 10.1038/nn.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel M, Marder E, Abbott LF. Activity-dependent current distributions in model neurons. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:11308–11312. doi: 10.1073/pnas.91.24.11308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman S. NeuroTribes: The Legacy of Autism and the Future of Neurodiversity. New York, New York: Avery: Penguin Random House; 2015. [Google Scholar]
- Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. discussion 8086-8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Splawski I, Yoo DS, Stotz SC, Cherry A, Clapham DE, Keating MT. CACNA1H mutations in autism spectrum disorders. The Journal of biological chemistry. 2006;281:22085–22091. doi: 10.1074/jbc.M603316200. [DOI] [PubMed] [Google Scholar]
- Stark E, Roux L, Eichler R, Senzai Y, Royer S, Buzsaki G. Pyramidal cell-interneuron interactions underlie hippocampal ripple oscillations. Neuron. 2014;83:467–480. doi: 10.1016/j.neuron.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic S, Bassell GJ, Mihailescu MR. G quadruplex RNA structures in PSD-95 mRNA: potential regulators of miR-125a seed binding site accessibility. RNA. 2015;21:48–60. doi: 10.1261/rna.046722.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Levy WB. Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci. 1982;2:284–291. doi: 10.1523/JNEUROSCI.02-03-00284.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- The Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Thompson BA, Tremblay V, Lin G, Bochar DA. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Molecular and cellular biology. 2008;28:3894–3904. doi: 10.1128/MCB.00322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Voineagu I, Pasca SP, Won H, Chandran V, Horvath S, Dolmetsch RE, Geschwind DH. Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy syndrome. Genome Med. 2014;6:75. doi: 10.1186/s13073-014-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro R, Konyukh M, Delorme R, Leblond C, Chaste P, Fauchereau F, Coleman M, Leboyer M, Gillberg C, Bourgeron T. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26:363–372. doi: 10.1016/j.tig.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488:647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman R, Moshe SL, Rapin I. Convulsing toward the pathophysiology of autism. Brain Dev. 2009;31:95–103. doi: 10.1016/j.braindev.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner G, Partington M, Kerr B, Mangelsdorf M, Gecz J. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am J Med Genet. 2002;112:405–411. doi: 10.1002/ajmg.10714. [DOI] [PubMed] [Google Scholar]
- Turner TN, Sharma K, Oh EC, Liu YP, Collins RL, Sosa MX, Auer DR, Brand H, Sanders SJ, Moreno-De-Luca D, et al. Loss of delta-catenin function in severe autism. Nature. 2015;520:51–56. doi: 10.1038/nature14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Ueda S, Fujimoto S, Hiramoto K, Negishi M, Katoh H. Dock4 regulates dendritic development in hippocampal neurons. J Neurosci Res. 2008;86:3052–3061. doi: 10.1002/jnr.21763. [DOI] [PubMed] [Google Scholar]
- Uzunova G, Hollander E, Shepherd J. The role of ionotropic glutamate receptors in childhood neurodevelopmental disorders: autism spectrum disorders and fragile x syndrome. Curr Neuropharmacol. 2014;12:71–98. doi: 10.2174/1570159X113116660046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varea O, Martin-de-Saavedra MD, Kopeikina KJ, Schurmann B, Fleming HJ, Fawcett-Patel JM, Bach A, Jang S, Peles E, Kim E, et al. Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:6176–6181. doi: 10.1073/pnas.1423205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJMH, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DPA, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a Gene (Fmr-1) Containing a Cgg Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile-X Syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Vogt D, Cho KK, Lee AT, Sohal VS, Rubenstein JL. The parvalbumin/somatostatin ratio is increased in Pten mutant mice and by human PTEN ASD alleles. Cell Rep. 2015;11:944–956. doi: 10.1016/j.celrep.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt D, Hunt RF, Mandal S, Sandberg M, Silberberg SN, Nagasawa T, Yang Z, Baraban SC, Rubenstein JL. Lhx6 directly regulates Arx and CXCR7 to determine cortical interneuron fate and laminar position. Neuron. 2014;82:350–364. doi: 10.1016/j.neuron.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulto-van Silfhout AT, Rajamanickam S, Jensik PJ, Vergult S, de Rocker N, Newhall KJ, Raghavan R, Reardon SN, Jarrett K, McIntyre T, et al. Mutations affecting the SAND domain of DEAF1 cause intellectual disability with severe speech impairment and behavioral problems. American journal of human genetics. 2014;94:649–661. doi: 10.1016/j.ajhg.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltes R, Duketis E, Knapp M, Anney RJ, Huguet G, Schlitt S, Jarczok TA, Sachse M, Kampfer LM, Kleinbock T, et al. Common variants in genes of the postsynaptic FMRP signalling pathway are risk factors for autism spectrum disorders. Hum Genet. 2014;133:781–792. doi: 10.1007/s00439-013-1416-y. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H, Evans J, Clarke A, Pelka GJ, Tam PP, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. American journal of human genetics. 2004;75:1079–1093. doi: 10.1086/426462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg P, Flames N, Sawa H, Garriga G, Hobert O. The SWI/SNF chromatin remodeling complex selectively affects multiple aspects of serotonergic neuron differentiation. Genetics. 2013;194:189–198. doi: 10.1534/genetics.112.148742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, Reichert J, Buxbaum JD, Meisler MH. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Molecular psychiatry. 2003;8:186–194. doi: 10.1038/sj.mp.4001241. [DOI] [PubMed] [Google Scholar]
- Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, Zondag S, Toriello HV, Magenis RE, Elsea SH. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. American journal of human genetics. 2010;87:219–228. doi: 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, State MW. Autism spectrum disorders: from genes to neurobiology. Curr Opin Neurobiol. 2015;30:92–99. doi: 10.1016/j.conb.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondolowski J, Dickman D. Emerging links between homeostatic synaptic plasticity and neurological disease. Front Cell Neurosci. 2013;7:223. doi: 10.3389/fncel.2013.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Jia M, Ruan Y, Liu J, Guo Y, Shuang M, Gong X, Zhang Y, Yang X, Zhang D. Positive association of the oxytocin receptor gene (OXTR) with autism in the Chinese Han population. Biol Psychiatry. 2005;58:74–77. doi: 10.1016/j.biopsych.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Xue M, Atallah BV, Scanziani M. Equalizing excitation-inhibition ratios across visual cortical neurons. Nature. 2014;511:596–600. doi: 10.1038/nature13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimizu T, Pan JQ, Mungenast AE, Madison JM, Su S, Ketterman J, Ongur D, McPhie D, Cohen B, Perlis R, et al. Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Molecular psychiatry. 2015;20:284. doi: 10.1038/mp.2014.181. [DOI] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nature neuroscience. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nature neuroscience. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- Yucel G, Altindag B, Gomez-Ospina N, Rana A, Panagiotakos G, Lara MF, Dolmetsch R, Oro AE. State-dependent signaling by Cav1.2 regulates hair follicle stem cell function. Genes Dev. 2013;27:1217–1222. doi: 10.1101/gad.216556.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- Zhang F. CRISPR-Cas9: Prospects and Challenges. Hum Gene Ther. 2015;26:409–410. doi: 10.1089/hum.2015.29002.fzh. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hou LF, Klann E, Nelson DL. Altered Hippocampal Synaptic Plasticity in the Fmr1 Gene Family Knockout Mouse Models. Journal of Neurophysiology. 2009;101:2572–2580. doi: 10.1152/jn.90558.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]