

Abstract
Integrating LC-MS/MS molecular networking and bioassay-guided fractionation enabled the targeted isolation of a new and bioactive cyclic octapeptide, samoamide A (1), from a sample of cf. Symploca sp. collected in American Samoa. The structure of 1 was established by detailed 1D and 2D NMR experiments, HRESIMS data, and chemical degradation/chromatographic (e.g., Marfey’s analysis) studies. Pure compound 1 was shown to have in vitro cytotoxic activity against several human cancer cell lines in both traditional cell culture and zone inhibition bioassays. Although there was no particular selectivity between the cell lines tested for samoamide A, the most potent activity was observed against H460 human non-small cell lung cancer cells (IC50 = 1.1 μM). Molecular modeling studies suggested that one possible mechanism of action for 1 is the inhibition of the enzyme dipeptidyl peptidase (CD26, DPP4) at a reported allosteric binding site, which could lead to many downstream pharmacological effects. However, this interaction was moderate when tested in vitro at up to 10 μM, and only resulted in about 16% peptidase inhibition. Combining bioassay screening with the cheminformatics strategy of LC-MS/MS molecular networking as a discovery tool expedited the targeted isolation of a natural product possessing both a novel chemical structure and a desired biological activity.
Graphical Abstract
Natural products research has had a profound impact on drug discovery and development leading to a high percentage of drugs used in human medicine, as recently reviewed in a meta-analysis of new small-molecule drug approvals by worldwide regulatory agencies from 1981 to 2014.1 An even higher percentage of cancer chemotherapeutic agents approved during this period were natural products, semi-synthetic derivatives thereof, or synthetic compounds with a natural product pharmacophore.1,2 Several reasons for the historical success of natural products-based drug discovery research have been advanced in the literature. One of these is the existence of ethnobotanical knowledge (in particular for use of plant extracts in many systems of traditional medicine) to guide the selection of candidate species for study.3,4 Another concept is that natural products are particularly well suited for interaction with medically relevant targets due to their production within a biological context, such as by providing the producing organisms with ecological or evolutionary advantages.5 In this regard, natural products have much greater molecular complexity and drug-like characteristics compared to compounds produced entirely by chemical synthesis, and especially those deriving from combinatorial chemistry techniques.6–8
At the same time, the field of natural products research is undergoing revitalization through the utilization and development of new tools and techniques such as genome mining, compound activity mapping and high content biological screening along with integrated bioinformatics, as well as molecular networking, principal component analysis and other cheminformatics approaches.9–13 The improved access to DNA sequence information of prokaryotes has indicated that a staggering number of uncharacterized biosynthetic gene clusters are present in these organisms, and that there is much molecular diversity left to discover and explore for biologically valuable properties.14 In turn, this has encouraged many researchers to pursue a “bottom-up” genetics-based discovery program.15 However, several major challenges exist in this latter strategy, such as gaining physical access to the encoded compound and making rational the discovery and isolation of NPs with a desired pharmacological activity.16 As a result, there are no approved drugs to date that were discovered through a gene-guided approach. Rather, at the present time, innovation to the “top down” approach to pharmaceutical lead discovery appears to be a more efficient and productive strategy.15
A recent advance in the field of NP research with broad implications to metabolite dereplication, based on the greatly increased availability of LC-MS/MS instrumentation in NP research laboratories, has been the MS/MS-based Molecular Networks algorithm.10,17 This platform associates MS/MS spectra based on similar mass fragmentation patterns, with the underlying concept that structurally related molecules will fragment in similar ways to give analogous patterns. This tool, available on the Global Natural Products Social Molecular Networking (GNPS at http://gnps.ucsd.edu) Web site, allows users to include their newly described and structurally annotated data sets into a publicly available database, thereby enabling future compound dereplication against authentic, structurally annotated MS/MS spectra. In addition, new molecules that are related to known substances in the database can rapidly be assigned to specific structural families, thereby accelerating the discovery and characterization process.
In this current report, 10 field identified collections of the marine cyanobacterial genus Symploca spp. were evaluated using integrated MS/MS based molecular networking and cancer cell cytotoxicity. The genus Symploca has been especially productive in yielding a diversity of highly biologically active natural products, such as the tubulin polymerization inhibitor dolastatin 10,18,19 a derivative of which is now an approved drug,20 the histone deacetylase inhibitors largazole and santacruzamate A, the potent cathepsin E inhibitor symplocin A, the chymotrypsin inhibitor symplocamide A, and the antitumor agent symplostatin 1.18,21–26 However, it has recently been shown that some marine cyanobacteria that are morphologically characterized as belonging to the genus Symploca form a distinct clade by phylogenetic analysis, and it is expected that these will be described as a new but related genus in the near future.13 Thus, extracts obtained from field identified Symploca spp. represent a promising source for the discovery of new natural products with interesting biological activities. Herein described is a proof of concept that integration of bioassay screening with a cheminformatics-based dereplication strategy using LC-MS/MS molecular networking enables the targeted isolation of novel chemical entities that possess desired biological properties. Moreover, this approach efficiently bypasses the iterative bioassay steps employed in traditional bioactivity-guided fractionation efforts and successfully avoids the re-isolation of known molecules.
RESULTS AND DISCUSSION
Two field-identified collections of Symploca spp. from American Samoa, one from Saipan, six from Panama, and one grown in the lab from the Pasteur culture collection (PCC8002) were sonicated individually in CH2Cl2/MeOH to afford 10 crude Symploca extracts. These were initially separated by silica gel vacuum liquid chromatography (VLC) and subjected to untargeted LC-MS/MS analysis. A Molecular Network was constructed from these 10 samples using the Global Natural Product Social Molecular Networking (GNPS) platform. Three significant conclusions could be drawn from this dataset. First, several families of molecules were shown to be distributed across all of the samples tested, and these were chlorophyll derivatives that have been previously annotated in the spectral libraries. Next, several network clusters were distributed across only a few of the collections, and will be evaluated in due course. However, an initial analysis of some of these suggested that they may be known compounds or analogues thereof, such as the bastimolides,27 dolastatins,19,25,28 and viequeamides.29 Finally, a large number of network clusters and single agents with no matches to other metabolites were shown to arise from single collections. The full molecular network (Figure 1) highlights examples of each of the three described cluster types, and the chemical and biochemical investigation of one organism-specific cluster is described in detail herein.
Figure 1.

LC-MS/MS derived molecular network of extracts and systematically produced VLC fractions from ten Symploca spp. with tentatively identified annotations and examples of clusters correlating to molecules (e.g. chlorophylls) with ubiquitous distribution, (e.g. bastimolides, dolastatin 10, viequeamide B, and unknowns) of intermediate distribution, and (e.g. m/z 911 metabolite = compound 1, and yet-undescribed molecules) that have species-specific occurrence. Single-node clusters were pruned for visualization purposes. Node sizes are scaled to signal intensity, and the cluster containing 1 has been further expanded for legibility. Different node colors represent annotations of different source organisms. See expanded version in Figure S13, Supporting Information.
In addition to the above chemical analysis, the crude extracts and VLC subfractions from these Symploca spp. were subjected to in vitro cancer cell cytotoxicity testing. It was envisioned that the combination of biological activity with chemical novelty as indicated by molecular networking and spectral library searching could yield a new chemotype with in vitro cytotoxicity to cancer cells. Only fractions with greater than 75% cancer cell toxicity at the test concentration of 1 μg/mL were considered further, and these samples were additionally filtered by the results of the molecular networking. For example, fractions that were suggested by this latter technique to contain known cytotoxins such as dolastatin 10 and its structural analogues were excluded from further study. Furthermore, molecules distributed within the VLC subfractions of the same extract were excluded if they were present in inactive fractions. It was hypothesized that such selective exclusion criteria would allow for the chemical targeting of a structurally novel and biologically active molecule prior to structure determination, and efficiently guide isolation efforts without the need for iterative biological testing in association with further purification.
Thus, efforts were focused on a network cluster with a compound of interest at m/z 911 from the polar H fraction (25% MeOH/EtOAc elution on silica VLC) of an American Samoan Symploca sp. extract 2228. Its MS/MS spectrum was unrelated to any compound present in the GNPS library, and fraction 2228H possessed 88% in vitro cytotoxicity at 1 μg/mL to H-460 human non-small cell lung cancer cells. Further query of the Dictionary of Natural Products (http://dnp.chemnetbase.com) and MarinLit (http://pubs.rsc.org/marinlit) databases yielded no reasonable candidate molecules for the m/z 911 metabolite. Thus, the chemical identity of this likely new metabolite was pursued for isolation efforts from active fraction 2228H using LC-MS/MS as a guide.
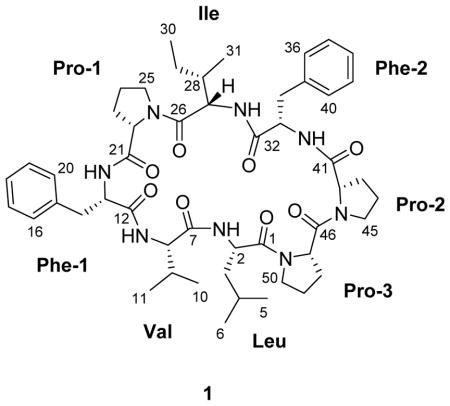
Repeated normal- and reversed-phase silica gel column chromatography followed by HPLC of this fraction yielded 8.2 mg of pure compound 1 (named samoamide A, 0.013% of the cyanobacterial mass). The molecular formula of 1 was determined to be C50H70N8O8 based on the protonated and sodiated pseudomolecular ion peaks in the HRESIMS at m/z 911.54187 (calcd for C50H71N8O8+, 911.53894) and 933.52322 (calcd for C50H70N8O8Na+, 933.52088), respectively. The 1H NMR spectrum exhibited characteristic signs for a polypeptide-type structure, and this was supported by fragmentation in the MS/MS spectrum. Overlapped signals in the 1H NMR spectrum of 1 were resolved using a combination of 1H-13C HSQC and 1H-1H TOCSY experiments. Based on the molecular formula of 1, samoamide A possessed 20 degrees of unsaturation. Eight of these were revealed as ester/amide-type carbonyls from 13C NMR resonances between δC 170–173, and eight more were present as two sets of phenyl rings as shown by 1H NMR resonances between δH 7.15–7.33. From the HSQC experiment, it was obvious that four resonances between δH 7.58 and 7.83 could be attributed to amide protons, as was a fifth at δH 5.81. Further examination of the 1H, 13C and TOCSY NMR spectra revealed the presence of three proline residues along with one isoleucine, leucine, and valine moiety.
The amino acid residues of 1 could clearly be distinguished due to characteristic patterns of TOCSY and COSY correlations with amide NH and α-methine protons. For instance, the isoleucine residue had unobstructed signals in the 1H NMR spectrum from an amide NH proton at δH 7.67 and an α-methine proton at δH 4.68 that were correlated in the TOCSY spectrum to each other and to a β-methine proton (δH 1.71), diastereotopic γ-methylene protons (δH 1.61 and 1.17), as well as γ- and δ-methyl groups (δH 0.90 and 0.88, respectively). Other appropriate patterns were observed in the TOCSY spectrum from amide NH and α-methine protons (Table 1) for a leucine (β-methylene, γ-methine and δ-geminal methyl groups), three prolines (β-, γ-, and downfield δ-methylene groups), and a valine residue (β-methine and γ- geminal methyl groups). Correlations in the HMBC spectrum from methylene groups at δH 3.64 and 3.25 ppm (CH2-14) to δC 129.3 (C-16 & 20) as well as δH 3.39 and 2.99 (CH2-34) to δC 128.9 (C-36 & 40) indicated that the two phenyl rings observed in the 1H NMR spectrum were each part of phenylalanine moieties. HMBC correlations, particularly from the amino acid NH, α, and δ (for proline) protons to adjacent residue carbonyl carbons led to the generation of two tetrapeptide subunits for the structure of 1 (Figure 2). The MS/MS fragmentation pattern observed for samoamide A suggested its macrocyclic structure, as the standard b7-b3 and y7-y3 cleavages both connected the two tetrapeptide subunits deduced from NMR, as well as overlapped after initial cleavage and charge residence at the two proline residues (Figure 3). Thus, the remaining four degrees of unsaturation were determined as rings from three proline residues and the overall macrocyclic structure of 1. Accordingly, the planar structure of 1 was resolved as cyclic Leu-Val-Phe(1)-Pro(1)-Ile-Phe(2)-Pro(2)-Pro(3) (cycLVFPIFPP); such a cyclic peptide is not present in any of the literature databases.
Table 1.
1H and 13C NMR Spectroscopic Data of Samoamide A (1) in CDCl3a
| residue | position | δC, type | δH, mult (J, Hz) | TOCSY |
|---|---|---|---|---|
| Leu | 1 | 170.55, C | ||
| 2 | 50.2, CH | 4.80 t (10.3) | 3–6, Leu-NH | |
| 3 | 41.2, CH2 | 1.94 m, 1.37 m | 2, 4–6, Leu-NH | |
| 4 | 25.6, CH | 1.73 m | 2, 3, 5, 6, Leu-NH | |
| 5 | 22.3, CH3 | 1.03 d (6.6) | 2–4, 6, Leu-NH | |
| 6 | 23.6, CH3 | 0.98 d (6.6) | 2–5, Leu-NH | |
| NH | 7.59 d (6.8) | 2–6 | ||
| Val | 7 | 170.64, C | ||
| 8 | 59.3, CH | 4.39 m | 9–11, Val-NH | |
| 9 | 32.1, CH | 1.91 m | 8, 10, 11, Val-NH | |
| 10 | 18.8, CH3 | 1.00 d (6.1) | 8, 9, 11, Val-NH | |
| 11 | 19.2, CH3 | 1.02 d (6.1) | 8–10, Val-NH | |
| NH | 7.81 d (8.4) | 8–11 | ||
| Phe-1 | 12 | 171.1, C | ||
| 13 | 60.4, CH | 4.00 m | 14, Phe-1-NH | |
| 14 | 35.5, CH2 | 3.64 m, 3.25 m | 13, Phe-1-NH | |
| 15 | 137.6, C | |||
| 16, 20 | 129.3, CH | 7.16 d (7.4) | 17–19 | |
| 17, 19 | 129.1b, CH | 7.31 mb | 16, 18, 20 | |
| 18 | 127.3b, CH | 7.23 mb | 16, 17, 19, 20 | |
| NH | 5.81 d (5.8) | 13, 14 | ||
| Pro-1 | 21 | 172.7, C | ||
| 22 | 62.7, CH | 3.66 m | 23–25 | |
| 23 | 29.9, CH2 | 2.02 m, 1.88 m | 22, 24, 25 | |
| 24 | 25.1, CH2 | 2.09 m, 1.97 m | 22, 23, 25 | |
| 25 | 48.8, CH2 | 3.93 m, 3.68 m | 22–24 | |
| Ile | 26 | 173.1, C | ||
| 27 | 54.9, CH | 4.68 m | 28–31, Ile-NH | |
| 28 | 38.1, CH | 1.71 m | 27, 29-3, Ile-NH | |
| 29 | 24.3, CH2 | 1.61 m, 1.17 m | 27, 28, 30, 31, Ile-NH | |
| 30 | 10.9, CH3 | 0.90 d (6.5) | 27–29, 31, Ile-NH | |
| 31 | 15.6, CH3 | 0.88 d (5.9) | 27–30, Ile-NH | |
| NH | 7.67 d (9.7) | 27–30 | ||
| Phe-2 | 32 | 171.9, C | ||
| 33 | 57.9, CH | 4.49 m | 34, Phe-2-NH | |
| 34 | 38.5, CH2 | 3.39 dd (13.9, 2.6), 2.99 m | 33, Phe-2-NH | |
| 35 | 136.9, C | |||
| 36, 40 | 128.9, CH | 7.18 d (7.7) | 37–39 | |
| 37, 39 | 128.8b, CH | 7.30 mb | 36, 38, 40 | |
| 38 | 127.2b, CH | 7.24 mb | 36, 37, 39, 40 | |
| NH | 7.60 d (6.6) | 33, 34 | ||
| Pro-2 | 41 | 170.8, C | ||
| 42 | 60.9, CH | 4.12 m | 43–45 | |
| 43 | 31.8, CH2 | 2.18 m, 1.82 m | 42, 44, 45 | |
| 44 | 21.3, CH2 | 1.48 m, 0.58 m | 42, 43, 45 | |
| 45 | 46.7, CH2 | 3.23 m, 3.00 m | 42–44 | |
| Pro-3 | 46 | 170.4, C | ||
| 47 | 59.3, CH | 4.10 m | 48–50 | |
| 48 | 28.5, CH2 | 1.96 m, 1.76 m | 47, 49, 50 | |
| 49 | 25.3, CH2 | 2.16 m, 1.81 m | 47, 48, 50 | |
| 50 | 47.4, CH2 | 3.70 m, 3.48 m | 47–49 |
Data recorded at 298 K, 600 MHz (1H) and 150 MHz (13C). Assignments supported by 2D NMR experiments.
Signals partially overlapped and may be interchanged.
Figure 2.

TOCSY and selected HMBC correlations used to determine the two planar tetrapeptide fragments of samoamide A (1). Bolded bonds represent correlated protons in the TOCSY spectrum. Arrows represent cross peaks from the 1H-13C HMBC spectrum.
Figure 3.
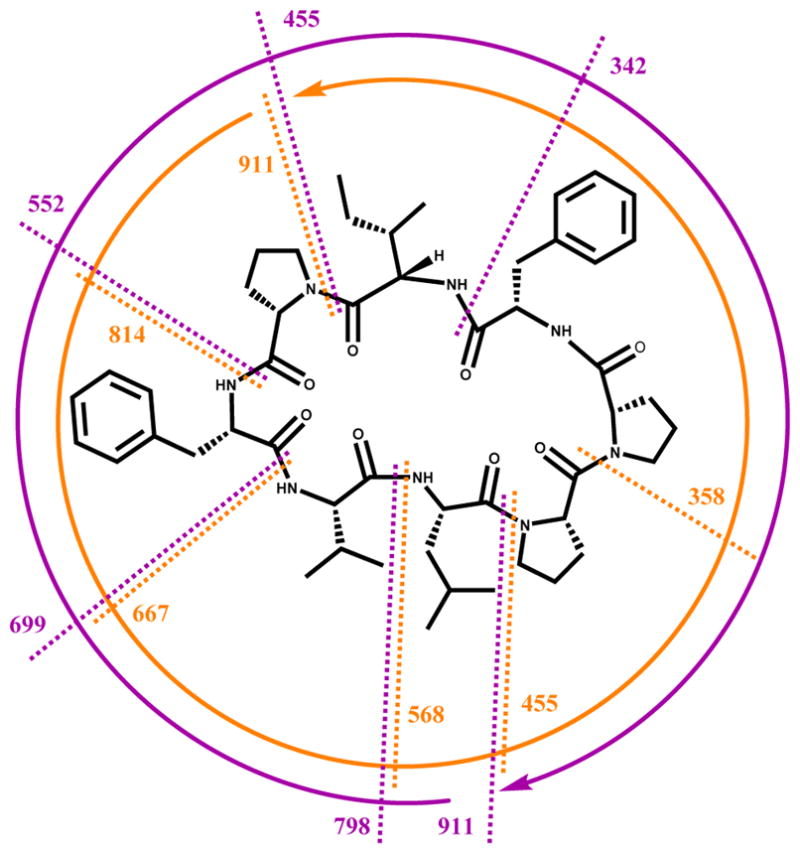
Structure of 1 with absolute configurations shown, annotated with observed MS/MS fragmentations. Standard b7-b3 cleavages are shown in purple, and y7-y3 cleavages in orange. Arrows represent direction of sequential fragmentation after initial ring opening adjacent to the prolyl nitrogen atoms during positive mode ESIMS.
Since all of the amino acids in samoamide A (1) were found to be proteinogenic and unmodified, a stereochemical investigation was completed using standard Marfey’s analysis by hydrolysis, chemical derivatization with D-FDAA, and LC-MS comparison against available standards.30,31 This revealed that all were of the unmodified L-configuration (Figure 3). The conformation of the three proline residues were determined by comparison of the 13C NMR shifts associated with the β and γ carbons of these residues according to a literature precedent.32 Based on the chemical shifts of these carbons as well as the differences in chemical shifts between the β and γ carbons (4.8 ppm for Pro-1, 10.5 ppm for Pro-2, and 3.2 ppm for Pro-3), they were assigned as being trans, cis, and trans, respectively. This completed the planar and stereostructure of samoamide A (1) as a structurally novel cyclic octapeptide.
Samoamide A (1) was tested against a series of cancer cell lines in vitro, including NCI H-460 human non-small lung cancer cells and HCT-116 human colorectal carcinoma cells. As 1 was active in both of these bioassays with an IC50 < 10 μM, it was further tested using a disk diffusion soft agar colony inhibition assay using several cancer cell lines, including H125 human lung adenocarcinoma, MCF7 human breast adenocarcinoma, LNCaP human prostate cancer, OVC5 human ovarian cancer, U251N human glioblastoma, PANC-1 human pancreatic carcinoma epithelial-like cells, CEM human acute lymphoblastic leukemia, and CFU-GM progenitor cells of human granulocytic and monocytic lineages.33 Samoamide A (1) was broadly cytotoxic to several cancer cell lines without any noteworthy selectivity (Table 2).
Table 2.
Results of in Vitro Cytotoxicity Testing on Samoamide A (1)a
| sample appliedb | zone diffusion inhibition (distance to perimeter, diffusion distance unitsc) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| H116 | H125 | MCF7 | LNCaP | OVC5 | U251N | PANC-1 | CEM | CFU-GM | |
| DMSO | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 (36 μmol) | 600 | 550 | 500 | 500 | 200 | 500 | 200 | 600 | 700 |
| 1 (9 μmol) | 200 | 300 | 300 | 300 | NT | 300 | NT | 350 | 400 |
IC50 = 1.1 μM to H460 cells and IC50 = 4.5 μM to H116 cells. Doxorubicin used as a standard, IC50 = 0.2 μM.
A total volume of 15 μL of 2.4 mM or 0.6 mM 1 was applied to filter disks and dried, per the protocol described by Refs. 33 and 42.
diffusion distance units were calculated by the formula [200 x (distance of inhibition in mm)/(diameter of the filter disk in mm)]. NT – Not tested.
In an attempt to understand the cytotoxicity observed for 1 in relationship to its chemical structure, the diprolyl moiety was a candidate as an especially distinctive structural feature. It was hypothesized that samoamide A may interact with dipeptidyl peptidase IV (DPP4; CD26), because this is the molecular target of the FDA-approved anti-hyperglycemic agent vildagliptin which was developed from a series of diprolyl nitrile lead compounds.34 Vildagliptin itself has been shown to prevent DPP4 from degrading glucagon-like peptide 1 (GLP-1), which in turn has a multitude of downstream effects including treatment of diabetes mellitus and osteoporosis.35 More recently, however, DPP4 was shown to be a cancer stem cell marker and potential therapeutic target, and vildagliptin has in vivo tumor suppressor activity when tested in murine colorectal cancer lung metasteses.36,37 Moreover, a set of high quality X-ray crystal structures for DPP4 in complex with validated small molecule inhibitors has been reported (PDB code 4N8D and 4N8E).38
Thus, an in silico computational docking study was conducted using 1 as a substrate for DPP4. This was completed using an energy minimized molecular model of 1 as a starting point from which to generate a conformer library to examine in docking studies with the crystal structure of DPP4. The conformation of this molecular model was supported by the occurrence of the same trans, cis, trans configuration for the three proline residues of 1 as were determined experimentally above by NMR spectroscopy. However, contrary to the initial hypothesis and contrary to the binding site of vildagliptin, samoamide A was not anticipated to interact with the active site of this enzyme. Rather, 1 was predicted to interact with DPP4 at the allosteric inhibition site reported from the X-ray crystal structure for this enzyme (Figure 4).38 When tested in vitro, samoamide A (1) demonstrated weak DPP4 inhibition activity at 10 and 3.33 μM, of 15.7% and 6.8% inhibition, respectively, which were the highest concentrations tested. No DPP4 inhibitory activity was observed at the lower dose concentrations of 1 tested. It is thus likely that 1 has one or more other biological targets responsible for its cytotoxic activity.
Figure 4.

Results of in silico docking of 1 with DPP4. Panel A – predicted interaction of a generated samoamide A (1) conformer library with the surface-colored allosteric inhibition site. Panel B – predicted molecular interactions of a low energy conformer at the location shown in panel A; red bond lines denote predicted solvent exposure outside of the interaction cleft of the enzyme. Panel C – legend generated from Molecular Operating Environment (MOE) Software for panel B.
Although all of the amino acids in samoamide A (1) are of unmodified structure, it is not clear if it biosynthetically derives from a non-ribosomal peptide synthase (NRPS) or ribosomally synthesized and post-translationally modified peptide (RiPP) pathway. Being that it has been reported that the coding for diprolyl motifs (XPP/PPX) can inhibit ribosomal translation with a difficult diprolyl peptide bond formation, it should not be inherently assumed that the RiPP pathway would be the more likely of the two, in this case.39 The diprolyl moiety observed in 1 is relatively rare in natural products, but has been found in the cyanobacterial metabolites dolastatin 15 and the wewakpeptins, both of which are believed to be products of NRPS biosynthesis.40,41 A DNA extraction and amplification of the preserved Symploca sp. ASI-16JUL14-3 specimen was attempted, but, unfortunately, failed to produce DNA of high enough quality for sequencing and analysis for NRPS or RiPP pathways.
Altogether, the early integration of cheminformatics with bioassay data in this study allowed for the targeted isolation of the new cytotoxic natural product, samoamide A (1), from an American Samoan cf. Symploca sp. This proof of concept supports the idea that combining these types of approaches in natural products research can increase efficiency, reduce redundancy, and generally accelerate drug discovery programs. This technique for target prioritization, along with other recently demonstrated and upcoming technical developments, modernizes and reinvigorates the field of natural products research. Samoamide A (1) was shown in this study, after purification, to be broadly cytotoxic to several different human cancer cell lines in vitro. The diprolyl moiety contained within the structure of 1 led to the generation of a hypothetical molecular target for this molecule, namely DPP4. In situ docking studies supported the notion that compound 1 could interact with DPP4, but surprisingly also suggested that this would occur at an allosteric inhibition site and not at the active site of the enzyme. This interaction was validated in an in vitro enzymatic assay of DPP4 inhibition, but since only weak activity was observed at a 10 μM concentration of 1, it is likely that one or more additional biological targets of this molecule exist in cancer cells. Further studies will be necessary to validate the in vivo interaction between samoamide A (1) with DPP4 and its downstream signaling, or to further investigate the potential of this molecule as a cytotoxic agent.
EXPERIMENTAL SECTION
General Experimental Procedures
A JASCO P-2000 polarimeter (JASCO Analytical Instruments, Easton, MD, USA) was used to measure optical rotations at 25 °C. Ultraviolet and visible (UV-Vis) spectra were recorded using a Beckman Coulter DU 800 spectrophotometer (Beckman Coulter Life Sciences, Indianapolis, IN, USA). Infrared (IR) spectra were obtained on a Thermo Nicolet IT 100 FT-IR (Thermo Fisher Scientific, Waltham, MA, USA). A Bruker Avance III 600 NMR spectrometer equipped with a Bruker cryoplatform and a 5 mm inverse detection triple resonance (H-C/N/D) cryoprobe with z-gradients was used to record NMR data at 298 K using standard Bruker pulse sequences. Nominal mass resolution LC-MS data were collected using an HPLC comprising a Thermo Finnigan Surveyor PDA Plus Detector, Autosampler Plus, and LC Pump Plus coupled to an LCQ Advantage Plus mass spectrometer (all Thermo Fisher Scientific), with a Phenomenex Kinetex C18 100 x 4.6 mm x 5 μm analytical column (Phenomenex, Torrance, CA, USA) installed. Exact mass HRESIMS data were recorded using an Agilent 6530 Accurate-Mass QTOF mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) in positive ion mode. Preparative HPLC was performed using a Kinetex C18 150 x 10.0 mm x 5 μm semi-preparative column (Phenomenex) connected to an HPLC comprising a Thermo Dionex UltiMate 3000 Pump, RS Autosampler, RS Diode Array Detector, and Automated Fraction Collector (all Thermo Fisher Scientific). HPLC-grade acetonitrile was purchased from Thermo Fisher Scientific, and HPLC-grade water was obtained by filtration using a Milli-Q Direct water purification system (Millipore, Billerica, MA, USA). Deuterated NMR solvents were purchased from Cambridge Isotope Laboratories (Tewksbury, MA, USA).
Organism Collection and Identification
Tuft-like colonies of this Symploca species were obtained in July, 2014 by snorkel diving in 1–3 m deep water in Vatia Bay, American Samoa (S 14°14′46.0″, W 170°40′22.2″). The colony-forming organism was identified morphologically as a black cf. Symploca species then encoded and vouchered as collection number ASI-16JUL14-3, available from WHG. The organism was preserved in 0.5 L of 1:1 seawater-isopropanol solution, transported to the laboratory in San Diego, CA, USA, and frozen at −20 °C until extraction.
Extraction, Molecular Networking, and Isolation
The raw biomass of cf. Symploca sp. ASI-16JUL14-3 (64.2 g) was filtered from 0.5 L of 1:1 seawater-isopropanol preservation solution and exhaustively extracted by sonication in 2:1 CH2Cl2-MeOH. The crude extract (2228; 0.5 g) was dried under vacuum and applied to a silica gel VLC column. This column was eluted to produce nine subfractions: A (elution by 100% hexanes), B (elution by 10% EtOAc/hexanes), C (elution by 20% EtOAc/hexanes), D (elution by 40% EtOAc/hexanes), E (elution by 60% EtOAc/hexanes), F (elution by 80% EtOAc/hexanes), G (elution by 100% EtOAc), H (elution by 25% MeOH/EtOAc), and I (elution by 100% MeOH). The sample yields from this chromatography were 2228A (51.7 mg), 2228B (163.7 mg), 2228C (88.5 mg), 2228D (24.9 mg), 2228E (11.9 mg), 2228F (7.8 mg), 2228G (11.1 mg), 2228H (23.0 mg), and 2228I (67.1 mg). Of these, fractions 2228E and 2228G demonstrated weak in vitro cytotoxicity to H-460 cells, 2228F was moderately cytotoxic, and 2228H was the most potently cytotoxic when tested at 1 and 10 μg/mL.
Concurrently, the crude extract and subfractions 2228A-I were analyzed by nominal mass LC-MS. Samples were each filtered over C18 SPE cartridges by application of 0.3 mg sample and elution with 1 mL CH3CN. A 10 μL aliquot of each sample was injected into the LC-MS and eluted at 0.7 mL/min by a gradient program of CH3CN/H2O (0.1% formic acid modifier): 30% for 5 min to 99% in 17 min, held for 3 min, to 30% in 1 min, held for 4 min, during which the mass spectrometer was set to observe m/z 190–2000 in positive ESI mode and with an automated full dependent MS/MS scan enabled. For the purpose of dereplication and analytical comparison, the nine other cf. Symploca spp. that had been previously extracted and separated on VLC by identical conditions as 2228 were similarly processed by SPE and analyzed by LC-MS. All chromatograms were converted digitally to .mzxml files using freely available MSConvert software (www.proteowizard.sourceforge.net) The crude extract, nine A-I subfractions, and one blank injection were submitted to GNPS molecular networking (http://gnps.ucsd.edu) per cf. Symploca sample (110 LCMS files in total). A molecular network was generated to interconnect MS/MS spectra from these samples along with blank CH3CN injections for the purpose of background subtraction. A detailed integrated analysis of the molecular network and bioassay data led to the chemical targeting of samoamide A (1) based on the network cluster with its associated molecular mass (m/z 911).
Accordingly, fraction 2228H was selected for further purification efforts based on its composition and biological activity. This fraction (23.0 mg) was applied to a 5 g/20 mL Gracepure C18 max SPE cartridge in 30% MeOH/H2O and rinsed with the same solvent (breakthrough; 2228H.1; 3.1 mg), then successively eluted with 50% MeOH/H2O (2228H.2; 2.1 mg), 70% MeOH/H2O (2228H.3; 3.4 mg), 100% MeOH (2228H.4; 6.0 mg), 50% CH3CN/MeOH (2228H.5; 3.0 mg), and 100% CH3CN (2228H.6; 6.0 mg). Each fraction was analyzed by LC-MS using the analytical method described above for the parent fraction, and 2228H.3–5 were observed to contain the m/z 911 metabolite. These fractions were pooled and further separated by preparative HPLC using a Phenomenex Kinetex C18 150 x 100 mm x 5 μm column eluted at 4.0 mL/min with isocratic 50% CH3CN/H2O (0.1% formic acid modifier) for a duration of 30 min per run. The chromatogram was monitored by UV absorbance at 200 nm. Approximately 2.5 mg of 2228H.3–5 were injected per run, and fractions were pooled to yield pure samoamide A (1) (8.2 mg; tR = 17.5 min).
Samoamide A (1)
Amorphous light green solid; [α]25D −146.9 (c 0.1, MeOH) and −178.9 (c 0.1, CHCl3); UV (MeOH) λmax (log ε) 214 (4.98), 284 (3.74), 421 (3.12), 676 (2.55) nm; IR (film) νmax 3301, 2962, 2931, 2877, 1658, 1620, 1527, 1458, 1323, 1250, 1199, 1141, 756, 702 cm−1; 1H NMR and 13C NMR, see Table 1; HRESIMS m/z 911.54187 (calcd for C50H71N8O8+, 911.53894) and 933.52322 (calcd for C50H70N8O8Na+, 933.52088).
Chemical Degradation and Stereochemical Analysis
The amino acid residues from 0.75 mg (75 μL of a 10 mg/mL solution in CH3CN) of samoamide A (1) were hydrolyzed in a sealed vial at 110 °C in 600 μL of 6 M HCl for 16 h. The reaction mixture was cooled to room temperature and evaporated to dryness under a stream of dry nitrogen gas. The hydrolysate was then treated with 165 μL of 1 M NaHCO3 and 2.2 mg of D-FDAA in 2.2 mL of acetone at 40 °C for 1 h before the solution was neutralized with 165 μL of 1 M HCl. The D-FDAA derivatized hydrolysate of 1 was then evaporated to dryness under a stream of dry N2 gas. This material was reconstituted in 1 mL of 1:1 CH3CN/H2O and filtered through a 0.45 μm syringe filter. A 10 μL aliquot was taken and injected into an analytical LC-MS in ESI ± modes and compared against authentic amino acid standards that were previously D-FDAA derivatized. Retention times for the D-FDAA derivatized authentic amino acid standards were as follows: D-Leu (38.59 min), L-Leu (43.26 min), D-Ile (37.91 min), L-Ile (42.71 min), D-allo-Ile (38.28 min), L-allo-Ile (42.76 min), D-Val (33.51 min), L-Val (38.23 min), D-Phe (38.72 min), L-Phe (42.30 min), D-Pro (27.20 min), and L-Pro (28.83 min). The D-FDAA derivatized 1-hydrolysate peaks with the expected masses were observed at 43.38, 42.70, 38.47, 42.54, and 28.96 min, respectively, corresponding to L-Leu, L-Ile, L-Val, L-Phe, and L-Pro, respectively. Minimal racemization of the hydrolysates was observed in this experiment (≤ 5% by peak integration).
Molecular Modeling and In Silico Docking
Computational molecular modeling of the energy minimized structure of samoamide A (1) was performed using Spartan’10 (Wavefunction, Inc., Irvine, CA, USA). The resultant molecular coordinates were imported into the Molecular Operating Environment software (MOE; Chemical Computing Group, Montreal, Quebec, Canada) for conformation searching using default settings and energy calculation using OPLS-AA force fields to generate a small 29 molecule conformer library. MOE was also used to import the PDB crystallographic structure of human DPP4 (PDB code 4N8D), to determine tentative interaction sites using the site finder method (Receptor Atoms). The induced fit method was used to dock the conformer library with the 4N8D structure, annotate surface maps (VDW method), and highlight specific ligand-receptor interactions using standard protocols. All computational calculations were completed using an HP Elitebook 850 G1 laptop (Hewlett Packard, Palo Alto, CA, USA) running 64-bit Windows 7 OS, containing 8 GB ram, and with an Intel i7-4600U CPU @ 2.1 GHz.
In Vitro Cytotoxicity Test Protocols
Samples were evaluated for cytotoxicity to H-460 according to previously published protocols with the exception of the concentrations studied.29 Briefly, 180 μL of suspended cells were added to each well of several 96-well plates at 3.33 x 104 cells/mL in RPMI 1640 medium with 10% FBS and 1% penicillin-streptomycin. The plates were incubated overnight at 37 °C in a 5% CO2 chamber before application of test samples. Samples were dissolved in DMSO and diluted in RPMI 1640 medium without FBS to a final concentration of 1 or 10 μg/mL for crude samples, and then additional concentrations diluted to 0.1, 0.01, and 0.0001 μg/mL for pure compounds. Doxorubicin and DMSO in RPMI 1640 without FBS were used as positive and negative controls, respectively. Plates were incubated for 48 h and then stained with MTT (thiazolyl blue tetrazolium bromide 98%; Sigma Aldrich, St. Louis, MO, USA) prior to being measured on a ThermoElectron Multiskan Ascent plate reader (Thermo) at 630 and 570 nm. Dose-response curves were generated using nonlinear regression in GraphPad Prism computer software (GraphPad Software Inc., San Diego, CA, USA). Samples were also evaluated for cytotoxicity to H-116 human colorectal carcinoma cells according to a similar reported protocol.33
In Vitro Zone Inhibition Test Protocol
The disk diffusion soft agar colony inhibition assay was performed according to a published protocol,33,42 using several cancer cell lines including H125 human lung adenocarcinoma, MCF7 human breast adenocarcinoma, LNCaP human prostate cancer, OVC5 human ovarian cancer, U251N human glioblastoma, PANC-1 human pancreatic carcinoma epithelial-like cells, CEM human acute lymphoblastic leukemia, and CFU-GM progenitor cells of human granulocytic and monocytic lineages.
In Vitro Dipeptidyl Peptidase IV (DPP4; CD26) Inhibition Test Protocol
Samples were evaluated for inhibitory activity against human DPP4 enzyme in solution using a commercially available test kit and standard protocol (ABCam, Cambridge, MA, USA). Briefly, human recombinant DPP4 diluted in buffer (20 mM Tris-HCl, pH 8.0, containing 100 mM NaCl, and 1 mM EDTA) was added to a 96-well plate. Samples of 1 in DMSO were added to test wells to yield a final concentration of 10 μM and three-fold serial dilutions to 1.5 nM, in addition to negative control (DMSO) and positive control (100 μM sitagliptin). Additionally, a background was recorded in the absence of enzyme or any test sample. To all wells a 200 μM solution of tagged substrate (H-Gly-Pro AMC/aminomethylcoumarin) was added. After 30 min of incubation at 37 °C, the plate was excited at a wavelength of 355 nm and emissions were read out at 455 nm using a Molecular Devices Spectramax M2 plate reader (Molecular Devices, Sunnyvale, CA, USA). Each experimental condition was tested in triplicate.
Supplementary Material
Acknowledgments
We gratefully acknowledge the governments of Panama and American Samoa, as well as the Panama International Cooperative Biodiversity Group (Panama ICBG), for permission to collect and study marine samples. This research was partially supported by NIH grants R01 CA100851 (to F.A.V. and W.H.G.) and R01 GM107550 (to P.C.D. and W.H.G.). We appreciate Drs. B. Duggan and P. Jordan, both of UC San Diego, for, respectively, facilitating acquisition of NMR and HRESIMS data. C.B.N. acknowledges and appreciates financial support provided by a postdoctoral fellowship from an NCI/NIH Training Program in the Biochemistry of Growth Regulation and Oncogenesis (T32 CA009523). L.A. thanks the Brazilian National Council for Scientific and Technological Development for a pre-doctoral scholarship (CNPq 234850/2014-0).
Footnotes
DEDICATION
Dedicated to Professor Phil Crews, of the University of California, Santa Cruz, for his pioneering work on bioactive natural products.
Notes. The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.6b00907.
1H NMR, 13C NMR, 1H-1H COSY, 1H-1H TOCSY, 1H-13C HSQC and HMBC spectra for compound 1, nominal mass LC-MS/MS spectrum of 1, LC-MS chromatograms of Marfey’s analysis hydrolysis and derivatization products of 1, along with derivatized authentic amino acid standards, as well as molecular model coordinates for 1 and an expanded view of the LC-MS/MS molecular network used to generate Figure 1 (PDF).
References
- 1.Newman DJ, Cragg GM. J Nat Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ, Giddings LA. Phytochem Rev. 2014;13:123–137. [Google Scholar]
- 3.Gurib-Fakim A. Mol Asp Med. 2006;27:1–93. doi: 10.1016/j.mam.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Cordell GA, Colvard MD. J Nat Prod. 2012;75:514–525. doi: 10.1021/np200803m. [DOI] [PubMed] [Google Scholar]
- 5.Williams DH, Stone MJ, Hauck PR, Rahman SK. J Nat Prod. 1989;52:1189–1208. doi: 10.1021/np50066a001. [DOI] [PubMed] [Google Scholar]
- 6.Feher M, Schmidt JM. J Chem Inf Comput Sci. 2003;43:218–227. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- 7.Lachance H, Wetzel S, Kumar K, Waldmann H. J Med Chem. 2012;55:5989–6001. doi: 10.1021/jm300288g. [DOI] [PubMed] [Google Scholar]
- 8.Ortholand JY, Ganesan A. Curr Opin Chem Biol. 2004;8:271–280. doi: 10.1016/j.cbpa.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 9.Kellogg JJ, Todd DA, Egan JM, Raja HA, Oberlies NH, Kvalheim OM, Cech NB. J Nat Prod. 2016;79:376–386. doi: 10.1021/acs.jnatprod.5b01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang JY, Sanchez LM, Rath CM, Liu X, Boudreau PD, Bruns N, Glukhov E, Wodtke A, de Felicio R, Fenner A, Wong WR, Linington RG, Zhang L, Debonsi HM, Gerwick WH, Dorrestein PC. J Nat Prod. 2013;76:1686–1699. doi: 10.1021/np400413s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross AC, Gulland LES, Dorrestein PC, Moore BS. ACS Synth Biol. 2015;4:414–420. doi: 10.1021/sb500280q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurita KL, Glassey E, Linington RG. Proc Natl Acad Sci USA. 2015;112:11999–12004. doi: 10.1073/pnas.1507743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salvador-Reyes LA, Engene N, Paul VJ, Luesch H. J Nat Prod. 2015;78:486–492. doi: 10.1021/np500931q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J, Birren BW, Takano E, Sali A, Linington RG, Fischbach MA. Cell. 2014;158:412–421. doi: 10.1016/j.cell.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo Y, Cobb RE, Zhao H. Curr Opin Biotechnol. 2014;30:230–237. doi: 10.1016/j.copbio.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen PR, Chavarria KL, Fenical W, Moore BS, Ziemert N. J Ind Microbiol Biotechnol. 2014;41:203–209. doi: 10.1007/s10295-013-1353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, Porto C, Bouslimani A, Melnik AV, Meehan MJ, Liu W-T, Crüsemann M, Boudreau PD, Esquenazi E, Sandoval-Calderón M, Kersten RD, Pace LA, Quinn RA, Duncan KR, Hsu C-C, Floros DJ, Gavilan RG, Kleigrewe K, Northen T, Dutton RJ, Parrot D, Carlson EE, Aigle B, Michelsen CF, Jelsbak L, Sohlenkamp C, Pevzner P, Edlund A, McLean J, Piel J, Murphy BT, Gerwick L, Liaw C-C, Yang Y-L, Humpf H-U, Maansson M, Keyzers RA, Sims AC, Johnson AR, Sidebottom AM, Sedio BE, Klitgaard A, Larson CB, Boya PCA, Torres-Mendoza D, Gonzalez DJ, Silva DB, Marques LM, Demarque DP, Pociute E, O’Neill EC, Briand E, Helfrich EJN, Granatosky EA, Glukhov E, Ryffel F, Houson H, Mohimani H, Kharbush JJ, Zeng Y, Vorholt JA, Kurita KL, Charusanti P, McPhail KL, Nielsen KF, Vuong L, Elfeki M, Traxler MF, Engene N, Koyama N, Vining OB, Baric R, Silva RR, Mascuch SJ, Tomasi S, Jenkins S, Macherla V, Hoffman T, Agarwal V, Williams PG, Dai J, Neupane R, Gurr J, Rodríguez AMC, Lamsa A, Zhang C, Dorrestein K, Duggan BM, Almaliti J, Allard P-M, Phapale P, Nothias L-F, Alexandrov T, Litaudon M, Wolfender J-L, Kyle JE, Metz TO, Peryea T, Nguyen D-T, VanLeer D, Shinn P, Jadhav A, Müller R, Waters KM, Shi W, Liu X, Zhang L, Knight R, Jensen PR, Palsson BØ, Pogliano K, Linington RG, Gutiérrez M, Lopes NP, Gerwick WH, Moore BS, Dorrestein PC, Bandeira N. Nat Biotechnol. 2016;34:828–837. doi: 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luesch H, Moore RE, Paul VJ, Mooberry SL, Corbett TH. J Nat Prod. 2001;64:907–910. doi: 10.1021/np010049y. [DOI] [PubMed] [Google Scholar]
- 19.Pettit GR, Kamano Y, Herald CL, Tuinman AA, Boettner FE, Kizu H, Schmidt JM, Baczynskyj L, Tomer KB, Bontems RJ. J Am Chem Soc. 1987;109:6883–6885. [Google Scholar]
- 20.Senter PD, Sievers EL. Nat Biotechnol. 2012;30:631–637. doi: 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- 21.Taori K, Paul VJ, Luesch H. J Am Chem Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 22.Ying Y, Taori K, Kim H, Hong J, Luesch H. J Am Chem Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 23.Pavlik CM, Wong CYB, Ononye S, Lopez DD, Engene N, McPhail KL, Gerwick WH, Balunas MJ. J Nat Prod. 2013;76:2026–2033. doi: 10.1021/np400198r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molinski TF, Reynolds KA, Morinaka BI. J Nat Prod. 2012;75:425–431. doi: 10.1021/np200861n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrigan GG, Luesch H, Yoshida WY, Moore RE, Nagle DG, Paul VJ, Mooberry SL, Corbett TH, Valeriote FA. J Nat Prod. 1998;61:1075–1076. doi: 10.1021/np980321c. [DOI] [PubMed] [Google Scholar]
- 26.Linington RG, Edwards DJ, Shuman CF, McPhail KL, Matainaho T, Gerwick WH. J Nat Prod. 2008;71:22–27. doi: 10.1021/np070280x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao CL, Linington RG, Balunas MJ, Centeno A, Boudreau P, Zhang C, Engene N, Spadafora C, Mutka TS, Kyle DE, Gerwick L, Wang CY, Gerwick WH. J Org Chem. 2015;80:7849–7855. doi: 10.1021/acs.joc.5b01264. [DOI] [PubMed] [Google Scholar]
- 28.Luesch H, Yoshida WY, Moore RE, Paul VJ, Mooberry SL, Corbett TH. J Nat Prod. 2002;65:16–20. doi: 10.1021/np010317s. [DOI] [PubMed] [Google Scholar]
- 29.Boudreau PD, Byrum T, Liu WT, Dorrestein PC, Gerwick WH. J Nat Prod. 2012;75:1560–1570. doi: 10.1021/np300321b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marfey P. Carlsb Res Commun. 1984;49:591–596. [Google Scholar]
- 31.Bhushan R, Brückner H. Amino Acids. 2004;27:231–247. doi: 10.1007/s00726-004-0118-0. [DOI] [PubMed] [Google Scholar]
- 32.Schubert M, Labudde D, Oschkinat H, Schmieder P. J Biomol NMR. 2002;24:149–154. doi: 10.1023/a:1020997118364. [DOI] [PubMed] [Google Scholar]
- 33.Valeriote FA, Tenney K, Media J, Pietraszkiewicz H, Edelstein M, Johnson TA, Amagata T, Crews P. J Exp Ther Oncol. 2012;10:119–134. [PubMed] [Google Scholar]
- 34.Zhao G, Taunk PC, Magnin DR, Simpkins LM, Robl JA, Wang A, Robertson JG, Marcinkeviciene J, Sitkoff DF, Parker RA, Kirby MS, Hamann LG. Bioorg Med Chem Lett. 2005;15:3992–3995. doi: 10.1016/j.bmcl.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 35.Ceccarelli E, Guarino EG, Merlotti D, Patti A, Gennari L, Nuti R, Dotta F. Front Endocrinol. 2013;4 doi: 10.3389/fendo.2013.00073. Article 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies S, Beckenkamp A, Buffon A. Biomed Pharmacother. 2015;71:135–138. doi: 10.1016/j.biopha.2015.02.031. [DOI] [PubMed] [Google Scholar]
- 37.Jang JH, Baerts L, Waumans Y, De Meester I, Yamada Y, Limani P, Gil-Bazo I, Weder W, Jungraithmayr W. Clin Exp Metastasis. 2015;32:677–687. doi: 10.1007/s10585-015-9736-z. [DOI] [PubMed] [Google Scholar]
- 38.Namoto K, Sirockin F, Ostermann N, Gessier F, Flohr S, Sedrani R, Gerhartz B, Trappe J, Hassiepen U, Duttaroy A, Ferreira S, Sutton JM, Clark DE, Fenton G, Beswick M, Baeschlin DK. Bioorg Med Chem Lett. 2014;24:731–736. doi: 10.1016/j.bmcl.2013.12.118. [DOI] [PubMed] [Google Scholar]
- 39.Peil L, Starosta AL, Lassak J, Atkinson GC, Virumäe K, Spitzer M, Tenson T, Jung K, Remme J, Wilson DN. Proc Natl Acad Sci USA. 2013;110:15265–15270. doi: 10.1073/pnas.1310642110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han B, Goeger D, Maier CS, Gerwick WH. J Org Chem. 2005;70:3133–3139. doi: 10.1021/jo0478858. [DOI] [PubMed] [Google Scholar]
- 41.Pettit GR, Kamano Y, Dufresne C, Cerny RL, Herald CL, Schmidt JM. J Org Chem. 1989;54:6005–6006. [Google Scholar]
- 42.Vogel CV, Pietraszkiewicz H, Sabry OM, Gerwick WH, Valeriote FA, Vanderwal CD. Angew Chem. 2014;53:12205–12209. doi: 10.1002/anie.201407726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.